

Abstract
G-protein-coupled receptors (GPCRs) mediate most of our physiological responses to hormones, neurotransmitters and environmental stimulants, and so have great potential as therapeutic targets for a broad spectrum of diseases. They are also fascinating molecules from the perspective of membrane-protein structure and biology. Great progress has been made over the past three decades in understanding diverse GPCRs, from pharmacology to functional characterization in vivo. Recent high-resolution structural studies have provided insights into the molecular mechanisms of GPCR activation and constitutive activity.
The past two years have seen remarkable advances in the structural biology of G-protein-coupled receptors (GPCRs). Highlights have included solving the first crystal structures of ligand-activated GPCRs—the human β2 adrenergic receptor (β2AR), the avian β1AR and the human A2A adenosine receptor—as well as the structures of opsin and an active form of rhodopsin. These successes followed decades of effort by many laboratories across the world, and are of great interest from the perspectives of membrane-protein biophysics, cell biology, physiology and drug discovery.
GPCRs are the largest family of membrane proteins and mediate most cellular responses to hormones and neurotransmitters, as well as being responsible for vision, olfaction and taste. At the most basic level, all GPCRs are characterized by the presence of seven membrane-spanning α-helical segments separated by alternating intracellular and extracellular loop regions. GPCRs in vertebrates are commonly divided into five families on the basis of their sequence and structural similarity1: rhodopsin (family A), secretin (family B), glutamate (family C), adhesion and Frizzled/Taste2. The rhodopsin family is by far the largest and most diverse of these families, and members are characterized by conserved sequence motifs that imply shared structural features and activation mechanisms. Despite these similarities, individual GPCRs have unique combinations of signal-transduction activities involving multiple G-protein subtypes, as well as G-protein-independent signalling pathways and complex regulatory processes. Despite intensive academic and industrial research efforts over the past three decades, little is known about the structural basis of GPCR function. The crystal structures obtained in the past two years provide the first opportunity to understand how protein structure dictates the unique functional properties of these complex signalling molecules.
In this Review, we discuss the similarities and differences among the four known three-dimensional structures of GPCRs in their inactive states. The extracellular surfaces of these structures reveal the molecular underpinnings of antagonist and inverse-agonist ligand recognition. Differences in interactions involving highly conserved residues at the cytoplasmic surface help to explain the varying levels of agonist-independent basal G-protein coupling activity, or ‘constitutive activity’, among the receptors. We then discuss the recently obtained structures of opsin, which reveal in molecular detail several of the key conformational changes associated with GPCR activation. Finally, we address some of the remaining challenges in the structural biology of GPCRs that must be addressed to fully understand the molecular basis for the physiological function of these proteins.
Multifaceted functionality
Much of vertebrate physiology is based on GPCR signal transduction. As the receptors for hormones, neurotransmitters, ions, photons and other stimuli, GPCRs are among the essential nodes of communication between the internal and external environments of cells. The classical role of GPCRs is to couple the binding of agonists to the activation of specific heterotrimeric G proteins, leading to the modulation of downstream effector proteins. Taking the human β2AR as an example, the binding of adrenaline and noradrenaline to cells in the target tissues of sympathetic neurotransmission leads to the activation of the stimulatory subunit of the heterotrimeric G protein (Gαs), the stimulation of adenylyl cyclase, the accumulation of cyclic AMP (cAMP), the activation of cAMP-dependent protein kinase A (PKA) and the phosphorylation of proteins involved in muscle-cell contraction2 (Fig. 1). However, a wealth of research has shown that many GPCRs have much more complex signalling behaviour. For example, β2AR exhibits significant constitutive activity, which can be blocked by inverse agonists3,4. The β2AR couples to both Gαs and the inhibitory subunit (Gαi) in cardiac myocytes5, and can also signal through MAP kinase pathways in a G-protein-independent manner through arrestin6,7. Similarly, the process of GPCR desensitization involves multiple pathways, including receptor phosphorylation events, arrestin-mediated internalization into endosomes, receptor recycling and lysosomal degradation8,9. These activities are further complicated by factors such as GPCR oligomerization10, localization to specific membrane compartments11 and resulting differences in lipid-bilayer composition. Such multifaceted functional behaviour has been observed for many different GPCRs.
Figure 1. Signal transduction in G-protein-coupled receptors.

Diverse signalling pathways regulated by the type 2 beta adrenergic receptor (β2AR). The β2AR can activate two G proteins, Gαs and Gαi (part of the Gs and Gi heterotrimers, respectively), which differentially regulate adenylate cyclase. Adenylate cyclase generates cyclic AMP (cAMP), which activates protein kinase A (PKA), a kinase that regulates the activity of several cellular proteins including the L-type Ca2+ channel and the β2AR. cAMP second messenger levels are downregulated by specific phosphodiesterase proteins (PDEs). Activation of the β2AR also leads to phosphorylation by a G-protein-coupled receptor kinase (GRK) and subsequent coupling to arrestin. Arrestin is a signalling and regulatory protein that promotes the activation of extracellular signal-regulated kinases (ERK), prevents the activation of G proteins and promotes the internalization of the receptor through clathrin-coated pits. PKC, protein kinase C. The inset shows classification of ligand efficacy for GPCRs. Many GPCRs exhibit basal, agonist-independent activity. Inverse agonists inhibit this activity, and neutral antagonists have no effect. Agonists and partial agonists stimulate biological responses above the basal activity. Efficacy is not directly related to affinity; for example, a partial agonist can have a higher affinity for a GPCR than a full agonist.
How does this complex functional behavior reconcile with the biochemical and biophysical properties of GPCRs? The effect of a ligand on the structure and biophysical properties of a receptor, and hence on the biological response, is known as the ligand efficacy. Natural and synthetic ligands can be grouped into different efficacy classes (Fig. 1, inset): full agonists are capable of maximal receptor stimulation; partial agonists are unable to elicit full activity even at saturating concentrations; neutral antagonists have no effect on signalling activity but can prevent other ligands from binding to the receptor; and inverse agonists reduce the level of basal or constitutive activity below that of the unliganded receptor. The wide spectrum of ligand efficacies for individual GPCRs shows that efficient energy transfer between the binding pocket and the site of G-protein interaction is dependent on multiple interactions between receptor and hormone, and requires more than simply occupying the binding site. Further, biophysical studies on purified fluorescently labelled β2AR demonstrated that partial and full agonists containing different subsets of functional groups stabilize distinct conformational states by engaging with distinct subsets of conformational switches in the receptor12–14. These findings lead to a complex picture of GPCR activation in which a distinct conformation stabilized by a ligand’s structure determines the efficacy towards a specific pathway. Many GPCRs can stimulate multiple signalling systems, and specific ligands can have different relative efficacies to different pathways15. In the extreme case, even opposite activities for different signalling pathways are observed: for β2AR, agonists for the arrestin/MAP kinase pathway are also inverse agonists for the classical Gαs/cAMP/PKA pathway7,16. GPCRs are no longer thought to behave as simple two-state switches. Rather, they are more like molecular rheostats, able to sample a continuum of conformations with relatively closely spaced energies17. Specific ligands achieve varying efficacies for different signalling pathways by stabilizing particular sets of conformations that can interact with specific effectors.
The inactive states of four GPCRs
The first insights into the structure of GPCRs came from two-dimensional crystals of rhodopsin18,19. These structures revealed the general architecture of the seven transmembrane helices. However, given the conformational complexity of ligand-activated GPCRs, it is not surprising that it took so long to obtain three-dimensional crystal structures. As detailed in Box 1, a variety of different protein-modification and engineering approaches have contributed to recent advances in GPCR crystallography. We now have inactive-state structures of four GPCRs for comparison: human β2AR bound to the high-affinity inverse agonists carazolol20–22 and timolol23; avian β1AR bound to the antagonist cyanopindolol24; the human A2A adenosine receptor bound to the antagonist ZM241385 (ref. 25); and bovine rhodopsin26–28 containing the covalently bound inverse agonist 11-cis retinal. The superpositions of different receptors using the homologous transmembrane domains led to root mean squared deviation (r.m.s.d.) values of less than 3 Å. This degree of overlap indicates that these four proteins have a similar overall architecture, yet the divergences are still high enough to signify important differences in helical packing interactions (Fig. 2).
Box 1. Challenges in GPCR crystallography.
Summary of structural modifications required to obtain crystals of G-protein-coupled receptors (GPCRs)
| GPCR | Topology | Ligand (indicated by yellow oval) | Method of stabilization | Other modifications (indicated by red ovals and circles) |
|---|---|---|---|---|
| β2AR-Fab |
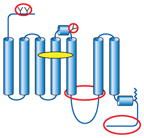
|
Inverse agonist for stabilization | Stabilized TM5/TM6 (transmembrane) region through binding to Fab, which improves crystal lattice-forming contacts | Truncated flexible C terminus (potential Ser/Thr phosphorylation sites) Removed N-linked glycosylation N-terminal affinity tag |
| β2AR-T4 lysozyme Adenosine A2A-T4 lysozyme |
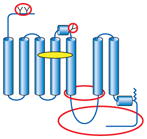
|
Inverse agonist or antagonist for stabilization | Stabilized TM5/TM6 region through T4L insertion, which improves crystal lattice-forming contacts | Truncated flexible ICL3 (intracellular loop) and C terminus (potential Ser/Thr phosphorylation sites) Removed N-linked glycosylation N- or C- terminal affinity tag |
| β1AR |
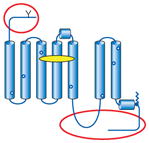
|
Antagonist for stabilization | Mutations to increase thermal stability and functional expression (indicated by blue circles) | Truncated flexible ICL3, N- and C-termini (glycosylation and Ser/Thr phosphorylation sites) Removed palmitoylation site C-terminal affinity tag |
| Rhodopsin/opsin |
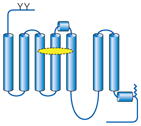
|
Crystal structures obtained with and without bound retinal | Native protein crystallizable without modifications | Not required |
The first major challenge in GPCR crystallography is that most GPCRs are expressed at low levels in native tissues. A suitable recombinant expression system must therefore be developed to generate natively folded membrane protein. So far only Sf9 and Hi5 insect cells and COS-1 mammalian cells64 have produced enough purified GPCR for structure determination (for bovine rhodopsin, a high level of expression in native rod-cell disc membranes allows purification from a natural source65).
The second major challenge is overcoming thermodynamic and proteolytic protein-stability problems (see the table above). GPCRs other than rhodopsin typically have poor thermal stability66 and are prone to proteolysis as a result of their disordered extramembranous loop regions. In recent successful structural efforts, methods used to enhance the thermal stability of the target GPCRs include stabilizing ligands20–22,25 (β2AR, β2AR-T4L and A2A-T4L), a combination of stabilizing mutations24,66 (β1AR), the addition of lipids during purification and crystallization20–22,25 (β2AR, β2AR-T4L and A2A-T4L), and having a high salt concentration25 (A2A-T4L). Methods to enhance proteolytic stability include the truncation of disordered regions20–22,25 (β2AR, β2AR-T4L, β1AR and A2A-T4L), fusion of a stable, well-folded protein domain at the third intracellular loop21,25 (β2AR-T4L and A2A-T4L) and complex formation with an antibody Fab fragment20,67 (β2AR). Structural studies on dark-state bovine rhodopsin did not require such modifications, but purification and crystallization of the protein from a recombinant source did benefit from the engineering of a stabilizing disulphide bond between extracellular loop regions64. Further, the crystals of opsin44,45 were formed at low pH, which is known to stabilize an active conformation of the retinal-free receptor47.
Beyond the purification of large quantities of homogeneous stable membrane protein, several recent successful structural efforts relied on modifications to coax GPCRs into crystals (see the table above). For detergent-solubilized membrane proteins in general, and GPCRs specifically, the absence of significant exposed polar surface area outside the micelle can be a major impediment to crystallization. This was a crucial motivation for both the antibody Fab complex approach20,67 and the T4L fusion strategy21 for the β2AR. In both these structures, as well as in the subsequent A2A-T4L structure, most of the lattice contacts stabilizing the crystals involved the bound antibody or the fused T4L domain. It remains to be seen how general these approaches are to other GPCRs, although the application of the T4L strategy to the A2A receptor25 is a promising sign that other similarly engineered receptor structures may follow. Finally, the β2AR/Fab complex and both T4L fusion-protein structures relied on lipid-mediated crystallogenesis, the former based on the bicelle methodology68,69 and the latter based on lipidic mesophase techniques70. Although the examples of rhodopsin and the mutation-stabilized β1AR show that these strategies are not absolutely necessary for crystal formation and X-ray structure determination of GPCRs, it is likely that most native receptors will not succumb easily to traditional methods of membrane-protein structural biology. The alternative engineering strategies described above are not without risk, namely that the introduction of modifications will alter or skew certain native features of the receptor. An example is seen in the β2AR-T4L structure21,22, where an arginine residue from T4 lysozyme forms a salt bridge with the mechanistically important Glu3268 from the receptor. Therefore, when these alternative strategies are applied to other GPCRs, it is important to rigorously characterize the modified proteins for native-like pharmacological and biophysical properties.
Figure 2. Comparison of four GPCR structures.

a, Bovine rhodopsin (purple), avian β1AR (orange) and human A2A adenosine receptor (green) are each superimposed on the human β2AR structure (blue). The extracellular loop 2 (ECL2), intracellular loop 2 (ICL2), cytoplasmic helix 8 (H8) and several of the transmembrane segments are indicated on one of the structures. The greatest diversity in these structures lies in the extracellular ends of the transmembrane helices and the connecting loops. b, Extracellular views of rhodopsin, the β2AR and the A2A adenosine receptor. The ligands are shown as spheres.
Extracellular surfaces and ligand-binding sites
As might be expected from the functional differences between the receptors, the most significant structural divergences lie in the extracellular loops and ligand-binding region (Fig. 2b). The second extracellular loop (ECL2) of rhodopsin forms a short β-sheet that caps the covalently bound 11-cis retinal, shielding the chromophore from bulk solvent and preventing Schiff base hydrolysis. Further, the glycosylated amino terminus of rhodopsin adopts a structured conformation at the extra-cellular apex of the protein that further shields the covalently bound ligand27,28. In contrast, the ECL2 regions of β1AR and β2AR contain a short α-helix that is stabilized by intra- and inter-loop disulphide bonds, and the cytoplasmic N-terminal regions are disordered21,22,24. oIn the A2A receptor, the ECL2 region lacks a predominant secondary structure, although the loop has multiple disulphide bonds that constrain the observed conformation and expose the ligand-binding cavity to extracellular bulk solvent25. Additional structures will be needed to confirm whether the open binding pocket is a general feature of GPCRs that recognize diffusible small-molecule ligands.
How similar are the ligand-binding pockets of these four receptors? The sites of carazolol, cyanopindolol and 11-cis retinal binding are partly overlapping in superpositions of β2AR, β1AR and rhodopsin, respectively (Fig. 3a). However, the overall positions of the ligands within the β2AR and β1AR structures are slightly more extracellular than 11-cis-retinal in rhodopsin. This difference in the positions of the ligands results in a significant difference in inverse-agonist–antagonist interactions with the residues Trp 2866.48 (human β2AR, with Ballesteros/Weinstein numbering29 in superscript) and Trp 3036.48 (avian β1AR), which are suggested to undergo key rotamer conformational transitions in GPCR activation, referred to as the ‘rotamer toggle switch’30.
Figure 3. Comparison of conserved regions of four GPCR structures.

a, The locations of bound ligands for the four superimposed receptor structures bovine rhodopsin (purple, bound to 11-cis retinal), avian β1AR (orange, bound to cyanopindolol), human A2A adenosine receptor (green, bound to ZM241385) and human β2AR (blue, bound to carazolol) are shown. W6.48 is the key residue of the rotamer toggle switch. TM, transmembrane segment. b, The ionic-lock residues at the cytoplasmic end of TM3 (R3.50 and E/D3.49), and TM6 (E6.30) are shown for the same four structures. R3.50 engages Y5.58 on TM5, rather than E6.30 on TM6 in the opsin ‘active state’. The rotameric position of E6.30 differs for the two β2AR structures. c, The location of several highly conserved residues around a cluster of water molecules (coloured spheres) is shown. These residues may be part of a common pathway for propagating conformational changes from the ligand-binding pocket to the G-protein coupling domains. Amino acids are numbered using the Ballesteros/Weinstein numbering system29, in which the number preceding the dot refers to the transmembrane helix on which an amino acid resides. The second number designates the position relative to the most highly conserved residue among family A GPCRs, numbered 50.
Partial agonism of β2AR can be achieved by ligands without engaging the toggle switch, but full agonism appears to require this conformational change14. The ionone ring of retinal makes direct contact with the analogous Trp residue in rhodopsin, whereas carazolol in β2AR and cyanopindolol in β1AR pack against aromatic residues that shield the residue from the binding site. The less direct link between inverse-agonist binding and the inactive conformation of the rotamer toggle switch in β2AR may help to explain the elevated basal activity of this receptor relative to rhodopsin. For both adrenergic receptors and rhodopsin, ligand binding is mediated by polar and hydrophobic contact residues from transmembrane helices 3, 5, 6 and 7 (TM3, TM5, TM6 and TM7). In contrast to the β2AR, β1AR and rhodopsin structures, the ligand ZM241385 binds to the A2A receptor in a mode that is roughly perpendicular to the bilayer plane, and the packing interactions with the protein, mostly with TM6 and TM7, extend all the way from the toggle switch Trp 2466.48 to the extracellular loops25. This comparison shows that, despite the highly conserved seven-transmembrane architecture, GPCRs can support a wide variety of ligand-binding modes that have differing degrees of interaction with regions involved in known conformational switches.
The structures of avian β1AR bound to cyanopindolol24 and human β2AR bound to carazolol21,22 have almost identical binding pockets. This finding is expected owing to the related function of the proteins and the almost complete conservation of binding-site contact residues31. This high conservation in the ligand-binding pocket is also observed in other subfamilies of GPCRs (such as dopamine, serotonin and histamine), and probably explains some of the difficulty in obtaining potent subtype-selective compounds in pharmaceutical discovery programs32. Nevertheless, subtype-specific binding affinities are observed for β1AR and β2AR33,34. These differences cannot be based primarily on the amino acids forming the binding pocket, but involve more subtle influences on the arrangement of these amino acids as a result of subtype-specific conformational preferences in more distant residues.
Cytoplasmic surfaces of the GPCR structures
At the cytoplasmic surface, a major structural difference between the ligand-activated GPCRs and rhodopsin lies in the ‘ionic lock’ between the highly conserved E/DRY motif on TM3 and a glutamate residue on TM6 (Fig. 3b). Conserved among all family A GPCRs, these amino acids form a network of polar interactions that bridges the two trans-membrane helices, stabilizing the inactive-state conformation35. For β2AR, mutation of these residues increases constitutive activity36,37, and biophysical studies have shown that both full and partial agonists can modulate the structure around the ionic lock38. This interaction network has been observed in dark-state rhodopsin crystals27,28, but the analogous polar interactions are broken in all the ligand-activated GPCR structures, β2AR, β1AR and A2A. The lack of an intact ionic lock in the crystal structures can be interpreted in two ways: either this interaction does not exist in the captured ligand-bound states, or the interaction is so weak that it is overcome energetically by various crystal packing forces. This observation is compatible with the findings that ligand-activated GPCRs generally have higher basal activity than rhodopsin39. Polar contact between adjacent acidic and basic residues on TM3 (E/D3.49 followed by R3.50 of the E/DRY motif; Fig. 3b) is maintained in all four inactive-state structures, and this interaction is also likely to be inhibitory to conformational changes leading to the active state (see below).
One common feature at the cytoplasmic surface of the structures is the chemical environment surrounding residues of the highly conserved NPXXY motif 40 (Fig. 3c). The cytoplasmic end of TM7, in which this motif is located, participates in key conformational changes associated with GPCR activation (see below). In all the structures, the proline in this motif causes a distortion in the α-helical structure, and the tyrosine faces into a pocket lined by TM2, TM3, TM6 and TM7. First observed in rhodopsin28,41 and later seen in the high-resolution structures of β2AR and A2A, networks of ordered water molecules in this region help to reinforce the helical deformation of TM7 and provide hydrogen-bonding partners to polar side chains. Although this ‘water pocket’ network is presumed to stabilize the inactive state, the relative ease of breaking these weakly favourable solvent-mediated interactions probably allows for rapid toggling to the active state when an agonist binds28,41,42.
A final difference between the structures at the cytoplasmic surface is found in the intracellular loop 2 region (ICL2), which includes a short α-helix in the β1AR and A2A structures that is absent in β2AR and rhodopsin. This structure serves as a platform for a hydrogen-bonding interaction of a conserved tyrosine (on ICL2) with the E/DRY motif (on TM3); its absence in β2AR could help to explain the higher relative basal activity of this receptor. Despite their differences, the four inactive-state GPCR structures are in close agreement. Figure 3c shows several of the most highly conserved residues in family A GPCRs, mapped onto the superimposed structures of β2AR, β1AR, A2A and rhodopsin (including the rotamer toggle switch tryptophan, the NPXXY motif and several prolines that induce structurally important helical deformations). The clustering of these residues in the cytoplasmic half of the transmembrane bundle reflects the basic conservation of mechanism across GPCRs, a remarkable structural affirmation of a hypothesis made more than 20 years ago43.
Active state of a GPCR in opsin crystals
The fundamental question of the mechanism for ligand-activated GPCRs remains: how does binding of an agonist, and the resulting changes in interactions at the ligand-binding pocket, lead to conformational changes that are propagated from the extracellular portion of the molecule to the cytoplasmic surface involved in G-protein binding. The recent structures of opsin provide clues to the transmembrane helix rearrangements that can be expected as a result of agonist binding44,45. Opsin is the retinal-free photoreceptor protein generated after photoactivation and Schiff base hydrolysis of rhodopsin. After photobleaching, rod photoreceptors exhibit residual activity that is presumed to result from basal activity of the unliganded state of rhodopsin46. On the basis of biochemical and infrared spectroscopic characterization, opsin at low pH is thought to be stabilized in an active state that resembles metarhodopsin II47,48.
In the crystal structure of opsin at low pH44, there are several subtle changes in the conformations of binding-pocket residues, relative to rhodopsin. Most importantly, the side chain of Trp 2656.48 (the toggle switch) moves into space previously occupied by the ionone ring of retinal, and there is only weak electron density for the Schiff base-forming Lys 2967.43 (on TM7). The interaction between Lys 2967.43 and the Schiff base counterion Glu 1133.28 (on TM3) is broken, and the pocket becomes slightly wider than in rhodopsin. Recent solid-state nuclear magnetic resonance (NMR) studies provide evidence for conformational changes that disrupt a hydrogen-bond network between ECL2 and the extracellular ends of TM4, TM5 and TM6 in metarhodopsin II before the dissociation of retinal and the formation of opsin49.
More dramatic structural changes are observed at the cytoplasmic surface of the molecule. The cytoplasmic end of TM6 is shifted more than 6 Å outwards from the centre of the bundle relative to its position in the inactive state, and at the same time moves closer to TM5 (Fig. 4a, b). This rigid-body movement is consistent with previous biophysical studies of both rhodopsin50,51 and β2AR38. The new position of the cytoplasmic end of TM6 is stabilized by changes in several key interactions (Fig. 4b). Most importantly, the ionic lock is broken and new interactions are formed between Arg 1353.50 (of the ERY motif on TM3) and Tyr 2235.58 (TM5), as well as between Glu 2476.30 (TM6) and Lys 2315.66 (TM5) (Fig. 4b). This rearrangement and engagement of the ionic-lock residues in new interactions is distinct from the merely broken state of the ionic lock seen in the ligand-activated GPCRs. Additionally, Tyr 3067.53 from the NPXXY motif on TM7 undergoes a conformational change and inserts into space occupied by TM6 in dark-state rhodopsin, stabilizing the active conformation. The end result of the changes from inactive rhodopsin to active-state opsin is the creation of a cavity between TM3, TM5 and TM6 in which the G protein transducin can bind (Fig. 4c).
Figure 4. The structure of opsin obtained at low pH represents an active form of rhodopsin.

a, A comparison of the cytoplasmic surface of β2AR (blue), rhodopsin (purple) and opsin (green). With the exception of transmembrane segment 5 (TM5), β2AR is more similar to rhodopsin than to opsin. b, Differences between rhosopsin and opsin in interactions between conserved amino acids, including those of the ionic lock. c, Complex between opsin and a peptide representing the carboxyl terminus of the G protein transducin.
The structure of opsin bound to a carboxy-terminal peptide of transducin demonstrates that this cleft on the receptor does indeed provide the interaction surface for the most crucial binding epitope of the G protein45. Here the rearranged ionic-lock residues prove critical for the formation of the receptor–transducin peptide complex, notably where Arg 1353.50 of the ERY motif dissociates from Glu 1343.49 and forms the base of the peptide-binding cavity with stabilizing contacts from Tyr 2235.58 on TM5. The transducin-derived peptide adopts a C-capped α-helical structure and interacts with the receptor in an amphipathic manner: hydrophobic residues on one face of the transducin helix bind to a hydrophobic surface at the cytoplasmic ends of TM5 and TM6. The orientation of binding is enforced by a hydrogen-bonding network between the transducin C-cap and TM3, TM5 and helix 8 of opsin.
Considering the conserved three-dimensional structure and G-protein signalling mechanism between family A (rhodopsin family) GPCRs, it is reasonable to suppose that the activation of other GPCRs by diffusible ligands will be accompanied by similar changes in trans-membrane helix packing to those observed in the opsin structures. In fact, biophysical studies of β2AR are in good agreement with such a mechanism38. However, the question of how agonist binding far from the cytoplasmic surface leads to the expected packing rearrangements remains unanswered. In the β2AR–carazolol and β1AR–cyanopindolol complexes, the captured inactive-state conformations cannot allow for simultaneous contacts between known agonist-binding amino acids and both ends of the catecholamine scaffold21,24. The incompatibility between the inactive-state adrenergic-receptor structures and agonist binding is analogous to the fact that the retinal binding pocket in the dark state of rhodopsin cannot accommodate the photon-activated all-trans conformation of the chromophore27,28.
Using β2AR and β1AR as models, we conclude that conformational changes at the ligand-binding site must accompany agonist binding. One hypothesis is that the upper region of TM5, which contains several catechol-binding serines52,53, moves closer to TM3. Simultaneous engagement of the agonist by TM5–catechol hydrogen-bonding and TM3/TM7–amine polar contacts (also essential for agonist binding54) would facilitate changes in the packing of nearby aromatic amino acids that shield Trp 2686.48. In this manner, the binding of an agonist could be coupled to movements of the rotamer toggle switch. The resulting conformational change could then lead to tightly coupled packing rearrangements that propagate towards the cytoplasmic surface. This hypothesis is supported by the central and buried locations of residues in the adrenergic receptors whose mutation confers constitutive activity21 (‘CAM mutants’); disruption of these packing interactions would allow freer transmembrane helix motions in the absence of an agonist. In the case of the A2A receptor, the rotamer toggle switch is partly exposed at the base of the ligand-binding pocket and barely interacts with the buried furan ring of bound ZM241385 (ref. 25). The antagonist mainly contacts amino acids on TM5, TM6 and TM7, but the position on the adenine ring of ZM241385 to which the ribose moiety would be attached in the natural agonist adenosine orients the sugar towards TM3. Mutations at this precise region of TM3, analogous to an essential position for agonist-binding in β2AR and β1AR, have been shown to decrease agonist affinity to the A2A receptor55. Overall, it is less apparent for the A2A receptor how agonists might change the structure of the binding cavity, as seen in the inactive-state crystal structure. However, we can speculate that agonists with the ribose functional group would promote the engagement of TM3 residues, resulting in small changes in the relative transmembrane helix dispositions that could activate the rotamer toggle switch.
Future directions
What are the applications of these new GPCR structures, and what are the goals for future investigations? First, there is great interest in structural information to help guide GPCR drug discovery. Until recently, pictures of three-dimensional drug–receptor interactions could only be provided through speculative homology models based on rhodopsin56. For the GPCRs whose structures have now been solved, these modelling efforts have been shown to be imprecise at the level required by in silico drug designers. With the inactive-state structures of β2AR, β1AR and the A2A receptor, pharmaceutical chemists now have experimental data to guide the development of ligands for several active therapeutic targets. However, the value of these high-resolution structures for in silico screening may be limited. Recent molecular docking studies using the β2AR crystal structure as a template identified six new β2AR ligands that bound with affinities ranging from 9 nM to 4 μM; however, every compound exhibited inverse agonist activity. These results suggest that structures of inactive GPCRs will only be reliable for identifying compounds that stabilize the inactive state57.
In a broader sense, the success of these efforts proves that obtaining the crystal structures of GPCR–drug complexes, although still extremely challenging, is at least tractable. Nevertheless, the structures available represent only a small proportion of GPCRs, as implied by their relatively close phylogenetic relationships1. There are still no crystal structures for most of the main branches of the rhodopsin family, or for other GPCR families with large differences in architecture, such as the GABA or (γ-aminobutyric acid) mGluR receptors in family C. Validated drug targets are present throughout the GPCR phylogeny, making it vitally important to develop crystallization methods that can be applied to receptors distantly related to rhodopsin and the biogenic amines. The high-resolution crystallography of GPCRs will hopefully become as routine a tool for drug development as that of kinases.
Beyond the crystallization of more GPCRs, we must develop methods for acquiring structures of receptors bound to agonists. The opsin crystals, without bound retinal but prepared under low-pH activating conditions, have provided a molecular picture of a state resembling fully active metarhodopsin II. Similarly, agonist-bound receptor crystals may provide three-dimensional representations of the active states of other GPCRs. These structures will help clarify the conformational changes connecting the ligand-binding and G-protein-interaction sites, and lead to more precise mechanistic hypotheses. GPCR-targeted therapeutics include agonists as well as antagonists, so these structures will have a broader impact extending to medicinal chemistry and pharmacology. Given the conformational flexibility inherent to ligand-activated GPCRs and the greater heterogeneity exhibited by agonist-bound receptors58, stabilizing such a state will not be easy. The crystal structure of a photoactivated deprotonated intermediate of rhodopsin59 illustrates that a G-protein-interacting state of a GPCR may not be captured in a given crystal lattice, even with a covalent full agonist occupying the binding pocket. Indeed, the possibility of a deprotonated intermediate of rhodopsin in an inactive conformation was directly demonstrated by kinetic electron paramagnetic resonance (EPR) measurements60. Ultimately, the true active state of GPCRs will only be revealed through the co-crystallization of receptors with G proteins, which will also help to reveal how agonist binding is coupled to nucleotide exchange across the protein–protein interface. Such efforts will benefit from the predicted stabilization of a homogeneous agonist-bound receptor conformation in the ternary complex61, as well as the addition of a large soluble protein to participate in crystal-lattice formation. However, the complex dependency of this interaction on experimental conditions makes it difficult to trap a stable GPCR-G protein complex.
As important as the recent structures have been for GPCR research, crystallography has major limitations for characterizing and understanding these physiologically important receptors. As discussed above, GPCRs are inherently flexible proteins that are able to exhibit a spectrum of conformations depending on such factors as the presence of a bound ligand, the lipid environment and the presence of interacting proteins. The conformational dynamics of GPCRs are of more than academic interest: the stabilization of receptor states is the key to modulating GPCR function. To study the relationships between conformational states and the rates of interconversion between them, we need solution-based or membrane-compatible biophysical tools that make direct measurements of the relative positions of different receptor residues on a timescale consistent with the molecular motions. So far, fluorescence spectroscopy and EPR techniques have allowed the study of conformational changes for β2AR17 and rhodopsin62, respectively; however, the application of other methods, such as NMR spectroscopy, promises to greatly expand our knowledge of GPCR dynamics49,63. Important structural properties of GPCRs, such as oligomerization, are not effectively addressed by crystallographic structures, and biophysical techniques can potentially be harnessed to study these phenomena. Only a marriage of biophysical methods with high-resolution X-ray crystallography will provide a full structural understanding of GPCR function.
Acknowledgments
This work was supported by the US National Institute of General Medical Sciences (grant F32 GM082028 to D.M.R. and grant RO1-GM083118 to B. K.), the Lundbeck Foundation (to S.G.F.R.), the National Institute of Neurological Disorders and Stroke (grant RO1-NS28471 to B.K.) and the Mather Charitable Foundation (to B. K.).
Footnotes
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. This paper provides a comprehensive analysis of sequence relationships between G-protein-coupled receptors in the human genome. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman BB, Lefkowitz RJ. Adrenergic receptors in the heart. Annu Rev Physiol. 1982;44:475–484. doi: 10.1146/annurev.ph.44.030182.002355. [DOI] [PubMed] [Google Scholar]
- 3.Samama P, Pei G, Costa T, Cotecchia S, Lefkowitz RJ. Negative antagonists promote an inactive conformation of the beta 2-adrenergic receptor. Mol Pharmacol. 1994;45:390–394. [PubMed] [Google Scholar]
- 4.Chidiac P, Hebert TE, Valiquette M, Dennis M, Bouvier M. Inverse agonist activity of beta-adrenergic antagonists. Mol Pharmacol. 1994;45:490–499. [PubMed] [Google Scholar]
- 5.Xiao RP, Cheng H, Zhou YY, Kuschel M, Lakatta EG. Recent advances in cardiac beta(2)-adrenergic signal transduction. Circ Res. 1999;85:1092–1100. doi: 10.1161/01.res.85.11.1092. [DOI] [PubMed] [Google Scholar]
- 6.Shenoy SK, et al. Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 7.Azzi M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freedman NJ, Lefkowitz RJ. Desensitization of G protein-coupled receptors. Recent Prog Horm Res. 1996;51:319–351. discussion 352–353. [PubMed] [Google Scholar]
- 9.Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- 10.Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Insel PA, et al. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann NY Acad Sci. 2005;1047:166–172. doi: 10.1196/annals.1341.015. [DOI] [PubMed] [Google Scholar]
- 12.Ghanouni P, Steenhuis JJ, Farrens DL, Kobilka BK. Agonist-induced conformational changes in the G-protein-coupling domain of the beta 2 adrenergic receptor. Proc Natl Acad Sci USA. 2001;98:5997–6002. doi: 10.1073/pnas.101126198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swaminath G, et al. Sequential binding of agonists to the beta2 adrenoceptor. Kinetic evidence for intermediate conformational states. J Biol Chem. 2004;279:686–691. doi: 10.1074/jbc.M310888200. [DOI] [PubMed] [Google Scholar]
- 14.Swaminath G, et al. Probing the beta2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J Biol Chem. 2005;280:22165–22171. doi: 10.1074/jbc.M502352200. [DOI] [PubMed] [Google Scholar]
- 15.Galandrin S, Oligny-Longpre G, Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci. 2007;28:423–430. doi: 10.1016/j.tips.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Wisler JW, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci USA. 2007;104:16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Krebs A, Villa C, Edwards PC, Schertler GF. Characterisation of an improved two-dimensional p22121 crystal from bovine rhodopsin. J Mol Biol. 1998;282:991–1003. doi: 10.1006/jmbi.1998.2070. [DOI] [PubMed] [Google Scholar]
- 19.Schertler GF, Villa C, Henderson R. Projection structure of rhodopsin. Nature. 1993;362:770–772. doi: 10.1038/362770a0. This paper presents the first three-dimensional structure of a G-protein-coupled receptor using cryoelectron microscopy of two-dimensional crystals. [DOI] [PubMed] [Google Scholar]
- 20.Rasmussen SG, et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. This is the first reported three-dimensional crystal structure of a ligand-activated G-protein-coupled receptor. [DOI] [PubMed] [Google Scholar]
- 21.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into β 2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 22.Cherezov V, et al. High-resolution crystal structure of an engineered human β2-adrenergic G-protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanson MA, et al. A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warne T, et al. Structure of a β1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaakola VP, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palczewski K, et al. Crystal structure of rhodopsin: A G-protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. This paper presents the first three-dimensional crystal structure of a G-protein-coupled receptor, the visual photoreceptor rhodopsin. [DOI] [PubMed] [Google Scholar]
- 27.Okada T, et al. The retinal conformation and its environment in rhodopsin in light of a new 2.2 Å crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J Mol Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 29.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 30.Shi L, et al. β2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- 31.Horn F, et al. GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res. 2003;31:294–297. doi: 10.1093/nar/gkg103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baker JG. The selectivity of β-adrenoceptor antagonists at the human β1, β2 and β3 adrenoceptors. Br J Pharmacol. 2005;144:317–322. doi: 10.1038/sj.bjp.0706048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugimoto Y, et al. β1-selective agonist (−)-1-(3,4-dimethoxyphenetylamino)-3-(3,4-dihydroxy)-2-propanol [(−)-RO363] differentially interacts with key amino acids responsible for β1-selective binding in resting and active states. J Pharmacol Exp Ther. 2002;301:51–58. doi: 10.1124/jpet.301.1.51. [DOI] [PubMed] [Google Scholar]
- 35.Vogel R, et al. Functional role of the “ionic lock”—an interhelical hydrogen-bond network in family A heptahelical receptors. J Mol Biol. 2008;380:648–655. doi: 10.1016/j.jmb.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Ballesteros JA, et al. Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 37.Rasmussen SG, et al. Mutation of a highly conserved aspartic acid in the β2 adrenergic receptor: constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Mol Pharmacol. 1999;56:175–184. doi: 10.1124/mol.56.1.175. [DOI] [PubMed] [Google Scholar]
- 38.Yao X, et al. Coupling ligand structure to specific conformational switches in the β 2-adrenoceptor. Nature Chem Biol. 2006;2:417–422. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- 39.Bond RA, Ijzerman AP. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol Sci. 2006;27:92–96. doi: 10.1016/j.tips.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 40.Barak LS, Menard L, Ferguson SS, Colapietro AM, Caron MG. The conserved seven-transmembrane sequence NP(X)2,3Y of the G-protein-coupled receptor superfamily regulates multiple properties of the β2-adrenergic receptor. Biochemistry. 1995;34:15407–15414. doi: 10.1021/bi00047a003. [DOI] [PubMed] [Google Scholar]
- 41.Okada T, et al. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proc Natl Acad Sci USA. 2002;99:5982–5987. doi: 10.1073/pnas.082666399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pardo L, Deupi X, Dolker N, Lopez-Rodriguez ML, Campillo M. The role of internal water molecules in the structure and function of the rhodopsin family of G protein-coupled receptors. Chem Bio Chem. 2007;8:19–24. doi: 10.1002/cbic.200600429. [DOI] [PubMed] [Google Scholar]
- 43.Dixon RA, et al. Cloning of the gene and cDNA for mammalian β-adrenergic receptor and homology with rhodopsin. Nature. 1986;321:75–79. doi: 10.1038/321075a0. This paper reports the cloning of the first ligand-activated G-protein-coupled receptor. [DOI] [PubMed] [Google Scholar]
- 44.Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- 45.Scheerer P, et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. This paper presents the high-resolution stucture of an active-state G-protein-coupled receptor in complex with a G-protein peptide. [DOI] [PubMed] [Google Scholar]
- 46.Lamb TD, Pugh EN., Jr Dark adaptation and the retinoid cycle of vision. Prog Retin Eye Res. 2004;23:307–380. doi: 10.1016/j.preteyeres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 47.Vogel R, Siebert F. Conformations of the active and inactive states of opsin. J Biol Chem. 2001;276:38487–38493. doi: 10.1074/jbc.M105423200. [DOI] [PubMed] [Google Scholar]
- 48.Cohen GB, Oprian DD, Robinson PR. Mechanism of activation and inactivation of opsin: role of Glu113 and Lys296. Biochemistry. 1992;31:12592–12601. doi: 10.1021/bi00165a008. [DOI] [PubMed] [Google Scholar]
- 49.Ahuja S, et al. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nature Struct Mol Biol. 2009;16:168–175. doi: 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. This is the first biophysical study to demonstrate movement of transmembrane segment 6 upon activation of rhodopsin. [DOI] [PubMed] [Google Scholar]
- 51.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP, Hubbell WL. High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci USA. 2008;105:7439–7444. doi: 10.1073/pnas.0802515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RA. Identification of two serine residues involved in agonist activation of the β-adrenergic receptor. J Biol Chem. 1989;264:13572–13578. [PubMed] [Google Scholar]
- 53.Liapakis G, et al. The forgotten serine. A critical role for Ser-2035.42 in ligand binding to and activation of the β2-adrenergic receptor. J Biol Chem. 2000;275:37779–37788. doi: 10.1074/jbc.M002092200. [DOI] [PubMed] [Google Scholar]
- 54.Strader CD, et al. Conserved aspartic acid residues 79 and 113 of the β-adrenergic receptor have different roles in receptor function. J Biol Chem. 1988;263:10267–10271. [PubMed] [Google Scholar]
- 55.Jiang Q, Lee BX, Glashofer M, van Rhee AM, Jacobson KA. Mutagenesis reveals structure-activity parallels between human A2A adenosine receptors and biogenic amine G protein-coupled receptors. J Med Chem. 1997;40:2588–2595. doi: 10.1021/jm970084v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patny A, Desai PV, Avery MA. Homology modeling of G-protein-coupled receptors and implications in drug design. Curr Med Chem. 2006;13:1667–1691. doi: 10.2174/092986706777442002. [DOI] [PubMed] [Google Scholar]
- 57.Kolb P, et al. Structure-based discovery of β2-adrenergic receptor ligands. Proc Natl Acad Sci USA. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghanouni P, et al. Functionally different agonists induce distinct conformations in the G protein coupling domain of the β2 adrenergic receptor. J Biol Chem. 2001;276:24433–24436. doi: 10.1074/jbc.C100162200. [DOI] [PubMed] [Google Scholar]
- 59.Salom D, et al. Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc Natl Acad Sci USA. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Knierim B, Hofmann KP, Ernst OP, Hubbell WL. Sequence of late molecular events in the activation of rhodopsin. Proc Natl Acad Sci USA. 2007;104:20290–20295. doi: 10.1073/pnas.0710393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- 62.Hubbell WL, Altenbach C, Hubbell CM, Khorana HG. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv Protein Chem. 2003;63:243–290. doi: 10.1016/s0065-3233(03)63010-x. [DOI] [PubMed] [Google Scholar]
- 63.Werner K, Richter C, Klein-Seetharaman J, Schwalbe H. Isotope labeling of mammalian GPCRs in HEK293 cells and characterization of the C-terminus of bovine rhodopsin by high resolution liquid NMR spectroscopy. J Biomol NMR. 2008;40:49–53. doi: 10.1007/s10858-007-9205-3. [DOI] [PubMed] [Google Scholar]
- 64.Standfuss J, et al. Crystal structure of a thermally stable rhodopsin mutant. J Mol Biol. 2007;372:1179–1188. doi: 10.1016/j.jmb.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Okada T, et al. X-ray diffraction analysis of three-dimensional crystals of bovine rhodopsin obtained from mixed micelles. J Struct Biol. 2000;130:73–80. doi: 10.1006/jsbi.1999.4209. [DOI] [PubMed] [Google Scholar]
- 66.Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci USA. 2008;105:877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Day PW, et al. A monoclonal antibody for G protein-coupled receptor crystallography. Nature Methods. 2007;4:927–929. doi: 10.1038/nmeth1112. [DOI] [PubMed] [Google Scholar]
- 68.Faham S, Bowie JU. Bicelle crystallization: a new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure. J Mol Biol. 2002;316:1–6. doi: 10.1006/jmbi.2001.5295. [DOI] [PubMed] [Google Scholar]
- 69.Faham S, et al. Crystallization of bacteriorhodopsin from bicelle formulations at room temperature. Protein Sci. 2005;14:836–840. doi: 10.1110/ps.041167605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Caffrey M. Crystallizing membrane proteins for structure determination: use of lipidic mesophases. Annu Rev Biophys. 2008:38. doi: 10.1146/annurev.biophys.050708.133655. [DOI] [PubMed] [Google Scholar]