

Abstract
MicroRNA (miRNA) may be potential biomarkers of Alzheimer’s disease (AD). The objective of this investigation was to demonstrate that miRNAs in human brain or biofluids are differentially expressed according to disease status, tissue type, neuritic plaque score or Braak stage. Post-mortem brain (PMB) miRNA were profiled using arrays and validated using quantitative RT-PCR (qRT-PCR). Five qRT-PCR-validated miRNAs were measured in an independent sample of PMB, cerebrospinal fluid and plasma from the same subjects. Plasma miR-15a was found to be associated with plaque score in the independent sample. In conclusion, miRNA present in human biofluids may offer utility as biomarkers of AD.
Keywords: Amyloid, biomarker, Braak stage, miR-15a, miR-370, miR-328, miR-138, miR 132, plaque
INTRODUCTION
Alzheimer’s disease (AD) is an age-associated dementia pathologically characterized by the progressive accumulation of extracellular neuritic and Aβ plaques as well as intracellular neurofibrillary tangles. The stereotypic progression of these pathologic changes hallmarks of the disease and are used, at the time of autopsy, to establish a definitive diagnosis of AD (Hyman et al., 2012). Evidence suggests that asymptomatic AD-specific pathogenesis begins with plaque accumulation followed by tau tangle accumulation and neurodegeneration (Forlenza et al., 2010; Humpel, 2011; Jack, 2012; Trojanowski et al., 2010). However, while the greatest attributable risk for dementia is underlying AD pathologic changes (i.e. neurofibrillary tangles and neuritic plaques), other pathologic changes, particularly vascular disease and Lewy body disease, are also significant contributors to dementia in the elderly (Schneider et al., 2007; Sonnen et al., 2007; White et al., 2005). More importantly, in a large number of dementia cases, there are multiple pathologic changes contributing to the dementia syndrome (Schneider et al., 2007; Sonnen et al., 2007; White et al., 2005).
Taken together, these reports suggest that to effectively treat dementia, it will be vital to monitor the neurodegenerative process with markers that are specific to the neuropathologic changes present. Currently, the most robust AD biomarker is the combined decrease in Aβ42 and increase in phosphorylated tau levels in cerebrospinal fluid (CSF) (Mulder et al., 2010; van Harten et al., 2011). However, CSF markers of AD are only best able to discern dementia type when patients are also clinically cognitively impaired. Ultimately, the ideal AD specific biomarker will identify the neurodegenerative process before cognitive decline begins (Forlenza et al., 2010; Humpel, 2011; Jack, 2012; Trojanowski et al., 2010).
Cancer research has described the advantages of microRNA (miRNA) as markers of disease pathology (Zen & Zhang, 2012; Zhu & Fan, 2011) where specific miRNA correlate with tumor type and show utility for monitoring tumor regression after treatment (Kong et al., 2012; Mitchell et al., 2008). MiRNA are short 22 nucleotide endogenous noncoding RNA molecules that regulate gene expression post-transcriptionally. Although their modes of action are complex, miRNAs generally act as post-transcriptional repressors through translational inhibition and messenger RNA (mRNA) cleavage, but they also may be involved in epigenetic mechanisms of promoter silencing (Breving & Esquela-Kerscher, 2010; Das et al., 2013; Huntzinger & Izaurralde, 2011; Malumbres, 2013; Zhao et al., 2009). In addition, activation or enhancement of gene translation has been described (Mortensen et al., 2011; Srikantan et al., 2011). Animal models, post-mortem brain (PMB) and cell lines have been utilized to characterize the association between miRNA expression in aging (Smith-Vikos & Slack, 2012) and neurodegenerative disease (Delay et al., 2012; Mehler & Mattick, 2006; Nelson et al., 2008a). MiRNAs have been described as particularly important in the synapse where extracellular membrane microvesicles carry miRNA and play a major role in the nervous system under both physiological and pathological conditions (Goldie & Cairns, 2012; Lai & Breakefield, 2012; Olde Loohuis et al., 2012).
Evidence suggests that miRNA play a major role in the development and function of the central nervous system (Delay et al., 2012; Mehler & Mattick, 2006; Nelson et al., 2008a). MiRNA expression in AD is an emerging field where differences in miRNA levels in AD CSF or brain compared to controls has been described (Cogswell et al., 2008; Nunez-Iglesias et al., 2010; Schipper et al., 2007; Sethi & Lukiw, 2009; Wang et al., 2011). Differential miRNA expression has been described in AD compared to controls using blood mononuclear cells (Schipper et al., 2007) or cortex (Nunez-Iglesias et al., 2010; Sethi & Lukiw, 2009; Wang et al., 2011). CSF and hippocampus (HP) miRNA levels have been described in patients grouped into different Braak stages (Cogswell et al., 2008). Little is known about differences in miRNA levels between AD and other neurodegenerative diseases suggesting that additional studies of miRNA levels and neurodegenerative disease are required to demonstrate the utility of miRNA as biomarkers of AD pathology. To the best of our knowledge, a correlation between autopsy brain, live subject CSF, plasma miRNA levels (from the same subjects) and neuritic plaque score or Braak stage in multiple neurodegenerative diseases has not been described.
The first aim of this exploratory investigation was to identify potential miRNA biomarkers of AD-specific neuropathology using both PMB profiling arrays and quantitative RT-PCR (qRT-PCR). The second aim was to confirm the findings of the first aim in an independent sample of PMB using qRT-PCR. The third aim was to determine if these potential miRNA biomarkers are detectable in clinically relevant biofluids, specifically CSF or plasma. MiRNA profiling of AD compared to control HP miRNA levels and subsequent qRT-PCR validation in both the same sample population and an independent replication sample population revealed plasma miR-15a as a putative marker of neuritic plaque.
METHODS
Brain, CSF and plasma
Tissues used for this investigation were obtained from the University of Washington Alzheimer’s Disease Research Center (UW ADRC) following informed consent approved by the University of Washington institutional review board. Cerebrospinal fluid (CSF) and plasma were obtained during life and cerebellum (CB) and hippocampus (HP) were obtained at autopsy. All tissues were immediately frozen and stored at −80 °C. Patients were volunteers in the UW ADRC, where they were diagnosed during life (McKhann et al., 1984) and confirmed by post-mortem neuropathologic examination to have AD or another neurodegenerative disease according to standard criteria (1997). For the purposes of this study, neuropathologic AD was defined by the presence of a Consortium to Establish a Registry for Alzheimer Disease (CERAD) neuritic plaque score of B or C and a Braak neurofibrillary tangle stage IV or greater (Mirra et al., 1991). Control individuals were volunteers in the UWADRC and were never diagnosed with a central nervous system disorder, including dementia, and upon post-mortem histopathological examination did not meet criteria for an AD diagnosis. All autopsy material had a post-mortem interval less than or equal to 9 hours to limit RNA and protein degradation of the brain samples. Use of human tissue was approved by the University of Washington Institutional Review Board.
All CSF samples were collected in the morning after an overnight fast using the Sprotte 24-g traumatic spinal needle with the patient in either the lateral decubitus or sitting position (Peskind et al., 2005, 2006). Samples were aliquoted at the bedside and frozen immediately on dry ice and stored at −80 °C until assayed. All samples used were from comparable fractions of lumbar puncture, 20–25 ml, to limit variability from rostrocaudal concentration gradients. CSF samples with more than 10 RBCs/μl were excluded from the study. Whole blood was collected from subjects in ethylenediaminetetraacetic acid-coated tubes and centrifuged at 1500 × g for 15 min (4 °C). Plasma was then transferred to sterile polypropylene tubes on ice and centrifuged again at 3200 × g for 15 min (4 °C) to remove platelets. Platelet-free plasma samples were then aliquoted into 1ml per tube, flash frozen and stored at −80 °C within 90 min of blood collection. Tissue samples remained frozen at −80 °C 1–4 years before RNA extraction. CSF, plasma and brain miRNA have been previously reported to remain stable under these conditions (Mraz et al., 2009; Weber et al., 2010).
CSF, plasma and brain miRNA have been previously reported to remain stable under these conditions (Mraz et al., 2009; Weber et al., 2010).
RNA extraction and miRNA qRT-PCR
RNA was isolated from (1) 3mg of brain tissue, (2) 250 μl of CSF or (3) 250 μl of plasma using the AllPrep DNA/RNA Mini Kit with a modification that utilized Qiagen DNA elimination columns to deplete DNA content (Qiagen, Valencia, CA). In addition, a slight modification in the protocol utilized 100% ethanol for capturing small RNAs according to manufacturer’s instructions (Qiagen). Only total RNA samples with a A260/A280 ratio of 1.9–2.1 were utilized in this analysis. Samples were also tested for quality and integrity using the RNA FlashGel™ System (Lonza, Basel, Switzerland). Total RNA was also DNase treated using the TURBO DNA-free™ Kit (Applied Biosystems, Austin, TX) to further deplete DNA content.
For miRNA profiling of brain tissue, DNase-treated total RNA was pooled according to disease status and brain region: (1) Control CB, (2) Control HP, (3) AD CB and (4) AD HP (Figure 1). Three μls of equal concentrations (1.35 ng/μl) from each subject sample of DNase-treated total RNA was pooled to equal n=21 subject samples per array. Four ng of DNase-treated total RNA pools were reverse-transcribed using Megaplex RT primers human pool A (Applied Biosystems, Austin, TX) as well as minus RT for detection of DNA amplification. The RT primers human pool A contains specific stem-loop primers for 377 human miRNAs, three small housekeeping RNAs (RNU44, RNU48 and MammU6) and one negative control (ath-miR159a) and are all based on miRBase v. 10.1. A pre-amplification step using equal amounts from each pool (2.5 μl) of RT or minus RT product was added to enrich for human pool A array-specific miRNA using Megaplex PreAmp Primers pool A (Applied Biosystems). Nine microliter of the resulting pre-amplification complementary DNA (cDNA) plus 450 μl of TaqManUniversal Master Mix, no AmpErase, UNG (Applied Biosystems) was transferred to a TaqMan Human miRNA A Array v2.0 (Applied Biosystems), and quantitative PCR was performed using an Applied Biosystems 7900HT Sequence Detection system. Cycling conditions were 50 °C for 2 min, 94.5 °C for 10 min, 40 cycles of 97 °C for 30 s and 59.7 °C for 1 min. Cycle threshold (CT) values were recorded with SDS version 2.3 software (Applied Biosystems, Austin, TX).
Figure 1.

Study design flow chart
To limit low efficiency miRNA and because reliable relative quantification (RQ) requires at least 2 arrays without low efficiency miRNA, miRNA with CT values ≥ 32 for three or more of the arrays were considered beyond the limit of detection (a CT value of 35 represents a single molecule template detection) and were excluded from the qRT-PCR validation step (phase II). Values of ≥ 32 are represented as gray on the miRNome heat map (Supplemental Table 1). For example, array miR-220 results for control CB, control HP and AD CB were 40 CT, whereas AD HP was 29 CT. All mi-RNA-220 values were removed from further analysis (Supplemental Table 1). Please see the discussion section for limitations of this method of exclusion. Because miRNA microarray has high intra-platform repeatability and comparability to qRT-PCR when using reagents from the same company and when using the same RNA extraction sample, to reduce variability between the array platform and the qRT-PCR validation step, the first stage validation qRT-PCR step utilized the same concentration and total RNA extraction product from each individual subject sample (as used in the pooled samples from phase I) as well as the same protocol and reagents (Applied Biosystems) as described above (Nelson et al., 2008b; Sato et al., 2009). Briefly, total RNA (extracted from each brain sample; controls CB and HP; n=21 subjects, AD CB and HP; n=21 patients; Figure 1) is DNase treated; DNase-treated total RNA is reverse transcribed from each brain sample. The resulting cDNA from each brain sample was then pre-amplified as described above, and qRT-PCR was performed in triplicate. Triplicate mean CT values are provided in Supplemental Table 2. Phase III validation analyzes individual subjects samples from an independent sample (controls: n=3, AD: n=3, other neurodegenerative disease: n=6; Figure 1) and uses this same qRT-PCR procedure. Triplicate mean CT values for the phase III independent sample are provided in Supplemental Table 6.
Statistical analysis
Two RQ methods were used for analysis of array data and qRTPCR data of phases I and II. (1) Standard RQ was calculated by normalizing CT values to endogenous controls (RNU44 and RNU48) to calculate ΔCT values. ΔΔCT were calculated by subtracting one array miRNA ΔCT from another (i.e. ΔCTADHP −ΔCTControl HP=ΔΔCT). RQ ΔCT were calculated as 2−ΔΔCT. (2) Alternatively, CT values were subtracted from 41, and a RQ ratio was calculated to provide a fold difference value (i.e. AD HP 41-CT/control HP 41-CT) (see Supplemental Tables for all data). At least three miRNA were selected (mined) from each RQ ratio quartile for phase II qRTPCR validation. To limit variability introduced by using endogenous controls that vary by tissue type (e.g. RNU44 and RNU48 were not detected in phase III CSF and plasma) and disease status as reported by others (Carlsson et al., 2010; Chang et al., 2010; Davoren et al., 2008) and commonly presented as relative to control; statistical comparison data were not normalized to endogenous control values unless otherwise noted. Instead, CT values were subtracted from 41 (41-CT) so that low CT values, which represent high miRNA levels, are represented as high 41-CT values. Comparison of array data 2−ΔΔCT quartiles compared to the 41-CT quartiles demonstrates similarity for both RQ methods and demonstrates feasibility of using 41-CT (Supplemental Table 1). Since endogenous controls (RNU44 and RNU48) were not detected in CSF or plasma, all data across all phases of the study are presented as 41-CT. CT, ΔCTs and ΔΔCTs data (phase I CT values were normalized to the RNU44, RNU48 and MammU6; phase II CT data were normalized to RNU44 and RNU48 and phase III CT data are normalized to miR-132) are available in the Supplemental Tables. All statistical test results (p values) are available in the Supplemental Tables.
Disease status was dichotomized (AD versus controls based on criteria outlined above) when applicable. Standard t-tests were used to test for significant differences in qRT-PCR miRNA levels. Receiver operating characteristic (ROC) was used to test for sensitivity and specificity of each miRNA (Supplemental Table 4). Linear regression was used to determine correlation between miRNA level and disease status, neuritic plaque score or Braak Stage (Supplemental Table 5). MiRNA with p values <0.05 for both linear regression and ROC analyses were chosen for further validation in an independent sample. False discovery rates were taken into account using Bonferroni correction for multiple comparisons and are noted in the text, figures and tables. Statistical analyses and graph production were performed utilizing SPSS version 13 (SPSS, Los Angeles, CA) and Prism version 3.03 (GraphPad Software, La Jolla, CA).
RESULTS
Phase I miRNA profiling
The investigation included four phases (Figure 1). Phase I was the miRNA profiling phase where samples were pooled from 21 AD patients or 21 control subject samples of CB or HP and loaded onto four miRNA arrays to provide an AD miRNome for mining of AD-specific miRNA. The study design is described in Figure 1. The population sample post-mortem brain interval, Braak stage and plaque score are described in Table 1 and in more detail in Supplemental Table 9.
Table 1.
Population description.
| PHASE I AND II SAMPLE | PHASE III SAMPLE | ||||||
|---|---|---|---|---|---|---|---|
| Controls | AD | Controls | AD | Othera | |||
| n= | 21 | 21 | p Value | 3 | 3 | 6 | p Value |
| Age mean (SD) | 87 (5.59) | 81.4 (7.24) | 0.008 | 86 (13.9) | 72 (13.0) | 69.17 (12.0) | 0.095 |
| APOE ε4 | |||||||
| ε4+ | 4 | 14 | 1 | 3 | 3 | ||
| ε4− | 17 | 7 | 0.002 | 2 | 0 | 3 | 0.875 |
| Gender | |||||||
| Males | 11 | 10 | 0 | 3 | 5 | ||
| Females | 10 | 11 | 0.758 | 3 | 0 | 1 | 0.025 |
| Braak stage | |||||||
| 0 | 0 | 0 | 0 | 0 | 1 | ||
| I | 4 | 0 | 0 | 0 | 0 | ||
| II | 10 | 0 | 0 | 0 | 2 | ||
| III | 6 | 0 | 3 | 0 | 1 | ||
| IV | 1 | 2 | 0 | 0 | 1 | ||
| V | 0 | 5 | 0 | 1 | 1 | ||
| VI | 0 | 14 | <0.0001 | 0 | 2 | 0 | 0.521 |
| Neuritic plaque score | |||||||
| Absent | 6 | 0 | 1 | 0 | 1 | ||
| Sparse | 10 | 0 | 2 | 0 | 2 | ||
| Moderate | 5 | 6 | 0 | 0 | 0 | ||
| Frequent | 0 | 5 | <0.0001 | 0 | 3 | 3 | 0.397 |
| Post-mortem interval | |||||||
| Mean | 4.7 | 5.1 | 0.453 | 4.9 | 5.2 | 4.5 | 0.756 |
| Minimum | 2.3 | 2.2 | 3.3 | 3.5 | 3.0 | ||
| Maximum | 7.3 | 8.3 | 6.0 | 8.0 | 9.3 | ||
| RNA 260/280; mean (SD) | |||||||
| Cerebellum | 2.05 (0.068) | 2.06 (0.189) | 0.921 | 2.04 (0.10) | 2.04 (0.23) | 2.07 (0.12) | 0.7218 |
| Hippocampus | 2.06 (0.117) | 2.08 (0.216) | 0.717 | 2.03 (0.04) | 2.00 (0.00) | 2.01(0.07) | 0.7518 |
| Cerebrospinal Fluid | – | – | 2.83 (1.89) | 2.00 (0.00) | 2.17 (0.47) | 0.5707 | |
| Plasma | – | – | 2.00 (0.00) | 2.00 (0.00) | 2.50 (1.00) | 0.5328 | |
Lewy body variant AD (n=2), dementia with Lewy bodies (n=1), frontaltemporal dementia (n=2), Parkinson’s disease dementia (n=1).
To limit low efficiency miRNA and provide RQ of miRNA expression by disease status and brain region, miRNA array results were restricted to miRNA data that were positive (<32 CT) for at least two arrays (control CB, control HP, AD CB or AD HP array pools) (Supplemental Table 1). There were 215 miRNA with CT<32 for at least two arrays (Supplemental Table 1; Figure 1). These 215 miRNA were divided into fold difference quartiles according to each RQ ratio (AD HP/control HP, AD HP/AD CB, control HP/control CB, and AD CB/control CB). MiRNA profiling quartile fold differences represent (1) high, (2) high moderate, (3) low moderate and (4) low. Quartiles are represented in a heat map where green is increased and red is decreased miRNA levels (Supplemental Table 1; Figure 2). CT, ΔCTs and ΔΔCTs, where data are normalized to endogenous controls, are available in Supplemental Table 1.
Figure 2.
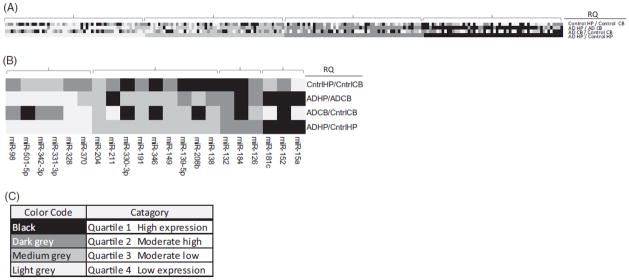
Phase I microRNA array heat maps. Panel A: heat map for relative quantification (RQ: RQ=(41-CT array 1)/(41-CT array 2)). RQ values are color coded according to quartiles for each RQ category (n=215 miRNA) and are arranged according to RQ AD HP/Control HP quartiles (brackets). Panel B: heat map shows n=21 selected candidate miRNA. At least three candidate miRNA from each AD HP/control HP RQ quartile were randomly selected for phase II validation (Panel B). Panel C: color codes for each quartile.
To validate miRNA expression differences between AD HP and control HP, 21 miRNAs were selected for phase II analyses, where at least three miRNAs were randomly chosen from each RQ AD HP/control HP ratio quartile. A greater proportion of miRNA were chosen from the low moderate quartiles using the hypothesis that moderately low miRNA may be both more easily detectable in phase III CSF and plasma than low miRNA as well as more important in regulation of AD relevant miRNA expression since low miRNA levels may result in up-regulation of AD-relevant genes (i.e. APP). CSF and plasma have been reported to have lower concentrations of miRNA than other tissues (Weber et al., 2010).
Phase II miRNA qRT-PCR first stage validation
In phase II, phase I miRNA were validated using the same phase I population sample. However, in phase II, miRNA levels were measured from each individual subject, instead of a pooled sample, where each AD patient or control subject CB and HP sample was analyzed. MiRNA expression levels were measured using miRNA qRT-PCR. CT, ΔCTs and ΔΔCTs, where data are normalized to endogenous controls, are available in Supplemental Tables.
To determine if miRNA level significantly predicts disease status, plaque score or Braak stage; sensitivity and specificity were evaluated for all 21 miRNA identified in phase I (Supplemental Table 4). Five miRNA (miR-370, miR-328, miR-138, miR-132 and miR-15a) had significant ROC as indicated by c-statistic (AUC) for high sensitivity and specificity for either disease status, plaque score or Braak stage (Supplemental Table 4). MiR-15a was significantly correlated with plaque score (Figure 3, Panel A) and miR-370 with Braak stage (Figure 3, Panel B) in linear regression models. Other miRNA were only marginally significantly correlated, in linear regression and Spearman correlation analyses, with plaque score or Braak stage and did not remain significant after correction for multiple comparisons (i.e. miR-328, miR-138 and miR-132; Supplemental Table 5).
Figure 3.
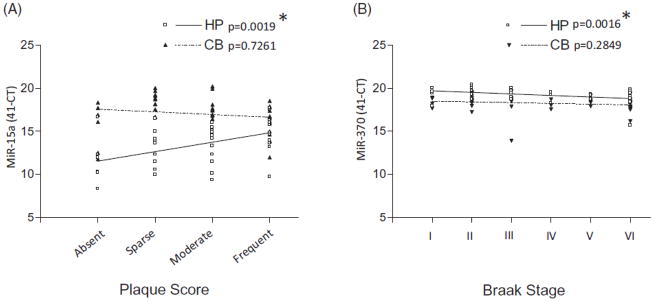
Phase II hippocampus (HP) or cerebellum (CB) miRNA qRT-PCR level correlation with neuritic plaque score or Braak stage. Of five miRNAs (miR-370, miR-328, miR-138, miR-132 and miR-15a) that had significant sensitivity, specificity or correlation with disease status, plaque score or Braak stage, two (miR-15a and miR-370) were significantly correlated with either plaque score (HP miR-15a, linear regression p value, 0.0019, Spearman correlation p value, 0.0028, Panel A) or Braak stage (HP miR-370, linear regression p value, 0.0016, Spearman correlation p value, 0.0021, Panel B). *Remains significant after Bonferroni correction for multiple comparisons.
Phase III miRNA qRT-PCR independent sample validation
In phase III, miRNA expression found to be significantly associated with disease status, plaque score or Braak stage in phase II for linear regression, Spearman correlation and ROC analyses (p value, <0.05 without correction for multiple comparisons) were chosen for further validation in a small independent replication sample. These miRNA (miR-370, miR-328, miR-138, miR-132 and miR-15a) were tested for expression in CB, HP, CSF and plasma. The population sample included; cognitively normal control subjects (n=3), AD patients (n =3) and other neurodegenerative disease patients (Lewy body variant AD (n =v2), dementia with Lewy bodies (n =1), frontotemporal dementia and Parkinson’s disease dementia (n=1) (Table 1; Supplemental Table 9). Since endogenous controls (RNU44 and RNU48) were not detected in CSF or plasma, all data across all phase of the study are presented as 41-CT. However, miR-132 was also used to normalize phase III data. CT, ΔCTs and ΔΔCTs data and analyses (where data is normalized to miR-132) are available in Supplemental Tables 6 and 8. All statistical test results for phase III are available in Supplemental Table 7.
All five miRNAs were detected in the independent sample CB and HP. CB and HP miRNA levels were not significantly correlated with each other (Supplemental Figure 1, Panel A). Three miRNA were detected in plasma and CSF (miR-328, miR-132 and miR-15a), but those levels were not significantly correlated with each other (Supplemental Figure 1, Panel B). CSF and plasma levels were not significantly different when comparing the mean of all three miRNAs for each tissue (p value: 0.1232; data not shown). HP miR-15a levels were non-significantly correlated with CSF (p value: 0.0630) and plasma levels (p value: 0.0865) (Supplemental Figure 1, Panels C and D, respectively).
MiR-15a HP levels were marginally correlated with plaque score (p value: 0.0928) (Figure 4, Panel A). Plasma miR-15a levels correlated with plaque score (linear regression p value: 0.0118; remains significant after multiple comparison correction; Spearman correlation p value: 0.0294) (Figure 4, Panel A) and Braak stage (linear regression p value: 0.0446; does not remain significant after multiple comparison correction) (Supplemental Figure 2, Panel A). Plasma miR-15a levels are differentially expressed in AD compared to other neurodegenerative diseases (Figure 4, Panel B; p value: 0.0473, not significant after multiple comparison correction) and in low compared to high plaque score subjects (Figure 4, Panel C; p value: 0.0467) but not between low and high Braak stage (Supplemental Figure 2, Panel B; p value: 0.2167). All phase III linear regression and Spearman correlation p values are available in Supplemental Table 7.
Figure 4.

Phase III CB, HP, CSF and plasma miR-15a levels in controls, AD and other neurodegenerative diseases (n=12). Plasma miR-15a levels significantly positively correlate with plaque score (linear regression p value, 0.0118, Spearman correlation p value, 0.0294) (Panel A). There is a difference in plasma miR-15a levels between; AD and other neurodegenerative disease (p value, 0.0473) (Panel B). Significance does not remain after taking into account multiple comparisons. There is a significant difference in plasma miR-15a levels between low and high plaque score subjects (p value, 0.0467) (Panel C). *Remains significant after Bonferroni correction for multiple comparisons.
RQ (RQ=2−ΔΔCT) data are presented for each phase of the investigation to demonstrate that a similar differential tissue expression of miR-15a was observed in each phase (Figure 5). Phase III CT data were normalized to miR-132, and the fold difference between tissues were calculated (MiR-132 was used since the endogenous controls available on the arrays of phase I were not detected in CSF or plasma). RQ results from the pooled arrays of phase I, qRT-PCR of phase II and qRT-PCR of phase III are shown in Figure 5. There is a large fold difference between AD HP and control HP for miR-15a for the phase I pooled array assay (Figure 5, Panel A), the phase II validation qRT-PCR assay (Figure 5, Panel B) and for the phase III-independent replication sample qRTPCR assay (Figure 5, Panel C). Phase III qRT-PCR of the three miRNA detected in CSF and plasma shows miR-15a fold difference is greatest between AD plasma and other neurodegenerative disease plasma (Figure 5, Panel D). ΔCT and RQ values (normalized to miR-132) are presented in supplemental Table 8.
Figure 5.

Relative quantification results for 5 miRNA from the pooled arrays of phase I, qRT-PCR phase II and qRT-PCR phase III. Data were normalized to miR-132, which was present in all tissues; cerebellum (CB), hippocampus (HP), cerebrospinal fluid (CSF) and plasma. Data are presented as a relative quantification (RQ)¼2 –DDCT, which represents the fold difference between tissue type. There is a large fold difference between AD HP and control HP for miR-15a for the phase I pooled array assay (Panel A), the phase II validation qRT-PCR assay (Panel B) and for the phase III independent replication sample qRT-PCR (Panel C). Phase III qRT-PCR of the 3 miRNA detected in CSF and plasma shows miR-15a fold difference as greatest between AD plasma and other neurodegenerative disease plasma (Panel D).
miRNA expression in human cell lines
AD HP has been reported to contain less neurons relative to astroglial populations than cognitively normal controls (West et al., 2004). Since five miRNAs were found to be associated with AD brain pathology, we hypothesized that neuronal miRNA expression, not astroglia expression, accounts for the differential expression in controls compared to AD. To test whether these miRNA are expressed in neurons compared to astroglia, miRNA expression was measured in human cell lines (Supplemental Figure 3). All five miRNAs were tested for expression in three human glioblastoma or astrocytoma cell lines (U138, U118 and U87) and three human neuroblastoma cell lines (IMR32, CHP212 and SHSY5Y). All five miRNAs were found to be expressed in all cell lines except for miR-370, which was not expressed in the glioblastoma cell line U138 or U118 (Supplemental Figure 3, Panel A). MiR-15a showed a significantly lower expression in human neuroblastoma cell lines compared to glioblastoma/astrocytoma cell lines (Supplemental Figure 3, Panel B; p value: 0.0396).
Phase IV miRNA target gene prediction
To demonstrate that the five phase II miRNAs represent putative regulators of AD relevant genes, the prediction site www.microRNA.org was utilized (Betel et al., 2008, 2010; John et al., 2004). AD-relevant genes were entered as target mRNA. AD-relevant genes included genes involved in amyloid precursor protein (APP) metabolism, tau phosphorylation and the top 10 genes found to be associated withAD in GWAS (www.alzgene.org) (Supplemental Figure 4). MiR-15a and MiR-370 were predicted to target the highest number of AD-relevant genes. For example, miR-15a is predicted to target nine genes and miR-370 is predicted to target seven genes, while miR-328 and miR-132 are predicted to target four AD-relevant genes and miR-138 is predicted to target five AD-relevant genes (Supplemental Figure 4).
DISCUSSION
Given that miRNAs play a role in neural development, nervous system function and neurological diseases, it is reasonable to assume they may also serve as putative markers of neurodegeneration (Delay et al., 2012; Mehler & Mattick, 2006; Nelson et al., 2008a). The overall aim of this exploratory investigation was to determine if miRNAs in human biofluids are differentially expressed according to disease status, tissue type, CERAD plaque score (neuritic plaques) or Braak stage (neurofibrillary tangles). Plasma miR-15a was found to be associated with plaque score in both the phase II qRT-PCR validation sample and in the phase III small independent sample. The main finding of this investigation was that some candidate miRNA identified in AD PMB are detectable in human biofluid during life and associated with the hallmarks of AD, neuritic plaques and neurofibrillary tangles. To our knowledge, this investigation is the first to evaluate correlations between CB, HP, CSF or plasma miRNA and plaque score or Braak stage in a sample of multiple neurodegenerative diseases. The results from this study suggest that it may be possible to use CSF or plasma miRNA levels to identify the hallmarks of AD neurodegeneration, neuritic plaques and neurofibrillary tangles.
In the first phase of our study, we found 215 miRNAs, of the 377 miRNAs profiled, to be expressed in the CB and HP of both AD and controls. MiRNA were grouped into quartiles according to AD HP relative to control HP fold difference. Both down-regulated and up-regulated AD HP miRNAs compared to control HP miRNAs were chosen for further analyses. A greater proportion of down-regulated miRNAs were chosen using the biologically based hypothesis that down-regulated miRNA may lead to up-regulation of pathological genes, such as, APP or PSEN1 (Figure 2, Panel B). However, it is an equally valid hypothesis that up-regulated miRNAs may inhibit AD-relevant gene expression and play an important role in AD; therefore, up-regulated miRNAs were also analyzed (Figure 2, Panel B).
In the second phase of this study, the same brain samples used in the first phase were further analyzed. MiRNA qRTPCR was performed on individual subject samples to validate the differential expression of 21 miRNA (Figure 2, Panel B). Of these 21 miRNA, five HP miRNA showed significant sensitivity and specificity for AD, plaque score or Braak stage, suggesting they are putative markers of AD pathology (Supplemental Tables 4 and 5).
Interestingly, phase III CB and HP levels of these five miRNAs were not significantly correlated with each other (Supplemental Figure 1, Panel A), while miR-15a showed lower levels in HP than in other tissues (Supplemental Figure 1, Panels A, C and D). These results implicate a tissue specific pattern of expression. Differential miRNA expression according to brain region is further supported by previous reports of differences in miRNA expression across brain regions (Cogswell et al., 2008). It is important to characterize differential brain region-specific miRNA expression in neurodegenerative disease since many neurodegenerative diseases show brain region specific patterns of pathology. In addition, defining tissue-specific miRNA patterns has proven useful in characterizing tumor-specific miRNA targets and biomarkers in cancer research (Kong et al., 2012; Mitchell et al., 2008; Zen & Zhang, 2012).
Linear regression and Spearman correlation analyses were used in phase II analyses to demonstrate that miRNA expression is associated with neuritic plaque score or Braak stage (Supplemental Table 5; Figures 3 and 4). A correlation was found between brain miRNA levels and either neuritic plaque score or Braak stage for only some of the 5 miRNA (Supplemental Table 5; Figure 4). MiR-370, miR-328 and miR-132 HP expression, but not CB expression, was significantly correlated with Braak stage (Supplemental Table 5 for all regression and Spearman correlation results; Figure 3, Panel B for miR-370; remains significant after multiple comparison correction). While miR-328, miR-138 and miR-132 did not remain significant after correction for multiple comparisons, MiR-15a HP expression was significantly correlated with neuritic plaque score (Figure 3, Panel A; remains significant after multiple comparison correction) and Braak stage (Figure 3, Panel B; does not remain significant after multiple comparison correction). MiR-15a CB expression correlation with Braak stage is not significant.
Interestingly, CB miR-15a appears to be normally expressed at higher levels than HP miR-15a, whereas under pathological conditions (high plaque score or Braak stage) CB and HP miR-15a expression level is increasingly similar (Figures 3, Panel A). Previous study suggests that CB shows very little, if any AD-related pathology in end-stage AD, compared to HP (Bobinski et al., 1997; Fukutani et al., 1995; Rossler et al., 2002). Therefore, in this study, CB was used as a control to be compared to HP. Indeed CB and HP miRNA did not correlate with each other for the 5 miRNA evaluated (Supplemental Figure 1, Panel A) suggesting that these 5 miRNA associated with AD pathology in HP did not show similar expression patterns in CB.
Braak stage VI has the highest correlation with clinical dementia diagnosis (Abner et al., 2011), and HP structure and cell type content is drastically affected where neurons are lost and substituted cell populations include primarily glia and astrocytes (Fuller et al., 2010; Wharton et al., 2009). Thus, since brain pathology at the time of death (end-stage disease) is likely to be different than at the time of CSF of plasma draw during life, it follows that CSF or plasma miRNA collected during life is an unlikely marker of miRNA present in the brain at autopsy. However, this exploratory investigation provides evidence that PMB miRNA, that correlates with brain pathology, may also be detectable in CSF or plasma during life suggesting that certain miRNA may be a feasible clinical biomarkers of AD pathology. Even though miRNA identified in this investigation were also found to be expressed in multiple human cell lines, including glia, astrocytes or neurons (Supplemental Figure 3), it is unknown whether the source of CSF or plasma miRNA are from cells in the brain or from another source. Interestingly, regardless of the cell source, it appears that one miRNA, miR-15a, is detectable in a clinically applicable biofluid and correlates with AD-specific neuropathology. Further analysis of CSF or plasma miRNA in independent samples using the miRNA array data presented here may yield additional AD specific miRNA. Furthermore, since only 377 miRNAs were tested in phase I of this investigation and over 1000 miRNAs have now been reported, many additional miRNAs remain to be evaluated as potential markers of AD.
Interestingly, CSF and plasma levels of these five miRNAs did not correlate with each other further suggesting differences in expression of these miRNA according to tissue type. Thus, given that brain includes a multitude of brain cell types, including blood-derived cells in addition to neurons or glial cells, we wondered if these five miRNA are expressed in human neurons or astroglia. To test whether these miRNA are expressed in neurons or astroglia, miRNA expression was measured in three human neuronal cell lines and three human astrocytes and glial cell lines (Supplemental Figure 3). Most of these miRNA (miR-370 was not expressed in U118 or U138) were expressed in the human cell lines tested suggesting that these miRNA are not expressed only in blood cells, but may also be expressed in human neurons and astroglia. However, the source of these five miRNAs in CSF and plasma is unknown and could be peripherally derived.
Others have reported patterns of miRNA expression in cognitively normal subjects and in AD brain, with both similar and contrasting results between studies (Cogswell et al., 2008; Nunez-Iglesias et al., 2010; Wang et al., 2011). It has been proposed that contrasting results between studies can be attributed to the tissue, platform and sample heterogeneity, emphasizing the importance of replication samples for validation (Hebert & Nelson, 2012). In this study, the same miRNA qRT-PCR platform was utilized to measure brain miRNA levels in two independent samples in an attempt to account for tissue, platform and sample heterogeneity.
To our knowledge, our study is the first to use an independent validation sample that includes multiple neurodegenerative diseases as well as PMB brain samples, plasma and CSF samples from the same subjects. The disadvantage of this approach was that the number of subjects available that fit these criteria was limited and led to small sample size limiting the power to detect low expressing miRNA and increasing the probability of false negatives. However, a power calculation of the phase II miRNA qRT-PCR levels with an alpha error level of 5% and a beta error level of 20% suggests that only three samples are necessary to detect a difference between groups suggesting a low probability of false negatives.
Despite small sample size, miR-15a levels are both detectable and differentially expressed according to plaque score in the qRT-PCR validation sample of phase II (Figure 3) as well as in the independent sample of phase III (Figure 4) where miR-15a levels positively correlate with neuritic plaque score. In addition, phase III HP miR-15a levels were marginally correlated with plasma miR-15a and CSF miR- 15a (Figure 4). Plasma miR-15a levels were highest in AD and the high plaque group compared to other neurodegenerative diseases and low plaque group, respectively. In addition, miR-15a shows a similar pattern of high relative tissue expression across all phases of this study (Figure 5). Taken together, these results suggest that higher plasma miR-15a levels may be a feasible marker of both high HP miR-15a levels and high neuritic plaque.
Results presented here should be approached with caution given that miR-370, miR-328, miR-138 and miR-132 were differentially expressed in AD HP compared to control HP in phase I and II analyses of this study, but similar results were not significant in phase III analyses. In addition, miR-370 and miR-138 were not detected in CSF or plasma in our independent study sample, whereas, in contrast, others have reported miR-370 in plasma (Gao et al., 2012) and miR-138 in CSF (Cogswell et al., 2008). Furthermore, in contrast, to the results presented in this study, a previous study describes human temporal cortex miR-15a levels as negatively correlated with neuritic plaque (Wang et al., 2011). As previously described, these contrasts may be attributable to differences in tissue, platform, data analysis methods and population sample (Hebert & Nelson, 2012).
Interestingly, miR-370 has been reported to be associated with lipid metabolism, coronary artery disease as well as cell proliferation of epithelial and neuronal cell types (Chang et al., 2012; Gao et al., 2012; Garcia-Orti et al., 2012; Iliopoulos et al., 2010; Liu et al., 2012; Wu et al., 2012). MiR-328 has been described to modulate the expression of mouse beta-amyloid precursor protein-converting enzyme 1(Boissonneault et al., 2009). MiR-138 appears to play a role in the response to peripheral nerve injury in mice (Adilakshmi et al., 2012) and in dendritic spine morphogenesis (Siegel et al., 2009). There have been multiple reports of miRNA- 132 involvement in neuronal development and maintenance, while miR-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy (Edbauer et al., 2010; Kawashima et al., 2010; Luikart et al., 2011; Magill et al., 2010; Nudelman et al., 2010; Pathania et al., 2012; Remenyi et al., 2010; Shaltiel et al., 2012; Smith et al., 2011). Thus, given reports by others and the phase II results described in this study, miR-370, miR-328, miR-138 and miR-132 may warrant further investigation.
Human miR-15a is located in a cluster at chromosome position 13q14 (Lagos-Quintana et al., 2001), and deletions of this region have been described to be affected in more than half of chronic lymphocytic leukemia cases and accelerate the proliferation of both human and mouse B-cells (Calin et al., 2002; Klein et al., 2010). This miRNA cluster appears to function as a tumor suppressor (Aqeilan et al., 2010; Bonci et al., 2008). In addition, cell proliferation inhibition by this miRNA cluster has been reported in both lymphoid and non-lymphoid tissue (Bonci et al., 2008). These findings suggest that miR-15a plays a major role in cell proliferation.
Furthermore, miR-15a is predicted to target nine AD-relevant genes, and miR-370 is predicted to target seven genes (Supplemental Figure 4). The other miRNA described in this study are predicted to target four AD-relevant genes suggesting that these miRNA may play a role in regulation of genes that play a role in AD (Supplemental Figure 4). However, the direct impact of these miRNA on the regulation of AD genes remains to be determined.
A limitation of this exploratory investigation is that miRNA tested on the arrays that were not expressed in all four conditions were eliminated from phase II and phase III analyses. This method may have led to elimination of important miRNA markers from the analysis. For example, miRNA exclusively expressed in the AD HP (i.e. miR-220; Supplemental Figure 4) may still be specific markers of AD; however, these miRNAs were not tested in the present investigation. In addition, a limitation to measuring PMB miRNA candidates in biofluids from CSF or plasma obtained from subjects while they were living is that miRNA may not represent the population of miRNA present, in some cases, many years before autopsy. Thus, further studies are needed to demonstrate that miRNA expressed in HP, but not expressed in other regions of the brain, correlate with plaque score or Braak stage, and are detectable in plasma or CSF.
Furthermore, a larger independent set of CSF or plasma samples from a clinically characterized cohort that shows that miR-15a or other miRNA can distinguish AD, mildly cognitively impaired, cognitively normal subjects that progress to AD or other neurodegenerative diseases from cognitively normal controls is required to provide definitive evidence that miRNAs are reliable biomarkers of AD specific neurodegeneration.
In summary, MiR-15a expression was found to be significantly correlated with increasing neuritic plaque score in HP. In addition, plasma miR-15a was significantly different in AD compared to other neurodegenerative diseases and for low plaque compared to high plaque subjects. Taken together, these results implicate plasma miR-15a level as a putative marker of HP neuritic plaque pathology. In addition, these results suggest that a multi-phase approach to biomarker discovery can yield candidate markers of AD in human biofluid and emphasizes the feasibility of further study.
CONCLUSION
This exploratory investigation takes a multi-phase approach to detect miRNA differentially expressed according to AD specific pathology. Five miRNAs were identified to be associated with disease, neuritic plaque score or Braak stage suggesting that these miRNA represent feasible markers of brain pathology. An independent sample provided proof-of-principle that plasma miRNA levels correlate with brain pathology. MiRNA present in biofluids may be a feasible marker of neuropathologic changes of AD and ultimately might even provide a means to detect the presence of AD pathology prior clinical symptom onset.
Supplementary Material
Acknowledgments
This work is supported, in part, by the U.S. Department of Veterans Affairs, Office of Research and Development Clinical Research and Development Program, the Biomedical Laboratory Research Program and NIH Grants: P50AG005136-27, P50NS062684-01A1, P50 NS062684 and K99AG034214-02/R00AG034214-03.
Footnotes
Declaration of interest: The authors report no declarations of interest
References
- Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- Abner EL, Kryscio RJ, Schmitt FA, et al. “End-stage” neurofibrillary tangle pathology in preclinical Alzheimer’s disease: fact or fiction? J Alzheimers Dis. 2011;25:445–53. doi: 10.3233/JAD-2011-101980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adilakshmi T, Sudol I, Tapinos N. Combinatorial action of miRNAs regulates transcriptional and post-transcriptional gene silencing following in vivo PNS injury. PLoS One. 2012;7:e39674. doi: 10.1371/journal.pone.0039674. (1–14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqeilan RI, Calin GA, Croce CM. miR-15a and miR-16-1 in cancer: discovery, function and future perspectives. Cell Death Differ. 2010;17:215–20. doi: 10.1038/cdd.2009.69. [DOI] [PubMed] [Google Scholar]
- Betel D, Koppal A, Agius P, et al. Comprehensive modeling of microRNA targets predicts functional non-conserved and noncanonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. (1–14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D, Koppal A, Agius P, et al. Comprehensive modeling of microRNA targets predicts functional non-conserved and noncanonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. (1–14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D, Wilson M, Gabow A, et al. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–53. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobinski M, Wegiel J, Tarnawski M, et al. Relationships between regional neuronal loss and neurofibrillary changes in the hippocampal formation and duration and severity of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:414–20. doi: 10.1097/00005072-199704000-00010. [DOI] [PubMed] [Google Scholar]
- Boissonneault V, Plante I, Rivest S, Provost P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem. 2009;284:1971–81. doi: 10.1074/jbc.M807530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci D, Coppola V, Musumeci M, et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–7. doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- Breving K, Esquela-Kerscher A. The complexities of microRNA regulation: mirandering around the rules. Int J Biochem Cell Biol. 2010;42:1316–29. doi: 10.1016/j.biocel.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J, Helenius G, Karlsson M, et al. Validation of suitable endogenous control genes for expression studies of miRNA in prostate cancer tissues. Cancer Genet Cytogenet. 2010;202:71–5. doi: 10.1016/j.cancergencyto.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Chang KH, Mestdagh P, Vandesompele J, et al. MicroRNA expression profiling to identify and validate reference genes for relative quantification in colorectal cancer. BMC Cancer. 2010;10:173. doi: 10.1186/1471-2407-10-173. (1–13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KW, Chu TH, Gong NR, et al. miR-370 modulates insulin receptor substrate-1 expression and inhibits the tumor phenotypes of oral carcinoma. Oral Dis. 2012;10:1–9. doi: 10.1111/odi.12046. [DOI] [PubMed] [Google Scholar]
- Cogswell JP, Ward J, Taylor IA, et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis. 2008;14:27–41. doi: 10.3233/jad-2008-14103. [DOI] [PubMed] [Google Scholar]
- Das S, Bryan K, Buckley PG, et al. Modulation of neuroblastoma disease pathogenesis by an extensive network of epigenetically regulated microRNAs. Oncogene. 2013;32:2927–36. doi: 10.1038/onc.2012.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoren PA, McNeill RE, Lowery AJ, et al. Identification of suitable endogenous control genes for microRNA gene expression analysis in human breast cancer. BMC Mol Biol. 2008;9:76. doi: 10.1186/1471-2199-9-76. (1–11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delay C, Mandemakers W, Hebert SS. MicroRNAs in Alzheimer’s disease. Neurobiol Dis. 2012;46:285–90. doi: 10.1016/j.nbd.2012.01.003. [DOI] [PubMed] [Google Scholar]
- Edbauer D, Neilson JR, Foster KA, et al. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron. 2010;65:373–84. doi: 10.1016/j.neuron.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlenza OV, Diniz BS, Gattaz WF. Diagnosis and biomarkers of predementia in Alzheimer’s disease. BMC Med. 2010;8:89. doi: 10.1186/1741-7015-8-89. (1–14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukutani Y, Kobayashi K, Nakamura I, et al. Neurons, intracellular and extracellular neurofibrillary tangles in subdivisions of the hippocampal cortex in normal ageing and Alzheimer’s disease. Neurosci Lett. 1995;200:57–60. doi: 10.1016/0304-3940(95)12083-g. [DOI] [PubMed] [Google Scholar]
- Fuller S, Steele M, Munch G. Activated astroglia during chronic inflammation in Alzheimer’s disease – do they neglect their neurosupportive roles? Mutat Res. 2010;690:40–9. doi: 10.1016/j.mrfmmm.2009.08.016. [DOI] [PubMed] [Google Scholar]
- Gao W, He HW, Wang ZM, et al. Plasma levels of lipometabolism-related miR-122 and miR-370 are increased in patients with hyperlipidemia and associated with coronary artery disease. Lipids Health Dis. 2012;11:55. doi: 10.1186/1476-511X-11-55. (1–8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Orti L, Cristobal I, Cirauqui C, et al. Integration of SNP and mRNA arrays with microRNA profiling reveals that MiR-370 is upregulated and targets NF1 in acute myeloid leukemia. PLoS One. 2012;7:e47717. doi: 10.1371/journal.pone.0047717. (1–9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldie BJ, Cairns MJ. Post-transcriptional trafficking and regulation of neuronal gene expression. Mol Neurobiol. 2012;45:99–108. doi: 10.1007/s12035-011-8222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert SS, Nelson PT. Studying microRNAs in the brain: technical lessons learned from the first ten years. Exp Neurol. 2012;235:397–401. doi: 10.1016/j.expneurol.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humpel C. Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol. 2011;29:26–32. doi: 10.1016/j.tibtech.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos D, Drosatos K, Hiyama Y, et al. MicroRNA-370 controls the expression of microRNA-122 and Cpt1alpha and affects lipid metabolism. J Lipid Res. 2010;51:1513–23. doi: 10.1194/jlr.M004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR., Jr Alzheimer disease: new concepts on its neurobiology and the clinical role imaging will play. Radiology. 2012;263:344–61. doi: 10.1148/radiol.12110433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, et al. Human microRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. (1862–79) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima H, Numakawa T, Kumamaru E, et al. Glucocorticoid attenuates brain-derived neurotrophic factor-dependent upregulation of glutamate receptors via the suppression of microRNA-132 expression. Neuroscience. 2010;165:1301–11. doi: 10.1016/j.neuroscience.2009.11.057. [DOI] [PubMed] [Google Scholar]
- Klein U, Lia M, Crespo M, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Kong YW, Ferland-McCollough D, Jackson TJ, Bushell M. microRNAs in cancer management. Lancet Oncol. 2012;13:e249–58. doi: 10.1016/S1470-2045(12)70073-6. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–8. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lai CP, Breakefield XO. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front Physiol. 2012;3:228. doi: 10.3389/fphys.2012.00228. (1–14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DZ, Ander BP, Tian Y, et al. Integrated analysis of mRNA and microRNA expression in mature neurons, neural progenitor cells and neuroblastoma cells. Gene. 2012;495:120–7. doi: 10.1016/j.gene.2011.12.041. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Bensen AL, Washburn EK, et al. miR-132 mediates the integration of newborn neurons into the adult dentate gyrus. PLoS One. 2011;6:e19077. doi: 10.1371/journal.pone.0019077. (1–14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill ST, Cambronne XA, Luikart BW, et al. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci U S A. 2010;107:20382–7. doi: 10.1073/pnas.1015691107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M. miRNAs and cancer: an epigenetics view. Mol Aspects Med. 2013;34:863–74. doi: 10.1016/j.mam.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Mehler MF, Mattick JS. Non-coding RNAs in the nervous system. J Physiol. 2006;575:333–41. doi: 10.1113/jphysiol.2006.113191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA. 2008;105:10513–18. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen RD, Serra M, Steitz JA, Vasudevan S. Posttranscriptional activation of gene expression in Xenopus laevis oocytes by microRNA-protein complexes (microRNPs) Proc Natl Acad Sci USA. 2011;108:8281–6. doi: 10.1073/pnas.1105401108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mraz M, Malinova K, Mayer J, Pospisilova S. MicroRNA isolation and stability in stored RNA samples. Biochem Biophys Res Commun. 2009;390:1–4. doi: 10.1016/j.bbrc.2009.09.061. [DOI] [PubMed] [Google Scholar]
- Mulder C, Verwey NA, van der Flier WM, et al. Amyloid-beta (1–42), total tau, and phosphorylated tau as cerebrospinal fluid biomarkers for the diagnosis of Alzheimer disease. Clin Chem. 2010;56:248–53. doi: 10.1373/clinchem.2009.130518. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Wang WX, Rajeev BW. MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol. 2008a;18:130–8. doi: 10.1111/j.1750-3639.2007.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Wang WX, Wilfred BR, Tang G. Technical variables in high-throughput miRNA expression profiling: much work remains to be done. Biochim Biophys Acta. 2008b;1779:758–65. doi: 10.1016/j.bbagrm.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudelman AS, DiRocco DP, Lambert TJ, et al. Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus. 2010;20:492–8. doi: 10.1002/hipo.20646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez-Iglesias J, Liu CC, Morgan TE, et al. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS One. 2010;5:e8898. doi: 10.1371/journal.pone.0008898. (1–9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olde Loohuis NF, Kos A, Martens GJ, et al. MicroRNA networks direct neuronal development and plasticity. Cell Mol Life Sci. 2012;69:89–102. doi: 10.1007/s00018-011-0788-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M, Torres-Reveron J, Yan L, et al. miR-132 Enhances dendritic morphogenesis, spine density, synaptic integration, and survival of newborn olfactory bulb neurons. PLoS One. 2012;7:e38174. doi: 10.1371/journal.pone.0038174. (1–10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peskind ER, Riekse R, Quinn JF, et al. Safety and acceptability of the research lumbar puncture. Alzheimer Dis Assoc Disord. 2005;19:220–5. doi: 10.1097/01.wad.0000194014.43575.fd. [DOI] [PubMed] [Google Scholar]
- Peskind ER, Li G, Shofer J, et al. Age and apolipoprotein E*4 allele effects on cerebrospinal fluid beta-amyloid 42 in adults with normal cognition. Arch Neurol. 2006;63:936–9. doi: 10.1001/archneur.63.7.936. [DOI] [PubMed] [Google Scholar]
- Remenyi J, Hunter CJ, Cole C, et al. Regulation of the miR-212/132 locus by MSK1 and CREB in response to neurotrophins. Biochem J. 2010;428:281–91. doi: 10.1042/BJ20100024. [DOI] [PubMed] [Google Scholar]
- Rossler M, Zarski R, Bohl J, Ohm TG. Stage-dependent and sector-specific neuronal loss in hippocampus during Alzheimer’s disease. Acta Neuropathol. 2002;103:363–9. doi: 10.1007/s00401-001-0475-7. [DOI] [PubMed] [Google Scholar]
- Sato F, Tsuchiya S, Terasawa K, Tsujimoto G. Intra-platform repeatability and inter-platform comparability of microRNA microarray technology. PLoS One. 2009;4:e5540. doi: 10.1371/journal.pone.0005540. (1–12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipper HM, Maes OC, Chertkow HM, Wang E. MicroRNA expression in Alzheimer blood mononuclear cells. Gene Regul Syst Bio. 2007;1:263–74. doi: 10.4137/grsb.s361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- Sethi P, Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neurosci Lett. 2009;459:100–4. doi: 10.1016/j.neulet.2009.04.052. [DOI] [PubMed] [Google Scholar]
- Shaltiel G, Hanan M, Wolf Y, et al. Hippocampal microRNA-132 mediates stress-inducible cognitive deficits through its acetylcholinesterase target. Brain Struct Funct. 2012;218:59–72. doi: 10.1007/s00429-011-0376-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel G, Obernosterer G, Fiore R, et al. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat Cell Biol. 2009;11:705–16. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Vikos T, Slack FJ. MicroRNAs and their roles in aging. J Cell Sci. 2012;125:7–17. doi: 10.1242/jcs.099200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PY, Delay C, Girard J, et al. MicroRNA-132 loss is associated with tau exon 10 inclusion in progressive supranuclear palsy. Hum Mol Genet. 2011;20:4016–24. doi: 10.1093/hmg/ddr330. [DOI] [PubMed] [Google Scholar]
- Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–13. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- Srikantan S, Marasa BS, Becker KG, et al. Paradoxical microRNAs: individual gene repressors, global translation enhancers. Cell Cycle. 2011;10:751–9. doi: 10.4161/cc.10.5.14825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, Vandeerstichele H, Korecka M, et al. Update on the biomarker core of the Alzheimer’s Disease Neuroimaging Initiative subjects. Alzheimers Dement. 2010;6:230–8. doi: 10.1016/j.jalz.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Harten AC, Kester MI, Visser PJ, et al. Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin Chem Lab Med. 2011;49:353–66. doi: 10.1515/CCLM.2011.086. [DOI] [PubMed] [Google Scholar]
- Wang WX, Huang Q, Hu Y, et al. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: white matter versus gray matter. Acta Neuropathol. 2011;121:193–205. doi: 10.1007/s00401-010-0756-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JA, Baxter DH, Zhang S, et al. The microRNA spectrum in 12 body fluids. Clin Chem. 2010;56:1733–41. doi: 10.1373/clinchem.2010.147405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Kawas CH, Stewart WF, et al. Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol Aging. 2004;25:1205–12. doi: 10.1016/j.neurobiolaging.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Wharton SB, O’Callaghan JP, Savva GM, et al. Population variation in glial fibrillary acidic protein levels in brain ageing:relationship to Alzheimer-type pathology and dementia. Dement Geriatr Cogn Disord. 2009;27:465–73. doi: 10.1159/000217729. [DOI] [PubMed] [Google Scholar]
- White L, Small BJ, Petrovitch H, et al. Recent clinical-pathologic research on the causes of dementia in late life: update from the Honolulu-Asia Aging Study. J Geriatr Psychiatry Neurol. 2005;18:224–7. doi: 10.1177/0891988705281872. [DOI] [PubMed] [Google Scholar]
- Wu Z, Sun H, Zeng W, et al. Upregulation of mircoRNA-370 induces proliferation in human prostate cancer cells by downregulating the transcription factor FOXO1. PLoS One. 2012;7:e45825. doi: 10.1371/journal.pone.0045825. (1–11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zen K, Zhang CY. Circulating microRNAs: a novel class of biomarkers to diagnose and monitor human cancers. Med Res Rev. 2012;32:326–48. doi: 10.1002/med.20215. [DOI] [PubMed] [Google Scholar]
- Zhao BJ, Sun DG, Zhang M, et al. Identification of aberrant promoter methylation of EDNRB gene in esophageal squamous cell carcinoma. Dis Esophagus. 2009;22:55–61. doi: 10.1111/j.1442-2050.2008.00848.x. [DOI] [PubMed] [Google Scholar]
- Zhu H, Fan GC. Extracellular/circulating microRNAs and their potential role in cardiovascular disease. Am J Cardiovasc Dis. 2011;1:138–49. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.