

Abstract
We report a phenomenon wherein induction of cell death by a variety of means in wing imaginal discs of Drosophila larvae resulted in the activation of an anti-apoptotic microRNA, bantam. Cells in the vicinity of dying cells also become harder to kill by ionizing radiation (IR)-induced apoptosis. Both ban activation and increased protection from IR required receptor tyrosine kinase Tie, which we identified in a genetic screen for modifiers of ban. tie mutants were hypersensitive to radiation, and radiation sensitivity of tie mutants was rescued by increased ban gene dosage. We propose that dying cells activate ban in surviving cells through Tie to make the latter cells harder to kill, thereby preserving tissues and ensuring organism survival. The protective effect we report differs from classical radiation bystander effect in which neighbors of irradiated cells become more prone to death. The protective effect also differs from the previously described effect of dying cells that results in proliferation of nearby cells in Drosophila larval discs. If conserved in mammals, a phenomenon in which dying cells make the rest harder to kill by IR could have implications for treatments that involve the sequential use of cytotoxic agents and radiation therapy.
Author Summary
In multicellular organisms where cells exist in the context of other cells, the behavior of one affects the others. The consequences of such interactions include not just cell fate choices but also life and death decisions. In the wing primordia of Drosophila melanogaster larvae, dying cells release mitogenic signals that stimulate the neighbors to proliferate. Such an effect is proposed to compensate for cell loss and help regenerate the tissue. We report here that, in the same experimental system, dying cells activate a pro-survival microRNA, bantam, in surviving cells. This results in increased protection from the killing effect of ionizing radiation (IR). Activation of ban requires tie, which encodes a receptor tyrosine kinase. tie and ban mutant larvae are hypersensitive to killing by IR, suggesting that the responses described here are important for organismal survival following radiation exposure.
Introduction
In metazoa where cells exist in the context of other cells, the behavior of one affects the others. The consequences of such interactions include not just cell fate choices but also life and death decisions. In wing imaginal discs of Drosophila melanogaster larvae, dying cells release mitogenic signals [1], [2], [3]. Signaling from dying cells, or dying cells kept alive by the caspase inhibitor p35 (the so-called ‘undead’ cells), in wing discs operate through activation of Wingless (Drosophila Wnt) and JNK, and through repression of the tumor suppressor Salvador/Warts/Hippo pathway. A crosstalk between JNK and Hpo has also been reported [1]. The consequences on the neighbors include increased number of cells in S phase and activation of targets of Yki, a transcription factor that is normally repressed by Hpo signaling [3]. Mitogenic signals from dying cells results in increased proliferation of neighbors, which is proposed to compensate for cell loss and help regenerate the disc.
A target of Yki is bantam microRNA [4], but ban was not examined in above-described studies. ban was first uncovered in a genetic screen for promoters of tissue growth when overexpressed in Drosophila [5]. Further study found a role for ban in both preventing apoptosis and promoting proliferation [6]. A key target of ban in apoptosis is hid, a Drosophila ortholog of mammalian SMAC/Diablo proteins. These proteins antagonize DIAP1 to liberate active caspases and allow apoptosis. Hid is pro-apoptotic; repression of Hid by ban via binding sites in hid 3′UTR curbs apoptosis [6].
Since the initial characterization of ban, the role of this miRNA has expanded to include coordinating differentiation and proliferation in neural and glial lineages, cell fate decisions in germ line stem cells, in circadian rhythm, and in ecdyson hormone production [7], [8], [9], [10], [11]. In these and other contexts, ban is regulated by a number of transcriptional factors and signaling pathways including, Hpo/Yki, Wg, Myc, Mad, Notch and Htx [6], [12], [13], [14], [15]. The regulatory region of ban gene is likely to be complex and substantial; p-element insertions more than 10 kb away from ban sequences produce ban phenotypes [5].
The experimental evidence in Drosophila that dying cells promote proliferation presaged by several years the experimental evidence for a similar but mechanistically different phenomenon in mammals. A response called ‘Phoenix Rising’ occurs in mice after cell killing by ionizing radiation. Here, the activity of Caspase 3 and 7 is required in dying cells and mediates the release of prostaglandin E2, a stimulator of cell proliferation [16]. These signals act non-autonomously to stimulate proliferation and tissue regeneration. A follow-up study in mice found a requirement for Caspase 3 in tumor regeneration after radiation treatment [17]. Not all consequences on neighboring cells are protective or mitogenic. In the classical ‘radiation bystander effect’, seen in cell culture and in mice, the effect of irradiated cells on the neighbors is destructive, making the latter more prone to death [18], [19], [20]. There is evidence for a soluble signal; media from irradiated cells can induce the bystander effect on naïve cells. Inhibitors of the bystander effect include antioxidants [21], suggesting that oxidative stress and energy metabolism may be involved in radiation bystander effect.
We showed previously that ban activity increased after exposure to ionizing radiation (IR) in wing imaginal discs of Drosophila larvae [22]. IR-induced increase in ban activity required caspase activity: expression of a viral caspase inhibitor, p35, or mutations in p53 that reduced and delayed the onset of caspase activation attenuated ban activation. We noted that while IR-induced cell death was scattered throughout the disc, ban activation was homogeneous. This suggested a non-cell-autonomous component in activation of ban. The current study came out of our efforts to understand how ban is activated in response to IR. We identified Drosophila tie, which encodes a receptor tyrosine kinase of VGFR/PDGFR family, as an important mediator of IR-induced changes in ban. Previous knowledge of Tie function in Drosophila was limited to long range signaling for border cell migration during oogenesis [23]. We report here that Tie was needed to activate ban in response to cell death. One consequence of ban activation, we found, was that remaining cells were harder to kill by IR.
Results
bantam activation in response to cell killing by IR or clonal cell death
To detect ban activity, we used a published GFP transgene that is expressed from the tubulin promoter and includes 2 perfect ban target sequences in its 3′UTR, allowing for repression by ban [6]. We reproduced our published data that ban activity increased in wing discs after exposure to IR [22] (Fig. S1). A published control sensor that lacks sequences for ban binding did not show a change in GFP under the same conditions (Fig. S1 ‘control sensor’).
Next, we investigated whether the ban sensor was responsive to cell killing by another method; we were concerned that although only some cells died in irradiated discs, all were exposed to a death-inducing DNA-damaging agent that might have caused changes in ban. FLP-recombinase mediated ‘flip-out’ method was used to express GAL4 in random, scattered cells in 3rd instar larval wing discs. GAL4 then drove the expression of pro-apoptotic genes, hid and reaper, to result in cell kill. In these experiments, FLP was driven from a heat shock promoter using a brief (10–20′) heat pulse at 37°C. Constitutive expression of GAL80ts from a tubulin promoter kept GAL4 repressed, and allowed clones to form. To induce cell death, GAL80 was inactivated by an incubation at 29°C for 8–12 h (10′ heat shock and 8 h incubation at 29° shown in Fig. 1). Cells expressing GAL4 were marked with RFP. In irradiated discs, a drop in GFP from the sensor was most prominent at >20 h after robust cell death [22]; this was expected because GFP has a half-life of 26 h [24]. Therefore, we monitored the sensor 22–24 h after de-repression of GAL4 (Fig. 1G). We found that GFP was reduced in discs with Hid/Rpr clones compared to discs with RFP-only clones (Fig. 1C vs. D). In discs with flip-out clones, surviving cells had not been exposed to a death-inducing DNA-damaging agent. Yet, these also showed a drop in sensor, reflecting ban activation. Co-expression of p35 with Hid/Rpr in clones prevented the decrease in GFP (Figure S1). This is in agreement with our published finding that p35 also prevented a decrease in the GFP ban sensor in irradiated discs [22].
Figure 1. Activation of ban after clonal induction of cell death in wing discs.
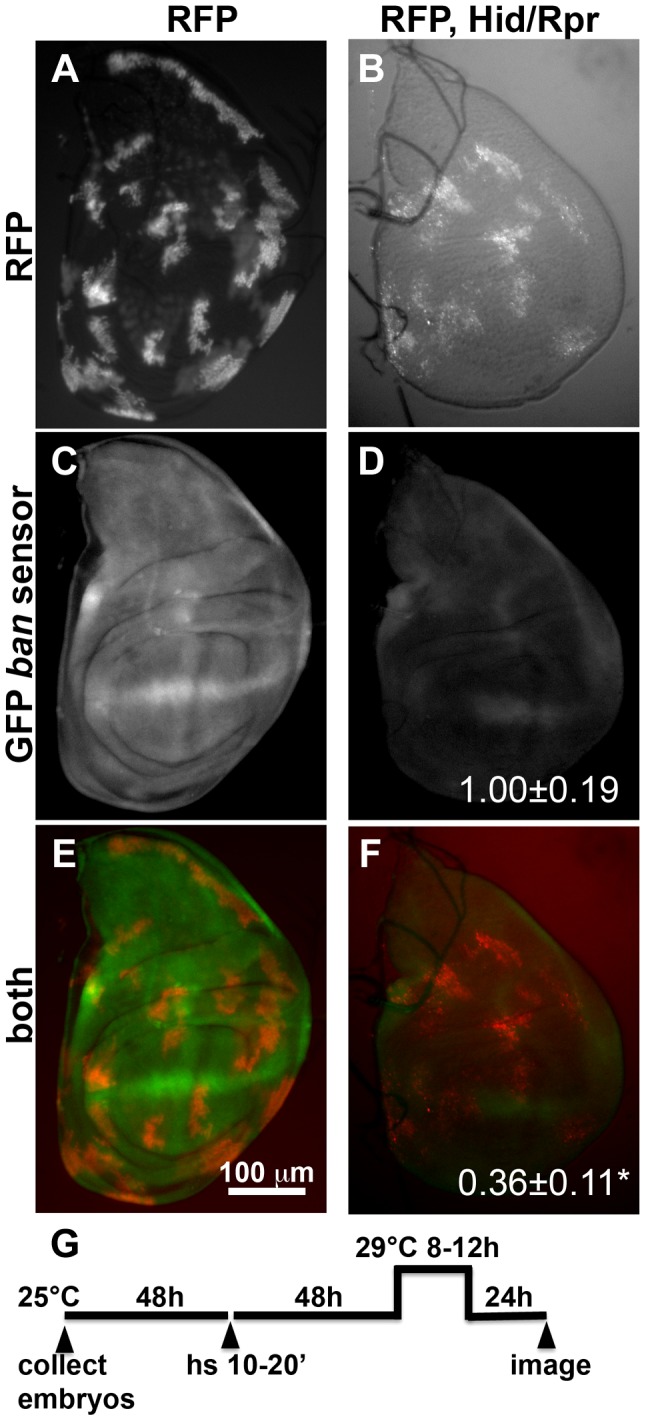
The larvae were heat-shocked (hs) to induce FLP-mediated excision of intervening sequences to allow GAL4 expression from the actin promoter. After heat-shock, actin-GAL4 was kept repressed with tubulin-GAL80ts to allow clones to form. A shift to 29°C inactivated GAL80 and de-repressed GAL4, which drove the expression of UAS-hid, UAS-rpr and the UAS-RFP clonal marker. (A, C, E) Wing discs from a control RFP only larva. (B, D, F) Wing discs from an RFP, hid, rpr larva. The discs were imaged live for RFP and GFP. (G) shows the timeline followed. GFP images in C and D were acquired and processed using identical parameters to allow comparison. RFP was weaker in Hid/Rpr clones than in control clones; therefore RFP images such as those in B were visualized with increased brightness. The mean GFP signal was quantified for each disc, normalized to the average for controls (RFP only) discs and shown on panels D and F. *p<0.001. ‘RFP’ = hs-FLP/Y; GFP ban sensor/+;tub-GAL80ts/Act>>GAL4, UAS-RFP. ‘RFP, Hid/Rpr’ carries the same transgenes and has UAS-hid, UAS-rpr instead of Y. ‘h’ = hour or hours.
bantam activation in response to cell killing in the ptc domain
In irradiated wing discs or in wing discs with Hid/Rpr clones, cell death was scattered but GFP ban sensor decreased throughout the disc. We infer that at least some instances of ban activation occurred in non-apoptotic cells, that is, non-autonomously. To address this idea more rigorously, we killed cells in a known, defined, and invariant location in wing discs.
Expression of a dsRNA against de2f1 under the control of patched-GAL4 (to be called ‘ptc4>dE2f1RNAi’) reduced E2F1 protein levels and the expression of an E2F1 target reporter in a stripe of anterior compartment cells along the Anterior/Posterior (A/P) compartment boundary of wing discs [25]. And the corresponding region in the adult wings was reduced in size. Mutations in dE2F1 caused cell-autonomous apoptosis [26]. Therefore, we asked whether depletion of dE2F1 by RNAi also killed cells. We detected little apoptosis in wing discs of ptc4>dE2f1RNAi 3rd instar larvae raised at 25°C (Fig. 2), even though we could see the expression of a UAS-eGFP reporter in the same larvae (Fig. S2). Presumably, RNAi-mediated knockdown of dE2F1 was sub-optimal under these conditions. A temperature-dependence in the effect of ptc4>dE2f1RNAi on the size of the inter-vein region of the adult wing was noted before [25]. Therefore, in an attempt to enhance the killing effect of dE2f1RNAi, we shifted the larvae from 25°C to 29°C, at 72 h after egg deposition (AED). We observed robust caspase activity in the ptc domain 24 hours (h) after the temperature shift (Fig. 2C). Active caspase staining was stronger in the pouch than the rest of the disc; therefore, we focused on the pouch in subsequent analyses of the consequence of cell death. Similar results were seen by shifting larvae at 96 h AED instead of 72 h. At these times, ptc domain had already narrowed to a vertical stripe [Fig. S2 in [27]; Fig. S2].
Figure 2. Changes in GFP ban sensor in discs with ptc-GAL4-driven cell death.
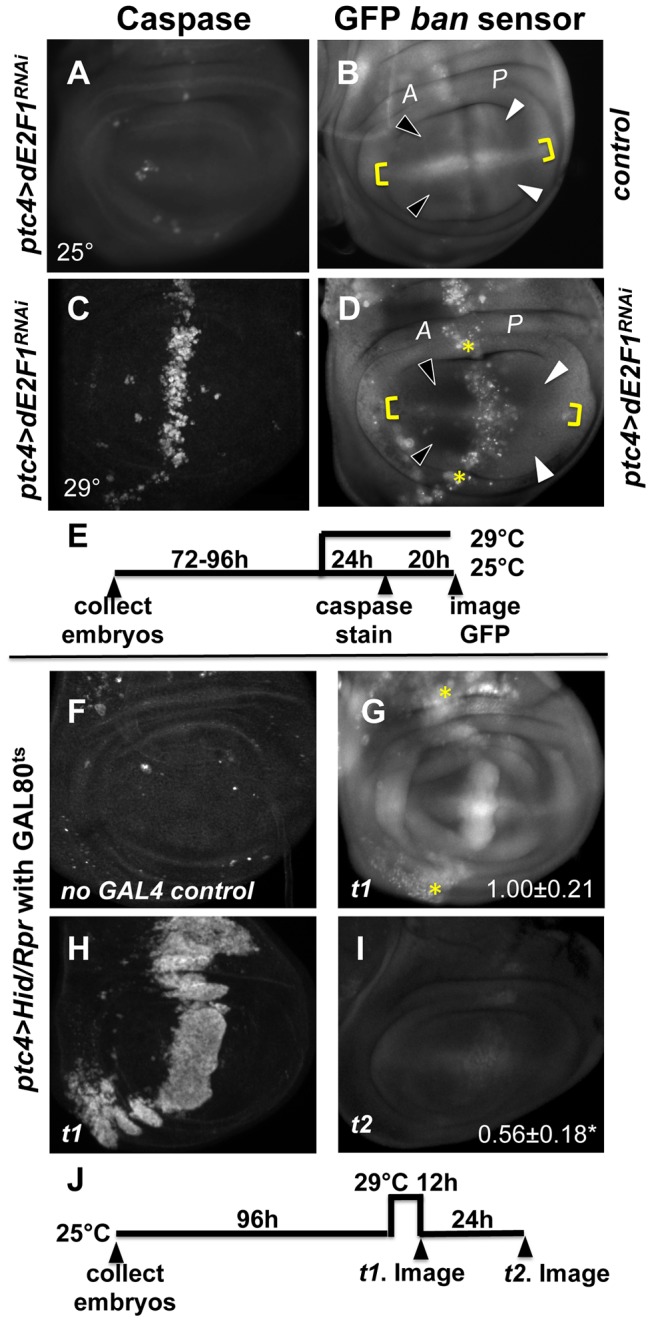
(A–D) Larvae carrying one copy each of ptc-GAL4, UAS-ds RNA against dE2F1 (ptc4>dE2F1RNAi) and the GFP ban sensor were raised at 25°C for 72–96 hours and either maintained at the same temperature (A, B) or shifted to 29°C for 24 h (C, D). Wing imaginal discs were fixed and stained for cleaved Caspase 3 (A, C) or imaged live for GFP ban sensor (B, D). (E) shows the timeline followed. Embryos were collected for 4–6 hours. A = Anterior; P = Posterior. Cell death was minimal at 25°C (A) but prominent after the temperature shift (C). Cells anterior to the ptc domain (black arrowheads) showed lower GFP compared to cells posterior (white arrowheads) in (D) but not in (B). The ratio of GFP fluorescence in A and P compartment for each disc was quantified using Image J, and the averages are shown in Table 1. Only the pouch regions were considered. (F–I) Larvae carrying one copy each of ptc-GAL4, UAS-hid, UAS-rpr, tub-GAL80ts and the GFP ban sensor were raised at 25°C for 96 hours and shifted to 29°C to inactivate GAL80. (J) shows the timeline followed. Embryos were collected for 4–6 hours. Wing imaginal discs were fixed and stained for cleaved Caspase 3 (F, H) or imaged live for GFP ban sensor (G, I). Cell death was absent in ‘no GAL4’ controls (F) but robust after de-repression of GAL4 (H). ban sensor was bright immediately after de-repression of GAL4 for 12 hours (G, t1 time point), but was reduced throughout such discs 24 hours later (I, t2 time point). The mean GFP signal was quantified for each disc, normalized to the average for discs at t1 and shown on panels G and I. Dying cells in these discs sometimes, but not always, displayed elevated GFP (* in D and G); we do not know the reason.
To see if ptc4>dE2f1RNAi-induced cell death resulted in changes in ban activity, we monitored GFP ban sensor after an additional 20 h to allow for the half-life of GFP, that is, at 44 h after temperature shift. In wing discs from control larvae without ptc4>dE2f1RNAi, pouch cells in the Anterior (A) and the Posterior (P) compartments showed similar GFP signals, producing an A/P ratio of 1 (Fig. 2B, quantified in Table 1). A stripe of cells at the dorsal/ventral (D/V) boundary showed high sensor signal (low ban activity; between brackets). This was noted before and represents repression of ban by N/wg along the D/V boundary [6], [15]. In wing discs from ptc4>dE2f1RNAi larvae, cells anterior to the ptc domain showed lower GFP signal than cells posterior to the ptc domain (Fig. 2D, black vs. white arrowheads; quantified in Table 1). This reduction was small, reducing the A/P ratio by about 20%, but was reproducible and bore physiological consequences as described later. The P compartment of some but not all ptc4>dE2f1RNAi discs also showed signs of reduced GFP (Fig. 2D and B, white arrows). But this difference was weaker than the change in the A compartment (thus producing A/P ratios <1 in discs with ptc4>dE2f1RNAi) and not reproducible. Shifting larvae to 29°C at 96 h AED instead of 72 h produced similar results. We conclude that cell death in the ptc domain resulted in a localized decrease in GFP ban sensor, reflecting activation of ban.
Table 1. Mean fluorescence in the wing pouch (Anterior normalized to Posterior).
| assay | genotype | A/P | STD | p value | p value relative to | n |
| GFP | ban sensor/+ | 1.00 | ±0.08 | <0.001 | PE3/ban sensor | 8 |
| GFP | PE3/ban sensor | 0.80 | ±0.09 | N/A | N/A | 12 |
| GFP | PE3/control sensor | 1.03 | ±0.10 | <0.001 | PE3/ban sensor | 8 |
| GFP | PE3/+; tie{Df}/+ | 0.90 | ±0.09 | <0.05 | PE3/ban sensor | 11 |
| caspase | +/+ (yw) | 1.03 | ±0.14 | <0.001 | PE3/+ | 8 |
| caspase | PE3/+ | 0.60 | ±0.11 | N/A | N/A | 25 |
| caspase | PE3/+; ban{D1}/+ | 0.86 | ±0.18 | <0.001 | PE3/+ | 6 |
| caspase | PE3/banA; ban{ D1}/+ | 0.60 | ±0.15 | 0.002 | PE3/+; ban{ D1}/+ | 15 |
| caspase | PE3/+; tie{Df}/+ | 0.87 | ±0.18 | <0.001 | PE3/+ | 10 |
| caspase | PE3/banA; tie{Df}/+ | 0.67 | ±0.08 | <0.01 | PE3/+; tie{Df}/+ | 9 |
| caspase | Pvf1{EP1624}; PE3/+ | 0.86 | ±0.12 | <0.001 | PE3/+ | 14 |
| TUNEL | +/+ (yw) | 0.95 | ±0.22 | <0.001 | PE3/+ | 7 |
| TUNEL | PE3/+ | 0.47 | ±0.16 | N/A | N/A | 11 |
N = number of discs examined. ‘PE3’ = ptc-GAL4>UAS-dsRNA against dE2F1.
Expression of UAS-Hid/Rpr from the ptc-GAL4 driver also produced cell death. In fact, we needed to keep GAL4 repressed with GAL80ts in order to allow survival to 3rd instar. De-repression of GAL4 by incubation at 29°C for 12 h was sufficient to induce robust cell death in the ptc domain (Fig. 2H). This was followed by a decrease in GFP throughout the disc 24 h later (Fig. 2I). We conclude that with more robust cell death in the ptc domain, a greater area of the disc activated ban.
In some wing discs expressing ptc4>dE2f1RNAi, bright specks of GFP were seen in the ptc domain as cells underwent apoptosis (e.g. * in Fig. 2D). Elevated GFP was also seen in the dying cells in ptc4>Hid/Rpr discs immediately after GAL4 de-repression (* in Fig. 2G) but not at later times (Fig. 2I), and rarely in irradiated discs with cell death (Fig. S1). We do not know the reason for elevated GFP in dying cells in some of these cases, but we emphasize that a decrease in the sensor occurred in the survivors outside the domain of cell death in all cases, which we focused on in subsequent sections.
Apoptosis in the ptc domain accompanied resistance to IR in the surviving cells
ban is anti-apoptotic and acts by repressing Hid expression [6]. hid, along with rpr and skl are induced by radiation in a p53-dependent manner (e.g. [28], [29]). Hid, Rpr and Skl antagonize DIAP1 to activate caspases and induce apoptosis. Reduction of hid gene dosage by half has been shown to reduce IR-induced apoptosis [28], [29].
If ban was activated in the surviving cells in the experiments described above, the former may show altered sensitivity to apoptotic-inducing stimuli. To test this possibility, we irradiated larvae with ptc-GAL4-driven cell death and monitored IR-induced cell death using an antibody against cleaved Caspase 3. In y1w1118 control discs (Fig. 3B), there was robust cell death in both A and P compartments of the wing pouch. Along the D/V boundary, low ban activity correlated with high caspase activity as expected (brackets).
Figure 3. Cells in discs with prior cell death are resistant to IR-induced cell death.
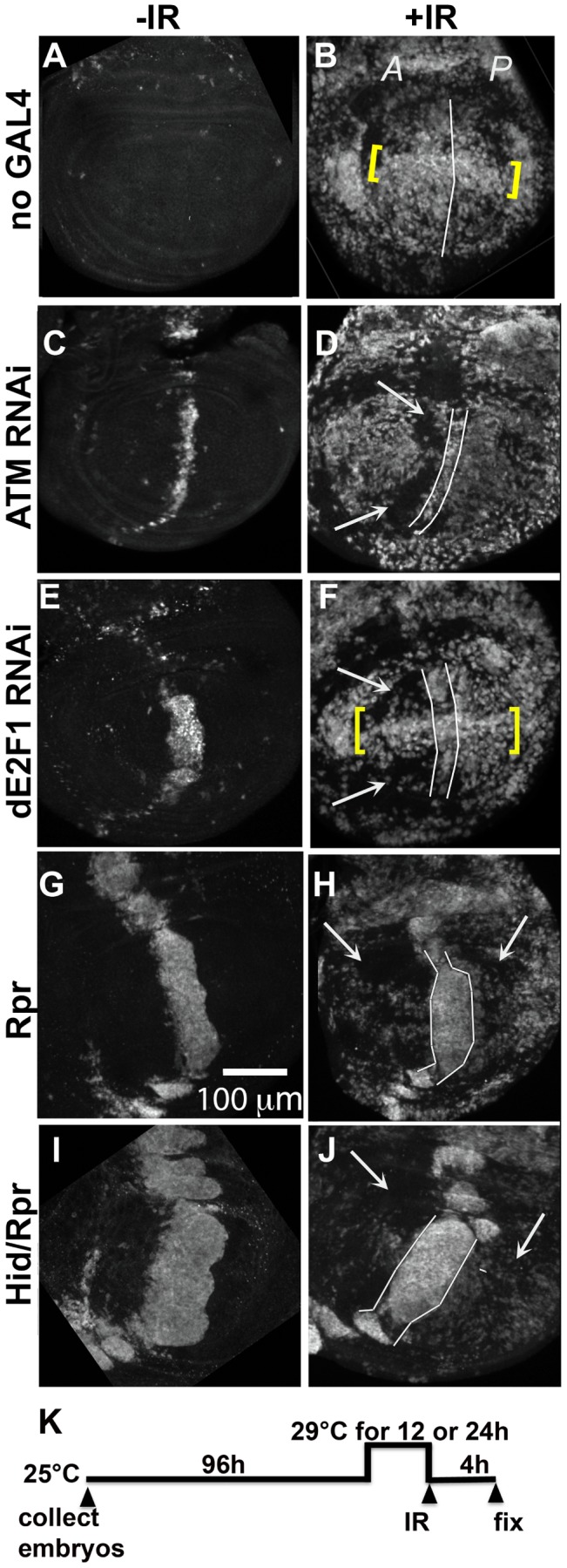
Wing imaginal discs were extirpated from third instar larvae 4(-IR) or 4000 R (+IR) of X-rays, fixed and stained for cleaved Caspase 3. Only the pouch regions are shown. A = Anterior. P = Posterior. (A–B) control discs without GAL4. (B) IR-induced caspase staining was similar in the A and P halves of the pouch in y1w1118 discs. Cells along the D/V boundary where ban sensor is high, reflecting low ban activity, were more sensitive to IR-induced death (yellow brackets). (C–F) discs from larvae with one copy of ptc-GAL4 and one copy of UAS-ds RNA against ATM (C,D) or dE2F1 (E, F). Areas with reduced IR-induced caspase activity (arrows) were found anterior to the domain of cell death induced by ptc-GAL4 (between lines). Cells along the D/V boundary were refractory to the protection (yellow brackets in F). (G–J) discs from larvae with one copy of ptc-GAL4 and one copy of UAS-rpr (G, H) or UAS-hid, UAS-rpr (I, J). These larvae also carried one copy of tub-GAL80ts. The protected areas (arrows) included most of the A compartment and parts of the P compartment in (H). The protected area was even greater in (J). We note that death in the ptc domain in Hid/Rpr discs was wider in –IR discs than in +IR discs. These cells seemed to protrude from the disc after irradiation, which could explain the narrowing of the domain. (K) shows the timeline followed. Embryos were collected for 4–6 hours. The duration of incubation at 29°C was 24 hours for ATM or dE2F1 RNAi larvae and 12 hours for Rpr or Hid/Rpr larvae.
To test for any protective effects of cell death, we used two additional death-inducing constructs. Loss of Drosophila ATM, encoded by telomere fusion, resulted in cell autonomous apoptosis due to telomere dysfunction and activation of the DNA damage responses [30]. This method removed complications due to reduced dE2F1. Cell killing by ATMRNAi, we found, was not as robust as cell killing by dE2f1RNAi (Fig. 3C vs. E; Fig. S2). We also used UAS-Rpr to induce death by ptc-GAL4. As in the case of UAS-Hid/Rpr, UAS-Rpr also had to be kept repressed with GAL80ts to allow larval survival to 3rd instar and was able to induce cell death with a 12 h de-repression of GAL4. But the resulting cell death was less robust than UAS-Hid/Rpr under the same experimental conditions (Fig. 3 G vs. I).
Irradiation of these discs showed that prior cell death in the ptc domain accompanied a reduction in IR-induced caspase activation (Fig. 3D, F, H and J arrows). The protected area was localized in D and F but expanded in H and J, showing an increase with increasing level of prior death. In discs expressing dE2f1RNAi (Fig. 3F) or Hid/Rpr (Fig. 3J), the protected area corresponded to the area with reduced ban sensor (Fig. 2D and I). We conclude that prior cell death in the ptc domain resulted in ban activation and protection from IR-induced cell death in the rest of the disc, in a dose-dependent manner. Cells in the same compartment as the dying cells (A compartment) were the first to benefit but the protective effect was able to reach the P compartment with increased ptc-GAL4-driven cell death in the case of Rpr and Hid/Rpr (compare Fig 3J to B).
To rule out the possibility that the protection is due simply to disruptions in the ptc domain, we induced cell death in random clones; this too resulted in protection (Fig. 4, described in detail below). To rule out unwanted consequences of depleting dE2F1 earlier in development, we repeated the experiments with ptc4>dE2f1RNAi but used GAL80tts to repress GAL4 until 96 h AED. GAL4 was then de-repressed for 24 h and protection from IR-induced apoptosis assayed (Fig. 5D). The results were indistinguishable from what we saw without GAL80 (Fig. 3F). We also ruled out the possibility that dE2f1RNAi in the ptc domain altered cell proliferation in the pouch to interfere with IR-induced cell death; we saw no change in the mitotic index in A and P compartments in discs with ptc4>dE2f1RNAi (Fig. S3). We also ruled out the possibility that death in the ptc domain interfered with developmental signaling; we saw no change in Dpp-lac Z reporter expression in control and ptc4>dE2f1RNAi discs (Fig. S4).
Figure 4. Cell death and protection in discs with clonal Hid/Rpr expression.

The larvae were generated as described for Figure 1 and irradiated with 4000R of X-rays after de-repression of GAL4. Wing imaginal discs were extirpated 4 h after irradiation and fixed and stained for DNA (A, E) and cleaved active Caspase 3 (B, F). RFP clonal marker (C, G) was used to mark the clonal boundaries in the images. (D, H) show merged images. (I) shows the timeline followed. * shows areas outside the clone that display robust caspase activation in RFP only discs (top row) but not in RFP, Hid/Rpr discs (bottom row).
Figure 5. The effect of ptc>dE2F1RNAi is modified in different genetic backgrounds.

Wing imaginal discs from larvae carrying one copy each of ptc4-GAL4 and UAS-dsRNA against dE2F1 in different mutant backgrounds were fixed and stained for cleaved Caspase 3 (as indicated) or processed for TUNEL (F) at 4 h after irradiation with 4000R of X-rays. The discs were also stained for DNA. DNA stained images were used to locate the pouch (within the dashed line), the A/P boundary (solid vertical line) and the ptc domain (between vertical lines). UAS-GFP (green) in (D) confirmed the location of the ptc domain. Yellow brackets indicate apoptosis along the presumed D/V boundary in some panels. The experimental timeline in Fig. 3 K was followed, using a 24 h incubation at 29°C to de-repress GAL4. Embryos were collected for 4–6 hours. The caspase or TUNEL signal from A and P compartments of the pouch, exclusive of the ptc domain, were quantified from images such as these and shown in Table 1. (A–B) y1w1118 discs reproduced from Fig. 3B, for comparison. (C-P) All larvae carried one copy each of ptc-GAL4 and UAS-dsRNA against dE2F1. The larva in (C,D) also carried one copy each of tub-GAL80ts and UAS-GFP. Additional genotypes are indicated next to the panels. banA = UAS-banA.
Death in Hid/Rpr flip-out clones also resulted in protection from IR
Death within the ptc domain accompanied increased ban activity and protection from IR-induced cell death outside the ptc domain. Death in Hid/Rpr clones also increased ban activity outside the clones (Fig. 1). Therefore, we asked if Hid/Rpr clones also resulted in protection from IR-induced cell death. Discs like those in Fig. 1C and D were irradiated and IR-induced caspase activity was monitored. We found robust caspase activation in areas outside RFP-only clones (Fig. 4A–D). In contrast, areas outside RFP-marked Hid/Rpr clones showed reduced caspase activation (Fig. 4E–H). We conclude that Hid/Rpr clones also resulted in protection from IR-induced death and that the protected area included cells outside the clones.
The protective effect of ptc-GAL4-induced death is sensitive to ban gene dosage
Wing discs expressing ptc4>dE2f1RNAi showed an intermediate level of protection in the A compartment (Fig. 3F and 5D). The P compartments in these discs showed similar level of caspase activation as the P compartments of controls (Fig. 5B vs. D); the average mean caspase fluorescence in the P compartments was 182±32 arbitrary units for ptc4>dE2f1RNAi and 163±16 for yw in two different experiments. In contrast, the A compartment of ptc4>dE2f1RNAi pouches show reduced caspase stain compared to P cells in the same disc (Fig. 5D). This allowed us to use the P compartment as an internal control to account for variations in antibody staining, and quantify the reduction in the A compartment in terms of reduced A/P ratio. The ptc domain was visible in the DNA images as a ‘depression’ as dead cells were extruded from the cell layer (Fig. 5C). Therefore, we used the DNA images as a guide to locate A and P compartments. The normalized A/P ratio for caspase fluorescence in ptc4>dE2f1RNAi discs was 0.60±0.11 and was significantly different from the controls (Table 1). TUNEL assay produced similar results (Fig. 5F and Table 1).
This quantitative assay for protection allowed us to test the role of other factors. Co-expression of caspase inhibitor p35 in dying cells attenuated the protective effect (Table 1). This is in agreement with our data that p35 abolished changes in ban sensor after irradiation [22] and in discs with Hid/Rpr clones (Fig. S2). Reduction of ban gene dosage using heterozygotes of a null allele, banD1, attenuated the protection (Fig. 5H and Table 1); this was consistent with the observation that ban was activated in the protected regions (Fig. 2D). One copy of the UAS-banA transgene, which encodes the primary transcript and rescued the lethality and growth defects of ban null mutants without a GAL4 driver [6], also rescued the protection in banD1/+ (Fig. 5J and Table 1).
We also addressed the role of ban in cell death-induced protection in ptc4>Hid/Rpr discs. In initial experiments, we were unable to see significant effects of banD1/+ using a protocol in which GAL4 was de-repressed for 12 h (Fig. 3K). We reasoned this could be because the protective effect of Hid/Rpr was too strong to overcome by removing just one copy of ban. Removing both copies of ban was not an option because we found before that homozygous ban mutant cells in the wing disc underwent spontaneous apoptosis [22]. To get around this problem, we de-repressed ptc4>Hid/Rpr for 6 h instead of 12 h (Fig. 6). This reduced cell death in the ptc domain to the level seen after de-repressing ptc4>Rpr for 12 h (compare Fig.6B to Fig. 3G). The protective effect was also reduced (Fig. 6D) and resembled the protection with Rpr de-repressed for 12 h (Fig. 3H) than Hid/Rpr de-repressed for 12 h (Fig. 3J). Importantly, banD1 heterozygosity further compromised the protection such that we could see caspase activation throughout A and P compartments (Fig. 6F arrows, compare to arrows in 6D). The protection was not completely abolished, however. This could be because protective mechanisms could still act on the remaining copy of ban. We conclude that protection by ptc4>Hid/Rpr was also sensitive to ban gene dosage.
Figure 6. Protection by Hid/Rpr-induced cell death is sensitive to ban gene dosage.
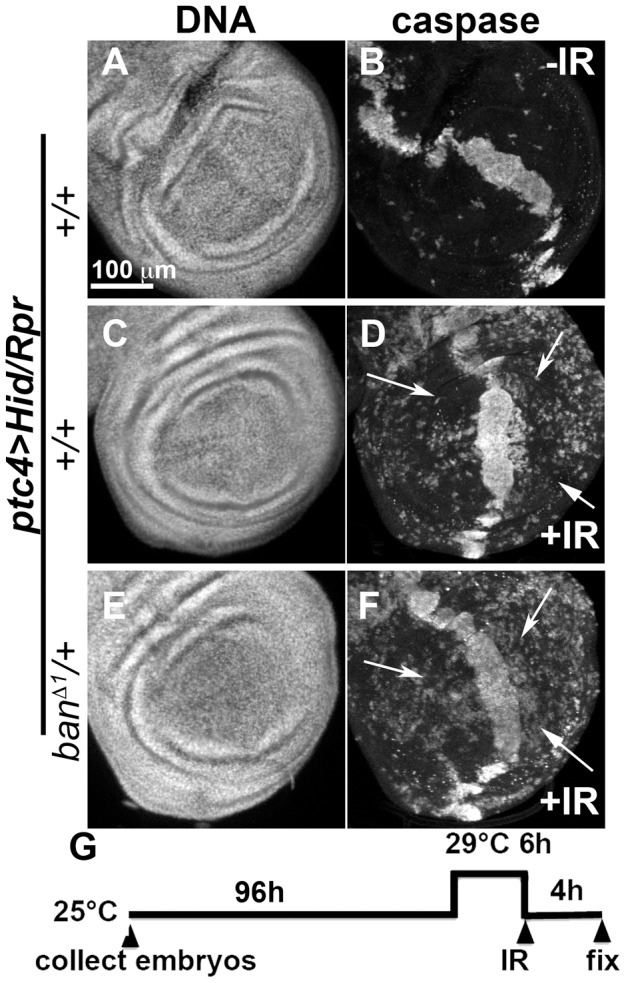
Wing imaginal discs from larvae carrying one copy each of ptc4-GAL4, UAS-hid, UAS-rpr and tub-GAL80ts were fixed and stained for DNA (A, C, E) and cleaved Caspase 3 (B, D, F) at 4 h after irradiation with 0 (-IR) or 4000R (+IR) of X-rays. Additional genotypes were as indicated. (G) shows the timeline followed. In otherwise wild type background (D), areas outside the ptc domain showed reduced caspase staining reflecting protection in response to ptc4>Hid/Rpr (arrows). In contrast, the corresponding areas in ban/+ discs showed caspase activity (F, arrows).
A screen for modifiers of ban identifies tie
To better understand the role of ban in radiation responses, we performed a forward genetic screen for modifiers of a ban phenotype. ban mutant larvae are hypersensitive to IR but this sensitivity can be rescued by reducing the gene dosage of hid, a known target of ban [22]. Using a similar experimental protocol, we sought to identify dominant modifiers of IR sensitivity of ban mutant larvae. We used 4000R of X-rays because this dose resulted in an intermediate level of lethality in ban mutants such that both suppressors and enhancers may be uncovered.
Chromosomal deficiencies (Df) used in the screen, on their own, could show IR sensitivity and modify ban through simple additive effects. To exclude these, we measured the % survival of ban mutants and each Df, and used these to compute the expected survival for an additive interaction (Fig. S5A). We then identified ban/Df combinations that produced observed survival that was significantly different from the expected, by X2 analysis. We focused on deficiencies that showed consistent and statistically significant effect on both ban1170 and banEP3622 alleles. Of 78 Df that cover 85% of chromosome 3L and part of 3R, 3 were significant modifiers. Df(3L)9028 and Df(3L)6115 enhanced and Df(3L)6086 suppressed the IR sensitivity of ban mutants. We focused our efforts on Df(3L)9028 because it deleted a single gene, tie (Fig. S5B). tie encodes a Drosophila homolog of mammalian Tie (Tyrosine kinase with Ig and epidermal growth factor homology domain) [31]. We confirmed the deletion by PCR of genomic DNA (data not shown), and also by q-RT-PCR for tie expression (Fig. S5C). Df(3L)9028 is homozygous viable.
The data from the screen suggested that tie mutants were IR sensitive. We confirmed this using three additional alleles; larvae homozygous of all four alleles show significant and reproducible sensitivity to X-rays (Fig. 7A). Importantly, all three alleles show genetic interaction with ban seen with Df(3L)9028; each allele, in heterozygous state, reduced the radiation survival of ban1170 heterozygous larvae to levels below what was expected for an additive effect between two mutants (Fig. 7B, data not shown). If tie regulates ban to promote survival after irradiation, increasing ban gene dosage may rescue radiation survival in tie mutants. We found that UAS-banA did rescue the survival in irradiated tie heterozygous larvae (Fig. 7C), but did not change the survival of irradiated w1118 controls in the same experiment (p = 0.6).
Figure 7. tie interacts with ban to promote radiation survival.

Larvae were irradiated at 96±2 h after egg deposition with 4000R of X-rays. Percent eclosion was determined by counting full and empty pupal cases 10 days after irradiation. Error bar = 1 SEM. (A) Homozygous tie mutants were more radiation sensitive than w1118 controls (p<0.05). N = 106 to 494 pupae per genotype in at least three independent experiments. tie alleles were Df(3L)Exel9028 and transponson insertion alleles in Fig. S5: tiee03394 or ‘03394’, tiec098 or ‘co98’ and tiee02680 or ‘e02680’. (B) tie and ban showed genetic interaction in radiation survival. The observed eclosion of double heterozygotes (tie/ban1170) is shown for each allele of tie. The % eclosion expected from an additive effect between ban1170/+ and tie/+ was computed from observed % eclosion for each allele and shown as solid horizontal bars above each tie allele. In all cases, the observed eclosion was lower than the expected. The difference between expected and observed were significantly different for the three tie alleles shown (X2 test, p<0.05). N = 94–536 pupae in at least three independent experiments per genotype. (C) A UAS-ban transgene ‘banA’ rescued the radiation sensitivity of Df(3L)Exel9028 (‘Df’) and tiee03394 heterozygotes. N = 359–634 pupae in at least six experiments per genotype.
tie is needed for IR-induced changes in ban
The data described so far are consistent with tie acting upstream of or parallel to ban to confer survival after irradiation. To distinguish between these possibilities, we investigated whether tie was needed for IR-induced changes in ban. We found that while GFP ban sensor decreased after irradiation in control larvae, reflecting activation of ban (‘wt’ in Fig. 8C, F and I), this decrease was attenuated in tie mutants. All three allelic combination of tie studied showed this defect, with Df(3L)9028 homozygotes showing the greatest defect, i.e. the smallest change in GFP between –IR and +IR discs (Fig. 8C). In q-RT-PCR analysis, tie was the only gene whose expression was reduced in mutants used here; neither of the flanking genes showed a consistent change in expression (Fig. S5C). In addition, a UAS-tie transgene rescued the change in ban sensor in Df(3L)9028 homozygotes after irradiation (Fig. 8J-L). Similar to UAS-banA [6], the rescue occurred without GAL4 (see also Discussion). Collectively, these data support the idea that tie is needed to activate ban after irradiation.
Figure 8. tie is needed for IR-induced changes in GFP ban sensor.

(A–I) Third instar larvae were irradiated at 96±2 hr after egg deposition with 0R (-IR) or 4000R (+IR) of X-rays and wing discs imaged live 24 h later. Images were acquired and treated identically. The mean GFP signal was quantified for each disc using Image J, and the averages are shown for each genotype/treatment. Age-matched sensor only controls (i.e. wild type for tie) were included in each experiment (images not shown here but see Fig. S1). (A–C) Wing discs from homozygotes for Df(3L)Exel2098 that removes only tie (‘tie [17]’). N = 31, 28, 31 and 31 discs (left to right) in three independent experiments. (D–F) Wing imaginal discs from homozygotes for tiee03394. N = 26, 27, 26 and 25 discs (left to right) in three independent experiments. (G–I) Wing imaginal discs from trans-heterozygotes tiee03394/Df(3L)Exel2098. N = 29, 29, 30 and 30 discs (left to right) in three independent experiments. (J–L) Wing imaginal discs from Df(3L)Exel2098 homozygotes that carry one copy of a UAS-tie transgene. The data are from 5 discs per sample in two independent experiments. The larvae carried two copies of the GFP sensor in A-I and one copy in J-L. Error bar = ±1SEM. p-values reflect a comparison of two samples at the ends of each bracket. (M) tie is needed for IR-induced changes in ban levels. Quantitative RT-PCR was used to quantify mature ban miRNA. The values were normalized to an internal a-tubulin control, and expressed as fold change from un-irradiated w1118 controls (‘w2-’ set at 1). ‘-‘ = no IR; ‘+’ = 4000R. ‘w’ = w1118; Df’ = homozygotes of Df(3L)Exel2098 that removes only tie; ‘3394’ = tiee03394 homozygotes. * = difference compared to w2-. ** and *** = significance compared to w2+. * = p <0.01; ** = p <0.001; *** = p < 10−6. Student's t-test was used to determine significance.
Previous work showed that IR-induced caspase activity was needed for ban activation following irradiation [22]. Therefore, it is possible that tie mutants could not activate ban because they could not activate caspases. We do not favor this possibility, however, because we found that wing discs from tie mutant larvae were capable of activating caspases after irradiation, as were tie homozygous mutant clones of cells (Fig. S6). We conclude that tie is not required for IR-induced apoptosis. Because both tie and caspase activity were required to activate ban but tie was not required to activate caspase activation, we conclude that tie acts after caspase activation to activate ban.
To address how ban was activated, we monitored mature ban miRNA levels by q-RT-PCR. ban increased 2-fold at 2 h after irradiation and returned to control levels by 18 h (Fig. 8M). This timing was consistent with the change in GFP ban sensor at 20+ hours after irradiation, given that the half-life of GFP is 26 h. The IR-induced increase in ban was abolished in homozygotes of two tie alleles examined, suggesting that tie was required to increase ban levels after IR. We conclude that tie-dependent increase in ban activity after IR was due, at least in part, to a tie-dependent increase in ban levels.
The protective effect of ptc-GAL4-induced death is sensitive to tie gene dosage
Using the ptc4>dE2f1RNAi system, we addressed the role of tie in the protective effect of dying cells. Wing discs from tie Df(3L)9028 heterozygotes showed an attenuation of both ban activation, as seen by changes in the GFP ban sensor (Fig. S7 and Table 1), and the protective effect of cell death in the ptc domain (Fig. 5L, Table 1). This defect was rescued by one copy of UAS-banA (Fig. 5N and Table 1). We conclude that tie was required to activate ban and to confer protection in response to ptc-GAL4-induced cell death. Haplo-insufficiency of tie in these experiments was consistent with the identification of tie as a dominant modifier of ban in our screen.
Pvf1, potential ligand for Tie, is up-regulated by IR and contributes to the protection
The ligands for mammalian Tie-2 are Angiopoietin 1 and 2. Although Drosophila genome includes five predicted Ang homologs [31], there is no evidence for these being ligands for Drosophila Tie. Instead, there is evidence for a Drosophila PDGF/VGF-like protein, Pvf1, in border cell migration, a process in which Tie is known to function [23]. We found that mRNA for Pvf1, Pvf2 and CG10359, which encodes one of the putative Ang homologs, were induced at 2 h after IR in wing imaginal discs ([32]; Supplemental Table 1). In p53 mutants, where caspase activation and apoptosis after irradiation were reduced and delayed [29], these genes were no longer induced. An independent RNAseq analysis confirmed these results (our unpublished data). In these experiments, mRNA levels for tie, another Pvf homolog, Pvf3, and four remaining predicted Ang homologs were unchanged (Table S1 and data not shown). Analysis of Pvf1 and Pvf2 expression using enhancer trap lines corroborated these results; we found that both genes were induced in the ptc domain after expression of ptc4>dE2f1RNAi (Figure S8).
These data suggest that cell death induced Pvf1, Pvf2 and/or CG10359 to activate Tie. To address this possibility, we monitored the protective effect of ptc4>dE2f1RNAi but in Pvf1 and Pvf2 mutants and a chromosomal deficiency that deletes CG10359. The results with Pvf2 and CG10359 were negative (not shown), but a hemizygote of a Pvf1 allele, shown before to express no detectable mRNA or protein [33], showed reduced protection (Fig. 5P and Table 1).
Discussion
We document a previously unknown phenomenon in wing imaginal discs of Drosophila larvae; dying cells protected nearby cells from death. We found that killing cells by any one of three methods __ ptc-GAL4-driven expression of dE2F1RNAi or pro-apoptotic genes hid and rpr, exposure to ionizing radiation (IR) and clonal induction of Hid/Rpr __ activated an anti-apoptotic microRNA, bantam. Death by ptc-GAL4 or clonal expression of Hid/Rpr also made surviving cells more resistant to killing by IR. The protective effect was sensitive to ban gene dosage. We have named this phenomenon ‘Mahakali effect’, after the Hindu goddess of death who protects her followers. Mahakali effect differs from classical radiation ‘bystander effect’ in which byproducts from cell corpses make surviving cells more prone to death [18], [19], [20]. The Mahakali effect appears to operate in a non-cell-autonomous fashion. Disc-wide protection by ptc4>Rpr and Hid/Rpr that included even cells in the P compartment that did not express ptc, provides the strongest evidence for non-autonomy. This idea is supported by the finding that IR-induced caspase activation was reduced in cells outside Hid/Rpr flip-out clones.
A recent paper describes a non-autonomous induction of apoptosis by apoptotic cells [34]. We do not believe these results necessarily contradict what is reported here. Most of the experiments in the published work used undead cells kept alive by p35; Mahakali effect is seen without p35. Non-autonomous apoptosis was assayed at, typically, 3–4 days after induction of undead cells; we detect Mahakali effect 6 hr after cell death induction using similar death-inducing stimuli (Hid/Rpr). It would be interesting to see how long Mahakali effect persists and whether non-autonomous apoptosis, occurring at longer time points, also produces Mahakali effects of its own. Another recent paper describes tissue regeneration after massive cell ablation in wing discs [35]. It would also be interesting to see if the Mahakali effect operates among regenerating cells.
The data shown here suggest that the basic components of the Mahakali effect are caspase activity in dying cells (because expression in dying cells of p35, an inhibitor of effector caspases, blocked ban activation), ban (because ban activation resulted from cell death and the protective effect was sensitive to ban gene dosage), and tie (because tie was required to activate ban and the protective effect was sensitive to tie gene dosage). We propose a model in which caspase activity in dying cells acts through Tie to cause non-autonomous activation of ban and the Mahakali effect (Figure 9). A validated target of ban in apoptosis inhibition is hid, whose 3′UTR includes 4 potential ban binding sites. We have shown previously that a GFP sensor with hid 3′UTR is reduced after IR [22], reflecting repression of hid by ban. Deletion of two potential ban-binding sites in the hid 3′UTR abolished the IR-induced changes in GFP.
Figure 9. A model for the Mahakali effect.
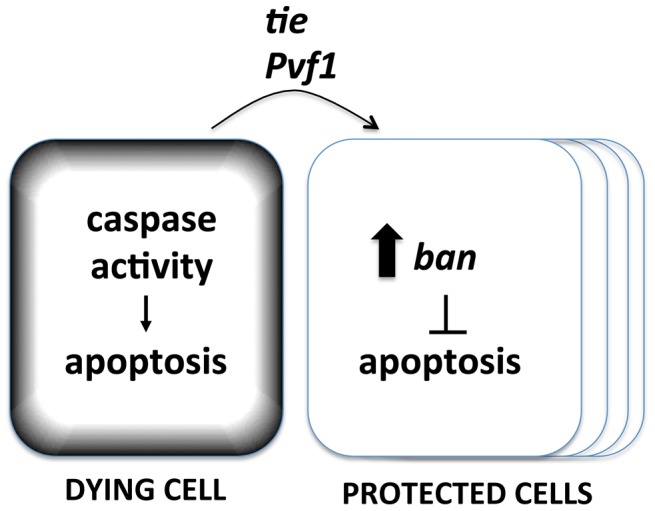
Known genetic determinants are shown.
The Mahakali effect differs in two ways from previously described effects of dead/dying cells in wing discs. First, the Mahakali effect extended further than previously reported signaling from dead/dying cells. In the extreme case of ptc4>Hid/Rpr, the protection reached as far as the edge of the disc. This distance, on of order of 100 or more mm (scale bar in Fig. 3) is comparable to the distance of border cell migration [36], in which Tie is known to function. In contrast, the mitogenic effect that occurs through JNK/Wingless in response to undead cells in the wing disc is seen up to 5 cells away [2]. Activation of proliferation through the Hpo/Yki axis also spans 3–5 cells away. This can be seen as activation of Yki targets such as DIAP1 [3]. We could reproduce this result: ptc4>dE2f1RNAi activated a Yki target, DIAP1, but only within or close to the ptc domain (Fig. S4). YkiB5 allele, which disrupts cell death-induced proliferation [3], did not alter the Mahakali effect (Fig. S9), further supporting the idea that the two effects are different. Second, ban activation in response to cell death was sensitive to the caspase inhibitor p35. In contrast, the mitogenic effect of dying cells in wing imaginal discs is not sensitive to p35 [2], [37], [38], [39], [40], [41]. We note that the mitogenic effect of dying cells is inhibited by p35 in the differentiating posterior region of eye imaginal discs [42], which is similar to what we saw for ban activation in the wing discs.
We found that tie was required for IR-induced activation of ban and for larval survival after irradiation. There were similarities as well as differences in the role of ban and tie. tie mutants were IR-sensitive (this study), as are viable alleles of ban [22]. Tissue-specific overexpression of ban results in abnormal growth [5]; we found that 6 independent UAS-tie transgenic lines were lethal when driven by actin-GAL4 (data not shown). Thus, too much ban or tie has consequences. On the other hand, reducing tie or ban gene dosage by half attenuated the Mahakali effect. Thus, too little ban or tie also has consequences. In fact, UAS-ban or UAS-tie without a GAL4-driver was sufficient to rescue ban and tie mutant phenotypes [[6] and this study]. Thus, intermediate levels of expression may be important for the function of these genes.
The biggest difference between ban and tie, of course, was that while tie homozygous larvae were viable (this study), ban homozygous larvae are lethal [6]. tie became necessary only after radiation exposure. This suggests that tie was needed to regulate ban not during normal development but after radiation exposure. How is IR and cell death linked to Tie? We found that mRNA for Pvf1, a ligand for Tie in border cell migration, was induced by IR and that this induction appeared to be dependent on cell death (abolished in p53 mutants). Pvf1EP1624 mutants that are mRNA and protein null [33], also showed reduced Mahakali effect. The degree of reduction was significant but not back to the level seen in control discs without ptc4>dE2f1RNAi, suggesting the involvement of additional ligands or mechanisms for Tie activation. In agreement, we could not see ban activation or the Mahakali effect after overproduction of Pvf1 (data not shown). Pvf1 was necessary but insufficient to produce these effects without cell death.
Tie activated ban, at least in part by increasing ban levels. How IR and caspase activity promotes Pvf1 expression and how Tie activity increases ban levels will be key questions to address in the future. Testing the role of known apoptosis regulators, such as Diap1, and signaling molecules, such as Wg, may help address these questions. We also plan to complete the genetic screen that identified Tie; it has the potential to identify additional components of the Mahakali effect.
Pvr, a PDGF/VEGF receptor homolog that function redundantly with Tie in border cell migration, also plays an anti-apoptotic role in embryonic hemocytes [43]. A recent study in wing discs found that Pvr is activated in neighbors of dying cells in a JNK-dependent manner, to result in cytoskeletal changes that allow the engulfment of the dead cell by the neighbor [44]. It is interesting that two PDGF/VEGF receptor homologs that function redundantly in cell migration during oogenesis may also play non-redundant roles in non-autonomous responses to cell death in wing discs.
Cancer therapy routinely comprises the application of two or more cytotoxic agents (taxol and radiation, for example) to cancer cells. A phenomenon in which cell killing by one agent influence resistance to the second agent is, therefore, of potential clinical significance. The bulk of our analysis focused on protection from IR-induced cell death. But we also have preliminary indication that the Mahakali effect can also protect against cell death induced by maytansinol (Fig. S10), a microtubule depolymerizing agent with relevance to cancer therapy that we found before to induce cell death in Drosophila wing discs [45]. An important question is whether a phenomenon like Mahakali effect exists in mammals and acts as a survival mechanism in response to cell death. Ang-1, a ligand for mammalian Tie-2, is a pro-survival factor for endothelial cells during serum deprivation and after irradiation in cell culture models [46], [47], [48]. Interestingly, Ang1 is produced not by endothelial cells but by neighbors, at least in cell culture [49]. Based on these data, we think it possible that radiation exposure results in Ang1 production by dead/dying cells that promote the survival of endothelial cells via Tie-2. Consistent, an Ang-1 derivative that is a potent activator of Tie-2 has been shown to protect endothelial cells from radiation-induced apoptosis [50].
Materials and Methods
Fly stocks
ban mutants: ban 1170, ban EP3622, ban D1 [5].
tie mutants:y1 w1118; PBac{3HPy+}TieC098 (Bloomington #16280); PBAC{RB}Tiee03394 (Harvard Exelixis Collection); PBAC{RB}Tiee02680 (Harvard Exelixis Collection); w1118; Df(3L)Exel9028, PBac{RB5.WH5}Exel9028 (Bloomington #7925).
Pvf1 mutant: pvf1EP1624 [33].
Clonal induction of GAL4: hs-FLP22; 20.X/T(2;3) CyO Tb/Act<FRT>GAL4>UAS-RFP; Generated using w1118; P{GAL4-Act5C(FRT.CD2).P}S, P{UAS-RFP.W}3/TM3, Sb1 (Bloomington#30558) and crossed to UAS-hid, rpr on Chr I [51].
Mitotic clones: FRT80B-Df(3L)Exel9028 crossed to FRT80B-Ubi-GFP
Other stocks: ptc-GAL4 and UAS-dsRNA against dE2F1 or ‘PE3’ on Chr II [25]; ban sensor ‘20.X’ on Chr II [6]; Dpp-lac Z reporter, P{BS3.0}H1-1, cn1; ry506 (Bloomington#5527); DIAP1-lac Z reporter [52]; Yki5B [53]; UAS-ATMRNAi on Chr III (VDRC stock #22502); Ptub-GAL80ts on Chr III (Bloomington stock center); Pvf1 enhancer trap, w1118 Mi{ET1}Pvf1MB01242 (Bloomington#23032); Pvf2 enhancer trap, w1118 Mi{ET1}Pvf2MB03230 (Bloomington#24055).
tie rescue construct
To clone a Tie cDNA, total RNA was isolated from whole Sevelin pupae 2-3 days post-white pupa stage, reverse transcribed (RT primer: ATGCGCTGCACGCCTAAATCA3), PCR-amplified with tie specific primers (amplification primers:5′ CGTGTGTGTATGTGTGTGTCG and
3′ GGGTAGGGGTTGGCTCAGTCA), and cloned into a pCR-XL-Topo vector (Invitrogen). Tie cDNA was sub-cloned into pUAST and injected into w1118 embryos to make transgenic stocks at a commercial facility (Best Gene, Inc.). The integrity of the cDNA was verified by DNA sequence analysis after each cloning steps. Eight independent lines on Chromosome II and III were obtained. The data shown is with one Ch II line.
Irradiation
Larvae in food were irradiated in a Faxitron Cabinet X-ray System Model RX-650 (Lincolnshire, IL) at 115 kv and 5.33 rad/sec. Irradiated larvae were incubated at 25°C for indicated amounts of time before dissection.
Antibody, TUNEL, X-gal and Acridine Orange staining
Cleaved Caspase 3 (1∶100, rabbit polyclonal, Cell Signaling Cat# 3661) was used as described before [29]. Phospho-Histone H3 (Upstate Biotech, 1∶1000) and Engrailed (1∶500, Developmental Hybridoma Bank Cat#4D9) were used as described before [54]. Secondary antibodies were used at 1∶500 (Jackson). For TUNEL, wing discs were dissected in PBS, fixed in 4% para-formaldehyde in PBS for 20 minutes, and washed three times in 0.3% Triton X-100/PBS for at least 20 minutes total. The discs were permeabilized overnight at 4°C in 0.3% Triton X-100/PBS, followed by three washes in 0.3% Triton X-100/PBS for at least 10 minutes total. The discs were processed using an Apoptag Red kit (Millipore), according to manufacturer's instructions. For X-gal staining, wing discs were extirpated in PBS and fixed in PBS +4% formaldehyde for 10 min. The discs were washed three times for 5 min each in PBT (PBS +0.2% Tween 20). The discs were stained overnight in 1 mg/ml X-gal in staining solution (50 ml contains: 0.684 ml 1 M Na2HPO4, 0.316 ml 1 M Na2PO4, 1.5 ml 5 M NaCl, 0.05 ml 1 M MgCl2, 0.065 g K4Fe(II)(CN)6, 0.051 g K3Fe(II)(CN)6, 0.15 ml Triton X 100, 47 ml H20). The discs were washed in PBT. The discs were counter-stained with 10 ug/ml Hoechst33258 in PBT or PBTx (0.1%TritonX-100) for 2 min, washed 3 times, and mounted on glass slides in Fluoromount G (SouthernBiotech).
Acridine Orange staining was as described before [29]. AO is excluded from live cells and has been shown to specifically stain apoptosis but not necrotic cells in Drosophila [55].
Image analysis
To quantify the GFP sensor, wing imaginal discs were extirpated in PBS, mounted between a glass slide and a glass coverslip, and imaged live. For mean GFP signal per disc, the images were acquired on a Leica DMR fluorescence microscope using Slidebook (Intelligence Imagine), and compiled in Photoshop. For relative GFP signal between different parts of the same disc, images were acquired using a Perkin Elmers spinning disc confocal on a Leica DMR microscope. 26–36 Z sections 1 mm apart were collected and collapsed by maximal projection using Image J (NIH opensource), and mean GFP signal for defined areas measured. Anterior signals were normalized by dividing with the posterior signals and averaged for each sample.
To quantify caspase and TUNEL, discs were imaged on a Perkin Elmers spinning disc confocal attached to a Leica DMR microscope. 26–36 Z-sections 1 mm apart were collected per disc and collapsed using ‘maximum projection’ in Image J. Collapsed images were corrected for background using the ‘threshold’ function in Image J. The mean fluorescence from the area of interest was measured and averaged for all discs in a sample. To quantify mitotic indices, cells showing phosphor-Histone H3 stain were manually counted.
Quantitative RT-PCR
Larvae were irradiated at 96±2 hr AEL. Between 15–20 wing discs were dissected per sample per time point and flash frozen in PBS using liquid nitrogen. Total RNA was isolated using Invitrogen TRIzol kit according to the manufacturer's instructions, and treated with DNase I (Amplification Grade, Invitrogen). RT reactions were performed with Superscript III (Invitrogen) according to the manufacturers instructions, using primers to amplify NT1, tie, CG11353 or a-tubulin mRNA as control. The primers used were:
tie:
5′ GGCGACGGGAAAGCCGAAA
3′ GGTGCGACGAGCAGCCAACA
NT1:
5′ GGCGGATGAGGGATTGCGCC
3′ TGCCAAACATCATGCGAACCTGT
CG11353:
5′ AGCGCGGCATACTCGGCAAA
3′ GGTCTTTGGACGCCGCGACA
a-Tub84B (a-tubulin):
5′TCCAATCGCAACAAAAAATTCA
3′ TCGTTTTCGTATGCTTTTCAGTGT
For mature bantam miRNA, custom primers were purchased and used according to manufacturer's instructions (TaqMan, MicroRNA Assay, Applied Biosystems).
Q-PCR was performed using 1 X SYBR Green Mix (Applied Biosystems) and 4 ng of each cDNA for 35 cycles using the indicated primers. Standard curves using 0.01-20 ng of cDNA pools were used. Plates were read in an Applied Biosystems 7900HT Real-time PCR instrument (Absolute Quantification Method). Values were normalized to those of a-tubulin.
Statistical analysis
p-values were calculated using 2-tailed Student's t-test except in the screen that used X2 tests.
Supporting Information
Activation of ban after induction of cell death in wing discs. (relates to Figure 1). (A, B) Wing discs were isolated 4 hours (h) after exposure to 0 R (-IR) or 4000 R (+IR) of X-rays, fixed and stained for cleaved Caspase 3. (C–F) GFP images of wing discs from larva carrying the ban sensor (C, D) or the control sensor that lacked ban binding sites (E, F). Wing discs were extirpated and imaged live from larvae 24 h after irradiation with 0 R (-IR) or 4000 R (+IR) of X-rays. (G) Mean GFP signal for each disc was quantified from images such as those in (C–F) and normalized to –IR values. ‘ctrl’ = control sensor. (H, I) Wing discs with clones expressing RFP (H) or RFP, Hid/Rpr, and caspase inhibitor p35 (I) were imaged live 48 h after heat-shock induction of GAL4. Larvae were heat-shocked at 37°C for 10 min at 2 days after egg collection. ‘RFP’ = hs-FLP/Y; GFP ban sensor/+;UAS-p35/Act>>GAL4, UAS-RFP. ‘RFP, Hid/Rpr,p35’ carried the same transgenes and had UAS-hid, UAS-rpr instead of Y.
(PDF)
Maturation of the ptc domain and comparison of cell killing due to E2F1 RNAi and ATM RNAi. (relates to Figures 2 and 3). (A–C) Wing discs from larvae carrying one copy each of ptc-GAL4, UAS-dsRNA against dE2F1 and UAS-GFP, at 72 h (A) or 96 h (B, C) after egg deposition (AED) at 25°C. The ptc strip did not span the wing pouch at 72 h AED and narrowed further by 96 h AED. The larva in (C) carried a copy of tub-GAL80ts that repressed GAL4 and GFP expression at this temperature. Scale bar in (C) applies to (A–C). (D–F) Wing discs were extirpated from third instar larvae and stained with the vital dye acridine orange. (D) A wing disc from a ptc4>dE2F1RNAi larvae raised at 25°C before shifting to 29°C for 24 h. Robust cell death was apparent at this time after temperature shift. (E, F) Wing disc from larvae expressing dsRNA against ATM under the control of ptc-GAL4. Larvae were raised at 25°C before shifting to 29°C for 24 h (e) or 48 h (D). Cell death was induced only after longer temperature shift and, even then, was not as robust as in ptc4>dE2F1RNAi discs. Scale bar in (F) applies to (D–F).
(PDF)
Mitotic Indices in anterior and posterior compartments are similar. (relates to Figure 3). Wing imaginal discs were fixed and stained for DNA (A, B) and for phosphorylated histone H3 (pH3) as a mitotic marker (C, D). DNA stain was used as a guide to circle the pouch and to mark the Anterior/Posterior boundary. Mitotic index was computed by manually counting pH3-positive cells and normalizing by the area measured using Image J. Mitotic index of the Anterior was divided by the mitotic index of the Posterior compartment for each disc and shown in the graph in (E). N = 8 in two experiments for CyO discs. The averages are indicated with horizontal bars for each sample. The numbers are not significantly different from each other (p = 0.37).
(PDF)
Expression of Dpp-lacZ and DIAP1-lacZ reporters in discs with cell death (relates to Figure 3). Wing discs were extirpated from feeding third instar larvae and stained to detect β-galactocidase. Larvae were maintained at 25°C for 4 days and shifted to 29°C for 24 h before dissection. Larvae carried either the CyO balancer (A and C) or transgenes for ptc-GAL4 and UAS-dsRNA against dE2F1 (B and D). The larvae also carried a Dpp-lacZ reporter (A and B) or a DIAP1-lacZ reporter (C and D). The stripe of Dpp-lacZ expression remained even in discs in which some cells had been killed in the ptc domain (B), and looked similar to Dpp-lacZ expression in CyO controls (A). ptc4>dE2F1RNAi induced the expression of DIAP1 (D) compared to CyO controls (C). Note that induction of DIAP1 was confined to within or proximity of the ptc domain and did not spread to the entire anterior compartment of the pouch.
(PDF)
A screen for modifiers of radiation sensitivity of ban mutants (relates to Figure 7). (A) A screen for modifiers of ban was designed to identify deficiencies that dominantly modulated the radiation sensitivity of ban mutants. Larvae were exposed to 4000 R of X-rays at 96±4 h after egg deposition. Percent eclosion was determined by counting full and empty pupal cases 10 days after irradiation. To calculate the expected survival from additive effects of ban and the deficiency (Df), banL1170/TM6 Tb or banEP3622/TM6 Tb virgin females were crossed to +/+ males and Df/TM6 Tb males were crossed to +/+ virgin females. Percent eclosions for Tb+ pupae (ban/+ and Df/+) were multiplied with each other to get the expected survival. For example, if % eclosion for ban/+ and Df/+ were 60% and 60%, the expected survival would be 0.6×0.6 or 36%. To measure the observed survival, ban/TM6 Tb virgin females were crossed to Df/TM6 Tb males. X2 analysis (1 df) was used to calculate the significance of the difference between observed and expected survival, using a cut of value of p<0.001. (B) tie locus on 3R is shown to scale. Predicted and known genes are blue bars. NT1 encodes Neurotrophin 1, which has a role in axonal activity. tie transcript is in orange; boxes are exons and lines are introns. Transposon insertion sites for three tie alleles used in this study are indicated with red triangles. tiee03394 and tiee02680 are p-element insertions into the second intron and tieCO98 is a p-element insertion into the predicted 3′UTR. The deficiency used in this study removes the region shown as a red bar. All information are from Flybase (FB2012_02, released 03/02/12). (C) mRNA expression levels of tie and two flanking genes, CG11353 and NT1, in wild type and tie mutants were quantified by q-RT-PCR. The average and standard error from three independent experiments are shown. The data have been normalized to expression in wild type (w1118 or ‘w’). tie alleles were Df(3L)Exel2098 and tiee03394.
(PDF)
tie mutants can induce cell death after irradiation (relates to Figure 8). (A–C) Wing imaginal discs were extirpated from third instar larvae at 4 or 20 h after exposure to 4000R of X-rays, fixed and stained with an antibody against cleaved (active) Caspase 3 and for DNA. To image discs, Z-sections were acquired on a spinning disc confocal microscope, collapsed and shown. Representative images from the 4 h time point are shown. (D–E) Mean fluorescence for each disc was measured using Image J software and normalized to the average mean fluorescence of w1118 control discs. ‘Df’ = homozygotes of Df(3L)Exel2098; ‘e03394’ = tiee03394 homozygotes. Error bar = ±1SEM. N = 15–23 discs per genotype for each time point in two independent experiments. (F–I) Homozygous mutant clones of tie Df(3L)Exel2098 (no GFP) showed robust caspase activity 4 h and 24 h after exposure to 4000R of X-rays. Larvae were irradiated 48 h after heat shock to induce FLP recombinase. Homozygous mutant clones lack GFP, heterozygotes have one copy of GFP and homozygous wild type sister clones have two copies of GFP.
(PDF)
Changes in the GFP ban sensor is sensitive to tie gene dosage (relates to Table 1). To image discs, Z-sections were acquired on a spinning disc confocal microscope, collapsed and shown. Brackets indicate bright GFP along the D/V boundary. (A) A control disc from a larva carrying the GFP ban sensor that had been subjected to the experimental protocol in (C). GFP from the ban sensor was equivalent in the A (black arrowheads) and the P (white arrowheads) compartments of the wing pouch. (B) In tie Df/+ discs with cell death due to ptc4>dE2F1RNAi, the reduction of GFP in the A compartment was not obvious (A/P ratio quantified in Table 1). A few speckles of dying cells with bright GFP were visible in the ptc domain (* in B).
(PDF)
The expression of Pvf1 and Pvf2 enhancer traps is induced by ptc4>dE2F1RNAi (relates to Figure 5). Wing imaginal discs were extirpated from third instar larvae 24 h after a temperature shift to 29°C, and imaged live for GFP. The larvae carried GFP enhancer traps for Pvf1 (A, B) and Pvf2 (C, D). The larvae were heterozygous for the CyO-RFP balancer (A, C) or ptc4>dE2F1RNAi (B, D). Induction of GFP from the enhancer traps was observed in the ptc domain where the dying cells are. N = 10 for each genotype/panel in two independent experiments.
(PDF)
Yki heterozygotes still show protection from IR-induced cell death in the ptc4>dE2F1RNAi background (relates to Figures 3 & 4). Wing imaginal discs were extirpated from third instar larvae 4 h after exposure to 4000R of X-rays, fixed and stained for DNA and with an antibody against cleaved Caspase 3. The larvae are heterozygous for ptc4>dE2F1RNAi and ykiB5. DNA stained images are used to discern the location of the pouch (within the dashed line), anterior/posterior boundary (solid vertical line) and the ptc domain (dotted vertical line). The horizontal stripe of cell death along the dorsal/ventral boundary is also seen here. Importantly, caspase-active cells are fewer in the anterior half than in the posterior half.
(PDF)
Cell death in the ptc domain protects against maytansinol. Larvae were generated to express ptc-GAL4>UAS-Rpr as shown in Figure 3K. Immediately after de-repressing GAL4 for 12 h, maytansinol (NSC292222, Developmental Therapeutics Program, NCI) in DMSO was added to the food to a final concentration of 2 μM (and 0.1% DMSO). Wing discs were extirpated 24 hours after drug addition, fixed and stained for DNA and active Caspse 3. (A, B) male siblings that lack the UAS-rpr transgene served as controls. Cell death in response to maytansinol was equivalent in the A and P compartments. (C, D) female experimental larvae showed cell death in the ptc domain (between vertical lines). Caspase activity in the A compartment was lower than in the P compartment. We note that the depression in the DNA stain in the ptc domain was not visible in these discs. Maytansinol depolymerizes microtubule, suggesting that microtubules are necessary for the protrusion of dead cells.
(PDF)
Changes in mRNA levels at 2 hours after IR. The data for pvf1, pvf2 and CG10359 are from published supplemental microarray data in Ref#30. The data for tie and pvf3 are from an RNAseq dataset (our unpublished data).
(PDF)
Acknowledgments
Drosophila stocks were obtained from the laboratories of Jun-yuan Ji, Nicholas Dyson, Georg Halder, Kristin Scott, Denise Montell and Steve Cohen, and from Bloomington, Harvard, and Vienna stock centers.
Funding Statement
This work was supported by an NIH grant to TTS (RO1 GM87276). AB was supported in part by a NIH pre-doctoral training grant. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sun G, Irvine KD (2011) Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Developmental biology 350: 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ryoo HD, Gorenc T, Steller H (2004) Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Developmental cell 7: 491–501. [DOI] [PubMed] [Google Scholar]
- 3. Grusche FA, Degoutin JL, Richardson HE, Harvey KF (2011) The Salvador/Warts/Hippo pathway controls regenerative tissue growth in Drosophila melanogaster. Developmental biology 350: 255–266. [DOI] [PubMed] [Google Scholar]
- 4. Thompson BJ, Cohen SM (2006) The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell 126: 767–774. [DOI] [PubMed] [Google Scholar]
- 5. Hipfner DR, Weigmann K, Cohen SM (2002) The bantam gene regulates Drosophila growth. Genetics 161: 1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM (2003) bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113: 25–36. [DOI] [PubMed] [Google Scholar]
- 7. Boulan L, Martin D, Milan M (2013) bantam miRNA promotes systemic growth by connecting insulin signaling and ecdysone production. Current biology : CB 23: 473–478. [DOI] [PubMed] [Google Scholar]
- 8. Kadener S, Menet JS, Sugino K, Horwich MD, Weissbein U, et al. (2009) A role for microRNAs in the Drosophila circadian clock. Genes & development 23: 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Parrish JZ, Xu P, Kim CC, Jan LY, Jan YN (2009) The microRNA bantam functions in epithelial cells to regulate scaling growth of dendrite arbors in drosophila sensory neurons. Neuron 63: 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shcherbata HR, Ward EJ, Fischer KA, Yu JY, Reynolds SH, et al. (2007) Stage-specific differences in the requirements for germline stem cell maintenance in the Drosophila ovary. Cell stem cell 1: 698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang Y, Xu S, Xia L, Wang J, Wen S, et al. (2009) The bantam microRNA is associated with drosophila fragile X mental retardation protein and regulates the fate of germline stem cells. PLoS genetics 5: e1000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peng HW, Slattery M, Mann RS (2009) Transcription factor choice in the Hippo signaling pathway: homothorax and yorkie regulation of the microRNA bantam in the progenitor domain of the Drosophila eye imaginal disc. Genes & development 23: 2307–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oh H, Irvine KD (2011) Cooperative regulation of growth by Yorkie and Mad through bantam. Developmental cell 20: 109–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Herranz H, Perez L, Martin FA, Milan M (2008) A Wingless and Notch double-repression mechanism regulates G1-S transition in the Drosophila wing. The EMBO journal 27: 1633–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Becam I, Rafel N, Hong X, Cohen SM, Milan M (2011) Notch-mediated repression of bantam miRNA contributes to boundary formation in the Drosophila wing. Development 138: 3781–3789. [DOI] [PubMed] [Google Scholar]
- 16. Li F, Huang Q, Chen J, Peng Y, Roop DR, et al. (2010) Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Science signaling 3: ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang Q, Li F, Liu X, Li W, Shi W, et al. (2011) Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nature medicine 17: 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mothersill C, Seymour C (2006) Radiation-induced bystander effects: evidence for an adaptive response to low dose exposures? Dose-response : a publication of International Hormesis Society 4: 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mothersill C, Seymour CB (2006) Radiation-induced bystander effects and the DNA paradigm: an “out of field” perspective. Mutation research 597: 5–10. [DOI] [PubMed] [Google Scholar]
- 20. Singh H, Saroya R, Smith R, Mantha R, Guindon L, et al. (2011) Radiation induced bystander effects in mice given low doses of radiation in vivo. Dose-response : a publication of International Hormesis Society 9: 225–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mothersill C, Stamato TD, Perez ML, Cummins R, Mooney R, et al. (2000) Involvement of energy metabolism in the production of 'bystander effects' by radiation. British journal of cancer 82: 1740–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jaklevic B, Uyetake L, Wichmann A, Bilak A, English CN, et al. (2008) Modulation of ionizing radiation-induced apoptosis by bantam microRNA in Drosophila. Developmental biology 320: 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang X, Bo J, Bridges T, Dugan KD, Pan TC, et al. (2006) Analysis of cell migration using whole-genome expression profiling of migratory cells in the Drosophila ovary. Developmental cell 10: 483–495. [DOI] [PubMed] [Google Scholar]
- 24. Corish P, Tyler-Smith C (1999) Attenuation of green fluorescent protein half-life in mammalian cells. Protein engineering 12: 1035–1040. [DOI] [PubMed] [Google Scholar]
- 25. Morris EJ, Ji JY, Yang F, Di Stefano L, Herr A, et al. (2008) E2F1 represses beta-catenin transcription and is antagonized by both pRB and CDK8. Nature 455: 552–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neufeld TP, de la Cruz AF, Johnston LA, Edgar BA (1998) Coordination of growth and cell division in the Drosophila wing. Cell 93: 1183–1193. [DOI] [PubMed] [Google Scholar]
- 27. Hamaratoglu F, de Lachapelle AM, Pyrowolakis G, Bergmann S, Affolter M (2011) Dpp signaling activity requires Pentagone to scale with tissue size in the growing Drosophila wing imaginal disc. PLoS biology 9: e1001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brodsky MH, Weinert BT, Tsang G, Rong YS, McGinnis NM, et al. (2004) Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol 24: 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wichmann A, Jaklevic B, Su TT (2006) Ionizing radiation induces caspase-dependent but Chk2- and p53-independent cell death in Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United States of America 103: 9952–9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oikemus SR, McGinnis N, Queiroz-Machado J, Tukachinsky H, Takada S, et al. (2004) Drosophila atm/telomere fusion is required for telomeric localization of HP1 and telomere position effect. Genes & development 18: 1850–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dormer A, Beck G (2005) Evolutionary analysis of human vascular endothelial growth factor, angiopoietin, and tyrosine endothelial kinase involved in angiogenesis and immunity. In silico biology 5: 323–339. [PubMed] [Google Scholar]
- 32. van Bergeijk P, Heimiller J, Uyetake L, Su TT (2012) Genome-Wide Expression Analysis Identifies a Modulator of Ionizing Radiation-Induced p53-Independent Apoptosis in Drosophila melanogaster. PLoS ONE 7: e36539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P (2001) Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell 107: 17–26. [DOI] [PubMed] [Google Scholar]
- 34. Perez-Garijo A, Fuchs Y, Steller H (2013) Apoptotic cells can induce non-autonomous apoptosis through the TNF pathway. eLife 2: e01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herrera SC, Martin R, Morata G (2013) Tissue homeostasis in the wing disc of Drosophila melanogaster: immediate response to massive damage during development. PLoS genetics 9: e1003446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kokai E, Paldy FS, Somogyi K, Chougule A, Pal M, et al. (2012) CalpB modulates border cell migration in Drosophila egg chambers. BMC developmental biology 12: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huh JR, Guo M, Hay BA (2004) Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Current biology : CB 14: 1262–1266. [DOI] [PubMed] [Google Scholar]
- 38. Kondo S, Senoo-Matsuda N, Hiromi Y, Miura M (2006) DRONC coordinates cell death and compensatory proliferation. Molecular and cellular biology 26: 7258–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perez-Garijo A, Martin FA, Morata G (2004) Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 131: 5591–5598. [DOI] [PubMed] [Google Scholar]
- 40. Perez-Garijo A, Shlevkov E, Morata G (2009) The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development 136: 1169–1177. [DOI] [PubMed] [Google Scholar]
- 41. Wells BS, Yoshida E, Johnston LA (2006) Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol 16: 1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fan Y, Bergmann A (2008) Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Developmental Cell 14: 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bruckner K, Kockel L, Duchek P, Luque CM, Rorth P, et al. (2004) The PDGF/VEGF receptor controls blood cell survival in Drosophila. Developmental cell 7: 73–84. [DOI] [PubMed] [Google Scholar]
- 44. Ohsawa S, Sugimura K, Takino K, Xu T, Miyawaki A, et al. (2011) Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Developmental cell 20: 315–328. [DOI] [PubMed] [Google Scholar]
- 45. Edwards A, Gladstone M, Yoon P, Raben D, Frederick B, et al. (2011) Combinatorial effect of maytansinol and radiation in Drosophila and human cancer cells. Disease models & mechanisms 4: 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, et al. (1999) Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284: 1994–1998. [DOI] [PubMed] [Google Scholar]
- 47. Kwak HJ, So JN, Lee SJ, Kim I, Koh GY (1999) Angiopoietin-1 is an apoptosis survival factor for endothelial cells. FEBS letters 448: 249–253. [DOI] [PubMed] [Google Scholar]
- 48. Papapetropoulos A, Garcia-Cardena G, Dengler TJ, Maisonpierre PC, Yancopoulos GD, et al. (1999) Direct actions of angiopoietin-1 on human endothelium: evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors. Laboratory investigation; a journal of technical methods and pathology 79: 213–223. [PubMed] [Google Scholar]
- 49. Kim I, Kim HG, So JN, Kim JH, Kwak HJ, et al. (2000) Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-Kinase/Akt signal transduction pathway. Circulation research 86: 24–29. [DOI] [PubMed] [Google Scholar]
- 50. Cho CH, Kammerer RA, Lee HJ, Yasunaga K, Kim KT, et al. (2004) Designed angiopoietin-1 variant, COMP-Ang1, protects against radiation-induced endothelial cell apoptosis. Proceedings of the National Academy of Sciences of the United States of America 101: 5553–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou L, Schnitzler A, Agapite J, Schwartz LM, Steller H, et al. (1997) Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proceedings of the National Academy of Sciences of the United States of America 94: 5131–5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hay BA, Wassarman DA, Rubin GM (1995) Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell 83: 1253–1262. [DOI] [PubMed] [Google Scholar]
- 53. Hamaratoglu F, Gajewski K, Sansores-Garcia L, Morrison C, Tao C, et al. (2009) The Hippo tumor-suppressor pathway regulates apical-domain size in parallel to tissue growth. Journal of cell science 122: 2351–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wichmann A, Uyetake L, Su TT (2010) E2F1 and E2F2 have opposite effects on radiation-induced p53-independent apoptosis in Drosophila. Developmental biology 346: 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Abrams JM, White K, Fessler LI, Steller H (1993) Programmed cell death during Drosophila embryogenesis. Development 117: 29–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Activation of ban after induction of cell death in wing discs. (relates to Figure 1). (A, B) Wing discs were isolated 4 hours (h) after exposure to 0 R (-IR) or 4000 R (+IR) of X-rays, fixed and stained for cleaved Caspase 3. (C–F) GFP images of wing discs from larva carrying the ban sensor (C, D) or the control sensor that lacked ban binding sites (E, F). Wing discs were extirpated and imaged live from larvae 24 h after irradiation with 0 R (-IR) or 4000 R (+IR) of X-rays. (G) Mean GFP signal for each disc was quantified from images such as those in (C–F) and normalized to –IR values. ‘ctrl’ = control sensor. (H, I) Wing discs with clones expressing RFP (H) or RFP, Hid/Rpr, and caspase inhibitor p35 (I) were imaged live 48 h after heat-shock induction of GAL4. Larvae were heat-shocked at 37°C for 10 min at 2 days after egg collection. ‘RFP’ = hs-FLP/Y; GFP ban sensor/+;UAS-p35/Act>>GAL4, UAS-RFP. ‘RFP, Hid/Rpr,p35’ carried the same transgenes and had UAS-hid, UAS-rpr instead of Y.
(PDF)
Maturation of the ptc domain and comparison of cell killing due to E2F1 RNAi and ATM RNAi. (relates to Figures 2 and 3). (A–C) Wing discs from larvae carrying one copy each of ptc-GAL4, UAS-dsRNA against dE2F1 and UAS-GFP, at 72 h (A) or 96 h (B, C) after egg deposition (AED) at 25°C. The ptc strip did not span the wing pouch at 72 h AED and narrowed further by 96 h AED. The larva in (C) carried a copy of tub-GAL80ts that repressed GAL4 and GFP expression at this temperature. Scale bar in (C) applies to (A–C). (D–F) Wing discs were extirpated from third instar larvae and stained with the vital dye acridine orange. (D) A wing disc from a ptc4>dE2F1RNAi larvae raised at 25°C before shifting to 29°C for 24 h. Robust cell death was apparent at this time after temperature shift. (E, F) Wing disc from larvae expressing dsRNA against ATM under the control of ptc-GAL4. Larvae were raised at 25°C before shifting to 29°C for 24 h (e) or 48 h (D). Cell death was induced only after longer temperature shift and, even then, was not as robust as in ptc4>dE2F1RNAi discs. Scale bar in (F) applies to (D–F).
(PDF)
Mitotic Indices in anterior and posterior compartments are similar. (relates to Figure 3). Wing imaginal discs were fixed and stained for DNA (A, B) and for phosphorylated histone H3 (pH3) as a mitotic marker (C, D). DNA stain was used as a guide to circle the pouch and to mark the Anterior/Posterior boundary. Mitotic index was computed by manually counting pH3-positive cells and normalizing by the area measured using Image J. Mitotic index of the Anterior was divided by the mitotic index of the Posterior compartment for each disc and shown in the graph in (E). N = 8 in two experiments for CyO discs. The averages are indicated with horizontal bars for each sample. The numbers are not significantly different from each other (p = 0.37).
(PDF)
Expression of Dpp-lacZ and DIAP1-lacZ reporters in discs with cell death (relates to Figure 3). Wing discs were extirpated from feeding third instar larvae and stained to detect β-galactocidase. Larvae were maintained at 25°C for 4 days and shifted to 29°C for 24 h before dissection. Larvae carried either the CyO balancer (A and C) or transgenes for ptc-GAL4 and UAS-dsRNA against dE2F1 (B and D). The larvae also carried a Dpp-lacZ reporter (A and B) or a DIAP1-lacZ reporter (C and D). The stripe of Dpp-lacZ expression remained even in discs in which some cells had been killed in the ptc domain (B), and looked similar to Dpp-lacZ expression in CyO controls (A). ptc4>dE2F1RNAi induced the expression of DIAP1 (D) compared to CyO controls (C). Note that induction of DIAP1 was confined to within or proximity of the ptc domain and did not spread to the entire anterior compartment of the pouch.
(PDF)
A screen for modifiers of radiation sensitivity of ban mutants (relates to Figure 7). (A) A screen for modifiers of ban was designed to identify deficiencies that dominantly modulated the radiation sensitivity of ban mutants. Larvae were exposed to 4000 R of X-rays at 96±4 h after egg deposition. Percent eclosion was determined by counting full and empty pupal cases 10 days after irradiation. To calculate the expected survival from additive effects of ban and the deficiency (Df), banL1170/TM6 Tb or banEP3622/TM6 Tb virgin females were crossed to +/+ males and Df/TM6 Tb males were crossed to +/+ virgin females. Percent eclosions for Tb+ pupae (ban/+ and Df/+) were multiplied with each other to get the expected survival. For example, if % eclosion for ban/+ and Df/+ were 60% and 60%, the expected survival would be 0.6×0.6 or 36%. To measure the observed survival, ban/TM6 Tb virgin females were crossed to Df/TM6 Tb males. X2 analysis (1 df) was used to calculate the significance of the difference between observed and expected survival, using a cut of value of p<0.001. (B) tie locus on 3R is shown to scale. Predicted and known genes are blue bars. NT1 encodes Neurotrophin 1, which has a role in axonal activity. tie transcript is in orange; boxes are exons and lines are introns. Transposon insertion sites for three tie alleles used in this study are indicated with red triangles. tiee03394 and tiee02680 are p-element insertions into the second intron and tieCO98 is a p-element insertion into the predicted 3′UTR. The deficiency used in this study removes the region shown as a red bar. All information are from Flybase (FB2012_02, released 03/02/12). (C) mRNA expression levels of tie and two flanking genes, CG11353 and NT1, in wild type and tie mutants were quantified by q-RT-PCR. The average and standard error from three independent experiments are shown. The data have been normalized to expression in wild type (w1118 or ‘w’). tie alleles were Df(3L)Exel2098 and tiee03394.
(PDF)
tie mutants can induce cell death after irradiation (relates to Figure 8). (A–C) Wing imaginal discs were extirpated from third instar larvae at 4 or 20 h after exposure to 4000R of X-rays, fixed and stained with an antibody against cleaved (active) Caspase 3 and for DNA. To image discs, Z-sections were acquired on a spinning disc confocal microscope, collapsed and shown. Representative images from the 4 h time point are shown. (D–E) Mean fluorescence for each disc was measured using Image J software and normalized to the average mean fluorescence of w1118 control discs. ‘Df’ = homozygotes of Df(3L)Exel2098; ‘e03394’ = tiee03394 homozygotes. Error bar = ±1SEM. N = 15–23 discs per genotype for each time point in two independent experiments. (F–I) Homozygous mutant clones of tie Df(3L)Exel2098 (no GFP) showed robust caspase activity 4 h and 24 h after exposure to 4000R of X-rays. Larvae were irradiated 48 h after heat shock to induce FLP recombinase. Homozygous mutant clones lack GFP, heterozygotes have one copy of GFP and homozygous wild type sister clones have two copies of GFP.
(PDF)
Changes in the GFP ban sensor is sensitive to tie gene dosage (relates to Table 1). To image discs, Z-sections were acquired on a spinning disc confocal microscope, collapsed and shown. Brackets indicate bright GFP along the D/V boundary. (A) A control disc from a larva carrying the GFP ban sensor that had been subjected to the experimental protocol in (C). GFP from the ban sensor was equivalent in the A (black arrowheads) and the P (white arrowheads) compartments of the wing pouch. (B) In tie Df/+ discs with cell death due to ptc4>dE2F1RNAi, the reduction of GFP in the A compartment was not obvious (A/P ratio quantified in Table 1). A few speckles of dying cells with bright GFP were visible in the ptc domain (* in B).
(PDF)
The expression of Pvf1 and Pvf2 enhancer traps is induced by ptc4>dE2F1RNAi (relates to Figure 5). Wing imaginal discs were extirpated from third instar larvae 24 h after a temperature shift to 29°C, and imaged live for GFP. The larvae carried GFP enhancer traps for Pvf1 (A, B) and Pvf2 (C, D). The larvae were heterozygous for the CyO-RFP balancer (A, C) or ptc4>dE2F1RNAi (B, D). Induction of GFP from the enhancer traps was observed in the ptc domain where the dying cells are. N = 10 for each genotype/panel in two independent experiments.
(PDF)
Yki heterozygotes still show protection from IR-induced cell death in the ptc4>dE2F1RNAi background (relates to Figures 3 & 4). Wing imaginal discs were extirpated from third instar larvae 4 h after exposure to 4000R of X-rays, fixed and stained for DNA and with an antibody against cleaved Caspase 3. The larvae are heterozygous for ptc4>dE2F1RNAi and ykiB5. DNA stained images are used to discern the location of the pouch (within the dashed line), anterior/posterior boundary (solid vertical line) and the ptc domain (dotted vertical line). The horizontal stripe of cell death along the dorsal/ventral boundary is also seen here. Importantly, caspase-active cells are fewer in the anterior half than in the posterior half.
(PDF)
Cell death in the ptc domain protects against maytansinol. Larvae were generated to express ptc-GAL4>UAS-Rpr as shown in Figure 3K. Immediately after de-repressing GAL4 for 12 h, maytansinol (NSC292222, Developmental Therapeutics Program, NCI) in DMSO was added to the food to a final concentration of 2 μM (and 0.1% DMSO). Wing discs were extirpated 24 hours after drug addition, fixed and stained for DNA and active Caspse 3. (A, B) male siblings that lack the UAS-rpr transgene served as controls. Cell death in response to maytansinol was equivalent in the A and P compartments. (C, D) female experimental larvae showed cell death in the ptc domain (between vertical lines). Caspase activity in the A compartment was lower than in the P compartment. We note that the depression in the DNA stain in the ptc domain was not visible in these discs. Maytansinol depolymerizes microtubule, suggesting that microtubules are necessary for the protrusion of dead cells.
(PDF)
Changes in mRNA levels at 2 hours after IR. The data for pvf1, pvf2 and CG10359 are from published supplemental microarray data in Ref#30. The data for tie and pvf3 are from an RNAseq dataset (our unpublished data).
(PDF)