

The unfolded protein response PERK branch induces recruitment of misfolded proteins and the ubiquitin ligase HRD1 to the ER-derived quality control compartment (ERQC), a staging ground for ER-associated degradation (ERAD). This is accomplished by up-regulation of homocysteine-induced ER protein (Herp), which recruits the ERAD complex at the ERQC.
Abstract
A functional unfolded protein response (UPR) is essential for endoplasmic reticulum (ER)-associated degradation (ERAD) of misfolded secretory proteins, reflecting the fact that some level of UPR activation must exist under normal physiological conditions. A coordinator of the UPR and ERAD processes has long been sought. We previously showed that the PKR-like, ER-localized eukaryotic translation initiation factor 2α kinase branch of the UPR is required for the recruitment of misfolded proteins and the ubiquitin ligase HRD1 to the ER-derived quality control compartment (ERQC), a staging ground for ERAD. Here we show that homocysteine-induced ER protein (Herp), a protein highly upregulated by this UPR branch, is responsible for this compartmentalization. Herp localizes to the ERQC, and our results suggest that it recruits HRD1, which targets to ERAD the substrate presented by the OS-9 lectin at the ERQC. Predicted overall structural similarity of Herp to the ubiquitin-proteasome shuttle hHR23, but including a transmembrane hairpin, suggests that Herp may function as a hub for membrane association of ERAD machinery components, a key organizer of the ERAD complex.
INTRODUCTION
Despite numerous studies, the process of recruitment and delivery of misfolded secretory proteins for degradation at the cytosolic proteasomes is unclear. In previous articles we described how endoplasmic reticulum (ER)–associated degradation (ERAD) substrates accumulate in a specialized pericentriolar compartment, the ER-derived quality control compartment (ERQC), before delivery to the proteasomes (Kamhi-Nesher et al., 2001; Frenkel et al., 2004; Kondratyev et al., 2007). On accumulation of misfolded proteins, the lectin chaperones calnexin and calreticulin also concentrate in the ERQC, whereas other ER chaperones, such as heavy-chain-binding protein (BiP), protein disulfide isomerase, UDPGlc:glycoprotein glucosyltransferase, and the oxidoreductase ERp57, remain distributed throughout the ER (Kamhi-Nesher et al., 2001; Frenkel et al., 2004). ERAD machinery components (e.g., HRD1, p97, Derlin-1) are recruited to this compartment, making it a likely site for retrotranslocation (Kondratyev et al., 2007; Groisman et al., 2011). Of interest, the unfolded protein response (UPR) sensors PKR-like, ER-localized eukaryotic translation initiation factor 2α (eIF2α) kinase (PERK) and IRE1 also accumulate in the ERQC (Kondratyev et al., 2007) and are thus likely activated by physical segregation from BiP (Bertolotti et al., 2000; Walter and Ron, 2011). Paradoxically, PERK phosphorylation of eIF2α is needed for ERAD substrate accumulation at the ERQC (Kondratyev et al., 2007). Thus, in a positive feedback loop, eIF2α phosphorylation causes pericentriolar concentration at the ERQC, which in turn might amplify the UPR. Here we investigate which factors downstream of phosphorylated eIF2α are responsible for the compartmentalization. Phosphorylation of eIF2α causes general transient arrest in protein synthesis (Prostko et al., 1993; Harding et al., 1999; Walter and Ron, 2011) but also translational induction of the activating transcription factor 4 (ATF4; Harding et al., 2000) and possibly of other proteins. ATF4 transactivates downstream genes—C/EBP-homologous protein (CHOP; Ma et al., 2002) and homocysteine-induced ER protein (Herp; Kokame et al., 2000; Ma and Hendershot, 2004; Schulze et al., 2005). Herp, an ER membrane-bound protein, is one of the most highly up-regulated proteins in response to ER stress (Kokame et al., 2000; van Laar et al., 2000) and is coregulated by the PERK and the ATF6 branches of the UPR (Ma and Hendershot, 2004; Yamamoto et al., 2004). It plays a protective role in ER stress–induced apoptosis (Chan et al., 2004; Tuvia et al., 2007). Herp interacts with HRD1 and other ERAD components (Schulze et al., 2005; Okuda-Shimizu and Hendershot, 2007; Kny et al., 2011), ubiquitinated proteins, and proteasomes (Okuda-Shimizu and Hendershot, 2007). USA1, which binds HRD1 and Der1 and triggers oligomerization of HRD1 in Saccharomyces cerevisiae, is a suggested functional homologue of Herp, although it shares little homology (Horn et al., 2009; Carvalho et al., 2010). Here we show that, in mammalian cells, Herp is responsible for initiating ERAD by recruiting HRD1 and ERAD substrates at the ERQC.
RESULTS
Phosphorylated eIF2α and its downstream effector, Herp, are required for accumulation of an ERAD substrate in the ERQC
The ERQC is characterized by harboring accumulated ERAD substrates, for example, when their proteasomal degradation is inhibited (Figure 1, A, B, and D). The juxtanuclear accumulation is microtubule dependent (Supplemental Figure S1), close to but not colocalizing with the Golgi complex, and the chaperone BiP is mostly depleted from the ERQC (Figure 1, B and D; Kamhi-Nesher et al., 2001; Kondratyev et al., 2007). Therefore, juxtanuclear accumulation of a well-studied ERAD substrate, uncleaved precursor of asialoglycoprotein receptor H2a (Kondratyev et al., 2007; Avezov et al., 2008; Groisman et al., 2011), linked to monomeric red fluorescent protein (H2a-RFP), can serve as a marker of the ERQC. The accumulation can appear in a tight juxtanuclear pattern (Figure 1B) or relatively less restricted (Supplemental Figure S2A). We previously demonstrated that phosphorylation of eIF2α was sufficient for misfolded protein concentration in the ERQC; expression of a mutant that mimics constitutive phosphorylation, eIF2αS51D, had a similar effect (Kondratyev et al., 2007). Selective inhibition of phospho-eIF2α dephosphorylation with salubrinal (Sal; Boyce et al., 2005) showed a similar outcome (Figure 1, C and D). To test whether this effect was due to arrest in protein synthesis caused by phosphorylation of eIF2α (Harding et al., 2000), we incubated cells with a protein synthesis inhibitor, puromycin (Puro). This treatment did not cause accumulation of H2a-RFP in the ERQC but instead led to the appearance of widely distributed puncta (Figure 1E). H2a-RFP accumulation upon inhibition of its degradation by the proteasomal inhibitor lactacystin (Lac) was mostly unaffected by this relatively short incubation with Puro (Figure 1E, right).
FIGURE 1:

Inhibition of eIF2α-P dephosphorylation causes ERAD substrate targeting to the ERQC. (A–C) Juxtanuclear accumulation (typical of the ERQC) of the ERAD substrate H2a-RFP and not of BiP upon inhibition of proteasomes or eIF2α dephosphorylation. NIH 3T3 cells expressing H2a-RFP and the Golgi marker GalT-YFP and endogenous BiP stained with rabbit anti-BiP and Cy5-conjugated goat anti-rabbit IgG. At 24 h posttransfection the cells were treated with Lac (B, 17 μM) or Sal (C, 50 μM) for 4 h or left untreated (A). Representative images. Bar, 10 μm. (D) Top graph, percentage of cells with H2a-RFP accumulated in the juxtanuclear region (ERQC) as estimated by visual observation compared with those showing a disperse ER pattern in experiments similar to those in A–C. Graph bars represent averages of three independent experiments, with 130–200 cells counted in each; error bars, SE between experiments. *p (treated vs. untreated cells) < 0.008. A similar graph (bottom) of BiP distribution in the same cells shows no significant difference between treated and untreated samples. (E) Protein synthesis inhibition does not cause ERQC concentration of H2a-RFP. Similar observation of H2a-RFP as in A–C, except that cells were incubated with Puro (50 μg/ml) or both Puro and Lac for 3 h. (F) PERK pathway of the UPR. Accumulation of misfolded secretory proteins triggers phosphorylation and activation of PERK, which phosphorylates eIF2α, which inhibits general protein synthesis but activates preferential translation of ATF4 and possibly other proteins. Up-regulated ATF4 activates transcription of Herp, implicated in ERAD and CHOP proapoptotic, among other genes. Indicated are the activities of the phospho-eIF2α dephosphorylation inhibitor Sal, the protein synthesis inhibitor Puro, and the proteasome inhibitor Lac.
Phosphorylation of eIF2α also enhances ATF4 translation, regulating, among others, Herp and CHOP transcription (Figure 1F). To assess Herp requirement, we used a stable NIH 3T3 cell line, described before, with inducible expression of anti-Herp short hairpin RNA (shRNA; Okuda-Shimizu and Hendershot, 2007). The efficiency of the knockdown using this system was ∼50% (Supplemental Figure S2C). Under the conditions used, it did not cause any changes in cell morphology or viability. On Herp knockdown, H2a-RFP accumulation in the ERQC induced by proteasomal inhibition with Lac or MG-132 was strongly hindered as compared with control cells (Figure 2, B–D, and Supplemental Figure S2, A and B). In the absence of Lac, Herp knockdown increased somewhat the H2a-RFP level but remained in an ER pattern (Figure 2A). Because Herp is a short-lived proteasomal substrate, H2a-RFP accumulation in the ERQC could be caused by stabilization of Herp by proteasomal inhibition and not by inhibition of H2a-RFP degradation. However, accumulation of ERAD substrates in the ERQC is dependent on their levels and not on proteasomal inhibition (Kamhi-Nesher et al., 2001; Frenkel et al., 2004). Therefore, we overexpressed H2a-RFP and assessed the effect of Herp knockdown in untreated cells. Many cells overexpressing H2a-RFP showed accumulation in the ERQC, which was inhibited by Herp knockdown (Figure 2, E and F). In contrast with the Herp requirement, ERQC accumulation of H2a-RFP by Lac was unaffected in CHOP-null mouse embryonic fibroblast (MEF) cells (Figure 2, G and H). Taken together, the results suggest that ERAD substrate recruitment to the ERQC involves eIF2α phosphorylation and its downstream target, Herp.
FIGURE 2:
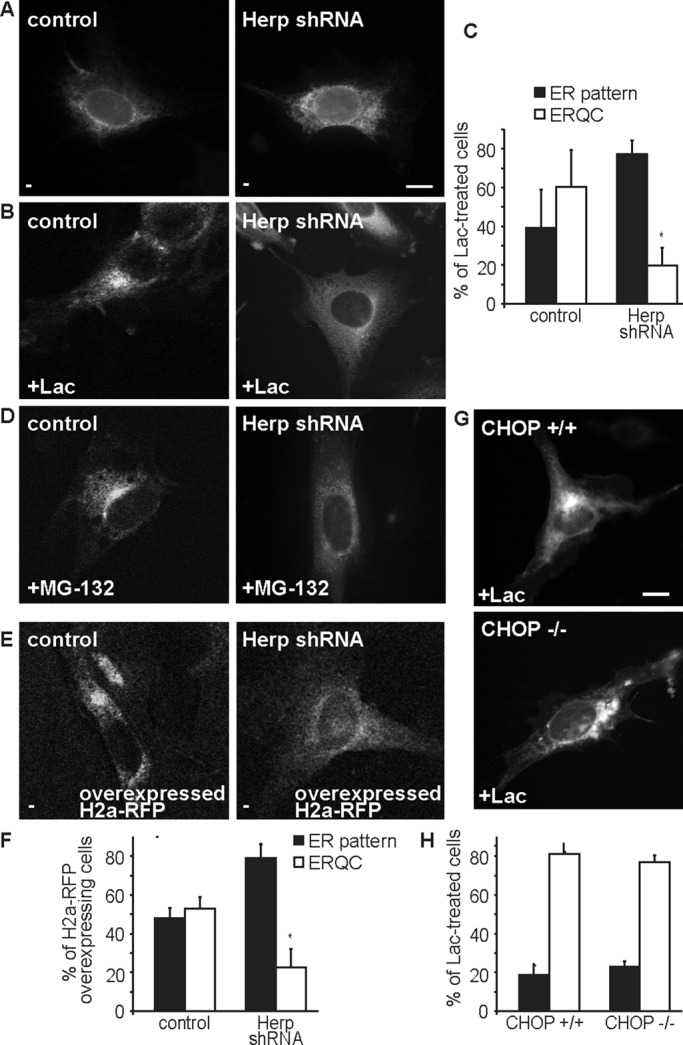
Herp, a downstream effector of eIF2α-P, is required for ERAD substrate targeting to the ERQC and CHOP is not. (A, B) Herp knockdown blocks ERQC targeting. H2a-RFP was expressed in NIH 3T3 cells stably expressing a doxycycline-inducible shRNA for Herp and in control cells, then incubated with 2 μg/ml Dox for 24 h, followed by incubation with or without Lac for 3 h. (C) Quantitation of H2a-RFP distribution in A and B. Bars in the graph represent averages of three independent experiments ± SE, 60 cells counted in each. *p (Herp shRNA vs. control cells) = 0.03. (D) Similar to A, but with cells incubated with MG-132 (40 μM) instead of Lac. (E, F) Similar to A and C but with untreated cells overexpressing H2a-RFP (three times more DNA was used in the transfection). *p (Herp shRNA vs. control cells) = 0.03. (G, H) CHOP knockout does not affect ERQC targeting. CHOP (−/−) MEF cells or control (+/+) cells were transiently transfected with H2a-RFP, grown for 48 h, and incubated with Lac for 3 h.
Herp is required for degradation of both glycosylated and nonglycosylated transmembrane ERAD substrates
Herp knockdown significantly inhibited H2a degradation and also that of an H2a mutant lacking all three of its glycosylation sites (H2aΔgly; Shenkman et al., 2013), as seen by pulse-chase analysis (Figure 3, A and B). Herp requirement was reported for degradation of several nonglycosylated ERAD substrates but not for all glycosylated substrates (Schulze et al., 2005; Okuda-Shimizu and Hendershot, 2007; Liang et al., 2008). Both H2a and H2aΔgly showed significant interaction with Herp (Figure 3C), which strongly increased upon proteasomal inhibition, suggesting that substrate molecules that are spared from degradation associate preferentially with Herp.
FIGURE 3:

Both a membrane glycoprotein ERAD substrate and its nonglycosylated mutant interact with Herp and are stabilized by Herp knockdown. (A) Pulse-chase analysis shows H2a stabilization by Herp shRNA. H2a was expressed by a retroviral vector in the cell lines used in Figure 2, A–E. After induction of Herp shRNA expression for 24 h the cells were pulse labeled for 20 min with [35S]cysteine and chased for the indicated times in complete medium. Shown is the phosphorimage of labeled H2a immunoprecipitated from cell lysates run on SDS–PAGE. The graph shows percentage of H2a remaining after chase relative to the pulse, from phosphorimager quantitations of the gels, average of three independent experiments ± SE. p (2h, Herp shRNA vs. control cells) = 0.02. (B) Similar to A but expressing a mutant H2a with all three glycosylation sites canceled (H2aΔgly). Shown is an experiment representative of four repeat experiments. (C) Herp coimmunoprecipitates with H2a and H2aΔgly, especially upon proteasome inhibition. NIH 3T3 cells expressing Herp-Flag and H2a, H2aΔgly, or a control with GFP (−), were incubated with or without MG-132 (40 μM) for 5 h and lysed. H2a immunoprecipitated from 90% of the lysates, and the remaining 10% were run on 12% SDS–PAGE and immunoblotted with anti-Herp and anti-H2a antibodies. The graph shows the relative amount of Herp-Flag that coimmunoprecipitated with H2a or H2aΔgly, normalized for the total amount of Herp-Flag and substrates, average of three independent experiments. p (H2aΔgly, + MG-132 vs. untreated) = 0.015. Note that endogenous Herp also coimmunoprecipitated with the substrates, but due to its low expression, the signal-to-noise ratio is much lower than for the exogenous Herp.
Herp localizes to the ERQC
Because upon proteasomal inhibition H2a accumulates in the ERQC (Figure 1) and is associated with Herp (Figure 3D), the prediction was that Herp also localizes to the ERQC. Indeed, endogenous Herp localized to the ERQC after its induction with tunicamycin (Tun) overnight followed by treatment for 3 h with Lac and colocalized substantially with H2a-RFP (Figure 4B). Individual treatments with Tun for only 3 h or with Lac gave a weaker but similar pattern (Supplemental Figure S3). Herp was in a very weak diffuse pattern in untreated NIH 3T3 cells (Figure 4A) and after induction was restricted to the ERQC, not colocalizing with a Golgi marker (Figure 4C). Similar results were obtained with a different approach; in iodixanol gradients Herp was barely detectable in untreated cells but colocalized with calnexin in the heavy fractions after induction with Tun and proteasomal inhibition. Herp appeared separate from the Golgi and from light fractions containing BiP and ERp57 (Figure 4D). Under these conditions, calnexin accumulates in the ERQC, but BiP and ERp57 remain mostly in the peripheral ER, with only a minor portion present in the ERQC (Frenkel et al., 2004; Kondratyev et al., 2007). The gradient showed much of BiP and ERp57 remaining in light fractions, although a lesser portion migrated together with Herp in the heavy, ERQC fractions. The rough ER partially overlapped with the ERQC, but it peaked in the heaviest fraction (fraction 11), where calnexin and Herp were relatively reduced.
FIGURE 4:

UPR up-regulated Herp localizes to the ERQC. (A, B) Similar to Figure 1, except that cells were incubated overnight without (A) or with (B) 5 μg/ml Tun followed by Lac for 3 h. The cells were fixed and stained with rabbit anti-Herp and Cy2-conjugated goat anti-rabbit IgG. (C) Similar to B, except for expression of the Golgi marker GalT-YFP; endogenous Herp was stained with rabbit anti-Herp and Cy5-conjugated goat anti-rabbit IgG. Bars, 10 μm. (D) Comigration of Herp and CNX at the ERQC. NIH 3T3 cells were incubated without (left) or with Tun (5 μg/ml) overnight, followed by MG-132 (40 μM) for 3 h. Cells were homogenized, and postnuclear supernatants were centrifuged on a 10–34% iodixanol gradient as detailed in Materials and Methods. Fractions of the gradients were run on SDS–PAGE and immunoblotted with the indicated antibodies. A representative experiment out of five repeat experiments is shown. Asterisks indicate nonspecific bands. Note that the gradients are optimized to resolve heavy fractions; light (smooth) ER fractions overlap with the Golgi. The graphs show quantitation of the immunoblots and density calculated from the refractive index of the fractions.
Herp is necessary and sufficient for HRD1 and ERAD substrate accumulation in the ERQC and substrate association with the OS-9 lectin
We wondered whether Herp is required for the localization of the ubiquitin ligase HRD1 and of other ERAD components. HRD1 localizes to the ERQC after accumulation of the ERAD substrate upon proteasomal inhibition (Kondratyev et al., 2007; Groisman et al., 2011; Figure 5A) compared with an ER pattern in untreated cells (Supplemental Figure S4A). On Herp knockdown, recruitment of HRD1 to the ERQC was blocked (Figure 5, C and E). The HRD1-SEL1L–associated lectin OS-9, a receptor for trimmed glycans that mark misfolded glycoproteins for ERAD (Hosokawa et al., 2009), also shows ERQC localization (Figure 5B). Surprisingly, OS-9 localization was not affected by Herp knockdown, whereas that of the ERAD substrate was (Figure 5, D and E). In contrast to HRD1, OS-9 is constitutively localized at the ERQC (Ron et al., 2011), as can be seen by immunofluorescence and in iodixanol gradients (Supplemental Figure S4, B and C).
FIGURE 5:

Herp is required for the targeting of HRD1 and the ERAD substrate to the ERQC and for substrate association with OS-9. (A–D) Herp knockdown blocks ERQC targeting of HRD1 but not of OS-9. H2a-RFP was expressed together with myc-HRD1 (A, C) or S-tagged OS-9.1/2 (B, D) in control (A, B) or Herp shRNA–expressing cells (C, D). Herp shRNA expression was induced with Dox for 24 h. Cells were then treated with Lac for 3 h. Mouse anti-tag antibodies and Cy2-conjugated secondary were used to visualize the tagged proteins. Representative images. Bars, 10 μm. (E) Bars are averages ± SE of three independent experiments from A–D (50–100 cells counted in each). *p (HRD1, shRNA vs. control cells) = 0.02, p (OS-9, shRNA vs. control cells) = 0.2 (NS). (F) Herp knockdown reduces, but not significantly, HRD1 coimmunoprecipitation with H2a. HRD1-myc was expressed with or without H2a in Herp shRNA–expressing cells induced as in C and D, and control cells were left untreated. H2a immunoprecipitates from 90% of cell lysates and the remaining 10% were run on 12% SDS–PAGE and immunoblotted with the indicated antibodies. The vertical line indicates elimination of an irrelevant lane. The graph shows the relative amount of HRD1-myc that coimmunoprecipitated with H2a, average of three independent experiments. This value was normalized to the total H2a and HRD1-myc levels. p (Herp shRNA vs. control cells) = 0.09 (NS). (G) Herp knockdown inhibits OS-9 coimmunoprecipitation with H2a. Similar to F but with expression of S-tagged OS-9.1/2 instead of HRD1-myc. ***p (Herp shRNA vs. control cells) = 0.0001.
We then investigated whether Herp is required for the association of the substrate with HRD1 and OS-9. Herp knockdown caused accumulation of H2a but a lesser increase in coimmunoprecipitated HRD1 (Figure 5F). However, this difference was not significant. The difference was much more pronounced for OS-9; the ERAD substrate coimmunoprecipitated significantly less with OS-9 upon Herp knockdown (Figure 5G). This is consistent with the segregation of H2a and OS-9 upon Herp knockdown (Figure 5D). Therefore, the presence of Herp allows accumulation of the ERAD substrate and of HRD1 at the ERQC, increased interaction with OS-9, and accelerated degradation. Taken together, the results suggest that Herp-mediated recruitment of HRD1 contributes to stabilize the interaction of the ERAD substrate with OS-9 and the retrotranslocation complex at the ERQC. On Herp knockdown, the membrane-bound substrate still associates to some degree with HRD1 in the peripheral ER but cannot be targeted to degradation because it cannot be delivered to a functional ERAD complex.
To assess the generality of our findings, we studied the effect of Herp knockdown on another well-established ERAD substrate, the type I membrane-bound glycoprotein BACE476. ERAD of BACE476 is not strictly dependent on HRD1; it can be targeted by either HRD1 or gp78 (Bernasconi et al., 2010). Herp knockdown similarly inhibited accumulation of H2a-RFP and BACE476 at the ERQC (Figure 6, A–E). Although interaction of BACE476 with HRD1 was very weak, it associated with OS-9. Similar to the results with H2a, Herp knockdown inhibited association of BACE476 with OS-9 and caused an increase in the total level of BACE476 (Figure 6F).
FIGURE 6:

Herp has a similar effect on the ERAD substrate BACE476. (A–E) Similar to Figure 5, A–E, but with cells expressing H2a-RFP together with BACE476-HA. **p (Herp shRNA vs. control cells) = 0.007. (F) Herp knockdown inhibits OS-9 coimmunoprecipitation with BACE476. Similar to Figure 5G but with expression of BACE476-HA instead of H2a. S-tagged OS-9 was precipitated with S-protein agarose, and coprecipitated BACE476-HA was immunoblotted. A representative experiment out of three is shown. The relative amount of BACE476 that coimmunoprecipitated with OS-9 is shown at the bottom.
We wondered whether a high concentration of Herp, as exists during the UPR, is sufficient to cause concentration of the ERAD substrates. Indeed, overexpression of Herp, at levels comparable to those reached by UPR induction (Figure 7B) caused accumulation of both glycosylated and nonglycosylated substrates in the ERQC (Figure 7A). This accumulation must be faster than substrate degradation. The accumulation was similar to that caused by a proteasome inhibitor, with the nonglycosylated substrate appearing in a broader pattern (Supplemental Figure S5). Overexpressed Herp-Flag accumulated in the ERQC but also in a more extended pattern than the ERAD substrate (Figure 7A). Consistent with functional Herp association with HRD1 (Schulze et al., 2005), Herp overexpression was also sufficient to cause accumulation of HRD1 (Figure 7, C and D). HRD1 accumulated in the region of the ERQC but also in other peripheral areas. We then analyzed whether Herp overexpression affects association of HRD1 and of OS-9 with the substrate. Herp overexpression reduced the total level of H2a and increased significantly the ratio of OS-9 coimmunoprecipitation, whereas no significant change was seen for HRD1 (Figure 7, E and F). Taken together, these results suggest that the substrate can associate with HRD1 both at low Herp levels (in the peripheral ER) and upon Herp overexpression (in the ERQC). In contrast, substrate association with OS-9 is much increased in parallel with substrate accumulation in the ERQC at high Herp levels.
FIGURE 7:

Herp overexpression is sufficient for ERAD substrate and HRD1 targeting to the ERQC and enhances substrate association with OS-9. (A) Overexpression of Herp is sufficient for H2a targeting to the ERQC. Cells expressing H2a-RFP or H2aΔgly together with Flag-tagged Herp were stained with mouse anti-Flag and FITC-conjugated secondary antibody; rabbit anti-H2a/Cy3-conjugated secondary was used for H2aΔgly detection. Bar, 10 μm. The graph shows quantitation of the experiments in Figure 7A and Supplemental Figure S5, average of three independent experiments ± SE, ∼70 cells counted in each. p (untreated vs. Lac-treated cells) < 0.008, p (untreated vs. Herp overexpression) < 0.0002. (B) Levels of overexpressed Herp are similar to levels of UPR-induced endogenous protein. NIH3T3 cells expressing or not Herp-Flag, treated with Tun (5 μg/ml) overnight or left untreated. (C, D) Overexpression of Herp is sufficient for HRD1 targeting to the ERQC. Cells transfected with HRD1-Myc without (C) or with (D) Herp-Flag. Mouse anti-Myc/Cy3-conjugated secondary was used for HRD1-Myc detection. The experiment was quantified as in A. p (untreated vs. Herp overexpression) = 0.05. Bar, 10 μm. (E) Herp overexpression does not affect HRD1 coimmunoprecipitation with H2a. HRD1-myc was expressed in HEK 293 cells with or without H2a and Herp-Flag. Samples were processed and results quantitated as in Figure 5F. A representative experiment out of three is shown. (F) Herp overexpression enhances OS-9 coimmunoprecipitation with H2a. Similar to E but with expression of S-tagged OS-9.1/2 instead of HRD1-myc. Weak nonspecific coimmunoprecipitation was subtracted for the quantitation. p (+Herp-Flag vs. untreated) = 0.004. Note that Herp overexpression accelerates the degradation, reducing the level of H2a, but there is no decrease in coimmunoprecipitated OS-9.
A model that can explain these results is that Herp recruits HRD1 to the ERQC and is necessary for assembly of functional retrotranslocation complexes that sequester the substrate (Figure 8A). Despite the fact that OS-9 does not require Herp to localize to the ERQC, it is likely unable to trap the substrate in this compartment due to its weak substrate binding (Ka = 0.2–0.3 mM; Hosokawa et al., 2009).
FIGURE 8:

Model of recruitment and organization of the ERAD complex at the ERQC by Herp. (A) Working model for Herp-mediated recruitment of an ERAD retrotranslocation complex at the ERQC. After trimming of two glucose residues and association with the chaperone calnexin, a misfolded glycoprotein is transported together with this chaperone to the ERQC (Kamhi-Nesher et al., 2001; Frenkel et al., 2004) and can recycle back to the peripheral ER. Trimming of the remaining glucose by glucosidase II and of three or four α1,2 mannose residues by ER mannosidase I and EDEM1 removes the glycoprotein from the calnexin folding cycle, determining its association with OS-9 (or XTP3-B; Groisman et al., 2011). Nonglycosylated misfolded proteins are also targeted by several of these components through protein–protein interactions (Shenkman et al., 2013). In the absence of Herp (left), the misfolded proteins can dissociate from OS-9 and recycle back to the peripheral ER. When Herp is present (right), it recruits HRD1 to the ERQC, which stabilizes binding of the misfolded proteins to the ERAD complex, including SEL1L, Derlin-1, ubiquilins (Ubqln), and the proteasome. The model is based on our present and previous results and those from others. Carbohydrate-binding pockets are highlighted. For simplicity, p97, UBX proteins, and other participants have not been included. (B) Scheme of the structure of Herp in comparison to hHR23A and hHR23B as predicted by the FFAS server and Monte Carlo simulations (Supplemental Figure S6). Domains are joined by unstructured linkers.
Herp is predicted to be weakly associated with the membrane by a hydrophobic hairpin and be similar in structure to hHR23
Given the importance of Herp in recruitment of ERAD substrates and machinery to the ERQC membrane, we sought to better understand its membrane association. Monte Carlo simulations strongly predicted a relatively unstable interaction of Herp with the membrane through a hairpin with most of the protein on the cytosolic side and only a short loop on the luminal side (Supplemental Figure S6 and Supplemental Information, Supplemental Bioinformatics). Fold recognition analysis using the FFAS03 fold recognition server (Jaroszewski et al., 2005) predicted that the three-dimensional structure of Herp has high similarity to hHR23A and hHR23B, proteins that stabilize xeroderma pigmentosum group C protein (XPC), which is involved in DNA damage repair. Through an ubiquitin-like (UBL) domain, hHR23B binds to proteasomes. Herp was also reported to bind to proteasomes (Okuda-Shimizu and Hendershot, 2007), but it is unclear whether this occurs through its UBL domain or possibly ubiquilins (Kim et al., 2008). The similarity along the entire length of the protein (except for the XPC-binding domain) suggests that in addition to the UBL domain, Herp also contains ubiquitin-associated (UBA) domains. Remarkably, the hydrophobic hairpin of Herp replaces the hHR23 XPC-binding domain (Figure 8B), and through this hairpin Herp could recruit HRD1 and indirectly the substrate to the ERQC membrane.
DISCUSSION
The results shown here, together with our previous findings (Kondratyev et al., 2007), suggest that Herp, which is induced by the PERK branch of the UPR, is essential for recruitment of the HRD1 complex in a specialized compartment, the ERQC, as a staging ground for ERAD. Herp is both necessary for this recruitment, as indicated by the knockdown experiments, and sufficient, as indicated by the effects of its overexpression. Although we cannot exclude that part of overexpressed Herp is misfolded and mislocalized, ERQC localization of endogenous Herp after its induction with Tun (Figure 4) would suggest that this is its natural fate. Tun would not affect directly Herp folding, as it is not a glycoprotein.
We had shown that the ERQC concentrates misfolded proteins and ERAD machinery, enhancing the efficiency of the degradation process and ensuring cell homeostasis and survival and therefore having a protective role (Kondratyev et al., 2007). This is consistent with the antiapoptotic role of Herp, which is in charge of this recruitment. In contrast, CHOP, also a downstream effector of the PERK pathway (Ma et al., 2002), is proapoptotic (Zinszner et al., 1998) and does not seem to be involved in the recruitment process (Figure 2).
Interaction of Herp with other ERAD components (Schulze et al., 2005; Okuda-Shimizu and Hendershot, 2007), proteasomes, and ubiquitinated substrates (Okuda-Shimizu and Hendershot, 2007) makes it a particularly good candidate for coordination at the ERQC of the UPR and ERAD processes, which are interdependent (Friedlander et al., 2000; Travers et al., 2000). Misfolded protein accumulation at the ERQC is not dependent on proteasomal inhibition but is on the levels of the protein (Figure 2E; Kamhi-Nesher et al., 2001; Frenkel et al., 2004) and eIF2α-P upon inhibition of its dephosphorylation (Figure 1C) and can also be achieved with a phosphomimetic mutant (eIF2a(S51D); Kondratyev et al., 2007). The accumulation is also triggered by overexpression of Herp (Figure 7). In resting cells, transient accumulation of unfolded protein molecules probably also causes temporary activation of the PERK pathway and induction of Herp at low routine levels of ER load, leading to dynamic protein recruitment to the pericentriolar ERQC. In other words, some level of UPR activation and Herp induction must occur under so-called physiological “unstressed” conditions and is a prerequisite for protein recruitment to the ERQC and for ERAD. The constitutive localization of OS-9 at the ERQC (Figure 5 and Supplemental Figure S4; Ron et al., 2011) points to the presence of the compartment in unstressed conditions and its recruitment of ERAD machinery components, as needed for protein disposal. This is consistent with the reported dynamic interaction of Herp with HRD1 (Kny et al., 2011). Retrotranslocation complexes might be transiently assembled only as needed. In addition, because Herp is unstable and degraded by the proteasomes, any situation that compromises proteasomal activity will increase its levels and lead to ERAD complex recruitment and compartmentalization. In fact, while our manuscript was in revision it was reported that accumulation of ERAD substrates can lead to Herp stabilization and induce its role in ERAD (Bernasconi et al., 2013).
The association of Herp with the substrate increases strongly upon proteasomal inhibition (Figure 3C). In contrast, there is a drastic reduction of interaction of the substrate with calnexin over time in the presence of MG-132 (Frenkel et al., 2004; Lynes et al., 2013). This suggests that unlike its interaction with calnexin, the substrate interacts with Herp at a late stage toward ERAD, likely at retrotranslocation sites that would be located at the ERQC (Figure 4). This seems to be true for both the glycosylated and nonglycosylated ERAD membrane-bound substrates that we studied, consistent with previously reported Herp involvement in ERAD of these two protein classes (Schulze et al., 2005; Liang et al., 2008; Gao et al., 2010). However, soluble glycosylated substrates might differ in part of this pathway (Okuda-Shimizu and Hendershot, 2007).
For ERAD of glycoproteins, trimming of mannose residues is essential for their binding to OS-9 and XTP3B and compartmentalization of the substrate (Hosokawa et al., 2009; Yamaguchi et al., 2010; Groisman et al., 2011). However, the trimming is not sufficient for the substrate to remain in the ERQC, as this compartmentalization does not occur upon knockdown of Herp. Our results suggest that it is the Herp-mediated recruitment of HRD1 that stabilizes the interaction of the ERAD substrate with the retrotranslocation complex at the ERQC.
Herp shows high predicted structural similarity to hHR23A and hHR23B. Besides stabilizing XPC, hHR23A and hHR23B also serve as proteasome shuttles. The XPC protein-adaptor domain of hHR23B can also bind PNGase, another protein involved in ERAD and responsible for glycoprotein deglycosylation before degradation (Zhao et al., 2006). Of interest, Herp is predicted to have a transmembrane hairpin instead of the XPC domain. Through the transmembrane hairpin Herp might recruit the ERAD complex to the ERQC membrane. The predicted UBA domains of Herp could, as in the case of hHR23, associate with the substrate after ubiquitination, although this awaits experimental confirmation.
As summarized in Figure 8A, the ability of Herp to assemble a complex containing HRD1, SEL1L, OS-9, Derlin-1 (Schulze et al., 2005), ubiquilins (Kim et al., 2008), proteasomes, and the ubiquitinated substrate (Okuda-Shimizu and Hendershot, 2007) suggests that it is a master organizer of the ERAD machinery.
MATERIALS AND METHODS
Materials
Rainbow 14C-labeled methylated protein standards were obtained from GE Healthcare (Little Chalfont, United Kingdom). Promix cell labeling mix ([35S]Met plus [35S]Cys), >1000 Ci/mmol, was from PerkinElmer (Boston, MA), protein A–Sepharose from Repligen (Needham, MA), and Lac from Calbiochem (La Jolla, CA). S-protein agarose was from Novagen (Gibbstown, NJ). Polyethylenimine (PEI) was from Polysciences (Eppelheim, Germany). Anti-mouse immunoglobulin G (IgG)–agarose, MG-132, Tun, Puro, nocodazole, Dox, Sal, Polybrene, and other common reagents were from Sigma-Aldrich (St. Louis, MO).
Plasmids and constructs
H2a and H2a mutated in its three glycosylation sites (H2aΔgly), subcloned in pCDNA1, were previously described (Kamhi-Nesher et al., 2001; Groisman et al., 2006), as were H2aG78R fused through its C-terminus to H2a-RFP (Kondratyev et al., 2007), FLAG-tagged Herp (Okuda-Shimizu and Hendershot, 2007), β1,3-galactosyltransferase (GalT)–yellow fluorescent protein (YFP), and myc-tagged human HRD1 (Groisman et al., 2006; Kondratyev et al., 2007; Avezov et al., 2008). Hemagglutinin (HA)-tagged BACE476 (Bernasconi et al., 2010) was a kind gift of M. Molinari (IRB, Bellinzona, Switzerland). S-tagged XTP3-B, S-tagged OS-9.1, and OS-9.2 (Christianson et al., 2008) were previously described (Shenkman et al., 2013). H2a subcloned using an EcoRI restriction site in MSCVpuro, H2aΔgly subcloned using EcoRI and BamH1 restriction sites in pMIG (Addgene, Cambridge, MA), and pCL-Eco Retrovirus Packaging Vector (Imgenex, San Diego, CA) were used for infections.
Antibodies
Rabbit polyclonal anti–H2 C-terminal and anti–N-terminal antibodies were described in earlier studies (Tolchinsky et al., 1996; Shenkman et al., 2000). Mouse monoclonal anti–H2 C-terminal antibody B9 was raised in our laboratory (Benyair et al., 2011). Other mouse monoclonals were as follows: anti-S tag from Novagen, anti-HA and anti-FLAG from Sigma-Aldrich, anti-c-Myc from Santa Cruz Biotechnology (Santa Cruz, CA), and anti–glyceraldehyde-3-phosphate dehydrogenase from Chemicon (Temecula, CA). Rabbit anti-calnexin, anti–OS-9, and goat anti-mouse IgG-agarose were from Sigma-Aldrich. Rabbit anti-ERp57 and anti-RPL26 were from Abcam (Cambridge, MA). Goat anti-mouse IgG conjugated to Cy2, goat anti-rabbit IgG-Cy3 and Cy5, and goat anti-rabbit and anti-mouse IgG conjugated to horseradish peroxidase were from Jackson ImmunoResearch Laboratories (West Grove, PA). Rabbit anti-Herp and anti-BiP were described before (Hendershot et al., 1995; Okuda-Shimizu and Hendershot, 2007). Rabbit anti-HRD1 was a kind gift of E. Wiertz (Leiden University, Leiden, Netherlands; Kikkert et al., 2004).
Cell culture, transfections, and infections
CHOP−/− MEFs (Zinszner et al., 1998) were a kind gift of David Ron (University of Cambridge, UK). MEF, NIH 3T3, HEK 293, and HEK 293T cells were grown in DMEM plus 10% fetal calf serum at 37°C under 5% CO2. Transient transfection of NIH 3T3 and CHOP−/− MEFs was performed using X-tremeGENE reagent (Roche, Indianapolis, IN) or a Microporator MP-100 (Digital Bio, Seoul, South Korea) according to the manufacturer's protocols or with PEI (Durocher et al., 2002) in a 1:3 ratio DNA:PEI in Opti-MEM. For pulse-chase experiments HEK 293T cells were transfected (calcium phosphate method) with pCL-Eco and H2a or H2aΔgly in the retroviral vectors (2:1 ratio). Medium was changed after 24 h, and after an additional 24 h the medium was collected, filtered (0.45 μm Whatman filter), and added to subconfluent NIH 3T3 cells. Polybrene (8 μg/ml) was added immediately, and experiments were performed 24–48 h later.
Inducible RNA interference
An NIH 3T3 stable cell line expressing tetracycline-inducible Herp shRNA was described previously (Okuda-Shimizu and Hendershot, 2007). The cells were grown with 3 μg/ml blasticidin S HCl (Invitrogen, Carlsbad, CA) and 3 μg/ml Puro. shRNA expression was induced with 2 μg/ml Dox for 24 h. The efficiency of the knockdown using this system was ∼50% in all experiments in which it was used.
[35S]Cys metabolic labeling, immunoprecipitation, SDS–PAGE, and quantitation
Subconfluent (90%) cell monolayers in 60-mm dishes were labeled with [35S]Cys, lysed, and immunoprecipitated with anti-H2 antibodies as described (Tolchinsky et al., 1996). SDS–PAGE was performed on 12% Laemmli gels and analyzed by fluorography or phosphorimager quantitation (Groisman et al., 2011).
Coimmunoprecipitation and immunoblotting
Cell lysis was done in 1% NP-40, 50 mM Tris/HCl (pH 8), 150 mM NaCl, and a protease inhibitor cocktail (Roche) for 30 min on ice. After pelleting of nuclei, lysates were immunoprecipitated overnight with rabbit or mouse anti–H2a C-terminal antibody using protein A–Sepharose or goat anti-mouse IgG–agarose respectively. Precipitation of S-tagged proteins was done with S-protein agarose. After three washes with lysis buffer diluted 1:5, bound proteins were eluted with sample buffer at 100°C for 5 min, except for samples with HRD1, which were heated at 65°C for 15 min. Immunoblotting and detection by enhanced chemiluminescence were done as described previously (Groisman et al., 2011).
Microsome isolation and gradient fractionation
Cells were incubated with Tun (5 μg/ml) overnight, followed by addition of MG-132 (40 μM) for another 3 h where indicated. After Dounce cell homogenization in an iso-osmotic buffer (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, and 250 mM sucrose), debris and nuclei were pelleted at 1000 × g at 4ºC for 20 min, and the supernatants were loaded on top of an iodixanol gradient (10–34%). Iodixanol solutions were prepared in 60 mM HEPES, pH 7.4, and 250 mM sucrose iso-osmotic buffer. The gradients were ultracentrifuged at 24,000 rpm (∼98,500 × g, Beckman SW41 rotor) for 16 h at 4ºC. Eleven fractions were collected from gradient top to bottom and subjected to immunoblotting. Density was calculated from the refractive index of the fractions.
Immunofluorescence and confocal microscopy
The procedures used were as described previously (Kamhi-Nesher et al., 2001; Avezov et al., 2008). Treatment of cells on coverslips with Lac (17 μM), Puro (50 μg/ml), or Sal (50 μM) was for 3–5 h at 37°C (CO2 incubator). Herp was induced with Tun (5 μg/ml) overnight followed by Lac (17 μM) plus Tun for another 3 h. Quantification of the percentage of cells with a protein in an ER pattern versus those with accumulation in the ERQC was done by counting cells showing each pattern as estimated by visual observation. The variation between observers was <10%.
Statistical analysis
Data are expressed as means ± SE. Student's t test (unpaired, two-tailed) was used to compare the two groups, and the p value was calculated in Prism 5 (GraphPad, La Jolla, CA). p < 0.05 is considered statistically significant.
Computational procedures
Conservation profile.
Homologous sequences were collected using FHMMER from the nr database. Redundant (>99% sequence identity) and fragmented sequences were removed. The sequences were aligned using the MAFFT program and then used to calculate the conservation profile using the ConSurf server (Ashkenazy et al., 2010).
Structural prediction and analysis.
To acquire preliminary structural information on the Herp protein, we exploited the secondary structure prediction algorithm PsiPred and several methods specifically designed to identify transmembrane (TM) segments, namely MEMSAT, HMMTOP, SPLIT4, and Octopus. Next we used the FFAS03 server (Jaroszewski et al., 2005) to explore remote similarity of Herp to proteins with known three-dimensional structure that may be used as templates.
Monte Carlo simulations.
We performed Monte Carlo simulations of the putative TM regions using the MCPep server (Gofman et al., 2012; http://bental.tau.ac.il/MCPep/). The membrane was represented as a smooth hydrophobic profile of native width 30 Å, corresponding to the hydrocarbon region. A negative surface charge was located on both sides of the membrane at a distance of 20 Å from the midplane. Its magnitude corresponded to the fraction of acidic lipids in the ER, that is, 22%. The membrane was embedded in an aqueous solution of 0.1 M monovalent salt and pH = 7, corresponding to physiological conditions.
Given the presence of the highly hydrophilic arginine in position 289, we deduced that the loop consisted of S288, R289, and F290 and modeled this region as a helix-loop-helix motif. Specifically, the first helix consisted of residues 261–277, the short loop included residues 288–290, and the second helix stretched between residues 291 and 313. This structure was used as the starting conformation for the Monte Carlo simulations. The second possible TM region, namely residues 361–391, was modeled as a canonical α-helix using the MCPep server utility.
Supplementary Material
Acknowledgments
We are grateful to David Ron, Maurizio Molinari, and Emmanuel Wiertz for reagents. This work was supported by grants from the Israel Science Foundation (1070/10) and German-Israeli Project Cooperation (Deutsch-Israelische Projektkooperation K 5-1).
Abbreviations used:
- BiP
heavy-chain-binding protein
- CHOP
C/EBP-homologous protein
- Dox
doxycycline
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- ERQC
ER-derived quality control compartment
- GalT
β1,3-galactosyltransferase
- Herp
homocysteine-induced ER protein
- Lac
lactacystin
- PERK
PKR-like ER-localized eIF2α kinase
- Puro
puromycin
- Sal
salubrinal
- UPR
unfolded protein response
Footnotes
*These authors contributed equally.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-06-0350) on January 29, 2014.
REFERENCES
- Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(suppl):W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avezov E, Frenkel Z, Ehrlich M, Herscovics A, Lederkremer GZ. Endoplasmic reticulum (ER) mannosidase I is compartmentalized and required for N-glycan trimming to Man5-6GlcNAc2 in glycoprotein ER-associated degradation. Mol Biol Cell. 2008;19:216–225. doi: 10.1091/mbc.E07-05-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benyair R, Kondratyev M, Veselkin E, Tolchinsky S, Shenkman M, Lurie Y, Lederkremer GZ. Constant serum levels of secreted asialoglycoprotein receptor sH2a and decrease with cirrhosis. World J Gastroenterol. 2011;17:5305–5309. doi: 10.3748/wjg.v17.i48.5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi R, Galli C, Calanca V, Nakajima T, Molinari M. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J Cell Biol. 2010;188:223–235. doi: 10.1083/jcb.200910042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi R, Galli C, Kokame K, Molinari M. Autoadaptive ER-associated degradation defines a preemptive unfolded protein response pathway. Mol Cell. 2013;52:783–793. doi: 10.1016/j.molcel.2013.10.016. [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Boyce M, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Stanley AM, Rapoport TA. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell. 2010;143:579–591. doi: 10.1016/j.cell.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SL, Fu W, Zhang P, Cheng A, Lee J, Kokame K, Mattson MP. Herp stabilizes neuronal Ca2 +homeostasis and mitochondrial function during endoplasmic reticulum stress. J Biol Chem. 2004;279:28733–28743. doi: 10.1074/jbc.M404272200. [DOI] [PubMed] [Google Scholar]
- Christianson JC, Shaler TA, Tyler RE, Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10:272–282. doi: 10.1038/ncb1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel Z, Shenkman M, Kondratyev M, Lederkremer GZ. Separate roles and different routing of calnexin and ERp57 in endoplasmic reticulum quality control revealed by interactions with asialoglycoprotein receptor chains. Mol Biol Cell. 2004;15:2133–2142. doi: 10.1091/mbc.E03-12-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2:379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- Gao H, et al. PRKCSH/80K-H, the protein mutated in polycystic liver disease, protects polycystin-2/TRPP2 against HERP-mediated degradation. Hum Mol Genet. 2010;19:16–24. doi: 10.1093/hmg/ddp463. [DOI] [PubMed] [Google Scholar]
- Gofman Y, Haliloglu T, Ben-Tal N. Monte Carlo simulations of peptide-membrane interactions with the MCPep web server. Nucleic Acids Res. 2012;40:W358–W363. doi: 10.1093/nar/gks577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman B, Avezov E, Lederkremer GZ. The E3 ubiquitin ligases HRD1 and SCFFbs2 recognize the protein moiety and sugar chains respectively of an ER-associated degradation substrate. Isr J Chem. 2006;46:189–196. [Google Scholar]
- Groisman B, Shenkman M, Ron E, Lederkremer GZ. Mannose trimming is required for delivery of a glycoprotein from EDEM1 to XTP3-B and to late endoplasmic reticulum-associated degradation steps. J Biol Chem. 2011;286:1292–1300. doi: 10.1074/jbc.M110.154849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Hendershot LM, Wei JY, Gaut JR, Lawson B, Freiden PJ, Murti KG. In vivo expression of mammalian BiP ATPase mutants causes disruption of the endoplasmic reticulum. Mol Biol Cell. 1995;6:283–296. doi: 10.1091/mbc.6.3.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn SC, Hanna J, Hirsch C, Volkwein C, Schutz A, Heinemann U, Sommer T, Jarosch E. Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol Cell. 2009;36:782–793. doi: 10.1016/j.molcel.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Kamiya Y, Kamiya D, Kato K, Nagata K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J Biol Chem. 2009;284:17061–17068. doi: 10.1074/jbc.M809725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroszewski L, Rychlewski L, Li Z, Li W, Godzik A. FFAS03: a server for profile–profile sequence alignments. Nucleic Acids Res. 2005;33:W284–W288. doi: 10.1093/nar/gki418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamhi-Nesher S, Shenkman M, Tolchinsky S, Fromm SV, Ehrlich R, Lederkremer GZ. A novel quality control compartment derived from the endoplasmic reticulum. Mol Biol Cell. 2001;12:1711–1723. doi: 10.1091/mbc.12.6.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van Voorden S, Thanedar S, Roitelman J, Chau V, Wiertz E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- Kim TY, Kim E, Yoon SK, Yoon JB. Herp enhances ER-associated protein degradation by recruiting ubiquilins. Biochem Biophys Res Commun. 2008;369:741–746. doi: 10.1016/j.bbrc.2008.02.086. [DOI] [PubMed] [Google Scholar]
- Kny M, Standera S, Hartmann-Petersen R, Kloetzel PM, Seeger M. Herp regulates Hrd1-mediated ubiquitylation in a ubiquitin-like domain-dependent manner. J Biol Chem. 2011;286:5151–5156. doi: 10.1074/jbc.M110.134551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokame K, Agarwala KL, Kato H, Miyata T. Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J Biol Chem. 2000;275:32846–32853. doi: 10.1074/jbc.M002063200. [DOI] [PubMed] [Google Scholar]
- Kondratyev M, Avezov E, Shenkman M, Groisman B, Lederkremer GZ. PERK-dependent compartmentalization of ERAD and unfolded protein response machineries during ER stress. Exp Cell Res. 2007;313:3395–3407. doi: 10.1016/j.yexcr.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Liang G, Li Q, Tang Y, Kokame K, Kikuchi T, Wu G, Chen XZ. Polycystin-2 is regulated by endoplasmic reticulum-associated degradation. Hum Mol Genet. 2008;17:1109–1119. doi: 10.1093/hmg/ddm383. [DOI] [PubMed] [Google Scholar]
- Lynes EM, et al. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2 +signaling. J Cell Sci. 2013;126:3893–3903. doi: 10.1242/jcs.125856. [DOI] [PubMed] [Google Scholar]
- Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. Herp is dually regulated by both the endoplasmic reticulum stress-specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. J Biol Chem. 2004;279:13792–13799. doi: 10.1074/jbc.M313724200. [DOI] [PubMed] [Google Scholar]
- Okuda-Shimizu Y, Hendershot LM. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prostko CR, Brostrom MA, Brostrom CO. Reversible phosphorylation of eukaryotic initiation factor 2 alpha in response to endoplasmic reticular signaling. Mol Cell Biochem. 1993;127–128:255–265. doi: 10.1007/BF01076776. [DOI] [PubMed] [Google Scholar]
- Ron E, Shenkman M, Groisman B, Izenshtein Y, Leitman J, Lederkremer GZ. Bypass of glycan-dependent glycoprotein delivery to ERAD by up-regulated EDEM1. Mol Biol Cell. 2011;22:3945–3954. doi: 10.1091/mbc.E10-12-0944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A, Standera S, Buerger E, Kikkert M, van Voorden S, Wiertz E, Koning F, Kloetzel PM, Seeger M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J Mol Biol. 2005;354:1021–1027. doi: 10.1016/j.jmb.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Shenkman M, Ehrlich M, Lederkremer GZ. Masking of an endoplasmic reticulum retention signal by its presence in the two subunits of the asialoglycoprotein receptor. J Biol Chem. 2000;275:2845–2851. doi: 10.1074/jbc.275.4.2845. [DOI] [PubMed] [Google Scholar]
- Shenkman M, Groisman B, Ron E, Avezov E, Hendershot LM, Lederkremer GZ. A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J Biol Chem. 2013;288:2167–2178. doi: 10.1074/jbc.M112.438275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolchinsky S, Yuk MH, Ayalon M, Lodish HF, Lederkremer GZ. Membrane-bound versus secreted forms of human asialoglycoprotein receptor subunits—role of a juxtamembrane pentapeptide. J Biol Chem. 1996;271:14496–14503. doi: 10.1074/jbc.271.24.14496. [DOI] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Tuvia S, Taglicht D, Erez O, Alroy I, Alchanati I, Bicoviski V, Dori-Bachash M, Ben-Avraham D, Reiss Y. The ubiquitin E3 ligase POSH regulates calcium homeostasis through spatial control of Herp. J Cell Biol. 2007;177:51–61. doi: 10.1083/jcb.200611036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Laar T, Schouten T, Hoogervorst E, van Eck M, van der Eb AJ, Terleth C. The novel MMS-inducible gene Mif1/KIAA0025 is a target of the unfolded protein response pathway. FEBS Lett. 2000;469:123–131. doi: 10.1016/s0014-5793(00)01253-9. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Yamaguchi D, Hu D, Matsumoto N, Yamamoto K. Human XTP3-B binds to {alpha}1-antitrypsin variant nullHong Kong via the C-terminal MRH domain in a glycan-dependent manner. Glycobiology. 2010;20:348–355. doi: 10.1093/glycob/cwp182. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. 2004;136:343–350. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- Zhao G, Zhou X, Wang L, Li G, Kisker C, Lennarz WJ, Schindelin H. Structure of the mouse peptide N-glycanase-HR23 complex suggests co-evolution of the endoplasmic reticulum-associated degradation and DNA repair pathways. J Biol Chem. 2006;281:13751–13761. doi: 10.1074/jbc.M600137200. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.