

Dynamic actin filament arrays localize to polygonal spaces between ER sheets and tubules and regulate lateral movement and transformations of sheets, that is, sheet persistence, and thereby the characteristic ER network architecture in cultured mammalian cells. Myosin 1c has a role in creating and/or maintaining ER-associated actin arrays.
Abstract
The endoplasmic reticulum (ER) comprises a dynamic three-dimensional (3D) network with diverse structural and functional domains. Proper ER operation requires an intricate balance within and between dynamics, morphology, and functions, but how these processes are coupled in cells has been unclear. Using live-cell imaging and 3D electron microscopy, we identify a specific subset of actin filaments localizing to polygons defined by ER sheets and tubules and describe a role for these actin arrays in ER sheet persistence and, thereby, in maintenance of the characteristic network architecture by showing that actin depolymerization leads to increased sheet fluctuation and transformations and results in small and less abundant sheet remnants and a defective ER network distribution. Furthermore, we identify myosin 1c localizing to the ER-associated actin filament arrays and reveal a novel role for myosin 1c in regulating these actin structures, as myosin 1c manipulations lead to loss of the actin filaments and to similar ER phenotype as observed after actin depolymerization. We propose that ER-associated actin filaments have a role in ER sheet persistence regulation and thus support the maintenance of sheets as a stationary subdomain of the dynamic ER network.
INTRODUCTION
The endoplasmic reticulum (ER) is a large, single-copy, membrane-bound organelle that is essential for synthesis, modification, and transport of membrane and secretory proteins, as well as several lipids, and is also the site for cytosolic calcium level regulation (Baumann and Walz, 2001). The ER comprises an elaborate three-dimensional (3D) network of diverse structural domains, including tubules with high membrane curvature, flat sheets, and parts that form contacts with nearly every other organelle (Friedman and Voeltz, 2011). To accommodate the vast range of functions, the ER network spreads throughout the cell, and its functions are distributed into structural subdomains according to their specific requirements. The tubule-associated ER functions include interactions that the ER network has with several other cell organelles (Friedman et al., 2010, 2013; Ryan et al., 2012; Asanov et al., 2013). On the other hand, based on the distribution of ER sheet-bound ribosomes (Puhka et al., 2012) and the direct correlation between ribosome-studded ER (RER) abundance and secretion activity of the cell (Wiest et al., 1990; Rajasekaran et al., 1993; Benyamini et al., 2009; Ueno et al., 2010), the synthesis, folding, and quality control of proteins are functions assigned especially to ER sheets.
Whereas integral membrane proteins are indispensable for ER structure, dynamics and network remodeling are accomplished through interactions with the cytoskeleton. The network architecture is defined by tubule and sheet persistence and the network-remodeling processes including transitions of sheet to tubules and vice versa and fission and fusion of subdomains. Different mammalian cell types vary greatly in their sheet and tubule proportions, as well as in sheet morphology, and show variation in structural transformations during cell division (Shibata et al., 2006; Puhka et al., 2012). In human hepatocyte–derived carcinoma (Huh-7) cells, for instance, the abundant ER sheets are heavily fenestrated, extend to the cell periphery, and are transformed into smaller and highly fenestrated sheet remnants during cell division (Puhka et al., 2012), whereas CHO-K1 cells have less abundant and small intact sheets and undergo complete sheet-to-tubule transformation during cell division (Puhka et al., 2007).
Tubules are stabilized by an assortment of membrane-bending proteins (Shibata et al., 2006; Voeltz et al., 2006; Hu et al., 2009), and their movement is accomplished through interactions with microtubules (MTs; Terasaki et al., 1986; Waterman-Storer and Salmon, 1998). The mechanisms used to form sheets or accomplish sheet transformations are largely unknown, and it has been suggested that sheets are formed spontaneously and do not require stabilization (Goyal and Blackstone, 2013). However, tubule-stabilizing proteins have been shown to provide high curvature at sheet edges (Shibata et al., 2010), and the relatively constant luminal thickness of ER sheets is maintained through proteins that form intraluminal bridges or cytoplasmic scaffolds (Klopfenstein et al., 2001; Benyamini et al., 2009). Other sheet-stabilizing mechanisms could involve the protein synthesis and folding machinery, consisting of a translocon complex with bound ribosome and luminal chaperones (Puhka et al., 2007, 2012).
Therefore the critical questions are how the functional subdomains are created and maintained and why the network is constantly being rearranged. To elucidate the interplay between ER dynamics, morphology, and functions, we need to learn more about the factors involved. Previous studies of ER dynamics have concentrated almost entirely on tubules. The most common tubule dynamics (ER sliding) involves nascent tubule growth along acetylated MTs, whereas the tip attachment complex mechanism (Waterman-Storer and Salmon, 1998) occurs only on nonacetylated MTs (Friedman et al., 2010). Although MTs are the major molecular tracks for ER tubule movement in animal cells, MTs and ER tubules do not have identical distribution in cells, and the ER can also interact with actin cytoskeleton in differentiated animal cells (Sanger et al., 1989; Baumann, 1992; Terasaki and Reese, 1994; Stürmer et al., 1995; Takagishi et al., 1996; Bridgman, 1999; Wang et al., 2002; Poteryaev et al., 2005; Wagner et al., 2011). Furthermore, actin-based mechanical linkages have been shown to enable ER spreading in mouse embryonic fibroblasts (Lynch et al., 2011). However, the mechanisms by which actin contributes to ER dynamics and morphology are unclear.
In the present study, we characterize the interplay between ER and the actin cytoskeleton in two mammalian cell lines. We show that an array of dynamic actin filaments localize in the polygons defined by the surrounding ER structures and propose that these filaments have a role in maintaining sheet–tubule balance and sheet structure by regulating ER sheet-remodeling events. We reveal a novel role for the unconventional motor protein myosin 1c in regulating these actin filament arrays. Similarly to actin depolymerization, myosin 1c depletion and dominant-negative expression of a mutated form of myosin 1c with abolished actin binding lead to loss of actin filament arrays and subsequent loss of ER sheets. We mimicked the naturally occurring relocation or loss of polygonal actin filament arrays by using an actin-depolymerizing drug and provide a mechanistic view of the interplay between ER and actin by showing that the loss of polygonal actin filament arrays allow ER sheets to move in a larger area and that the lateral sheet movement is accompanied by induction of sheet transformations. Thus, in addition to tubular associations with MTs, interactions with the actin cytoskeleton are essential to create and maintain ER sheet persistence and the characteristic architecture of the ER network in mammalian cells.
RESULTS
Actin and MT cytoskeleton depolymerization have opposite effects on ER sheet–tubule ratio
To study the effects of actin or MT cytoskeleton depolymerization on ER, we treated Huh-7 cells with latrunculin A or nocodazole, respectively, stained for endogenous luminal ER protein calreticulin, and imaged focusing at cell lamella where ER reside in one plane and sheets and tubules can be distinguished. In untreated cells, the ER was composed of tubules and abundant sheets extending to the cell periphery. Intersection quantification of light microscopy (LM) images taken from the lamella revealed that the sheet–tubule ratio was 67/33%. Consistent with previous studies (Terasaki et al., 1986), the depolymerization of nonacetylated MTs (Friedman et al., 2010) with 30-min nocodazole treatment shifted the ER structural balance toward sheets, resulting in 82/18% sheet–tubule ratio and retraction of the ER network toward the nuclear envelope (NE). After 15-min latrunculin A treatment, the amount of sheets at the cell periphery decreased (22/78% sheet–tubule ratio) and the ER network appeared more reticular (Figure 1, A and B). Similar effects were observed in Huh-7 cells overexpressing luminal ER marker (Supplemental Figure S1A) and HeLa cells immunolabeled for calreticulin (Supplemental Figure S2A). The relative fluorescence intensity of calreticulin was unaltered after nocodazole or latrunculin A treatment compared with control cells (Supplemental Figure S2B), suggesting that the structural changes observed after MT and actin depolymerization resulted from structural conversion between sheets and tubules and not from consumption of the ER.
FIGURE 1:

Actin and MT depolymerization have opposite effects on the ER sheet–tubule ratio in Huh-7 cells. (A) Wide-field image of fixed control and nocodazole- and latrunculin A–treated cells immunolabeled for calreticulin. Magnifications from the boxed areas (a and b) were Gaussian filtered. Tubules (arrows) and abundant sheets (arrowheads) are indicated. See also Supplemental Figures S1A and 2A. (B) Images were quantified by the intersection method, and results are expressed as relative proportions (percentage) of sheets and tubules. Error bars, ±SD. (C) Wide-field images and (D) relative fluorescence intensity measurements of fixed control and latrunculin A–treated cells stained with phalloidin. Long actin filaments (open arrowheads), filament arrays <5 μm (arrows) and foci (arrowheads) are shown with higher magnification in boxed regions (insets a and b). Error bars, ±SEM. See also Supplemental Figures S1, B and C, and S2, C and D. Bars, 10 μm.
It is noteworthy that cells remained attached to the substratum and their shape was not altered after 15 min of latrunculin A (Figure 1C). In untreated cells, phalloidin staining revealed long actin filaments, that is, cortical actin and stress fibers (open arrowheads), and, in addition, shorter, <5-μm-long actin filament arrays (arrows) and punctate-like foci (arrowheads; Figure 1C for Huh-7 and Supplemental Figure S2C for HeLa cells). The thickness or length of any of the structures was not constant, indicating that the observed actin arrays were composed of varying numbers of actin filaments. After latrunculin A treatment, longer actin filaments remained unaffected, whereas most of the shorter actin filament arrays collapsed into actin foci (Figure 1C for Huh-7 and Supplemental Figure S2C for HeLa cells), revealing a direct dynamic relation between arrays and foci and further suggesting that foci might be the initial polymerization sites. The effect of latrunculin A treatment on actin filaments and free monomeric actin can be monitored by phalloidin (Figure 1C) and DNaseI staining (Supplemental Figure S1B; Mannherz et al., 1980), respectively. The quantitation of the relative fluorescence intensity revealed a 34% decrease in filamentous actin and, correspondingly, 55% increase in monomeric actin upon latrunculin A treatment (Figure 1D and Supplemental Figure S1C). Similar results were obtained from experiments done in HeLa cells (Supplemental Figure S2D). In conclusion, the observed changes in ER morphology upon actin depolymerization (Figure 1A) are most likely due to collapse of actin filament arrays into foci, further suggesting involvement of a specific pool of actin in maintaining ER sheet–tubule balance
Actin depolymerization results in reduced sheets and unevenly distributed ER network
To analyze the effects of actin depolymerization on ER morphology in more detail, we prepared Huh-7 cells for serial block-face scanning electron microscopy (SB-EM) imaging (Puhka et al., 2012). The cells expressed ssHRP-KDEL in the ER lumen, allowing cytochemical ER staining to improve the contrast for semiautomated segmentation. ER models from control (Figure 2A and Supplemental Video S1) and latrunculin A–treated (Figure 2B and Supplemental Video S2) cells revealed that actin depolymerization leads to altered ER sheet and network morphology. First, the sheets became smaller (Figure 2Bc) and less abundant (Figure 2Ba) compared with highly fenestrated and large control sheets (Figure 2A, c and a), and second, although the ER network remained continuous, its distribution was uneven, and areas void of ER were frequently found (asterisk in Figure 2Bb). The 3D EM models covered an area extending from cell periphery to the NE and therefore enabled detailed structure identification within the whole 3D volume of the cell; they thus provided a more reliable visualization of the effects of the treatment compared with LM analysis. Owing to better resolution and reliable identification of tubules, we found that the portion of tubules was lower than quantitation of LM images indicated, suggesting that some profiles categorized as tubules in LM images after latrunculin A treatment (Figure 1, A and B) might actually originate from diminished sheets.
FIGURE 2:

Actin depolymerization–induced sheet remnants and uneven ER network distribution are revealed by 3D EM. (A) Control and (B) latrunculin A–treated Huh-7 cells transiently expressing ssHRP-KDEL were chemically fixed, cytochemically stained (dark precipitate), and imaged with SB-EM. (a) Model of the ER network (yellow) and the nuclear envelope (NE; blue), (b) one block face image; (c) a representative fenestrated sheet. Asterisk, area void of ER within unevenly distributed ER network; arrowheads, sheets; arrows, tubules. See Supplemental Videos S1 and S2 for the SB-EM data set and models. Bars, 1 μm (in perspective view [a] apply to center of image).
An array of actin filaments localizes to ER polygons and affects ER sheet transformations
To study the dynamic interplay between actin filament arrays and ER, we visualized ER and actin simultaneously in live Huh-7 cells. Live-cell imaging of ER sheets can be challenging, as they often reside at the densely occupied cell center and are large enough to occupy more than one imaging plane. Here we acquired videos of the ER from the thin lamella at the leading edge of the cells. The ER in these plate-like extensions resides mostly in one plane, allowing simultaneous imaging and identification of tubules and individual sheets. Live-cell confocal imaging of ER and actin revealed the close proximity and coinciding movement of ER and actin filament arrays within nonuniform polygons defined by the surrounding dynamic ER network of tubules and/or sheets (Figure 3A and Supplemental Video S3). Actin filament arrays and foci resembled the phalloidin-stained structures shown in Figure 1C and were found throughout the cell volume (Supplemental Figure S3), diverging from relatively longer actin filaments, that is, stress fibers or cortical actin, which had no clear interactions with the ER and mostly localized to cell bottom and cell periphery (Figure 3A and Supplemental Video S3). Similar results could be observed using another actin marker, LifeAct–red fluorescent protein (RFP; Figure 3A). It is noteworthy that the polygons are not fenestrations on ER sheets, as they are considerably smaller openings and beyond the resolution limit of conventional LM (Puhka et al., 2012).
FIGURE 3:

Actin filament arrays localize to polygons defined by the surrounding ER network and have a role in ER sheet movements in Huh-7 cells. (A) Confocal section of live cell coexpressing Hsp47-GFP (green) and mCherry-Actin (magenta) or LifeAct-RFP (magenta). Cortical actin (arrowheads), stress fibers (open arrowheads), and actin filament arrays and foci (arrows) localizing to the polygons (asterisks) are indicated. See also Supplemental Video S3. Time-lapse confocal frames of a cell coexpressing Hsp47-GFP (green) and mCherry-actin (magenta) show that (B) relocation and disappearance and (C) formation of actin arrays and the ER sheet dynamics are interdependent. In B, actin foci (arrow) relocate from polygon (1) to the adjacent polygon at 00:08, leading to subsequent polygon (1) closure at 00:11. Disappearance of actin foci (arrowhead) at 00:13 leads to subsequent polygon (2) closure. See also Supplemental Video S4. In C, arrowhead indicates formation of actin filament array leading to polygon (3) opening within the ER sheet at 00:09. See also Supplemental Figure S3 and Supplemental Video S5. Bars, 5 μm.
Our analysis of the movement of actin filament arrays and foci with respect to ER revealed that the natural relocation, disappearance, or formation of the actin structures frequently preceded polygon closure or opening, respectively, leading to subsequent ER sheet transformation (Figure 3, B and C). These results are consistent with previous studies showing that, at steady state, the polygonal ER network is balanced by ring closure (Lee and Chen, 1988) and that filling of a polygon within an ER sheet leads to larger sheets (Griffing, 2010). One characteristic transformation event that we frequently observed was membrane fusion leading to larger sheets (i.e., ring closure) followed closely by loss of associated actin filament (Figure 3B and Supplemental Video S4). We also observed that the formation of polygons in ER sheets followed the formation of an actin filament array (Figure 3C and Supplemental Video S5), indicating that the dynamics of actin arrays and ER sheet transformations are interdependent.
Myo1c localize to actin filament arrays and foci
Next we wanted to identify proteins participating in ER–actin interplay to gain a better understanding of the mechanism. We performed a screen in which >200 known functional human actin–binding proteins were depleted one by one and analyzed by LM to identify those with a role in ER morphology and/or dynamics. On the basis of the screen, we identified the unconventional motor protein myosin 1c (myo1c), whose depletion led to a strong ER phenotype and, in addition, affected the appearance of the actin filament arrays. It is noteworthy that while the library contained most of the known myosins, including other members of the myosin 1 family (a–f), the combined ER and actin filament array phenotype was observed only in conjunction with myo1c (screening data not shown). Myo1c has been suggested to link the actin cytoskeleton to cellular membranes (Novak et al., 1995) and localize to actin-rich sites of membrane ruffling and retraction at the cell periphery and to discrete punctate within the cytoplasm (Wagner et al., 1992; Wagner and Molitoris, 1997; Ruppert et al., 1995; Hokanson et al., 2006); therefore it was a good candidate for further studies. Immunolabeling of myo1c with respect to actin revealed that myo1c localized to the short actin filament arrays and foci throughout the cytoplasm (Figure 4A). Live-cell confocal imaging of the cell lamella demonstrated that the myo1c-positive actin filaments alternated between dynamic and static stages (Figure 4B and Supplemental Video S6). The appearance of myo1c–actin filaments changed substantially over time: the actin foci were observed to transform into actin filament arrays and vice versa, form plate-like actin structures, and undergo fusion and fission with other actin filaments and foci (Supplemental Video S6). Whereas the actin filament arrays and foci appearances were dynamic (i.e., their structure was constantly changing), the structures seemed to fluctuate in only a small area rather than to have directional movement (Figure 4B).
FIGURE 4:

Myo1c localizes and moves in conjunction with actin arrays in Huh-7 cells. (A) Wide-field image of a lamella of fixed cell expressing myo1c-2FLAG, immunolabeled with anti-FLAG antibody (magenta), and stained with phalloidin (green). Boxed areas are shown at higher magnification, portraying myo1c-positive actin filaments at the cell periphery (inset a) and in the cytoplasm (inset b). Occasional actin filaments negative for myo1c were found (inset c). Long actin filaments (open arrowheads) were not positive for myo1c. See also Supplemental Figures S2H and S3. (B) Time-lapse confocal frames (time scale, minutes:seconds) of cell coexpressing myo1c-EGFP (green) and mCherry-actin (magenta) taken from cell lamella show that the actin structures can be both dynamic (actin filament array; arrowhead) and static (actin foci; arrow) and that myo1c moves in conjunction with both actin filaments and foci. See also Supplemental Video S6. Bars, 10 μm (A), 5 μm (B).
To characterize the actin filament arrays further, we tested a number of proteins known to interact with actin, using immunofluorescence: the majority of actin filament arrays and foci were positive for cortactin (Supplemental Figure S4A). Quantification of the actin structures revealed that 97 ± 6.4% (SD) and 94 ± 3.8% (SD) of the actin filament arrays were positive for myo1c or cortactin, respectively. Immunolabeling of cortactin with respect to myo1c revealed that the proteins clearly decorate the same actin structures but not with identical labeling patterns (Supplemental Figure S4B). In addition, the following proteins did not show preference to actin filament arrays: stress fiber–associated proteins MHCIIA and MHCIIB (Vallenius, 2013); actin cross-linking and stress fiber–localizing protein α-actinin1 (Lazarides and Burridge, 1975); actin-stabilizing and conventional myosin II–binding stress fiber protein calponin (Strasser et al., 1993); tropomyosin (Gunning et al., 2008), which regulates actin filament association with several proteins; proteins associated with actin anchorage to the plasma membrane (PM), vinculin (Hazan et al., 1997) and moesin 1 (Gautreau et al., 2000); and a protein involved in receptor-independent endocytosis, caveolin (Pelkmans and Helenius, 2002).
Myo1c has a role in creating and/or maintaining actin filament arrays
We next wanted to study the relationship between myo1c and associated actin filament arrays. Based on wide-field LM, overexpression of myo1c in Huh-7 cells led to curved, bundled, and unorganized actin filament arrays compared with punctate or rather straight actin filament arrays in control cells (Figure 5, A and B). The overexpression of myo1c seemed to induce formation of actin filament arrays at the expense of disappearance of actin foci (Figure 5B). The depletion of myo1c, on the other hand, abolished actin filament arrays, and only actin foci were observed (Figures 5C and 6C). Together these results indicate that myo1c is required for formation of actin filament arrays.
FIGURE 5:

Myo1c regulates actin filament arrays in Huh-7 cells. Wide-field images of fixed cells expressing (A) CMVTag1 (control), (B) myo1c-2FLAG, (D) EGFP-myo1cΔABL, or (C) depleted with myo1c shRNAs were stained with phalloidin. Boxed areas are shown at higher magnification. (D) The EGFP-myo1cΔABL signal is observed as bright spots at the perinuclear area and as diffuse signal throughout the cytoplasm (inset a). See also Supplemental Figures S2E and S4. Bars, 10 μm.
FIGURE 6:

Myo1c manipulations lead to loss of sheets and ER network distribution deficiency in Huh-7 cells. (A) Wide-field image of myo1c overexpression–induced ER phenotype in fixed cell coexpressing Hsp47-GFP and 2FLAG-C1 (control) or myo1c-2FLAG (myo1c overexpression) and immunolabeled with anti-FLAG antibody (insets 1 and 2). Boxed areas (a and b) are shown at higher magnification. Sheets/interlinked sheet mass (arrowhead) and tubules (arrows) are indicated. See also Supplemental Figure S2F. (B) Live wide-field images of control (CMVTag1) or myo1c-depleted (shRNA) cells expressing Hsp47-GFP. ER tubules (arrows) and sheets (arrowheads) are indicated. See Supplemental Figures S2G and S6A for comparison. (C) Cells transfected with CMVTag1 (control) or pooled myo1c shRNAs (myo1c depletion) were analyzed by Western blotting with the indicated antibodies. See also Supplemental Figure S6B. (D) A cell coexpressing ssHRP-KDEL and pooled myo1c shRNAs was cytochemically stained (dark precipitate) and imaged with SB-EM (see Supplemental Video S7 for SB-EM data set and model). (a) Model of ER network (yellow) and NE (blue); (b) one block face image; (c) a representative sheet remnant. Sheet remnants (arrowheads) and tubules (arrow) are indicated. See Figure 2, A and B, for comparison. Bars, 5 μm (A), 10 μm (B), and 1 μm (D; in the perspective view [a] apply to center of image).
Next we created a green fluorescent protein (GFP)–tagged mutant myo1c construct with abolished actin binding (enhanced GFP [EGFP]–myo1cΔABL; see, e.g., Sasaki et al., 1999). Whereas wild-type (wt) myo1c localized heavily under the PM and to distinct foci at the cytoplasm (Figure 4A), EGFP-myo1cΔABL was observed as bright spots, likely protein aggregations, at the perinuclear area and as a diffuse signal throughout the cytoplasm (Figure 5D, inset a). In EGFP-myo1cΔABL–overexpressing cells, bright actin foci could be observed instead of arrays of actin filaments (Figure 5D), resembling latrunculin A–treated (Figure 1C) and myo1c-depleted (Figure 5C) cells, providing further evidence that myo1c, and more precisely the actin-binding domain, is essential for the creation of actin filament arrays. It is noteworthy that none of the aforementioned manipulations affected the overall shape of the cells, as the cells remained flat and attached to the substratum (Figure 5).
Based on our results, myo1c and cortactin decorated the same actin filament structures (Supplemental Figure S4). Furthermore, upon overexpression of EGFP-myo1cΔABL, the resulting actin foci remained positive for cortactin (Supplemental Figure S5A). Because cortactin is involved in both receptor-mediated (Cao et al., 2003) and non–receptor-mediated (Kelley and Weed, 2012) endocytosis, we next tested whether myo1c has a role in these endocytic events. To compare the efficiency of receptor- and non–receptor-mediated endocytosis in controls and myo1c-depleted Huh-7 cells, we measured the uptake of transferrin Alexa Fluor 647 and dextran Alexa Fluor 594, respectively (Supplemental Figure S5, B–E). The results show that myo1c depletion and the resulting alterations in actin filament appearance did not induce relevant changes in receptor- or non–receptor-mediated endocytosis. Together these results indicate that although myo1c partly localizes under the PM and to structures involved in endocytosis, it is most likely not involved in endocytosis, and its main function is instead to create and/or maintain actin filament arrays.
Myo1c manipulations affect ER sheet morphology and characteristic network distribution
So far, we have shown that both latrunculin A treatment and myo1c depletion abolish actin filament arrays, and actin depolymerization leads to subsequent loss of ER sheets. We next wanted to study the effects of myo1c manipulations on ER morphology. The overexpression of FLAG-tagged myo1c construct (myo1c-2FLAG) in Huh-7 (Figure 6A) and HeLa (Supplemental Figure S2F) cells shifted the ER sheet–tubule ratio toward sheets, with few tubules remaining and, instead of individual sheets, an interlinked sheet mass. In contrast, expression of 2FLAG-C1 construct alone did not induce changes in the ER (Figure 6A for Huh-7 and Supplemental Figure S2F for HeLa). The overexpression of myo1c saturated the natural binding sites, and strong accumulation of myo1c signal at the cell periphery and distinct punctae in the cytosol was observed. Expression of the control FLAG-construct (2FLAG-C1) was observed as diffuse staining in the cytosol (Figure 6A).
Wide-field live-cell imaging revealed that myo1c depletion led to uncharacteristic ER network appearance, with long tubules extending to the cell periphery and few degenerated sheets at the lamella (Figure 6, B and C). Morphologically, the phenotype resembled the ER in latrunculin A–treated cells (Figure 1A); that is, loss of sheets and induction of uneven network distribution was observed, although the magnitude of the change varied. The slight difference between observed ER phenotypes might result from very different treatment durations, 48 h for myo1c depletion and 15 min for latrunculin A. In general, drug treatments have a more global effect, affecting the whole cell culture uniformly, in comparison to protein depletion experiments, in which the level of depletion changes over time and varies from cell to cell. Furthermore, the myo1c depletion efficiency with short hairpin RNAs (shRNAs) varied and on average led to 60% depletion of myo1c (unpublished data). Myo1c depletion with target-specific pooled small interfering RNAs (siRNAs) led to a similar morphological phenotype (Supplemental Figure S6, A and B) as with shRNAs. We did not find any significant difference in protein synthesis and secretion rates between control, myo1c-depleted, and latrunculin A–treated cells after metabolic labeling for 20 min and 1- or 4-h chase (Supplemental Figure S7).
The myo1c-depletion-induced morphological changes were even more evident in 3D models obtained from SB-EM data sets. Control Huh-7 sheets were large and heavily fenestrated (Figure 2A and Supplemental Video S1), whereas in myo1c-depleted cells the sheets were smaller, unequally fenestrated, and less abundant (Figure 6D and Supplemental Video S7) resembling the actin-depolymerization phenotype (Figure 2B). The ER network remained continuous. Similar results were observed when myo1c was depleted using pooled siRNAs (Supplemental Figure S6C). Similar to the latrunculin A results, the 3D-EM results show that part of the reticular ER network appearance observed under wide-field LM resulted from interconnected consumed sheet remnants and tubules.
Our results showed that overexpression of EGFP-myo1cΔABL abolishes actin filament arrays, and so we next wanted to study the effect of EGFP-myo1cΔABL overexpression on ER. Based on confocal live-cell imaging, overexpression of the construct led to almost complete structural shift of ER toward a reticular network, as only few peripheral sheets were observed (Figure 7A), providing further support to the role of myo1c in maintenance of ER sheet–tubule balance through actin filament arrays.
FIGURE 7:

Myo1c actin-binding domain is crucial for proper ER phenotype in Huh-7 cells. (A) Confocal section of a live cell coexpressing Hsp47-mCherry (magenta) and EGFP- myo1cΔABL (green). Tubules (arrows) and the few remaining sheets (arrowheads) are indicated in the magnified image of the boxed area. See also Figure 5D and Supplemental Figure S2I. For rescue experiments, Huh-7 cells were transfected with Cy5-conjugated myo1c siRNA (blue) and, on the next day, siRNA-resistant construct (B) myo1c-EGFP (green) or (C) EGFP-myo1cΔABL (green) and immunolabeled for calreticulin (red). In B, tubules (arrows) and sheets/interlinked ER sheet mass (arrowheads) are indicated in the wide-field images of myo1c-depleted cell (outlined cell; D) and rescued cell (R). In C, the tubules (arrows) of nonrescued cells expressing EGFP-myo1cΔABL are indicated. See Figure 6A for comparison. Bars, 10 μm.
Although the off-target effects of myo1c depletion were addressed by using scrambled siRNA sequences and multiple siRNA target constructs, we designed rescue experiments using the expression of myo1c siRNA–resistant wt myo1c (myo1c-EGFP) and a mutant construct of myo1c with abolished actin-binding domain (EGFP-myo1cΔABL) to provide further control for the experiment. In myo1c-depleted Huh-7 cells (myo1c-siRNA-Cy5), expression of myo1c-EGFP led to a strong sheet-like ER phenotype (calreticulin-immunolabeled cell marked with R in Figure 7B) compared with the tubular phenotype (marked with D in Figure 7B) in nonrescued cells. Instead of leading to a normal ER phenotype (Figure 1A), the rescued phenotype resembled the sheet-like phenotype of the myo1c-overexpressed cells (shown in Figure 6A), showing the sensitivity of control of myo1c level in cells. In contrast, expression of EGFP-myo1cΔABL did not rescue the myo1c-depletion ER-phenotype, as the ER remained tubular (Figure 7C).
Depolymerization of ER-associated actin filament arrays increases sheet fluctuations and transformations
So far, we have shown that ER sheet dynamics is coupled to actin filament arrays and foci localizing to polygons and that disruption of actin filament arrays changes ER sheet–tubule balance and affects ER sheet size, abundance, and morphology. We next studied ER sheet dynamics and the role of ER-associated actin arrays in ER sheet transformations. As a tool to disrupt actin filament arrays, we used short latrunculin A treatment, as it provided a reliable and consistent tool to mimic the naturally occurring relocation or loss of actin filament arrays from ER polygons in real time (Figure 3, B and C). Huh-7 cells were imaged for 60 s (1 frame/s) with wide-field LM focusing at the cell lamella, and sheets with clear boundaries were randomly selected from the first acquired frame and observed for the acquisition time or until sheet transformation events (sheets transforming into tubules, undergoing sheet fission, or showing fusion events) were observed (Figure 8A), and these were then scored accordingly. The sheets were remarkably stable: 75% persisted for the analysis duration. On latrunculin A treatment, the portion of sheet transformations increased greater than threefold, as only 14% of the sheets persisted. Depolymerization of MTs by nocodazole did not have any effect on sheet persistence (Figure 8B). Sheet lifetime, counted from the first acquired frame, decreased from 53.7 ± 1.5 s (SEM; control) to 18.4 ± 2.8 s (SEM) in latrunculin A–treated cells. In accordance, actin depolymerization in HeLa affected sheet persistence and lifetime in a similar manner (proportion of persistent sheets in control, 49%; lifetime, 42.6 s ± 2.4 [SEM]; after latrunculin A treatment, 38% and 35.6 ± 3.1 s [SEM], respectively).
FIGURE 8:

Depolymerization of ER-associated actin filaments increases sheet transformations and sheet fluctuations in Huh-7 cells. (A) Wide-field time-lapse images of representative sheet (outlined) dynamics of cells expressing Hsp47-GFP. (B) Relative proportions (%) of persistent sheets vs. transforming sheets over the 60-s observation time (1 frame/s) in control and nocodazole- and latrunculin A–treated cells. (C) Still frame of a wide-field video subjected to center-of-mass analysis. The boxed area of a cell expressing Hsp47-GFP is shown at higher magnification on right, where the manually traced sheets and their calculated centroid (asterisks) are indicated. (D) Representative trajectories of sheet centroid movements over 60 s, plotted in a 1-μm2 area, of control and latrunculin A– and trichostatin A–treated cells and (E) the subgrouped centroid distances (μm) to the initial centroid position plotted against occurrence (%) and average distance (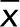 , μm ± SD) to the initial centroid position. The lateral movement increased significantly (**p < 0.05) with latrunculin A but not trichostatin A treatment compared with controls. Bars, 2.5 μm (A and magnified image in C), 10 μm (otherwise in C).
, μm ± SD) to the initial centroid position. The lateral movement increased significantly (**p < 0.05) with latrunculin A but not trichostatin A treatment compared with controls. Bars, 2.5 μm (A and magnified image in C), 10 μm (otherwise in C).
To study lateral sheet movement and velocity, we traced outlines of individual sheets manually throughout the 60-s wide-field acquisition (1 frame/s). Based on the calculated center-of-mass of the sheet (i.e., the centroid), the centroid's movement and velocity were quantified (Figure 8, C–E). The analysis was done only for persistent sheets to ensure that events that significantly increased or reduced sheet mass would not artificially shift the centroid. The observation duration was set to 60 s so that cell motility would not affect centroid movement and to ensure representative balance between persistent and transforming sheets. The analysis showed that in untreated cells, sheet movements did not exhibit clear directionality, as the sheets mainly fluctuated in a small area (Figure 8D). To analyze the effect of actin depolymerization and MT hyperacetylation on ER sheet movement, we treated cells with latrunculin A or trichostatin A, respectively. Trichostatin A increased the proportion of acetylated MTs (Supplemental Figure S8, A and B; Friedman et al., 2010) that act as tracks for ER tubule sliding. The average sheet velocity in latrunculin A– and trichostatin A–treated cells (0.11 ± 0.04 and 0.11 ± 0.03 μm/s [SD], respectively) remained roughly the same as in control (0.15 ± 0.23 μm/s [SD]). Remarkably, after latrunculin A treatment, the average sheet centroid distance to the initial centroid position increased significantly (p < 0.05), indicating that actin depolymerization allowed a greater lateral movement of sheets, whereas MT hyperacetylation had no effect on lateral movement (Figure 8E). The average distance to the initial centroid position () increased significantly in latrunculin A (0.43 ± 0.34 μm [SD]) but not trichostatin A treatment (0.30 ± 0.30 μm [SD]) compared with controls (0.27 ± 0.32 μm [SD]). When centroid distance measurements to the initial centroid position were grouped into categories against occurrence, the histogram revealed an increased frequency of events at further distances in latrunculin A–treated cells compared with controls or trichostatin A–treated cells (Figure 8E). To conclude, sheets are persistent and relatively static structures that fluctuate in a small area rather than relocate over longer distances, and therefore sheet dynamics differs from that of tubules, which typically show clear directionality and greater velocity. Together our results provide a mechanistic view of the interplay between ER and actin, suggesting that loss of actin filament arrays in polygons allows ER sheets to move in a larger area and that lateral movement is subsequently accompanied by increased sheet transformation.
DISCUSSION
Although the interplay between ER tubules and MTs has been well described, the maintenance of the other abundant ER subdomain—sheets—and the role of the actin cytoskeleton on ER structure have been obscure. Dreier and Rapoport (2000) reported in vitro formation of a reticular ER network independent of MTs and actin; however, it is unknown whether ER sheets were present. Furthermore, the interaction of actin cytoskeleton with ER has mainly been shown in specialized cells and functions. Here we found that the interplay between ER and actin cytoskeleton is essential for the persistence of ER sheets in cultured mammalian cells and thereby for the maintenance of overall ER network architecture. ER tubules represent the dynamic part of the ER, whereas sheets are persistent, and the balance between the structures contributes to the characteristic ER network architecture.
ER responds to actin depolymerization readily, as 15-min latrunculin A treatment led to decreased sheet–tubule ratio and unevenly distributed ER network. The effect was strong in a hepatoma cell line that has abundant sheets but was also observed in another commonly used epithelial cell line, HeLa. ER sheet dynamics was subjected to quantitative analysis. Results showed that actin depolymerization led to increased sheet fluctuation and transformations and, based on 3D-EM, consumption of large sheets into sheet remnants. However, actin depolymerization did not induce changes in the directionality or velocity of sheets, indicating that sheets do not move along the actin cytoskeleton as tubules move in conjunction with growing MTs, but instead that sheet repositioning to another area of the cell is likely to occur through new sheet formation. We propose that the role of interaction between sheets and actin filaments is to provide stability to sheets and control sheet transformations.
On the basis of live-cell imaging, we suggest that actin foci and filament arrays are the same structures at different stages of polymerization, as the foci could transform into actin filament arrays and vice versa, although some of the observed foci might, in fact, resemble actin filament arrays observed perpendicular to the imaging plane. Although the dynamic actin filament arrays responded readily to all actin manipulations, the foci were remarkably resistant to the treatments. Whether actin foci simply resemble actin-polymerization initiation sites remains to be resolved. Further identification of ER-associated actin filaments and foci was challenging not only because actin is the most abundant protein in the cell cytoplasm and >250 actin-binding proteins are known, but because actin filaments poorly endure EM sample preparation (Svitkina, 2013). We could exclude stress fibers because ER-associated actin filament arrays are dynamic, relatively short (<5 μm), and did not colocalize with commonly accepted stress fiber markers, as well as cortical actin because of its restricted localization at cell cortex. We identified two actin-binding proteins that associate with the actin filament arrays, namely myosin 1c and cortactin.
Myo1c is one of the eight members of the myosin 1 family of unconventional motor proteins and was previously reported to be involved in actin cytoskeleton dynamics and membrane protein anchoring in B lymphocytes (Maravillas-Montero et al., 2011) and to have a role in multiple signaling pathways (Bond et al., 2013). In addition, it has been shown that myo1 and actin filaments provide mechanical stability to microvillar ER (Pollard et al., 1991). Here we describe a novel role for myo1c in regulating ER-coupled actin filament arrays. Myo1c depletion or expression of EGFP-myo1c∆ABL resulted in reduction of the short actin filaments to punctate structures, and overexpression of wt myo1c induced unorganized, curved actin filaments, indicating that myo1c is required for creation and/or maintenance of ER-associated dynamic actin filament arrays. In accordance, myosin I has been shown to bind only the dynamic actin filament population and not tropomyosin-stabilized microfilaments (Tang and Ostap, 2001).
We propose a model in which, in addition to a role in creating and/or maintaining dynamic actin filaments, myo1c is also responsible for guiding forming actin filament arrays in the vicinity of ER through its lipid interaction. There are indications of straight electrostatic binding between the noncatalytic domains of myo1c and negatively charged phosphatidylinositol 4,5-bisphosphate (PIP2; Reizes et al., 1994; Bose et al., 2004; Hirono et al., 2004; Hokanson et al., 2006; Hokanson and Ostap, 2006), which in turn regulate actin filament assembly and disassembly (Sechi and Wehland, 2000; Diefenbach et al., 2002; Holt et al., 2002; Yin and Janmey, 2003). In agreement, myr2 (rat myosin I) has been shown to bind PIP2 (Barylko et al., 2005) and, based on immunoreactivity studies, associate with fractions enriched with ER and PM (Ruppert et al., 1995). It has been suggested that tethering of the myosin to actin polymerization sites might serve to trigger membrane remodeling of the associated organelles (Chhabra et al., 2009; Coudrier and Almeida, 2011; Korobova et al., 2013). The actual mechanism for ER–actin interplay remains open; whereas myo1c serves to localize actin filaments to the vicinity of ER, these filament arrays might form passive physical restriction barriers within the ER network, or yet-unidentified factors may be needed to mediate interaction between ER and actin filaments.
Because the cytoplasm is full of MT- and actin-binding proteins that provide identity to cytoskeletal filaments in response to various intracellular and extracellular cues, interplay between ER subdomains and different cytoskeletal elements provides a variety of regulatory possibilities. In addition to integral membrane proteins that support specific structural features, interaction with the cytoskeleton provides a more global and dynamic mechanism to control ER architecture in cells. MT-driven tubular ER is suited to hosting dynamic ER functions and provides a mechanism for rearranging the overall network distribution within the cell. In contrast, more stationary and persistent ER sheets might be more suitable for protein synthesis and quality control or other ER functions. The functional importance of ER network transformation is an important research question for the future.
MATERIALS AND METHODS
Cell culture, constructs, and transfection
Huh-7 (JCRB0403; Japanese Collection of Research Bioresources Cell Bank, Osaka, Japan) and HeLa (C3005-1; BD Biosciences Clontech, Palo Alto, CA) cells were cultured as previously described (Kuokkanen et al., 2007; Puhka et al., 2012). ssGFPKDEL and mCherry-actin were described previously (Kuokkanen et al., 2007; Vartiainen et al., 2007). ssHRP-KDEL, Hsp47-GFP-KDEL, LifeAct-RFP, and 2FLAG-C1 were described previously (Connolly et al., 1994; Kano et al., 2005; Riedl et al., 2008; Dopie et al., 2012), and provided by the respective laboratories. Mouse myo1c coding sequence was amplified by PCR using NIH 3T3–derived cDNA and subsequently cloned into XhoI and BamHI restriction sites of pGFP-C1 (Clontech), resulting in myo1c-EGFP-C1. EGFP sequence of pEGFP-N3 (Clontech) was replaced with 2xFLAG (DYKDDDDK), resulting in 2FLAG-N3 vector, which was used as a backbone for cloning myo1c coding sequence at XhoI and BamHI restriction sites, resulting in myo1c-2FLAG-N3. GFP in Hsp47-GFP-RDEL was replaced by mCherry by using PCR and subcloning into BamHI-NotI sites, creating Hsp47-mCherry. Cells were transfected with FuGENE HD (Roche, Indianapolis, IN; and Promega, Madison, WI) according to the manufacturer's instructions and incubated for 24–48 h. For cotransfection of two plasmids, a 1:2 (wt/wt) ratio was used. For cotransfection of four plasmids, 1:1:1:1 (wt/wt/wt/wt) was used. To obtain myo1c construct with abolished actin binding, we replaced a short stretch of Myo1c polypeptide chain spanning residues 323–330 (IIAKGEEL) with the flexible tripeptide AGA. Based on findings by several other groups (e.g., Sasaki et al., 1999), deletion of this region, termed the “myopathy loop,” leads to disruption of essential hydrophobic interactions between actin filaments and myosin head domain, thus preventing them from binding to each other. EGFP-myo1cΔABL was prepared by means of an overlap-extension PCR method using a pair of partially overlapping mutant oligonucleotides and primers flanking the myo1c coding sequence. The PCR product carrying the mutation was subsequently cloned into pEGFP-C1 vector using BamHI and XhoI restriction sites.
Antibodies and cytoskeleton manipulations
All reagents are from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Antibodies against calreticulin (PA3-900; Affinity BioReagents, Golden, CO), β-tubulin (2128; Cell Signaling, Danvers, MA), moesin 1 (3150; Cell Signaling), myosin1c (SC-130177; Santa Cruz Biotechnology, Dallas, TX), calponin (sc-28545; Santa Cruz Biotechnology), MHCIIA (PRB-440P; Covance, Princeton, NJ), MHCIIB (PRB-445P; Covance), cortactin (p80/85, clone 4F11/05-180; Millipore, Billerica, MA), α-actinin1 (A5044; Sigma-Aldrich), acetylated tubulin (clone 6-11B-1, T7451; Sigma-Aldrich), FLAG-tag (F1804; Sigma-Aldrich), tropomyosin (T2780; Sigma-Aldrich), vinculin (V9131; Sigma-Aldrich), caveolin (C 4490; Sigma-Aldrich) and anti-myosin Iβ (M3567-2000UL; Sigma-Aldrich) were used as primary antibodies. Secondary antibodies were rhodamine red-X–conjugated AffiniPure goat anti-mouse immunoglobulin G (IgG) + IgM (115-295-04; Jackson ImmunoResearch, West Grove, PA) and goat anti-rabbit IgG (111-295-144; Jackson ImmunoResearch). The 1.4-nm nanogold anti-mouse IgG (2002) for immuno-EM labeling was from Nanoprobes (Yaphank, NY), and goat anti-mouse and anti-rabbit horseradish peroxidase–conjugated antibodies (ab20043 and ab6721, respectively) for immunoblotting were from Abcam (Cambridge, United Kingdom). The F-actin probes Alexa Fluor 568 (A12380) and 488 (A12379) phalloidin and G-actin probe DNaseI Alexa 594 (deoxyribonuclease I, Alexa Fluor 594 Conjugate, D12372) were from Invitrogen (Carlsbad, CA). MT and actin cytoskeleton manipulations were done before fixation with 10 μM nocodazole (M1404-10MG; Sigma-Aldrich) for 30 min, 125 nM trichostatin A (T8552; Sigma-Aldrich) for 90 min, 1 μM latrunculin A (L12370; Invitrogen) for 15 min for cells to be fixed and 15–45 min for live-cell imaging, and 0.5 μM jasplakinolide (J7473; Molecular Probes, Eugene, OR) for 60 min.
Immunofluorescence staining and immunoblotting
Cells grown on glass coverslips were fixed with 4% paraformaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA), 0.1 mM MgCl2, and 0.1 mM CaCl2 in phosphate-buffered saline (PBS), quenched with 50 mM NH4Cl, permeabilized with 0.1% Triton X-100, and then blocked with 1% bovine serum albumin in PBS. When appropriate, the cells were then incubated consecutively with primary and secondary antibodies, diluted in blocking solution, or stained with phalloidin according to manufacturer's instructions. Samples were mounted in Mowiol (4-88) supplemented with Dabco (D-2522). Immunoblotting was done with indicated antibodies according to manufacturer's instructions and by using standard protocols.
Myo1c depletion
The myo1c target–specific shRNAs were obtained from the Genome Biology Unit, Biocenter Finland (www.biocenter.helsinki.fi/bi/gbu/kd/). The sequences of the shRNAs were CAACCTGACCGGAATCTC, TCCATCAACGACAAGAGT, and TCAAAGAATCCCATTATG. Huh-7 and HeLa cells were grown on class coverslips and cotransfected at a 1:4 ratio (wt/wt) with Hsp47-GFP and an inert plasmid (pCMV-Tag1; 211170; Agilent Technologies) or the pooled three myo1c target–specific shRNAs (1:1:1 ratio) for 24–48 h. For analysis of the effects of myo1c depletion on the actin cytoskeleton, the cells were then fixed and stained with phalloidin. For myo1c siRNA depletion experiments, a 10 nM pool of three target-specific 20- to 25-nt myosin1c siRNAs (sc-44604; Santa Cruz Biotechnology) and the corresponding concentration of nontargeting control siRNA with at least four mismatches to any human gene (ON-TARGET plus, D-001810-10-20; Thermo Scientific, Waltham, MA) were transfected with INTERFERin (Polyplus-transfection, Illkirch-Graffenstaden, France) according to manufacturer's instructions for 24 h, followed by transfection of Hsp47-GFP with FuGENE HD for additional 24 h.
Light microscopy
The live wide-field images were taken using a TILL imaging system equipped with PLANAPO 60×/1.45 oil objective with additional 1.6× magnification in an IX-71 inverted microscope (Olympus, Tokyo, Japan) and iXon 885 camera and Imago QE (Andor, Oxford Instruments, Abingdon, UK) with heating (37°C) and CO2 supply. Time-lapse videos were acquired with either TILL Photonics (FEI Company, Hillsboro, OR) or TILL Vision software, focusing at the cell lamella. Live-cell confocal images of Huh-7 were taken with an inverted TCS SP5II HCS A confocal (Leica, Solms, Germany) using an HCX PL APO (Leica) lambda blue 63.0×/1.2 W Corr/0.17 CS (water) objective, HyD detector (Leica) with either standard or BrightR detection mode, red (HeNe 633 nm/12 mW) and blue (Ar 488 nm/35 mW) laser lines, and DD 488/561beam splitter. Cells were grown on glass-bottom dishes (MatTek, Ashland, MA) and kept in their growth medium at 37ºC in 5% CO2 environment during imaging. Time-lapse videos were acquired from a single optical plane focusing at the cell lamella and recorded with LAS-AF software (Leica) using bidirectional scanning with 1000–1400 Hz speed, zoom 12, and line averaging 1–2, resulting in 80 × 80 nm–pixel size in the xy-direction and 0.600-μm pixel size in the z-direction. Wide-field images of fixed cells were taken with an upright Zeiss (Oberkochen, Germany) AxioImager M2 482 epifluorescence microscope equipped with 63×/Plan-Apochromat/1.40 oil/M27 and 483 AxioCam HRm camera (Zeiss). Images were acquired with AxioVision4 software (Zeiss). Live confocal stack of Huh-7/LifeAct-RFP was acquired with an SP2 AOBS (Leica) as described (Puhka et al., 2007).
Quantification of sheet–tubule ratio
The relative proportion of ER sheets and tubules was quantified from wide-field LM images of control and nocodazole- and latrunculin A–treated and fixed Huh-7 cells (n = 1172, 518, and 1510 structural hits from 10 cells, respectively) immunolabeled against endogenous calreticulin. Images were quantified by using the intersection method. Analysis was done for the ER residing at the cell lamella, where the network resides in one plane. A 4-mm-line grid was placed over pictures printed at 150× final magnification, and the number of intersections of sheets and tubules with lines was counted. The relative proportion of sheets and tubules was calculated as Nintersections in category/Ntotal intersections and presented as a percentage of total intersections. This procedure was performed in a blinded manner, and the results of each condition were averaged. Error bars are presented as SD.
Fluorescence intensity measurements
The effect of latrunculin A treatment on actin filaments and free monomeric actin was monitored by phalloidin and DNaseI staining. The relative fluorescence intensity of phalloidin and DNaseI was analyzed from randomized wide-field LM images (constant exposure time) of control and 15-min latrunculin A–treated Huh-7 and HeLa cells (n = 100 cells in each). The mean gray intensity of the staining and the background was measured with the MatLab im_browser Intensity-function (MatLab, MathWorks, Natick, MA). The normalized results present average values of the intensities with background reduction. Error bars are shown as SEM.
To quantify the total amount of ER, the relative fluorescence intensity of calreticulin in control, nocodazole, and latrunculin A treatments (n = 75 cells in each) was measured from randomized wide-field images of fixed HeLa cells (constant exposure time) with the Photoshop CS5.1 (Adobe, San Jose, CA) mean gray intensity tool. The results are shown as average ± SEM.
Quantitation of myo1c and cortactin-positive actin arrays
The percentage of actin filaments positive for myo1c (n = 579 from 10 cells) was quantified from Huh-7 cells transiently expressing myo1c-2FLAG-N3, immunostained with an antibody against FLAG, and stained with phalloidin–Alexa 568. The percentage of actin filaments positive for cortactin (n = 791 structures from 10 cells) was quantified from wide-field images of fixed Huh-7 immunolabeled against endogenous cortactin and stained with phalloidin–Alexa 568. Images were taken as described. A 25-μm2 area of the cell was randomly selected, and the total number of actin filaments and/or foci <5 μm was counted by using the PhotoShop CS5.1 count tool, after which the number of actin filament and/or foci positive either for cortactin or myo1c was calculated. Actin filaments >5 μm were omitted. The numbers are presented as percentage ± SD of actin filaments and/or foci positive for either cortactin or myo1c.
Endocytic assays
The efficiency of non–receptor-mediated endocytosis in Huh-7 control and myo1c-depleted cells was quantified by following the uptake of 0.05 μg/μl dextran–Alexa Fluor 594 (D-22913; Molecular Probes). Cells were transfected with Hsp47-GFP and CMVTag1 or three pooled myo1c shRNAs for 48 h. Cells were incubated for 5 min at 37ºC in 5% CO2 atmosphere, after which they were fixed and mounted. To quantify the rate of receptor-mediated endocytosis in Huh-7 control and myo1c-depleted cells, we measured the transferrin–Alexa Fluor 647 (T-23366; Molecular Probes) uptake as described previously (Bertling et al., 2004), with the exception that cells were not starved and the pulse time was 5 min before fixation and mounting. To estimate the efficiency of non–receptor-mediated and receptor-mediated endocytosis in control and myo1c-depleted cells (n = 50 in all samples), randomized wide-field images were collected with constant exposure time, images were changed into grayscale 8-bit format, and mean gray intensity was measured with ImageJ (MacBiophotonics, Bethesda, MD).
Rescue experiments
Myo1c-depletion phenotype rescue experiments were set up as follows. Huh-7 cells were transfected with either Cy5-tagged, human myo1c–targeted siRNA (AACCCGUCCAGUAUUUCAACA) or Qiagen AllStar negative control siRNA (1027281; Venlo, Limburg, Netherlands) at 20 nM concentration using INTERFERin. On the next day, siRNA-containing medium was removed, and cells were transfected with GFP-tagged, siRNA-resistant myo1c constructs, myo1c-EGFP or myo1c ΔABL, using Lipofectamine 2000 (Life Technologies, Carlsbad, CA). After 5 h of incubation, siRNA-containing medium was added back to the cells, and culturing continued for additional 24 h, after which the cells were fixed with 4% PFA for 15 min, permeabilized with 0.1% Triton X-100 for 5 min, and stained with antibody against calreticulin.
Persistence and center-of-mass analysis
To study the actin dependence of ER sheet transformations, the naturally occurring relocation or loss of polygonal actin filament arrays was mimicked with latrunculin A treatment, using nocodazole treatment as a control. Sheet persistence and lateral movement of the sheets and the sheet lifetime were assessed from 60-s-long wide-field videos (1 frame/s). The sheet lifetime and persistence were calculated by randomly selecting individual sheets from the first acquired frame and scoring the sheet dynamics according to the following characteristics and requirements. A persistent sheet was to stay connected to other sheets and/or tubules and not undergo any of the following transformation events: 1) sheet transformation to tubules—when the diameter of the observed sheet reached the diameter of tubules; 2) sheet fission—when the observed sheet split into two, with at least 30% of its area leaving, resulting in two sheets with clear boundaries; and 3) sheet fusion—when a connection larger than the diameter of a tubule was created between the observed sheet and another sheet. The sheet lifetime (seconds ± SEM) represents the average lifetime of a sheet during the 60-s acquisition and does not represent the total lifetime of sheets. The lifetime was calculated by observing individual sheets from first acquired frame until any of the transformation events occurred. The relative proportion (percentage) is shown as an average of 22 cells for control cells (n = 56 sheets), 31 cells for latrunculin A treatment (n = 50 sheets), and 11 cells for nocodazole treatment (23 sheets) in Huh-7, and of 28 cells (n = 81 sheets) for control and 24 cells (n = 50 sheets) for latrunculin A treatment in HeLa cells. All of the persistent Huh-7 sheets were further subjected to center-of-mass analysis: the outlines of individual sheets having branch points with other sheets and/or tubules were manually traced throughout the 60-s acquisition (MatLab; MathWorks). Shapes were used to calculate the coordinates of the center of mass (i.e., the centroid; value given by centroid property of the regionprops-function of MatLab). These coordinates were then used to calculate and the average velocity of the sheets (μm/s ± SD) and the centroid's average distance (μm ± SD) related to the initial centroid position () and trace the centroid movements (n = 50 sheets in 16 cells for trichostatin A treatment). Significance between control versus treated (latrunculin A or trichostatin A) was analyzed using nonparametric Mann–Whitney U test (PASW Statistics 18 software; SPSS, IBM, Chicago, IL). Trichostatin A treatment did not induce statistically relevant changes.
Specimen preparation for EM, microscopy, image analysis, and modeling
SB-EM samples were prepared as described previously (Puhka et al., 2012). The SB-EM images were acquired with an FEG-SEM Quanta 250 (FEI Company, Hillsboro, OR) equipped with a microtome (3View; Gatan, Pleasanton, CA) for serial imaging of block faces using a backscattered electron detector (Gatan). The cells were imaged with 2.5-kV beam voltage, spot size 3, and 0.3-Torr pressure. The images were processed and the models segmented using Microscopy Image Browser, a self-developed program under the MatLab environment. The visualization of models and rendering of videos were done in Amira (VSG; FEI Company). The videos were encoded using an H.264 encoder with Adobe Photoshop. The presented 3D models of ER and NE are shown in perspective view, and the bars apply to the center point of the image. In Figure 2, voxel size is 14.3 × 14.3 × 30 nm in A and 17.3 × 17.3 × 40 nm in B, and areas are 14.9 × 14.8 × 1.3 and 16.0 × 13.0 × 1.3 μm, respectively. In Figure 6D, voxel size is 14.6 × 14.6 × 30 nm, and area is 16.0 × 17.8 × 0.7 μm.
Statistical analysis and image processing
Statistical analysis of centroid distance relative to initial centroid position between Huh-7 control (n = 35) and latrunculin A–treated (n = 7) or trichostatin A–treated (n = 19) cells used nonparametric Mann–Whitney U test (PASW Statistics 18 software). The data were not normally distributed (confirmed with SPSS Boxblot). The two-tailed p value for latrunculin A treatment compared with control was 0.045 and was nonsignificant for trichostatin A. Brightness and contrast of fluorescent images were adjusted with Photoshop CS5.1, and all of the live confocal images, as well as the wide-field images, were Gaussian filtered (rotationally symmetric low-pass filter, hsize = [3; 3], σ = 0.6). Magenta and green pseudo coloring was used as indicated.
The screen against actin-binding proteins
The screen was performed using an shRNA-based knockdown library against 208 human genes for functional actin-binding proteins (Genome Biology Unit, Biocenter Finland). Depletion conditions were validated with the BCH Knockdown Setup Kit (www.biocenter.helsinki.fi/bi/gbu/kd/setupkit.html). For the screen, Huh-7 cells were grown on glass-bottom, removable chambered coverglass, 16-well format (Invitrogen), and cotransfected at 1:4 ratio (wt/wt) with Hsp47-GFP and an inert plasmid (pCMV-Tag1; 211170; Agilent Technologies) or the three pooled shRNAs (1:1:1 ratio). Individual targets of the shRNA-based knockdown library were depleted one by one for 48-h transfections with FuGENE HD). The ER dynamics at the cell lamella was studied using a wide-field TILL imaging system described earlier, collecting 5–10 videos of each sample (1 frame/s for 60 s). Controls and depletions (parallel) samples were then fixed and stained with phalloidin–Alexa 568 according to manufacturer's instructions. Fixed samples were screened for changes in ER morphology or actin cytoskeleton.
Analysis of protein synthesis and secretion rates
Huh-7 cells were transfected with CMVTag1 (control) or pooled myo1c shRNA for 48 h or treated with 1 μM latrunculin A (pretreatment for 15 min and during pulse and chase). After methionine/cysteine starvation for 20 min by incubation with methionine/cysteine-free medium supplemented with 2 mM l‑glutamine (Invitrogen), newly synthesized proteins were radiolabeled for 20 min at 37°C using methionine/cysteine-free medium supplemented with 2 mM l-glutamine and EasyTagT EXPRESS [35S]methionine/cysteine labeling mix (>1000 Ci/mmol; NEG772014M; PerkinElmer, Waltham, MA). Subsequently, cells were incubated in the chase medium containing an excess of unlabeled methionine/cysteine for 0, 1, or 4 h at 37°C. Cells were lysed on ice with PBS containing 0.5% Triton X-100, and secreted proteins in medium were concentrated by precipitation with trichloroacetic acid and resuspended in SDS sample buffer. Protein concentration of the cell lysates was measured using bicinchoninic acid protein assay (Thermo Scientific). The efficiency of myo1c depletion was assessed from separated cell lysates by SDS–PAGE and Western blotting with myo1c-specific antibody (as previously described). The myo1c depletion efficiency was 60–65% (quantitation not shown). To measure protein synthesis and secretion rates, lysate and medium samples were subjected to SDS–PAGE. Total cellular proteins were visualized by staining gels with Instant Blue (Expedeon, Cambridge, UK), and radiolabeled proteins were visualized by phosphorimaging. For phosphorimaging, gels were dried and exposed to clean phosphorimaging plates and develop the exposures after 10 d using an image analyzer equipped with lasers for detection of radioactive isotopes (FLA-8000; Nachoi-Fujikoshi, Greenwood, IN). Protein synthesis rates were assessed from cell lysate samples and calculated as the ratio of radiolabeled protein signal intensity measurements versus total protein. Densitometry analysis of raw files was performed using ImageJ software (National Institutes of Health, Bethesda, MD). Before the comparison, both sets of measurements were normalized to the control, 0-h time point. Protein secretion rates were calculated as a ratio of intensity quantifications of individual lanes between medium and cell lysate samples. Densitometry analysis of raw files from phosphorimaging scan was performed with use of AIDA image analysis software (Raytest, Langenzersdorf, Austria). Before the comparison, the cell lysate samples were normalized to the control, 0-h time point. Shapiro–Wilk test and F-test were used to compare the distribution of data with normal distribution and SDs between examined populations, respectively. Depending on the results, Student's t test or Mann–Whitney U test was used. Statistical significance cutoffs: *p ≤ 0.05, **p < 0.01, and ***p < 0.001. All statistical analyses were performed using Statistica software (Statsoft, Tulsa, OK).
Supplementary Material
Acknowledgments
We thank Mervi Lindman and Antti Salminen for assistance with EM and Kimmo Tanhuanpää and Mika Molin for technical assistance with live-cell imaging (Electron and Light Microscopy Units of the Institute of Biotechnology, University of Helsinki); Pekka Lappalainen (Institute of Biotechnology, University of Helsinki) for reagents and helpful discussions and comments on the manuscript; and Jaakko Saraste (University of Bergen, Bergen, Norway), Anette Pykäläinen, and Antti Aalto (University of California, San Diego, La Jolla, CA) for comments on the manuscript. This work was supported by the Academy of Finland (project 131650, to E.J.) and Biocenter Finland. M.J., O.R., and M.P. were students of the Viikki Graduate School in Molecular Biosciences, University of Helsinki. I.N. was supported by the Centre for International Mobility CIMO, Helsinki, and M.V. by a European Research Council Starting Grant.
Abbreviations used:
- LM
light microscopy
- MT
microtubule
- NE
nuclear envelope
- PM
plasma membrane
- SB-EM
serial block-face scanning electron microscopy
- wt
wild type
Footnotes
*Present address: Institute for Molecular Medicine Finland, 00014 University of Helsinki, Helsinki, Finland.
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-12-0712) on February 12, 2014.
REFERENCES
- Asanov A, Sherry R, Sampieri A, Vaca L. A relay mechanism between EB1 and APC facilitate STIM1 puncta assembly at endoplasmic reticulum-plasma membrane junctions. Cell Calcium. 2013;54:246–256. doi: 10.1016/j.ceca.2013.06.008. [DOI] [PubMed] [Google Scholar]
- Barylko B, Jung G, Albanesi JP. Structure, function, and regulation of myosin 1C. Acta Biochim Pol. 2005;52:373–380. [PubMed] [Google Scholar]
- Baumann O. Structural Interactions of actin-filaments and endoplasmic-reticulum in honeybee photoreceptor cells. Cell Tissue Res. 1992;268:71–79. doi: 10.1007/BF00331360. [DOI] [PubMed] [Google Scholar]
- Baumann O, Walz B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int Rev Cytol. 2001;205:149–214. doi: 10.1016/s0074-7696(01)05004-5. [DOI] [PubMed] [Google Scholar]
- Benyamini P, Webster P, Meyer DI. Knockdown of p180 eliminates the terminal differentiation of a secretory cell line. Mol Biol Cell. 2009;20:732–744. doi: 10.1091/mbc.E08-07-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertling E, Hotulainen P, Mattila PK, Matilainen T, Salminen M, Lappalainen P. Cyclase-associated protein 1 (CAP1) promotes cofilin-induced actin dynamics in mammalian nonmuscle cells. Mol Biol Cell. 2004;15:2324–2334. doi: 10.1091/mbc.E04-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond LM, Brandstaetter H, Kendrick-Jones J, Buss F. Functional roles for myosin 1c in cellular signaling pathways. Cell Signal. 2013;25:229–235. doi: 10.1016/j.cellsig.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose A, Robida S, Furcinitti PS, Chawla A, Fogarty K, Corvera S, Czech MP. Unconventional myosin Myo1c promotes membrane fusion in a regulated exocytic pathway. Mol Cell Biol. 2004;24:5447–5458. doi: 10.1128/MCB.24.12.5447-5458.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgman PC. Myosin Va movements in normal and dilute-lethal axons provide support for a dual filament motor complex. J Cell Biol. 1999;146:1045–1060. doi: 10.1083/jcb.146.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Orth JD, Chen J, Weller SG, Heuser JE, McNiven MA. Cortactin is a component of clathrin-coated pits and participates in receptor-mediated endocytosis. Mol Cell Biol. 2003;23:2162–2170. doi: 10.1128/MCB.23.6.2162-2170.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra ES, Ramabhadran V, Gerber SA, Higgs HN. INF2 is an endoplasmic reticulum-associated formin protein. J Cell Sci. 2009;122:1430–1440. doi: 10.1242/jcs.040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CN, Futter CE, Gibson A, Hopkins CR, Cutler DF. Transport into and out of the Golgi complex studied by transfecting cells with cDNAs encoding horseradish peroxidase. J Cell Biol. 1994;127:641–652. doi: 10.1083/jcb.127.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudrier E, Almeida CG. Myosin 1 controls membrane shape by coupling F-actin to membrane. Bioarchitecture. 2011;1:230–235. doi: 10.4161/bioa.18406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diefenbach TJ, Latham VM, Yimlamai D, Liu CA, Herman IM, Jay DG. Myosin 1c and myosin IIB serve opposing roles in lamellipodial dynamics of the neuronal growth cone. J Cell Biol. 2002;158:1207–1217. doi: 10.1083/jcb.200202028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dopie J, Skarp KP, Rajakyla EK, Tanhuanpaa K, Vartiainen MK. Active maintenance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci USA. 2012;109:E544–E552. doi: 10.1073/pnas.1118880109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier L, Rapoport TA. In vitro formation of the endoplasmic reticulum occurs independently of microtubules by a controlled fusion reaction. J Cell Biol. 2000;148:883–898. doi: 10.1083/jcb.148.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Dibenedetto JR, West M, Rowland AA, Voeltz GK. Endoplasmic reticulum-endosome contact increases as endosomes traffic and mature. Mol Biol Cell. 2013;24:1030–1040. doi: 10.1091/mbc.E12-10-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Voeltz GK. The ER in 3D: a multifunctional dynamic membrane network. Trends Cell Biol. 2011;21:709–717. doi: 10.1016/j.tcb.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, Voeltz GK. ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J Cell Biol. 2010;190:363–375. doi: 10.1083/jcb.200911024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautreau A, Louvard D, Arpin M. Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J Cell Biol. 2000;150:193–203. doi: 10.1083/jcb.150.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal U, Blackstone C. Untangling the web: mechanisms underlying ER network formation. Biochim Biophys Acta. 2013;1833:2492–2498. doi: 10.1016/j.bbamcr.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffing LR. Networking in the endoplasmic reticulum. Biochem Soc Trans. 2010;38:747–753. doi: 10.1042/BST0380747. [DOI] [PubMed] [Google Scholar]
- Gunning P, O'Neill G, Hardeman E. Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol Rev. 2008;88:1–35. doi: 10.1152/physrev.00001.2007. [DOI] [PubMed] [Google Scholar]
- Hazan RB, Kang L, Roe S, Borgen PI, Rimm DL. Vinculin is associated with the E-cadherin adhesion complex. J Biol Chem. 1997;272:32448–32453. doi: 10.1074/jbc.272.51.32448. [DOI] [PubMed] [Google Scholar]
- Hirono M, Denis CS, Richardson GP, Gillespie PG. Hair cells require phosphatidylinositol 4,5-bisphosphate for mechanical transduction and adaptation. Neuron. 2004;44:309–320. doi: 10.1016/j.neuron.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Hokanson DE, Laakso JM, Lin T, Sept D, Ostap EM. Myo1c binds phosphoinositides through a putative pleckstrin homology domain. Mol Biol Cell. 2006;17:4856–4865. doi: 10.1091/mbc.E06-05-0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokanson DE, Ostap EM. Myo1c binds tightly and specifically to phosphatidylinositol 4,5-bisphosphate and inositol 1,4,5-trisphosphate. Proc Natl Acad Sci USA. 2006;103:3118–3123. doi: 10.1073/pnas.0505685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JR, Gillespie SK, Provance DW, Shah K, Shokat KM, Corey DP, Mercer JA, Gillespie PG. A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell. 2002;108:371–381. doi: 10.1016/s0092-8674(02)00629-3. [DOI] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Zhu PP, Voss C, Rismanchi N, Prinz WA, Rapoport TA, Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano F, Kondo H, Yamamoto A, Kaneko Y, Uchiyama K, Hosokawa N, Nagata K, Murata M. NSF/SNAPs and p97/p47/VCIP135 are sequentially required for cell cycle-dependent reformation of the ER network. Genes Cells. 2005;10:989–999. doi: 10.1111/j.1365-2443.2005.00894.x. [DOI] [PubMed] [Google Scholar]
- Kelley LC, Weed SA. Cortactin is a substrate of activated Cdc42-associated kinase 1 (ACK1) during ligand-induced epidermal growth factor receptor downregulation. PLoS One. 2012;7:e44363. doi: 10.1371/journal.pone.0044363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfenstein DR, Klumperman J, Lustig A, Kammerer RA, Oorschot V, Hauri HP. Subdomain-specific localization of CLIMP-63 (p63) in the endoplasmic reticulum is mediated by its luminal alpha-helical segment. J Cell Biol. 2001;153:1287–1300. doi: 10.1083/jcb.153.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science. 2013;339:464–467. doi: 10.1126/science.1228360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuokkanen E, Smith W, Mäkinen M, Tuominen H, Puhka M, Jokitalo E, Duvet S, Berg T, Heikinheimo P. Characterization and subcellular localization of human neutral class II alpha-mannosidase [corrected] Glycobiology. 2007;17:1084–1093. doi: 10.1093/glycob/cwm083. [DOI] [PubMed] [Google Scholar]
- Lazarides E, Burridge K. Alpha-actinin: immunofluorescent localization of a muscle structural protein in nonmuscle cells. Cell. 1975;6:289–298. doi: 10.1016/0092-8674(75)90180-4. [DOI] [PubMed] [Google Scholar]
- Lee C, Chen LB. Dynamic behavior of endoplasmic reticulum in living cells. Cell. 1988;54:37–46. doi: 10.1016/0092-8674(88)90177-8. [DOI] [PubMed] [Google Scholar]
- Lynch CD, Gauthier NC, Biais N, Lazar AM, Roca-Cusachs P, Yu CH, Sheetz MP. Filamin depletion blocks endoplasmic spreading and destabilizes force-bearing adhesions. Mol Biol Cell. 2011;22:1263–1273. doi: 10.1091/mbc.E10-08-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannherz HG, Goody RS, Konrad M, Nowak E. The interaction of bovine pancreatic deoxyribonuclease I and skeletal muscle actin. Eur J Biochem. 1980;104:367–379. doi: 10.1111/j.1432-1033.1980.tb04437.x. [DOI] [PubMed] [Google Scholar]
- Maravillas-Montero JL, Gillespie PG, Patino-Lopez G, Shaw S, Santos-Argumedo L. Myosin 1c participates in B cell cytoskeleton rearrangements, is recruited to the immunologic synapse, and contributes to antigen presentation. J Immunol. 2011;187:3053–3063. doi: 10.4049/jimmunol.1004018. [DOI] [PubMed] [Google Scholar]
- Novak KD, Peterson MD, Reedy MC, Titus MA. Dictyostelium myosin I double mutants exhibit conditional defects in pinocytosis. J Cell Biol. 1995;131:1205–1221. doi: 10.1083/jcb.131.5.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkmans L, Helenius A. Endocytosis via caveolae. Traffic. 2002;3:311–320. doi: 10.1034/j.1600-0854.2002.30501.x. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Doberstein SK, Zot HG. Myosin-I. Annu Rev Physiol. 1991;53:653–681. doi: 10.1146/annurev.ph.53.030191.003253. [DOI] [PubMed] [Google Scholar]
- Poteryaev D, Squirrell JM, Campbell JM, White JG, Spang A. Involvement of the actin cytoskeleton and homotypic membrane fusion in ER dynamics in Caenorhabditis elegans. Mol Biol Cell. 2005;16:2139–2153. doi: 10.1091/mbc.E04-08-0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhka M, Joensuu M, Vihinen H, Belevich I, Jokitalo E. Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Mol Biol Cell. 2012;23:2424–2432. doi: 10.1091/mbc.E10-12-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhka M, Vihinen H, Joensuu M, Jokitalo E. Endoplasmic reticulum remains continuous and undergoes sheet-to-tubule transformation during cell division in mammalian cells. J Cell Biol. 2007;179:895–909. doi: 10.1083/jcb.200705112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran AK, Morimoto T, Hanzel DK, Rodriguez-Boulan E, Kreibich G. Structural reorganization of the rough endoplasmic reticulum without size expansion accounts for dexamethasone-induced secretory activity in AR42J cells. J Cell Sci. 1993;105:333–345. doi: 10.1242/jcs.105.2.333. [DOI] [PubMed] [Google Scholar]
- Reizes O, Barylko B, Li C, Sudhof TC, Albanesi JP. Domain structure of a mammalian myosin I beta. Proc Natl Acad Sci USA. 1994;91:6349–6353. doi: 10.1073/pnas.91.14.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5:605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppert C, Godel J, Muller RT, Kroschewski R, Reinhard J, Bahler M. Localization of the rat myosin I molecules myr 1 and myr 2 and in vivo targeting of their tail domains. J Cell Sci. 1995;108:3775–3786. doi: 10.1242/jcs.108.12.3775. [DOI] [PubMed] [Google Scholar]
- Ryan SD, Ferrier A, Kothary R. A novel role for the cytoskeletal linker protein dystonin in the maintenance of microtubule stability and the regulation of ER-Golgi transport. Bioarchitecture. 2012;2:2–5. doi: 10.4161/bioa.20302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger JM, Dome JS, Mittal B, Somlyo AV, Sanger JW. Dynamics of the endoplasmic reticulum in living non-muscle and muscle cells. Cell Motil Cytoskeleton. 1989;13:301–319. doi: 10.1002/cm.970130408. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Asukagawa H, Yasuda R, Hiratsuka T, Sutoh K. Deletion of the myopathy loop of Dictyostelium myosin II and its impact on motor functions. J Biol Chem. 1999;274:37840–37844. doi: 10.1074/jbc.274.53.37840. [DOI] [PubMed] [Google Scholar]
- Sechi AS, Wehland J. The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J Cell Sci. 2000;113:3685–3695. doi: 10.1242/jcs.113.21.3685. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Shemesh T, Prinz WA, Palazzo AF, Kozlov MM, Rapoport TA. Mechanisms determining the morphology of the peripheral ER. Cell. 2010;143:774–788. doi: 10.1016/j.cell.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Voeltz GK, Rapoport TA. Rough sheets and smooth tubules. Cell. 2006;126:435–439. doi: 10.1016/j.cell.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Strasser P, Gimona M, Moessler H, Herzog M, Small JV. Mammalian calponin. Identification and expression of genetic variants. FEBS Lett. 1993;330:13–18. doi: 10.1016/0014-5793(93)80909-e. [DOI] [PubMed] [Google Scholar]
- Stürmer K, Baumann O, Walz B. Actin-dependent light-induced translocation of mitochondria and ER cisternae in the photoreceptor cells of the locust Schistocerca gregaria. J Cell Sci. 1995;108:2273–2283. doi: 10.1242/jcs.108.6.2273. [DOI] [PubMed] [Google Scholar]
- Svitkina TM. Ultrastructure of protrusive actin filament arrays. Curr Opin Cell Biol. 2013;25:574–581. doi: 10.1016/j.ceb.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagishi Y, Oda S, Hayasaka S, Dekker-Ohno K, Shikata T, Inouye M, Yamamura H. The dilute-lethal (dl) gene attacks a Ca2+ store in the dendritic spine of Purkinje cells in mice. Neurosci Lett. 1996;215:169–172. doi: 10.1016/0304-3940(96)12967-0. [DOI] [PubMed] [Google Scholar]
- Tang N, Ostap EM. Motor domain-dependent localization of myo1b (myr-1) Curr Biol. 2001;11:1131–1135. doi: 10.1016/s0960-9822(01)00320-7. [DOI] [PubMed] [Google Scholar]
- Terasaki M, Chen LB, Fujiwara K. Microtubules and the endoplasmic reticulum are highly interdependent structures. J Cell Biol. 1986;103:1557–1568. doi: 10.1083/jcb.103.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki M, Reese TS. Interactions among endoplasmic reticulum, microtubules, and retrograde movements of the cell surface. Cell Motil Cytoskeleton. 1994;29:291–300. doi: 10.1002/cm.970290402. [DOI] [PubMed] [Google Scholar]
- Ueno T, Kaneko K, Katano H, Sato Y, Mazitschek R, Tanaka K, Hattori S, Irie S, Sata T, Ogawa-Goto K. Expansion of the trans-Golgi network following activated collagen secretion is supported by a coiled-coil microtubule-bundling protein, p180, on the ER. Exp Cell Res. 2010;316:329–340. doi: 10.1016/j.yexcr.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Vallenius T. Actin stress fibre subtypes in mesenchymal-migrating cells. Open Biol. 2013;3:130001. doi: 10.1098/rsob.130001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science. 2007;316:1749–1752. doi: 10.1126/science.1141084. [DOI] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Wagner MC, Barylko B, Albanesi JP. Tissue distribution and subcellular localization of mammalian myosin I. J Cell Biol. 1992;119:163–170. doi: 10.1083/jcb.119.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner MC, Molitoris BA. ATP depletion alters myosin I beta cellular location in LLC-PK1 cells. Am J Physiol. 1997;272:C1680–C1690. doi: 10.1152/ajpcell.1997.272.5.C1680. [DOI] [PubMed] [Google Scholar]
- Wagner W, Brenowitz SD, Hammer JA., 3rd Myosin-Va transports the endoplasmic reticulum into the dendritic spines of Purkinje neurons. Nat Cell Biol. 2011;13:40–48. doi: 10.1038/ncb2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mattson MP, Furukawa K. Endoplasmic reticulum calcium release is modulated by actin polymerization. J Neurochem. 2002;82:945–952. doi: 10.1046/j.1471-4159.2002.01059.x. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Salmon ED. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr Biol. 1998;8:798–806. doi: 10.1016/s0960-9822(98)70321-5. [DOI] [PubMed] [Google Scholar]
- Wiest DL, Burkhardt JK, Hester S, Hortsch M, Meyer DI, Argon Y. Membrane biogenesis during B cell differentiation: most endoplasmic reticulum proteins are expressed coordinately. J Cell Biol. 1990;110:1501–1511. doi: 10.1083/jcb.110.5.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.