

Fission yeast Rho1p exchange factor Rgf1p is relocalized to the cell nucleus during the stalled replication caused by hydroxyurea. The Cds1p checkpoint kinase, helped by the chaperone Rad24p, controls the nuclear accumulation of Rgf1p by inhibiting its nuclear export. Failure to do so produces inefficient recovery from replication arrest.
Abstract
Guanine nucleotide exchange factors control many aspects of cell morphogenesis by turning on Rho-GTPases. The fission yeast exchange factor Rgf1p (Rho gef1) specifically regulates Rho1p during polarized growth and localizes to cortical sites. Here we report that Rgf1p is relocalized to the cell nucleus during the stalled replication caused by hydroxyurea (HU). Import to the nucleus is mediated by a nuclear localization sequence at the N-terminus of Rgf1p, whereas release into the cytoplasm requires two leucine-rich nuclear export sequences at the C-terminus. Moreover, Rgf1p nuclear accumulation during replication arrest depends on the 14-3-3 chaperone Rad24p and the DNA replication checkpoint kinase Cds1p. Both proteins control the nuclear accumulation of Rgf1p by inhibition of its nuclear export. A mutant, Rgf1p-9A, that substitutes nine serine potential phosphorylation Cds1p sites for alanine fails to accumulate in the nucleus in response to replication stress, and this correlates with a severe defect in survival in the presence of HU. In conclusion, we propose that the regulation of Rgf1p could be part of the mechanism by which Cds1p and Rad24p promote survival in the presence of chronic replication stress. It will be of general interest to understand whether the same is true for homologues of Rgf1p in budding yeast and higher eukaryotes.
INTRODUCTION
How nuclear events are connected to cell polarization and morphogenesis is of great interest. Guanine nucleotide-exchange factors (GEFs) are directly responsible for the activation of the Rho-family GTPases in response to physical and chemical stimuli, and they ultimately regulate many cellular responses, such as polarized growth and movement. GEF proteins are characterized by a Dbl-homology (DH) domain, also called the RhoGEF domain, which contacts the Rho GTPase to catalyze nucleotide exchange, and an associated Pleckstrin homology (PH) domain, related to the binding of phosphoinositides and also important for GDP exchange. Most GEFs are divergent in regions outside the DH/PH module and contain additional protein–protein or lipid–protein interaction domains that presumably dictate unique cellular functions (Schmidt and Hall, 2002a; Rossman et al., 2005; Bos et al., 2007; Buchsbaum, 2007). These interactions and modifications can induce major changes in the localization and/or topology of GEFs, such as translocation to a specific compartment of the cell (Gulli and Peter, 2001), release from autoinhibition by a flanking domain or region (Schmidt and Hall, 2002a), or induction of allosteric changes in the catalytic domain (Russo et al., 2001).
Among these regulatory mechanisms, the subcellular localization of GEFs has been identified as an important factor in the ability of GTPases to function in different signaling pathways (Mor and Philips, 2006). Most Rho-GEFs localize either to the cytoplasm or to the plasma membrane (PM), and only a few of them are seen in the nucleus, and then under specific circumstances. Transfer between compartments may dictate the spatiotemporal activation of the GTPase, as has been well documented for Cdc24p-dependent Cdc42p differential recruitment during bud emergence and in response to pheromones (Park et al., 1997; Nern and Arkowitz, 2000; Shimada et al., 2000). In mammals, at least two RhoA-specific GEFs, Net1p and Ect2p, localize preferentially within the nucleus at steady state (Alberts and Treisman, 1998; Schmidt and Hall, 2002b; Chalamalasetty et al., 2006). Both Net1p and Ect2p contain nuclear localization sequences (NLSs) that are required for their targeting to the nucleus. Deletion of the NLS in Net1p promotes its redistribution to the cytoplasm, with the consequent activation of RhoA and the formation of stress fibers (Schmidt and Hall, 2002b). Ect2p localizes to the nucleus in interphase cells, but after nuclear envelope breakdown before mitosis it is released from the nucleus to activate the RhoA-mediated contraction of the actomyosin ring that drives cytokinesis (Tatsumoto et al., 1999; Chalamalasetty et al., 2006; Wolfe and Glotzer, 2009). In both situations, the nucleus seems to serve as a reservoir of the GEF that keeps it inactive and separate from GTPase. However, reports show that both GEFs, Net1p and Ect2p, could play a more active role in the nucleus. Net1p-knockdown cells fail to activate the nuclear RhoA fraction in response to ionizing radiation (Dubash et al., 2011), and Ect2p regulates epigenetic centromere maintenance by stabilizing newly incorporated CENP-A (a histone H3 variant that acts as the epigenetic mark defining centromere loci; Lagana et al., 2010).
We were interested in the spatial and temporal regulation of fission yeast Rho1p by its GEFs. Rho1p is a functional homologue of human RhoAp and budding yeast Rho1p (Nakano et al., 1997) and regulates cell integrity, coordinating cell wall biogenesis with the actin and microtubule cytoskeleton and polarized secretion (García et al., 2006b; Perez and Rincón, 2010). In fission yeast, three GEFs activate Rho1p: Rgf1p, Rgf2p, and Rgf3p (García et al., 2006b). Rgf1p localizes to the cell tips in interphase cells and the division septum in mitotic cells. It is not essential for viability, but it does play a role in cell integrity: 1) it activates the β-glucan synthase (GS) complex containing the catalytic subunit Bgs4p (Morrell-Falvey et al., 2005; Mutoh et al., 2005; García et al., 2006a) and 2) it signals upstream of the Pmk1p mitogen-activated protein kinase (MAPK) pathway (Garcia et al., 2009). In addition, Rgf1p regulates the growth pattern of fission yeast cells, being required for the actin reorganization necessary for cells to change from monopolar to bipolar growth during new end take-off (García et al., 2006a).
In this study, we investigated the molecular mechanisms of Rgf1p regulation by functional analysis of mutated versions of the protein. Our results suggest that Rgf1p, but not Rgf2p or Rgf3p, moves to the nucleus in response to the stalled replication caused by hydroxyurea (HU). HU causes deoxyribonucleoside triphosphate starvation by inactivating ribonucleotide reductase, and it blocks the progression of replication forks from early firing origins, thus activating the DNA replication checkpoint pathway (Boddy and Russell, 2001; Branzei and Foiani, 2009). In Schizosaccharomyces pombe, the central sensor of the DNA replication checkpoint pathway is Rad3p, the fission yeast homologue of ATR (Bentley et al., 1996). Rad3p phosphorylates and activates the checkpoint kinases Cds1p or Chk1p, depending on the stage of the cell cycle and the nature of the upstream signal. Replication stress and DNA damage inflicted during S phase lead to the activation of Cds1p (Murakami and Okayama, 1995; Lindsay et al., 1998; Brondello et al., 1999), whereas DNA damage activates Chk1p during the G2 phase (Walworth et al., 1993; Brondello et al., 1999). Once activated, Cds1p and Chk1p phosphorylate further-downstream targets to regulate cell-cycle progression and DNA repair mechanisms (Furuya and Carr, 2003; Kai and Wang, 2003).
We propose that Rgf1p would enter the nucleus in conditions of replication block caused by HU. Rgf1p accumulates in the nucleus in a manner dependent on the 14-3-3 chaperone Rad24p and the DNA replication checkpoint kinase Cds1p. Because the inhibition of nuclear export is a major mechanism allowing the nuclear accumulation of Rgf1p, and since Rad24p interacts with this protein, we favor a model for Rgf1p reorganization in which Cds1p and Rad24p specifically stabilize the active conformation of Rgf1p for nuclear accumulation. Moreover, our results suggest that Rgf1p is important for recovery from replication stress.
RESULTS
The Rgf1p mutated in the Dishevelled/Egl-10/Pleckstrin domain localizes to the nucleus
Besides the DH and PH domains typically found in Rho-GEFs, Rgf1p contains a Dishevelled, Egl-10, and Pleckstrin (DEP) domain close to its amino-terminal regulatory region and a Citron and NIK1-like kinase homology (CNH) domain at its carboxy terminus (Figure 1A). As a first step toward analyzing the function and regulation of Rgf1p, we transformed rgf1Δ mutant cells with integrative constructs encoding C-terminally green fluorescent protein (GFP)–tagged wild-type Rgf1p or mutated versions of Rgf1p under the control of its own promoter: Rgf1pΔN, lacking the first 303 amino acids (aa); Rgf1pΔDEP, lacking 26 internal aa at the DEP domain (436–462); Rgf1pΔPH, lacking 56 internal aa at the PH domain (881–937); and Rgf1pΔCNH, lacking the last 331 aa (Figure 1A). Rgf1p-GFP localized to sites of polarized growth, the poles and the septum (García et al., 2006a; Figure 1A). Of interest, when we monitored the localization of the mutated constructs we found a striking difference in their subcellular distributions. In this sense, Rgf1pΔDEP localized strongly to the nucleus (Figure 1A), as confirmed by colocalization with Hoechst-stained nuclear DNA (100% colocalization, n = 200 cells; Supplemental Figure S1A). Rgf1pΔN (lacking the first 303 aa) localized efficiently to the cortex but was not restricted to the cell tips. In addition, Rgf1pΔN also localized weakly inside the nucleus. For the Rgf1pΔPH and Rgf1pΔCNH constructs, the normal localization of Rgf1p at the two tips was disrupted, and the signal was mainly monopolar (Figure 1A). This effect was most evident in Rgf1pΔPH, where the mutated protein was exclusively localized to the growing tip (as assessed by calcofluor staining; unpublished data). Next we focused on the DEP domain, and we wondered whether a less dramatic perturbation of this domain might lead to a similar phenotype. We introduced double Pro substitutions (F444P T445P) into the predicted α1-helix of the DEP domain (Ballon et al., 2006) and tested it in the same way. These changes also caused a severe defect in Rgf1p localization: the F444P T445P mutation removed some of the Rgf1p-GFP from the poles and directed the protein to the nucleus (Figure 1A). In addition, a few cells showed the localization of Rgf1p to the spindle pole body (Figure 1A, yellow arrows). Thus both deletion and mutation of the DEP domain point to a new localization pattern for Rgf1p inside the nucleus.
FIGURE 1:

Deletion of the Rgf1p DEP domain directs the protein to the nucleus. (A) Schematic representation of the domain structure of the full-length Rgf1p (aa 1–1334) and the various deletion fragments and site-specific mutation generated. All constructs were expressed as proteins C-terminally tagged with GFP and integrated at the leu1 locus in an rgf1Δ background. The intracellular localization of the wild-type Rgf1-GFPp, Rgf1pΔN-GFP, Rgf1pΔDEP-GFP, Rgf1pFPTP-GFP, Rgf1pΔPH-GFP, and Rgf1pΔCNH-GFP was analyzed in cells grown to early log phase in YES medium. (B) The PH and CNH domains are essential for Rgf1p function in vivo. Caspofungin (Csp) hypersensitivity and the vic phenotype of the wild-type, rgf1Δ, Rgf1pΔN-GFP, Rgf1pΔDEP-GFP, Rgf1pFPTP-GFP, Rgf1pΔPH-GFP, and Rgf1pΔCNH-GFP strains were analyzed in plate assays. Cells were spotted onto YES plates without 1 μg/ml Csp (Cancidas) as serial dilutions (8 × 104 cells in the left row and then 4 × 104, 2 × 104, 2 × 103, 2 × 102, and 2 × 101 in each subsequent spot) and incubated at 28ºC for 3 d. For the vic phenotype, cells were spotted onto YES or YES plus 0.5 μg/ml FK506 and 0.2 M MgCl2 and incubated at 32ºC for 3 d. (C) Western blot analysis showing the level of the indicated GFP-tagged Rgf1p mutants. Anti-PSTAIR antibody against Cdc2p was used as a loading control. Molecular mass markers are shown on the right of the gel in kilodaltons.
We tested the functional relevance of each of the mutated domains. Only the Rgf1pΔPH and Rgf1pΔCNH strains were unable to grow in the presence of caspofungin (Csp), an antifungal agent that inhibits GS activity (Figure 1B), and they showed the vic phenotype (viable in the presence of immunosuppressant FK506 and chloride ion) characteristic of the knockout of the components of the Pmk1p MAPK cell integrity pathway (Figure 1B; Ma et al., 2006). This implies that both domains are essential for Rgf1p function in cell integrity (García et al., 2006a, 2009). As expected, cells carrying mutations in the PH and CNH domains grew in a monopolar manner, similar to cells of the null mutant (Supplemental Figure S1B). As a control, we also examined the levels of endogenous Rgf1p-GFP in different mutants. Figure 1C shows that all of the proteins were expressed at levels similar to that observed in wild-type cells, except for Rgf1pΔPH-GFP, which was less abundant. Taken together, these results suggest that both the PH and CNH domains are required for function of Rgf1p at the poles, whereas localization of mutated versions in the DEP domain could reveal an as-yet-unidentified function for Rgf1p in the nucleus.
Rgf1p has a functional NLS at the N-terminus
A search through the entire Rgf1p sequence revealed one potential NLS at the N-terminus of Rgf1p, close to the DEP domain (aa 403–412; Weis, 2003; http://www.pombase.org/). To investigate whether the NLS contributed to the nuclear localization of Rgf1p, we transformed the rgf1Δ mutant cells with integrative constructs encoding different N-terminal fragments of Rgf1p. As seen in Figure 2, Rgf1pN-302, a region encoding the first 302 aa of Rgf1p fused to GFP, was excluded from the nucleus, whereas Rgf1pN-535 (encoding the first 535 aa of Rgf1p) and Rgf1pN-302-535 (encoding residues 303–535) stained the nucleus intensely. Curiously, Rgf1pN-302 localized only faintly to the cortex (Figure 2, cells with a yellow arrow). The observed difference in the subcellular distribution among the three fragments seems to result from the presence of the NLS and the DEP domain in both the Rgf1pN-535 and the Rgf1pN-302-535 constructs. To demonstrate that the localization of the N-terminal Rgf1pN-302-535 fusion protein was directly related to the presence of an NLS, we mutated the NLS by replacing the four basic arginine residues (406NKRRRRI412) with alanine residues (406NKAAAAI412). Mutation of NLS led to the relocalization of Rgf1pN-302-535 in the cytoplasm (Figure 2). Therefore Rgf1pN-302-535 containing the NLS is sufficient to deliver an otherwise cytosolic passenger protein into the nucleus, whereas site-directed mutagenesis of the NLS (NKRRRRI) confirms that the NLS acts as an NLS motif.
FIGURE 2:
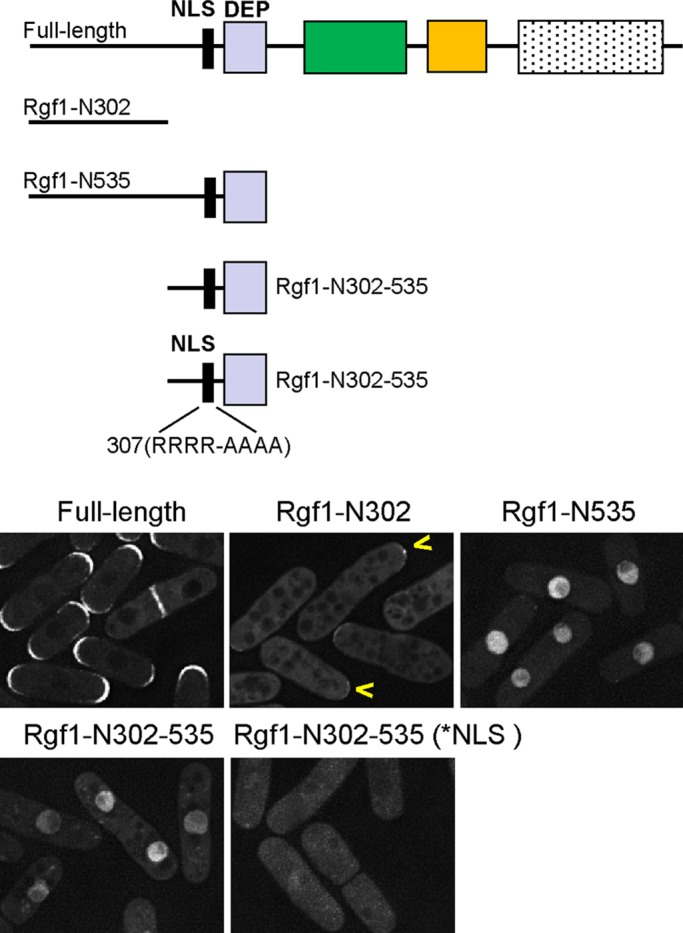
Rgf1p contains a functional NLS. Schematic representation of the full-length Rgf1p and several N-terminal constructs expressed as C-end GFP-fused proteins and integrated at the leu1 locus in an rgf1Δ background. The cellular locations of wild-type Rgf1p-GFP and the passenger GFP fused to N-fragments Rgf1p-N302-GFP, Rgf1p-N535-GFP, Rgf1p-302-535-GFP, and Rgf1p-302-535-NLS* were analyzed in cells grown to early log phase in YES medium.
Rgf1p localizes to the nucleus when DNA replication is inhibited by HU
Even though Rgf1p contains a functional NLS and mutations in its DEP domain lead the protein to become accumulated in the nucleus, throughout the cell cycle, Rgf1p-GFP localized to sites of polarized growth and was excluded from the nucleus in >95% of cells (Figures 1A, 2, and 3A; the nucleus appears darker than the cytoplasm in the wild-type Rgf1p-GFP images). We speculated that if the Rgf1p NLS is hidden by an intramolecular interaction that involves the DEP domain or the protein is bound to second partners, under stress situations, this might trigger a change that traps the protein inside the nucleus, mimicking the DEP mutations. Accordingly, we tested for the presence of Rgf1p-GFP in the nucleus after DNA damage, mating, osmotic stress, and heat shock. Most of these changes induced the transcription and new protein expression required for cell cycle arrest and cellular adaptation. We found that treatment with HU, but not other stresses, led to the translocation of Rgf1p into the nucleus (Figure 3A and Supplemental Figure S2A). HU is an inhibitor of the ribonucleotide reductase that blocks DNA replication through nucleotide deprivation, activating the replication checkpoint (Boddy and Russell, 2001).
FIGURE 3:
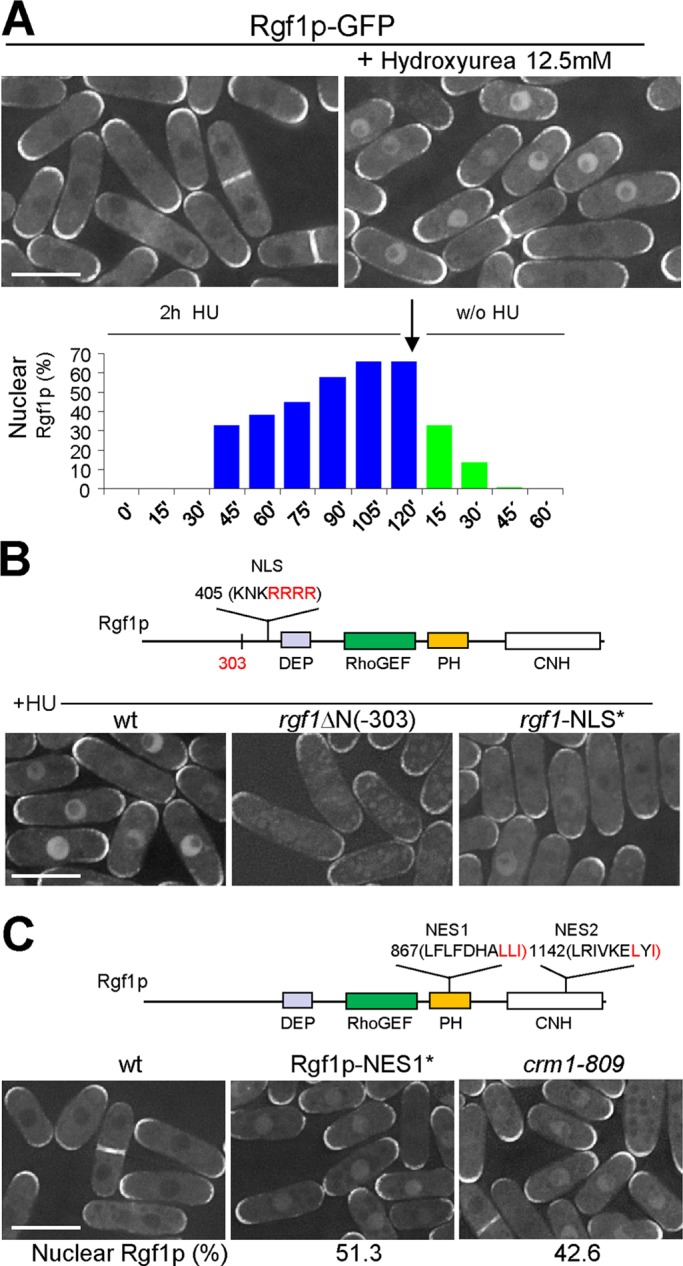
Rgf1p accumulates in the nucleus during HU-induced replication stress. (A) Asynchronous cultures of exponentially growing cells expressing a functional Rgf1p-GFP from its endogenous promoter (PG40) were treated for 2 h with HU (12.5 mM) at 28ºC and then released from the drug for another 1 h. Rgf1p localization was examined before and 2 h after the addition of HU (top), and the number of cells with nuclear Rgf1p was quantitated in live cells during and after treatment (bottom). Two hundred cells were analyzed per time point. (B) The NLS is required for the nuclear accumulation of Rgf1p in HU. Red amino acids were mutated to alanines to test the functionality of the NLS sequence. The intracellular localization of the wild-type Rgf1p, Rgf1pΔN-GFP, and Rgf1p-NLS*-GFP was analyzed in log-phase cells treated with 12.5 mM HU for 2 h at 28ºC. (C) The mutation in NES1 and the crm1-809 mutation cause the nuclear accumulation of Rgf1p-GFP in the absence of HU. Red amino acids were mutated to alanines to test the functionality of NES sequences. Photos for wild-type Rgf1p-GFP, Rgf1p-NES1*-GFP, and Rgf1p-GFP in crm1-809 exponentially growing cultures are shown, and quantification of the number of cells containing nucleus-accumulated Rgf1p was obtained as means of three independent experiments. Bar, 10 μM.
To study Rgf1p-GFP localization during replication stress in detail, we treated an asynchronous culture of Rgf1p-GFP with 12.5 mM HU. Cells with nuclear Rgf1p were first observed after 45 min in the presence of HU and rose to a maximum after 2 h (65–70% of the cells in the culture; Figure 3A). In addition, most cells in the asynchronous population (+HU) maintained Rgf1p at the poles or the septum (Figure 3A). This change in subcellular localization seemed to be specifically triggered by the action of the drug and was reversed as soon as the drug was removed (Figure 3A), suggesting the existence of a molecular mechanism that relates the nuclear accumulation of Rgf1p-GFP and the activation of the S-phase checkpoint response. Rgf1p is unique in this signaling process, since neither Rgf2p-GFP nor Rgf3p-GFP, the other two Rho1p-specific GEFs, moves to the nucleus in the presence of HU (Supplemental Figure S3).
We next used the mutated versions of Rgf1p labeled with GFP (Rgf1pΔN, Rgf1pΔPH, and Rgf1pΔCNH) to test which part of the Rgf1p was necessary for nuclear localization in the presence of HU. Only the Rgf1pΔN construct (lacking the first 303 aa) resulted in a partial loss of Rgf1p nuclear localization (Figure 3B). In addition, mutating the NLS in the full-length Rgf1p-GFP also led to a strong reduction in the relocalization of Rgf1p to the nucleus after HU treatment (8% in the mutated NLS as compared with 70% in the wild-type; n = 100; Figure 3B). Taken together, these results suggest that the nuclear accumulation in HU may depend on the N-terminus of Rgf1p.
Rgf1p has two nuclear export sequences involved in Rgf1p nuclear shuttling after HU treatment
In the foregoing we reported that Rgf1p moved out of the nucleus rapidly after HU wash off and was totally absent after 45 min (Figure 3A). One possible way for this distribution to be regulated is for Rgf1p to have a sequence(s) that causes the putative nuclear pool of the protein to be redistributed to the cytosol after treatment. In this regard, Rgf1p has two stretches of leucine-rich regions resembling the nuclear export sequence (NES) motifs that are sufficient for the nuclear export of particular nuclear/cytosolic shuttling proteins (Kutay and Güttinger, 2005). We focused our attention on NES1, an Rgf1p leucine-rich motif (857LFLFDHALLI867) that shows the greatest similarity to the NES consensus, and NES2 (1142LRIVKELYI1151), which contains another NES-like motif (Figure 3C). To test whether these regions had a NES function, we generated mutants in the C-terminal aliphatic amino acids of each: Rgf1p-GFP NES1 (LLI-AAA), in which L at 865, L at 866, and I at 867 were each changed to A (denoted NES1*), and Rgf1p-GFP NES2 (LYI-AYA), in which L at 1149 and I at 1151 were each changed to A (denoted NES2*; Wen et al., 1995). Even in the absence of HU, ∼55% cells in the NES1* mutant showed nuclear Rgf1p-GFP at 30ºC. These results indicate that NES1 (aa 857–867) functions as a conventional NES motif and suggest the possibility that Rgf1p could enter the nucleus transiently at some point of the cell cycle (Figure 3C). In the absence of stress, the Rgf1p mutated in NES2* (Rgf1-GFP NES2*) was not trapped in the nucleus (unpublished data). However, deletion of the last 189 aa at the C-end of the full-length protein (Rgf1pGFP-Δ189) led to the accumulation of the protein in the nucleus in unstressed cells, whereas in a similar construct, Rgf1pGFP-Δ131 was excluded (Supplemental Figure S4). Significantly, the NES2 sequence (1142LRIVKELYI1151) was present in the long protein but not in the short one. This observation suggests two important conclusions. One is confirmation that NES2 acts as an NES motif, and the other is that deletion of the C-terminal 189 aa allows Rgf1p to bypass the requirement for replication damage for its nuclear accumulation. Thus NES2 would have a weaker but still demonstrable role in export. To test for Rgf1p-GFP shuttling in another way, we used a crm1-809 mutant (Adachi and Yanagida, 1989). Crm1/Exportin1 is the cellular karyopherin receptor for proteins bearing a leucine-rich NES, and mutants harboring the thermolabile crm1-809 allele are blocked in the export of NES-containing proteins at the nonpermissive temperature (Fukuda et al., 1997; Ossareh-Nazari et al., 1997). Analysis of the crm1-809 cells by fluorescence microscopy revealed that in the absence of HU, ∼42% cells in the crm1-809 mutant showed nuclear Rgf1p-GFP at 30ºC (Figure 3C). Collectively these results indicate that in wild-type cells the nuclear export of Rgf1p-GFP after replication blockage probably occurs through the nuclear export receptor Crm1p and the Crm1p-dependent NES1 and NES2 of Rgf1p.
Rgf1p interacts functionally and physically with Rad24p and accumulates in the nucleus in response to DNA damage in a Rad24p-dependent manner
Next we sought to identify the molecular mechanisms involved in the nuclear localization of Rgf1p. To this end, we performed a yeast two-hybrid search. A total of six positive rad24+ clones were identified with the full-length Rgf1p (Figure 4A). Rad24p mediates nuclear localization of the Chk1 protein kinase upon DNA damage, whereas, under the same conditions, it leads to cytoplasmic sequestration of Cdc25p (Lopez-Girona et al., 1999; Zeng and Piwnica-Worms, 1999; Dunaway et al., 2005). We therefore addressed whether Rad24p might be important for controlling nuclear localization of Rgf1p upon DNA replication stress. To check this, we treated wild-type and rad24Δ cells expressing Rgf1p-GFP with HU or mock treated them. The localization of Rgf1p-GFP was similar in wild-type and untreated rad24Δ cells, in that Rgf1p-GFP was present at one or both poles in interphase and the septum during cytokinesis (Figure 4C). However, whereas the Rgf1p signal was clearly nuclear in HU-treated wild-type cells, Rgf1p persisted at the poles and septum in rad24Δ cells treated with HU (Figure 4C). In addition, immunoblotting experiments confirmed that Rgf1p-GFP levels were not altered in the rad24Δ mutant either with or without HU (unpublished data). Thus these studies strongly suggest that the steady nuclear localization of Rgf1p in response to replication stress would depend on Rad24p function.
FIGURE 4:

Rgf1p accumulation in the nucleus is Rad24p dependent. (A) Interaction between Rgf1p and Rad24p in yeast two-hybrid screening. Growth in –Leu/–Trp/–His media of yeast cotransformed with pBD-GAL4/rgf1+ (pRZ97) and the empty vector pADGAL4 (pGADT7) (lane 1), pAD-GAL4/rad24+ (pEM9), and the empty vector pBD-GAL4 (pGBKT7) (lane 2), or pAD-GAL4/rad24+ (pEM9) and pBD-GAL4/rgf1+ (pRZ97; lane 3), in yeast two-hybrid screenings. The results are representative of three separate cotransformation experiments. (B) GST pull-down assay showing the interaction of Rgf1p and Rad24p. Rad24p-GFP and Rad24p-GFP Rgf1p-GST cells grown to mid log phase were incubated in the presence or absence of 12.5 mM HU for 2.5 h and lysed under native conditions. The complexes precipitated with glutathione–sepharose beads were Western blotted and probed with anti-GFP and anti-GST antibodies to analyze Rad24p-GFP and GST-Rgf1p, respectively (IP). Whole-cell extract (Ext) fractions were assayed with anti-GFP and anti-GST antibodies. (C) Rgf1p-GFP localization in untreated and 12.5 mM HU–treated Rgf1p-GFP cells and rad24Δ Rgf1p-GFP cells. (D) The mutation in NES1 causes nuclear accumulation of Rgf1p-GFP in the absence of Rad24p. Photos for Rgf1p-NES1*-GFP and Rgf1p-NES1*-GFP in rad24Δ exponentially growing cultures. Quantification of the number of cells containing nuclear Rgf1p was performed on a mean of three independent experiments. (E) LMB treatment causes nuclear accumulation of Rgf1p-GFP in the absence of replication damage in wild-type and rad24Δ mutants. Wild-type Rgf1p-GFP and rad24Δ Rgf1p-GFP cells growing in YES medium were mock treated or treated with 100 ng/ml LMB for 30 min.
We also tested the localization of Rad24p-GFP in the absence of Rgf1p and failed to observed any differences: Rad24p was mainly cytoplasmic but was also detected in the nucleus, the spindle, and the septum in both strains, as shown previously (Mishra et al., 2005; unpublished data). We next wondered whether Rad24p might play a direct role in regulating Rgf1p localization, perhaps by binding to Rgf1p and escorting it to the nucleus. To check this, we constructed a strain in which the endogenous Rad24p was tagged with GFP and the endogenous Rgf1p was expressed with a C-terminal glutathione S-transferase (GST) tag. Rad24p-GFP was isolated on the Rgf1p-GST beads (Figure 4B). Curiously, the amount of Rgf1p associated with Rad24p was not substantially increased in response to HU (Figure 4B). These findings indicate that an important amount of Rgf1p may be bound to 14-3-3 proteins even in the absence of replication arrest. Similar conclusions were reported in studies of Cdc25p in fission yeast and mammalian cells (Peng et al., 1997; Lopez-Girona et al., 1999). The localization experiments shown here did not reveal at which step Rad24p performs its function with regard to Rgf1p. Rad24p could facilitate the nuclear accumulation of Rgf1p by, for example, opening up the conformation of Rgf1p to expose an NLS tethering the protein in the nucleus. Alternatively, Rad24p could indirectly inhibit the nuclear export of Rgf1p by hampering the access of Crm1p to the NES motif. To distinguish between these possibilities, we studied the nuclear accumulation of the Rgf1pGFP-NES1* mutant in a rad24Δ strain. As shown in Figure 3C, the mutation of NES1 bypassed the HU stress-signaling requirements for the nuclear accumulation of Rgf1p by preventing the export of GEF protein from the nucleus. In an rad24Δ background, ∼55% of the cells contained a clear nuclear concentration of Rgf1pGFP-NES1* (Figure 4D), suggesting that nuclear import is independent of Rad24p. To avoid the use of a mutated Rgf1p protein, we studied the nuclear localization of Rgf1p-GFP in wild-type and rad24Δ cells treated with leptomycin B (LMB). LMB binds Crm1p and inhibits export in S. pombe (Fukuda et al., 1997). After LMB treatment for 30 min, Rgf1p-GFP accumulated in the nucleus in nearly identical numbers in wild-type and rad24Δ cells (>95%; Figure 4E). Taken together, our results are consistent with the idea that Rad24p interferes with Rgf1p by hindering its exit from the nucleus and that the inhibition of nuclear export is a major mechanism allowing Rgf1p nuclear accumulation.
Replication stress triggers nuclear accumulation of Rgf1p dependent on Cds1p checkpoint kinase
Fission yeast have two genetically distinct checkpoint-signaling pathways that respond to DNA damage. Cds1p kinase mediates the intra-S checkpoint in response to stalled replication forks and DNA damage during the S phase, whereas the G2/M checkpoint is mediated by Chk1p and responds to strand breaks and other kinds of damage that may occur during the G2 phase (Figure 5A; McGowan and Russell, 2004; Lambert and Carr, 2005). To explore the relationship between Rgf1p and the checkpoint kinases, we studied Rgf1p-GFP localization in strains deleted for rad3, cds1, and chk1 (Boddy and Russell, 2001; Nyberg et al., 2002). In untreated cds1Δ rgf1-GFP and chk1Δ rgf1-GFP strains, Rgf1-GFP localization was indistinguishable from that of wild-type cells (Figure 5B). However, after 2 h of treatment with 12.5 mM HU the cds1Δ mutants displayed Rgf1p-GFP staining at the poles and the septum resembling that of untreated cells and clearly differing from chk1Δ rgf1-GFP, which exhibited nuclear staining similar to wild-type Rgf1p-GFP cells (Figure 5B). Of interest, in response to HU treatment, Rgf1p-GFP was excluded from the nucleus in rad3Δ mutants, confirming that Rgf1p-GFP nuclear accumulation depends on an active checkpoint. These results indicate that replication stress induced by HU triggers a Cds1p-dependent nuclear accumulation of Rgf1p and prompted us to wonder whether Cds1p could facilitate Rgf1p accumulation by helping its way in or by blocking its way out of the nucleus. Thus we studied the nuclear accumulation of the Rgf1pGFP-NES1* mutant in a cds1Δ strain and found that a high percentage of cds1Δ cells contained Rgf1pGFP-NES1* in the nucleus (unpublished data). Moreover, >95% of cds1Δ cells accumulated Rgf1p in the nucleus after LMB treatment, suggesting that nuclear import of Rgf1p is independent of Cds1p.
FIGURE 5:
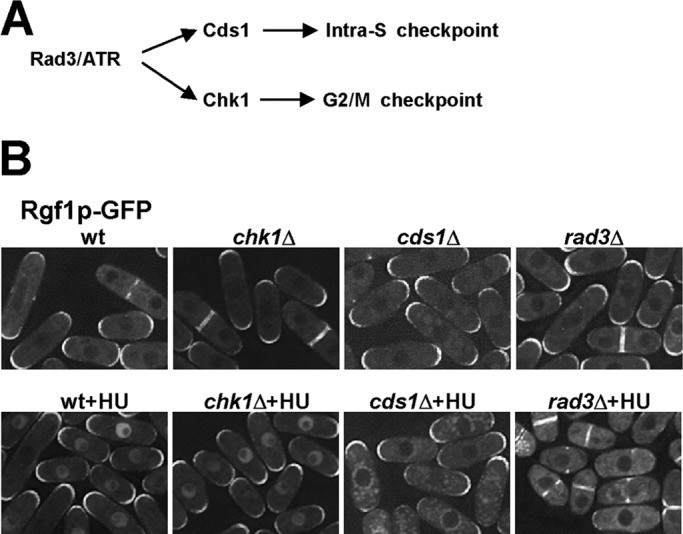
Nuclear accumulation of Rgf1p depends on the Cds1p checkpoint kinase. (A) Outline of checkpoint pathways in S. pombe. (B) Rgf1p-GFP localization in untreated or 12.5 mM HU–treated wild-type cells or cells lacking chk1+, cds1+, and rad3+. Whereas chk1 cells localize Rgf1p-GFP to the nucleus in response to HU, like wild-type cells, the protein remains in the cytoplasm in cells deleted for cds1+ or rad3+.
Rgf1p is required for survival in hydroxyurea, and this function is modulated by interaction with Cds1p/Rad24p
Rgf1p was imported to the nucleus after HU treatment, after which we examined the sensitivity of rgf1Δ cells to different concentrations of HU in a plate assay. The results clearly showed that rgf1Δ cells were unable to grow in the presence of 7.5 mM HU, whereas the wild-type cells grew in the same conditions (Figure 6A). For comparison, cell survival was also scored on the knockouts of the checkpoint kinases Cds1p and Chk1p. To test whether Rgf1p GEF activity was required for survival in HU, we checked a deletion mutation in the RhoGEF domain rgf1-PTTRΔ, which results in significantly reduced GEF activity toward Rho1p (Garcia et al., 2009). The rgf1-PTTRΔ mutant was hypersensitive to HU (Figure 6A) and completely nonfunctional in terms of Rgf1p nuclear relocalization after HU treatment (unpublished data). Moreover, cells of the rho1-596 thermosensitive mutant grew well at 32ºC (Viana et al., 2013) but were unable to grow in the same conditions in the presence of 5 mM HU (Supplemental Figure S5). These results suggest that the role of Rgf1p in survival under replication stress would be dependent on its interaction with Rho1p.
FIGURE 6:

Rgf1p is required for survival in HU. (A) For HU hypersensitivity, serial dilutions of the wild-type, rgf1Δ, rgf1ΔPTTR, cds1Δ, and chk1Δ cells were incubated at 28ºC on YES plates with no HU, 5 mM HU, or 7.5 mM HU (top). Bottom, the same type of analysis with the wild-type, rgf1Δ, Rgf1pΔN-GFP, Rgf1pΔDEP-GFP, Rgf1pFPTP-GFP, Rgf1pΔPH-GFP and Rgf1pΔCNH-GFP cells. (B) Nine putative Cds1p phosphorylation sites (RXXS) on Rgf1p are shown. Alanine substitution mutations of the nine Cds1p (S35A, S68A, S87A, S170A, S275A, S342A, S422A, S1085A, and S1322A) Rgf1p-9A sites, six Cds1p (S35A to S342A) Rgf1p-6A sites, and three Cds1p (S422A, S1085A, and S1322A) Rgf1p-3A sites were integrated chromosomally in the rgf1Δ deletion strain and expressed under the native promoter. Hypersensitivity to HU and Csp in the wild-type, rgf1Δ, Rgf1p-9A, Rgf1p-6A, and Rgf1p-3A is shown. (C) Localization of wild-type Rgf1p-GFP and the mutants rgf1Δ, Rgf1p-9A-GFP, and Rgf1-6A-GFP in untreated and 12.5 mM HU–treated cultures. Bottom, wild-type cells and Rgf1p-9A-GFP were treated with 100 ng/ml LMB for 30 min. (D) Proteins in cell extracts from Rgf1p-GFP– or Rgf1p-9A-GFP–expressing cells were separated by SDS–PAGE in the presence of 40 mM phostag and the proteins were detected by immunoblotting using anti-GFP antibodies. Closed arrow indicates major species observed in Rgf1-9A-GFP extracts. Open arrow indicates slower-migrating species observed in Rgf1p-GFP extracts. (E) GST pull-down assay showing the interaction of Rgf1p and Cds1p. Cells expressing endogenous Rgf1p-GFP and GST-Cds1p from a thiamine-regulated promoter (pREP4xGST-cds1+) or Rgf1p-GFP and the control plasmid (pREP4xGST) were grown in the absence of thiamine and incubated in the absence or presence of 12.5 mM HU for 2.5 h. The complexes precipitated with glutathione–sepharose beads were Western blotted and probed with anti-GFP antibodies to analyze Rgf1p-GFP in the immunoprecipitate (IP) and whole extract (Ext). (F) Interaction between Rgf1p and Rad24p was abolished by Rgf1p-9A. Rgf1p-GFP, Rgf1p-GFP Rad24p-GST, Rgf1p-9A-GFP, and Rgf1p-9A-GFP Rad24p-GST cells grown to mid log phase were incubated in the absence (left) or presence (right) of 12.5 mM HU for 2.5 h and lysed under native conditions. The complexes precipitated with glutathione–sepharose beads were Western blotted and probed with anti-GFP antibodies to analyze Rgf1p-GFP (IP). Whole-cell extract (Ext) fractions were assayed with anti-GFP and anti-GST antibodies.
We also wondered whether the Rgf1p mutations defective for nuclear accumulation of the protein in the presence of HU might be able to rescue the hypersensitivity of rgf1Δ cells to HU or not. As shown in Figure 6A, the Rgf1pΔN and the Rgf1pNLS (unpublished data) mutants, which showed reduced accumulation of Rgf1p in response to HU, were able to confer tolerance to HU on plates, whereas the Rgf1pΔPH and the Rgf1pΔCNH mutants, which were wild type for nuclear accumulation of the protein in HU but defective in polarity and cell integrity (Figure 1B), did not. Lack of correlation between nuclear accumulation and tolerance to the drug in these mutants could be explained in terms of the notion that N-terminal deletion and mutation in the NLS, although deficient for nuclear accumulation, are not excluded from the nucleus in the presence of the drug, whereas domains directly involved in catalysis are retained (Figures 1A and 3B).
The evidence linking Rgf1p and Cds1p/Rad24p suggested that Rgf1p might be an important target of regulation by Cds1p/Rad24p, controlling its localization and proficiency for survival in the presence of HU. To test this proposition, we attempted to identify a mutant that was functional but uncoupled for the regulation of Cds1p or Rad24p. Rgf1p contains a total of nine Cds1p consensus RXXS phosphorylation sites (where S becomes phosphorylated), which in its phosphorylated form is also the core consensus binding site for 14-3-3 proteins (Yaffe, 2002). At least three of these sites are phosphorylated in vivo (Gupta et al., 2013). Thus we mutated the nine phosphorylation sites to nonphosphorylatable alanine residues and expressed this Rgf1p-9A mutant tagged with GFP in cells lacking the endogenous rgf1+. In addition, we constructed another two mutants, Rgf1p-6A, with the six phosphorylatable sites clustered at the N-terminus changed to alanines, and Rgf1-3A, with the last putative phosphorylation site at the N-terminus and the two putative phosphorylation sites at the CNH domain changed to alanines (Figure 6B). We first examined whether the Rgf1p-9A-GFP was modified differently from wild-type Rgf1p-GFP. Cell lysates were prepared from either log-phase Rgf1p-9A-GFP or log-phase Rgf1p-GFP cells and analyzed by Western blotting. In phostag gels, additional species of Rgf1p-GFP that migrated with a slower electrophoretic mobility were observed, consistent with the idea of Rgf1p being phosphorylated (Figure 6D). These slower-migrating species were not seen in the Rgf1p-9A-GFP mutant (Figure 6D). Unlike the wild-type Rgf1p, the Rgf1p-9A protein was not accumulated in the nucleus after HU treatment (Figure 6C). Only treatment with LMB trapped the Rgf1p-9A protein in the nucleus (Figure 6C), suggesting that lack of phosphorylation regulates the exit of Rgf1p but does not impair its entrance. Most important, in an HU tolerance plate assay the Rgf1p-9A mutant strain behaved like the rgf1Δ strain, whereas it was wild type for growth in the presence of Csp (Figure 6B) and polarity (unpublished data). These data indicate that Rgf1p is probably phosphorylated by Cds1p or another kinase at one or multiple sites after HU treatment, allowing its nuclear retention. Failure of Cds1p/Rad24p to cause Rgf1p nuclear localization, as observed in Rgf1p-9A, correlates with a severe defect in survival from replication stress. Therefore regulation of Rgf1p could be part of the mechanism by which Cds1p promotes survival in the presence of HU. This regulation seems to be restricted to the N-terminus of the protein, since Rgf1p-6A is even more sensitive to HU than the fully dephosphorylated Rgf1p-9A mutant, and Rgf1-3A behaved like the wild-type protein (Figure 6B).
Having concluded that Cds1p/Rad24p are important for cytoplasmic retention of Rgf1p, we addressed the issue of whether there might be a physical interaction between Rgf1p and Cds1p, as observed before for Rgf1p and Rad24p. Accordingly, we performed a pull-down assay to check for Rgf1p-GFP/GST-Cds1p physical interaction. GST-Cds1p was induced and affinity purified from both untreated and checkpoint-activated cells, and blots were incubated with α-GFP antibody to detect Rgf1p-GFP. Our assays revealed an interaction between Cds1p and Rgf1p in both untreated cells and cells treated with 12.5 mM HU (Figure 6E). However, in the presence of HU the amount of Rgf1p was significantly higher. In addition, we noted the presence of a lower-mobility band in the extracts that expressed Cds1p, suggesting that the kinase binds and phosphorylates Rgf1p. In certain cases phosphorylation results in activation of the 14-3-3–binding sites (Lopez-Girona et al., 1999; Zeng and Piwnica-Worms, 1999). Thus we next assayed the ability of endogenous Rgf1p-9A-GFP to bind to endogenous Rad24p tagged with GST. Surprisingly, we found that Rgf1p-9A-GFP from cells treated with or without HU were unable to bind Rad24p-GST beads, although under identical conditions Rgf1p-GFP was able to bind Rad24p-GFP (Figure 6F). These experiments suggested that only a phosphorylated form of Rgf1p-GFP is able to bind Rad24p and this interaction is important for the protein to be retained inside the nucleus in a checkpoint-dependent manner and is therefore important for tolerance to replication stress.
Rgf1p is required for efficient recovery from replication arrest
HU is a ribonucleotide reductase inhibitor that stalls DNA replication after origin firing by dNTP depletion. To investigate the role of Rgf1p in survival in the presence of acute HU treatment, we compared the rgf1Δ mutants with the wild-type and cds1Δ strains for sensitivity to HU treatment for 2, 4, 6, and 8 h. Cells were treated in liquid medium with 12.5 mM HU and then plated onto HU-free plates. The rgf1Δ strain showed little if any sensitivity to the acute HU treatment, with >90% survival after 6 h treatment with HU, whereas <0.5% of the cds1Δ cells survived this treatment (Figure 7A). These results suggested that the sensitivity of rgf1Δ cells to HU requires long-term chronic exposure, a situation in which the cells have to pass through many replication cycles along the time of incubation.
FIGURE 7:

Rgf1p is required for efficient recovery from an HU-induced replication arrest. (A) Exponential cultures of wild-type, rgf1Δ, and cds1Δ cells were treated with 12.5 mM HU (time 0). At the indicated time points, samples were collected and washed free of HU, and viability was determined by colony formation on YES plates for 3 d at 28ºC. (B) Asynchronous cultures of wild-type and rgf1Δ cells grown at 28ºC were arrested with 12 mM HU. Cells were harvested at the time of HU addition (0) and at 2 h intervals thereafter, as indicated in the scheme. A portion of each culture was removed for photographing, and the rest of the cells were fixed and processed to analyze the DNA content by flow cytometry or stained with aniline blue to determine the number of septa (n > 200) for each time point. Data are means ±SD, n = 3. (C) Wild-type and rgf1Δ cells were photographed after 16 h of incubation at 28ºC on YES plates supplemented with 7.5 mM HU. White bar, 100 μm. (D) Asynchronous cultures of wild-type cells and rgf1Δ cells grown at 28ºC were treated with 12 mM HU for 3 h, released into fresh medium without HU, and then grown for another 4 h at 28ºC. Samples T0 and T1 were taken before and after HU treatment, and samples T3, T4, and T5 were taken as indicated in the scheme. A portion of each culture was removed for photographing, and the rest of the cells were processed as in B. (E) Quantitation of the fraction of nuclei containing more than one Rad52-YFP foci in asynchronously growing cells (0 h) or in cells treated with 12.5 mM HU for 2 and 4 h in wild type and rgf1Δ mutants (left). Asynchronous cultures of wild-type and rgf1Δ cells were arrested in 12.5 mM HU for 3 h and reinoculated into fresh medium without HU. Samples were taken at the indicated time points to quantify the fraction of nuclei containing more than one Rad52-YFP foci (middle). Right, nuclei containing Rad52-YFP in rgf1Δ and wild-type cells imaged 60 min after HU release.
To test this, we measured cell cycle progression in the presence of the drug. The wild-type and rgf1Δ cells were blocked in HU for 8 h, and DNA content, cell morphology, and septation index were assayed during arrest (Figure 7B). On treatment with HU, wild-type and rgf1Δ cells elongated and arrested with a 1C DNA content and completed bulk DNA replication after 8 h in HU (Figure 7B). However, we did see some differences between the two strains. In rgf1Δ cells, the septation index decreased after treatment but remained low even after 6 h at 30ºC (Figure 7B), showing that the mutant cells had not recovered from checkpoint arrest as soon as the wild-type cells. The results indicated that the rgf1Δ mutant cells were competent to activate the checkpoint as well as to resume replication during HU treatment. However, it seems that rgf1Δ cells reenter the cell cycle slowly in comparison with wild-type cells. If a similar delay occurred during each cell cycle for a period of growth of 3–4 d, the differences between the wild-type and rgf1Δ cells would be exacerbated. Figure 7C shows cells growing overnight (16 h) on 7.5 mM HU plates. The treatment led to a very elongated phenotype in the rgf1Δ mutant, whereas the wild-type cells were shorter and continued dividing under the same conditions (Figure 7C).
Next we measured cell cycle progression upon recovery from an HU block. We blocked cells for 3 h, released them into fresh medium without HU, and assayed their morphology and the DNA content during arrest and recovery (3 h arrest and 4 h recovery). The rgf1Δ cells showed a delay in septation (Figure 7D); in the wild-type cells the first septation peak occurred at T3 (2 h after drug release), whereas the rgf1Δ cells peaked for the first time at T5 (4 h after drug release). Fluorescence-activated cell sorting (FACS) analysis showed that in both strains bulk DNA synthesis was completed by 1–1.5 h after release (T3; Figure 7D). However, at T3 a high proportion of the rgf1Δ cells displayed an elongated phenotype, probably due to the presence of damaged or incorrectly replicated DNA (Figure 7D). These observations suggest that rgf1Δ cells recover from HU replication arrest inefficiently.
Many mutants with genome maintenance defects have elevated numbers of Rad22p/Rad52p foci (Meister et al., 2003; Noguchi et al., 2003). Rad52p concentrates in bright, visible foci at sites of double-strand break (DSB) repair and is essential for DSB repair by homologous recombination (HR). To test whether the elongated phenotype displayed by the rgf1Δ mutants after HU release was due to the presence of DNA lesions formed during replication in the presence of the drug, we analyzed the effect of rgf1Δ on the formation of Rad52p–yellow fluorescent protein (YFP) foci in the presence of HU and during recovery from HU arrest. In the absence of HU, 11% of a nonsynchronized wild-type population harbors a single Rad52p-YFP nuclear spot, which most likely corresponds to sites of postreplicative DNA repair (Meister et al., 2003). Only 1% of wild-type cells harbored more than one spot. These Rad52p foci appeared in G2 or very late S phase. In asynchronously growing rgf1Δ cells, we observed a slight reduction in the frequency of cells showing one Rad52p spot (9%) and a fourfold increase in the number of cells with several spots (4.24%; Figure 7E), even in the absence of HU. This suggested that loss of Rgf1p function leads to some spontaneous DNA damage or incompetence in facilitating its repair. When cells were exposed to HU for 2 h, the proportion of wild-type and rgf1Δ cells harboring multiple Rad52p-YFP foci increased slightly to 3.08 and 9.92%, respectively (Figure 7E). After HU release, the Rad52p-YFP foci persisted for at least 1 h in 40% of the rgf1Δ cells, whereas in wild-type strains Rad52p foci accumulated and peaked at 30 min after release (Figure 7E). In wild-type cells, the peak of Rad52p foci correlated with the completion of DNA replication, which, as judged by FACS analysis, took between 30 and 40 min (Figure 7D). In the rgf1Δ mutants these foci persisted after completion of replication but disappeared during the G2 phase, before mitosis (see elongated cells, Figure 7D, T3). From these observations, we conclude that rgf1Δ cells are significantly delayed in recovery from HU-induced replication arrest; however, most cells do eventually recover from conditions of acute HU exposure. Therefore the sensitivity of rgf1Δ cells to chronic HU exposure for several days may reflect defects deriving from multiple cycles of inefficient recovery.
DISCUSSION
Here we investigated the role of the Rho1p GEF protein Rgf1p during polarized growth by studying the localization of mutated versions of the Rgf1p-GFP. Unexpectedly, we found that mutants in the Rgf1p DEP domain (Dishevelled, Egl-10, and Pleckstrin) localized to the nucleus and that this domain was close to a canonical NLS. This was not an artifact, since we observed that the wild-type protein accumulated in the nucleus in response to the DNA replication arrest caused by HU. Accordingly, we outlined the overall mechanism for its nucleocytoplasmic shuttling (Figure 8). It is therefore tempting to speculate that there would be a strategic purpose underlying the accumulation of Rgf1p in the nucleus during the checkpoint. Replication arrest induced by HU leads to the Cds1p/Rad24p-dependent nuclear localization of Rgf1p. Failure of Cds1p/Rad24p to cause Rgf1p nuclear concentration, as observed in the Rgf1-9A mutant cells, was correlated with a severe defect in survival in the presence of replication arrest caused by HU. Thus regulation of Rgf1p appears to be a significant part of the mechanism by which Cds1/Rad24p promotes recovery from replication fork arrest.
FIGURE 8:
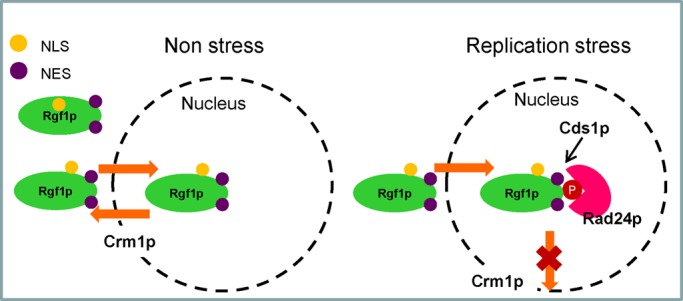
Possible mechanism for the nuclear accumulation of Rgf1p during replication stress in fission yeast. In nonstressed cells, Rgf1p enters the nucleus transiently. When cells are subject to replication stress, Rgf1p changes its conformation, probably by Cds1p phosphorylation, which allows its interaction with Rad24p. This remodeling would hide the NES, reducing its association with Crm1p and thus blocking its exit from the nucleus.
In a first series of experiments we examined the parameters that determine the subcellular localization and biological function of Rgf1p. The most important conclusion that can be drawn from the localization/function experiments is that Rgf1p is rapidly accumulated in the nucleus in cells under replication stress caused by HU. This is characteristic of Rgf1p, since neither Rgf2p nor Rgf3p was observed to undergo altered cellular localization in DNA replication–stressed cells (Supplemental Figure S3). Several lines of evidence support this view. First, a deletion or a point mutation that disrupts the structure of the DEP domain at the N-terminus induces the relocalization of Rgf1p to the nucleus. Second, Rgf1p contains one functional NLS near the DEP domain that mediates the import of the N-terminus into the nucleus. Mutation of this sequence leads to relocalization of the N-terminal fusion protein (Rgf1N-302-535) from the nucleus to the cytoplasm. Third, we found that HU triggers the relocalization of Rgf1p to the nucleus, thus promoting the same type of reorganization accomplished by manipulating the DEP domain at the N-terminus of the protein. This is highly specific to replication arrest, since only HU, but no other stresses, such as heat-shock or osmotic stress, will trigger nuclear localization.
After HU treatment, exit from the nucleus is mediated by two nuclear export signals, NES1 and NES2, and Crm1/exportin-1, which exports proteins containing a leucine-rich NES. Our observation that a fraction of cells in NES1* and crm1-809 mutants showed a nuclear localization of Rgf1-GFP in the absence of HU (Figure 3C) indicates that Rgf1p is actively exported from the nucleus. In addition, Rgf1-GFP accumulated inside the nucleus in 95% of cells treated with LMB (Figure 4E), consistent with the idea that Rgf1p continuously shuttles between the nucleus and the cytoplasm in a regular cell cycle.
How is Rgf1p transport into and out of the nucleus regulated?
In cycling cells, Rgf1p is localized to the cortex (the poles and the septum) and in most cells is excluded from the nucleus. However, upon activation of the DNA replication checkpoint in response to HU, Rgf1p was dispersed in the cell nucleus in early G2. This change of localization did not occur in rad3Δ, cds1Δ, or rad24Δ mutants, indicating that Rgf1p changes its subcellular localization in a checkpoint-dependent manner. Many cell-cycle regulators and an arsenal of enzymatic tools capable of remodeling and repairing DNA are controlled by checkpoint kinases in response to DNA stress (Branzei and Foiani, 2009; Ciccia and Elledge, 2010; Langerak and Russell, 2011). According to our results, Rgf1p is unique because it has the peculiarity of performing a function in response to replication stress that is apparently different from its role in morphogenesis during unperturbed cell cycles (García et al., 2006a, 2009), although this must be confirmed in the future. In budding yeast, Rom2p and Tus1p are the closest relatives to S. pombe Rgf1p, selectively activating different Rho1p-effector branches (Kono et al., 2008; Yoshida et al., 2009; Krause et al., 2012). Rom2p localizes to the growing bud surface and to the bud neck at cytokinesis, whereas Tus1p localizes only to the bud neck. However, it is unknown whether Rom2p or Tus1p is targeted to the nucleus in the presence of replication damage.
Here we defined the cis-acting elements required for the translocation of Rgf1p. The N-terminus, containing the NLS, is necessary for nuclear accumulation of the protein. In addition, GEF exchange activity is also responsible for activating its own nuclear accumulation. GEF activity could modulate Rgf1p interaction with trans-acting factors, represented by Rad24p and Cds1p, retaining the protein in the nucleus in response to DNA damage. Rad24p belongs to a family of highly conserved proteins with a broad range of unrelated functions (van Heusden and Steensma, 2006; Mohammad and Yaffe, 2009). Our results are consistent with the idea that Rad24p interferes with Rgf1p activity by hindering its exit from the nucleus. A similar mechanism can be invoked for nuclear localization of the Chk1 protein kinase upon DNA damage. Nuclear accumulation of Chk1p would thus result from stabilized interaction of Chk1p with Rad24p and the consequent reduction in its association with Crm1p (Dunaway et al., 2005).
A second binding partner of Rgf1p is the S-phase checkpoint kinase Cds1p (Murakami and Okayama, 1995; Lindsay et al., 1998). The evidence presented here shows that under replication stress, Cds1p triggers nuclear accumulation of the fission yeast Rgf1p by blocking its exit. Coimmunoprecipitation experiments showed that Rgf1p interacted with Cds1p, and overexpression of GST-Cds1p produced higher electrophoretic forms of Rgf1p (probably hyperphosphorylated; Figure 6E). Our results suggest that Rgf1p could be phosphorylated by Cds1p at several sites, but we were unable to detect differences in electrophoretic mobility in full-length Rgf1p-GFP with or without HU. This may be due to the low abundance of the phosphorylated forms with respect to the resident protein in the cortex. However, the behavior of the Rgf1p-9A mutant, mutated in the nine putative Cds1p consensus phosphorylation sites, strongly suggests that phosphorylation by Cds1p controls the localization of Rgf1p in the checkpoint response and is important for tolerance in the presence of chronic replication stress (Figure 6, B and C). Moreover, in a “pull-down” assay, Rgf1p-9A-GFP was unable to interact with endogenous GST-Rad24p, indicating that phosphorylation at one or several putative Cds1p sites also regulates binding of Rgf1p to Rad24p. In our working model, when cells were subject to replication stress, Rgf1p changed its conformation, probably by Cds1p phosphorylation, which promoted its interaction with Rad24p and consequent reduction in its association with Crm1p, blocking its exit from the nucleus (Figure 8). Of interest, nuclear localization of Rgf1ΔDEP and Rgf1N-535 (encoding the first 535 aa of Rgf1p) in unstressed cells was independent of Rad24p and Cds1p, suggesting that the DEP mutation might act by reducing Rgf1p association with Crm1p or blocking access of Crm1p to NES, favoring its nuclear retention.
What is the purpose of the accumulation of Rgf1p in the nucleus in response to a replication block?
The rgf1Δ mutants are not checkpoint defective, but from our analysis Rgf1p seems to be involved in efficient recovery from replicative arrest. In this sense, 1) rgf1Δ cells were significantly delayed in recovery from replication arrest, both in the presence of HU and after removal of the drug. This is best seen by looking at the first round of septation upon HU removal. Whereas the peak of septation in the wild type occurred after 2 h, in the mutant this was delayed to 4 h (Figure 7D). Thus the sensitivity to chronic exposure to the drug could be explained in terms of the existence of multiple rounds of inefficient recovery. In the acute treatment, only the first round of replication showed differences, but this was not reflected in the viability or size of the colonies of either strain after 4 d of growth (Figure 7A). 2) The rgf1Δ cells showed fourfold increase of nuclei with several spots of Rad52p, even in the absence of HU. Moreover, in the rgf1Δ cells recovering from HU block, the Rad52p foci persisted after the completion of replication but were resolved during a long G2 phase, before mitosis (Figure 7, D and E). Rad52p is a homologous recombination protein that loads the Rad51p recombinase at resected double-stranded DNA breaks and is also recruited to stalled replication forks, where it may stabilize structures by means of its strand-annealing activity. Thus Rgf1p may participate in Rad52p-dependent HR-mediated repair. Furthermore, in rgf1Δ cells, Rad52p foci were not accompanied by Chk1p activation, either in the presence or in the recovery from HU (unpublished data). Because no DNA damage signal was initiated, one explanation for the delay of these foci in disappearing is that in the absence of Rgf1p, recombination proteins bind stalled forks to generate recombination events that could be deleterious to bypass fork arrest (Ahn et al., 2005; Lambert et al., 2005).
We showed that the function of Rgf1p in tolerance to chronic HU depends on Cds1p/Rad24p. Cds1p can maintain the stability of stalled replication forks by regulating Mus81p-Eme1p nuclease, Rqh1p, a RecQ-family helicase involved in suppressing inappropriate recombination during replication, and Rad60, a protein required for recombinational repair during replication. This interaction reveals a direct link between Cds1p kinase and recombinational repair processes (Kai and Wang, 2003; Branzei and Foiani, 2009). Thus in the future it should be tested whether Rgf1p might play some role in processing blocked replication forks to prevent subsequent recombinogenic processes or whether Rgf1p's function in cytoskeleton dynamics might indirectly feed into these Rad52p-dependent processes. Another possibility is that Cds1p/Rad24p phosphorylation retains Rgf1p in the nucleus to facilitate sequestration of the protein until the cells decide to reenter the cell cycle. We showed that cells of the rho1-596 mutant, affected in the P-loop domain implicated in GTP binding, were sensitive to HU (Supplemental Figure S5). Moreover, a deletion mutation in the RhoGEF domain of Rgf1p caused sensitivity to HU. These results suggest an active role of Rho1p in survival under replication stress but not that the relocation of Rgf1p into the nucleus functions to sequester the protein from cell tips.
In conclusion, we uncovered a functional interaction between the chaperone Rad24p, the evolutionary conserved checkpoint kinase Cds1p, and the Rho1p GEF “morphogen” Rgf1p. In addition, Rgf1p appears to function in a checkpoint-dependent pathway in tolerance to replication stress. Rgf1p transiently shuttles in and out the nucleus during normal cell cycle progression. This basal role may be modified by interaction with Cds1p/Rad24p to promote the survival of lesions produced under conditions of chronic replication stress.
MATERIALS AND METHODS
General yeast methods
The S. pombe strains used in this study were constructed and maintained using standard techniques (Moreno et al., 1991) and are listed in Table 1. Cultures were grown in rich medium containing yeast extract plus supplements (YES), selective medium (MM) supplemented with the appropriate requirements, or sporulation medium (MEA). Crosses were performed by mixing appropriate strains directly on MEA plates. HU was filter sterilized and stored at −20°C in a stock solution (1 M) in H2O and was added to the medium after autoclaving. Caspofungin (Cancidas) was stored at −20°C in a stock solution (2.5 mg/ml) in H2O and was added to the media after autoclaving. Recombinant strains were obtained by tetrad analysis or the “random spore” method. For overexpression experiments using the nmt1 promoter, cells were grown in MM containing 15 μM thiamine up to logarithmic phase. Then the cells were harvested, washed three times with water, and inoculated in fresh medium (without thiamine) at an OD600 of 0.01 for 22 h.
TABLE 1:
S. pombe strains used in this work.
| Strain | Genotype |
|---|---|
| EM28 | h− rgf1::nat, leu1-32, ura4D18 |
| PG244a | h− leu1-32, ura4d18 |
| VT14 | h− rgf1::his3+, leu1-32, ade6M210, ura4D18, his3d1 |
| PG40 | h− rgf1::his3+, leu1-32::rgf1+-GFP:leu1+, his3d, ura4D18, ade6M210 |
| SM209 | h+ rgf1::his3+, leu1-32::rgf1+-GFP:leu1+, his3d, ura4D18, ade6M210 |
| SM48 | h− rgf1::his3+, leu1-32::rgf1-N2∆-GFP:leu1+, his3d1, ura4D18, ade6M210 |
| SM52 | h− rgf1::his3+, leu1-32::rgf1-DEP∆-GFP:leu1+, his3d1, ura4D18, ade6M210 |
| SM50 | h− rgf1::his3+, leu1-32::rgf1-FPTP-GFP:leu1+, his3d1, ura4D18, ade6M210 |
| SM15 | h− rgf1::his3+, leu1-32::rgf1-PH∆-GFP:leu1+, his3d1, ura4D18, ade6M210 |
| SM17 | h− rgf1::his3+, leu1-32::rgf1-CNH1∆-GFP:leu1+, his3d1, ura4D18, ade6M210 |
| SM99 | h− rgf1::his3+, leu1-32::rgf1302-GFP, ura4D18, ade6 M210 |
| SM101 | h− rgf1::his3+, leu1-32::rgf1535-GFP, his3d, ura4D18, ade6 M210 |
| EM56 | h− rgf1::his3+, leu1-32::rgf1302-535-GFP, his3d1, ura4D18, ade6 M210 |
| SM321 | h− rgf1::his3+, leu1-32:rgf1-GFP NLS4R-4A, his3d, ura4D18, ade6M210 |
| SM322 | h− rgf1::his3+, leu1-32::rgf1302-535-GFP-NLS4R-4A, his3d, ura4D18, ade6 M210 |
| SM335 | h− rgf1::his3+, leu1-32::rgf1+-GFP his3d, ura4D18, crm1-809 |
| SM302 | h− rgf1::his3+, leu1-32::rgf1-GFP-NES1, his3d, ura4D18, ade6M210 |
| EM50 | h− rgf1::his3+, leu1-32::rgf1-GFP-NES2, his3d, ura4D18, ade6M210 |
| SM330 | h− rad24-GFP:kan, leu1-32, ura4D18 |
| SM382 | h− rad24-GFP:kan, rgf1-GST:ura4+, leu1-32, ura4D18 |
| SM211 | h− rad24::ura4+, rgf1::his3+, leu1-32:rgf1+-GFP, his3d, ura4D18, ade6M210 |
| SM317 | h− rad3::ura4+, rgf1::his3+, leu1-32:rgf1+-GFP, his3d, ura4D18 |
| SM327 | h− cds1::ura4+, rgf1::his3+, leu1-32:rgf1+-GFP, his3d, ura4D18 |
| SM339 | h− chk1::ura4+, rgf1::his3+, leu1-32:rgf1+-GFP, his3d, ura4D18 |
| SM304b | h− chk1::ura4+, leu1-32, ura4D18 |
| SM305b | h− cds1::ura4+, leu1-32, ura4D18 |
| PG199 | h− rgf1::his3 his3DI leu1-32 ade6M210 ura4D18 leu1: rgf1+ (PTTRΔ)-GFP |
| SM19 | h− rgf1::his3+, leu1-32::rgf1-C189∆-GFP:leu1+, his3d1, ura4D18, ade6M210 |
| EM73 | h− rgf1-C131∆-GFP:ura4+, leu1-32, ura4D18 |
| SM394 | h− rgf1::his3+, leu1-32::rgf1-GFP-NES1, rad24::ura4+,his3d, ura4D18, ade6M210 |
| SM374 | h− rgf1::his3+, leu1-32::rgf1-GFP-NES1, cds1::ura4+,his3d, ura4D18, ade6M210 |
| SM616 | h− rgf1::nat, leu1-32::rgf1-fos9-GFP:leu1+, his3d1, ura4D18, ade6M210 |
| SM624 | h− rgf1::nat, leu1-32::rgf1-fos6-GFP:leu1+, his3d1, ura4D18, ade6M210 |
| SM620 | h− rgf1::nat, leu1-32::rgf1-fos3-GFP:leu1+, his3d1, ura4D18, ade6M210 |
| VT183 | h− rgf3::ura4+,leu1-32::rgf3+-8aGFP:leu1+, ura4D18 |
| SM213 | h+ leu1-32, ura4d18 +pAL-rgf2+ |
| SM680 | h− rad24-GST:kan, rgf1::nat, leu1-32::rgf1+-GFP |
| SM682 | h− rad24-GST:kan, rgf1::nat, leu1-32::rgf1-fos9-GFP:leu1+, his3d1, ura4D18 |
| SM308b | h− rad22-YFP:kan, leu1-32, ura4D18 |
| SM324 | h− rad22-YFP:kan, rgf1::his3+, leu1-32, ura4D18 |
| SM720a | h− leu1-32 ura4D-18 rho1-596:NatMX6 |
All strains were generated in this study, except as otherwise noted. For two-hybrid assays we used S. cerevisiae AH109 from Clontech (Takara, Mountain View, CA).
aPilar Perez (Instituto de Biología Funcional y Genómica, University of Salamanca, Salamanca, Spain).
bA. Bueno (Centro de Investigación del Cáncer, University of Salamanca, Salamanca, Spain).
Plasmid and strain construction
The mutated versions of Rgf1p were based on pAL-rgf1+-GFP (pGR45; GFP-tagged in a NotI site at the C-end; García et al., 2006a). To create Rgf1ΔN (pRZ83), pGR45 was modified by site-directed mutagenesis, introducing BglII sites at the ATG and 918 base pairs downstream of the start and then closing up the plasmid with BglII; for Rgf1ΔDEP (pRZ67), pGR45 was modified by site-directed mutagenesis with a loop oligonucleotide that eliminates base pairs from positions +1.310 to +1.386; for Rgf1FPTP (pRZ73), pGR45 was modified by site-directed mutagenesis, introducing a SmaI site 1.332 base pairs downstream of the start site; for Rgf1ΔPH (pRZ44), pGR41 was modified by site-directed mutagenesis with a loop oligonucleotide that eliminates base pairs from positions +2.643 to +2.856; for Rgf1ΔCNH-GFP (pRZ57), pGR45 was modified by site-directed mutagenesis by introducing a NotI site at 3.064 base pairs and then closing up the plasmid with NotI. From each plasmid, an EcoRI-EcoRI fragment containing the mutagenized Rgf1 open reading frame (ORF) and flanking sequences was subcloned into the integrative plasmid pJK148 and transformed into the rgf1Δ strain (VT14). Thus each construct was present as a single copy in the cell under the control of its own promoter. To create Rgf1-N302 (pSM12), pAL-rgf1+ (pGR41; García et al., 2006a) was modified by site-directed mutagenesis, introducing a NotI site +906 base pairs downstream of the ATG; the plasmid was closed with NotI, and the GFP epitope was inserted in-frame at the same site. For Rgf1-N535 (pSM14), pGR41 was modified by site-directed mutagenesis by introducing a NotI site at +1.581 base pairs and following the same steps as for pSM12. An EcoRI-EcoRI fragment containing the mutagenized Rgf1 ORF and flanking sequences was subcloned into the integrative plasmid pJK148 to create pSM21 and pSM22. For Rgf1-302-535 (pEM13), pSM22 was modified by site-directed mutagenesis, introducing BglII sites at the ATG and 918 base pairs downstream of the start and then closing up the plasmid with BglII. Rgf1-302-535 (*NLS)(pSM105) was based on pEM13 modified by site-directed mutagenesis with oligonucleotide NLSfw: 5′ctcgcctgttcataaaaatgctgctgcagctgctatatatgctgccttactttc3′. Full-length Rgf1p(*NLS) (pSM106) was based on pJK-rgf1+-GFP (pGR49) modified by site-directed mutagenesis with oligonucleotide NLSfw. Full-length Rgf1p-NES1* (pEM22) and Rgf1p-NES2* (pEM11) were based on pJK-rgf1+-GFP (pGR49) modified by site-directed mutagenesis with oligonucleotides NES1 5′gtttctctttgaccatgctgcggccgcagtaaaaccaaagactatt3′ or NES2 5′ccattgaggattgtcaaagaggcatatgcacccacggaatcaacttcg3′, respectively. pSM21 and pSM22, pEM13, pSM105, and the plasmids for the NLS*, NES1*, and NES2* mutations were linearized and transformed into the rgf1Δ strain (VT14). Rgf1p tagged with GST at the endogenous locus was obtained by replacing the C-end of Rgf1p with a cassette containing the C-end of Rgf1p fused to the GST, a ura4+ marker, and sequences from the Rgf1p 3′ untranslated region. To create the SM616 strain containing Rgf1p-9A, pGR45 was modified by site-directed mutagenesis to introduce S422A, S1085A, and S1322A, creating pSM135. A SacI-SacI fragment from pSM135 was eliminated and substituted by a SacI-SacI synthetic fragment containing the N-terminus mutations (S35A, S68A, S87A, S170A, S275A, and S342). As before, an EcoRI-EcoRI fragment containing the mutagenized Rgf1 ORF and flanking sequences was subcloned into the integrative plasmid pJK148 and transformed into the rgf1Δ strain (VT14). The strains SM624 containing Rgf1-6A (S35A, S68A, S87A, S170A, S275A, and S342) and SM620 carrying Rgf1-3A (S422A, S1085A, and S1322A) were derivatives of Rgf1-9A. The tagged strains Rad24-GST, Rad24-GFP, and Cds1-GFP were constructed using a PCR-based approach (Bähler et al., 1998) and confirmed by analytical PCR. We constructed pREP4x-GST-cds1+ based on the plasmid pREP1Cds1pGST kindly provided by A. Carr (University of Sussex, Brighton, United Kingdom; Lindsay et al., 1998).
Yeast two-hybrid analysis
For two-hybrid screen, the entire ORF of rgf1+ without intron was modified to introduce NdeI and SmaI flanking sites and was cloned into the same sites of pGBKT7 (Clontech) to express the GAL4 DNA-binding domain fused to the Rgf1 protein (pRZ97). Yeast two-hybrid screens were performed according to Clontech protocols. AH109 yeast strain expressing pBD-GAL4/Rgf1 (pRZ97) was transformed with pAD-GAL4 containing an S. pombe cDNA library (a gift of Stuart MacNeill, University of St Andrews, UK). Approximately 1 × 107 transformants were selected on synthetic dropout medium minus leucine, tryptophan, and histidine. cDNAs were rescued from positive clones using the DH10B strain of Escherichia coli, and the sequences of positive cDNAs in the prey vector were compared with the BLAST database (http://old.genedb.org/genedb/pombe/). Interaction between Rgf1 and Rad24 was further characterized by yeast two-hybrid assays. Full-length Rad24 was amplified by PCR from cDNA with NcoI and SmaI flanking sites and cloned into pGADT7 (Clontech) (pEM9). The full-length and partial fragments of Rgf1 fused to the GAL4 DNA-binding domain were expressed from pGBKT7.
Cell lysate preparation and pull-down assays
For cell lysate preparation, ∼3 ml of exponentially growing cells (OD = 0.8) were collected, washed once with water, and frozen together with acid-washed glass beads in liquid N2. Sample buffer, 150 μl (65 mM HCl-Tris, pH 6.8, 3% SDS, 10% glycerol, 5% β-mercaptoethanol, 50 mM NaF, 100 mM β-glycerol phosphate, 1 mM p-aminophenyl methanesulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 μg/ml pepstatin) was added, and cell homogenates were prepared in a Fast Prep FP120 device (Bio101 Savant; MP, Santa Ana, CA), boiled for 5 min, and cleared by centrifugation at 10,000 × g for 2 min. One-tenth of the supernatant was analyzed in a NuPAGE 3–8% Tris-acetate gel (Life Technologies, Carlsbad, CA), followed by immunoblotting with anti-GFP antibody JL-8 (BD Living Colors, Palo Alto, CA) and Cdc2 p34 (Y100.4) antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
For Rgf1p/Rad24p GST pull-down assays with cell extracts, Rgf1p-GST and Rad24p-GFP were expressed from their endogenous promoter. Cell homogenates from 50-ml cultures (OD = 0.6) were obtained using 200 μl of lysis buffer (50 mM HCl-Tris, pH 7.5, 200 mM NaCl, 2 mM EDTA, and 0.5% NP-40 containing 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 μg/ml pepstatin). Extracts were cleared by centrifugation at 1400 × g for 1.5 min, and ∼800 μg total protein was used in each immunoprecipitation in a final volume of 600 μl. The extracts were incubated with 20 μl of glutathione-Sepharose beads (GE Healthcare, Uppsala, Sweden) for 2 h at 4ºC in IP buffer (same as the lysis buffer without NP-40 and with 2% Triton X-100 instead). The beads were washed three times with IP buffer and once with IP buffer containing 500 mM NaCl and then resuspended in sample buffer and separated by 4–12% SDS–PAGE (Invitrogen). Proteins were transferred electrophoretically to an Immobilon-P membrane (Millipore, Bedford, MA), blotted with anti-GFP antibody JL-8 (BD Living Colors) to detect Rad24p-GFP, and conjugated anti–GST-horseradish peroxidase (HRP; Amersham Biosciences, Piscataway, NJ) to detect Rgf1-GST. Total protein levels were monitored in whole-cell extracts (10 μg of total protein) and used directly for Western blotting. For Rgf1pGFP/GST-Cds1p pull-down assays, wild-type cells expressing Rgf1p-GFP from its own promoter were transformed with either pREP4x-GST or pREP4x-GST-cds1+. Cds1 protein expression was induced by growing the cells in the absence of thiamine for 22 h at 28ºC. Cell homogenates from 50-ml cultures (OD = 0.6) were obtained as before. Proteins were blotted with anti-GFP antibody JL-8 (BD Living Colors) to detect Rgf1p-GFP and with conjugated anti–GST-HRP (Amersham Biosciences) to detect Cds1-GST.
Microscopy and image analysis
Cell samples were visualized using an Olympus IX71 microscope equipped with a personal Delta Vision system and a Photometrics CoolSnap HQ2 monochrome camera. Stacks of seven Z-series sections were acquired at 0.2-μm intervals. All fluorescence images are maximum two-dimensional projections of Z-series and were analyzed using deconvolution software from Applied Precision (Issaquah, WA). Measurements were made from micrographs using the ImageJ (National Institutes of Health, Bethesda, MD) or MetaMorph (Molecular Devices, Sunnyvale, CA) programs. For calcofluor, cells were harvested (1 ml), washed once, and resuspended in water with 20 μg/ml of calcofluor at room temperature.
Supplementary Material
Acknowledgments
We thank P. Perez, H. Valdivieso, J. C. Ribas, A. Bueno, S. Moreno, F. Chang, T. Toda, S. McNeill, and A. Carr for providing plasmids and strains. We acknowledge G. Ruiz de Garibay for construction of some of the plasmids. A. Bueno, C. Roncero, P. Perez, S. Moreno, and J. Encinar are acknowledged for comments and suggestions. P.G. was supported by a fellowship from the Junta de Castilla y León, and S.M. acknowledges support from a JAE-PreDoc fellowship granted by the Consejo Superior de Investigaciones Científicas, Spain. The text was revised by N. Skinner. This work was supported by grants BFU2008-00963/BMC and BFU2011-24683/BMC from the Comisión Interdepartamental de Ciencia y Tecnología, Spain, and GR231 from the Junta de Castilla y León, as well as by grant 10-0633 from the Swedish Cancer Fund to P.S. I.B.F.G. acknowledges the institutional support granted by the Ramón Areces Foundation during 2011–2012.
Abbreviations used:
- Csp
caspofungin
- DSBs
double strand breaks
- GEF
guanine nucleotide exchange factor
- GS
glucan synthase
- GST
glutathione S-sepharose
- HR
homologous recombination
- HU
hydroxyurea
- LMB
leptomycin B
- MAPK
mitogen-activated protein kinase
- NES
nuclear export sequence
- NLS
nuclear localization sequence
- PM
plasma membrane
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-11-0689) on January 29, 2014.
REFERENCES
- Adachi Y, Yanagida M. Higher order chromosome structure is affected by cold-sensitive mutations in a Schizosaccharomyces pombe gene crm1+ which encodes a 115-kD protein preferentially localized in the nucleus and at its periphery. J Cell Biol. 1989;108:1195–1207. doi: 10.1083/jcb.108.4.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn JS, Osman F, Whitby MC. Replication fork blockage by RTS1 at an ectopic site promotes recombination in fission yeast. EMBO J. 2005;24:2011–2023. doi: 10.1038/sj.emboj.7600670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts AS, Treisman R. Activation of RhoA and SAPK/JNK signalling pathways by the RhoA-specific exchange factor mNET1. EMBO J. 1998;17:4075–4085. doi: 10.1093/emboj/17.14.4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler J, Wu J-Q, Longtine MS, Shah NG, McKenzie A, III, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Ballon DR, Flanary P, Gladue PD, Konopka J, Dohlman HG, Thorner J. DEP-domain-mediated regulation of GPCR signaling responses. Cell. 2006;126:1079–1093. doi: 10.1016/j.cell.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Bentley NJ, Holtzman DA, Flaggs G, Keegan KS, DeMaggio A, Ford JC, Hoekstra M, Carr AM. The Schizosaccharomyces pombe rad3 checkpoint gene. EMBO J. 1996;15:6641–6651. [PMC free article] [PubMed] [Google Scholar]
- Boddy MN, Russell P. DNA replication checkpoint. Curr Biol. 2001;11:R953–R956. doi: 10.1016/s0960-9822(01)00572-3. [DOI] [PubMed] [Google Scholar]
- Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair (Amst) 2009;8:1038–1046. doi: 10.1016/j.dnarep.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Brondello J-M, Boddy MN, Furnari B, Russell P. basis for the checkpoint signal specificity that regulates Chk1 and Cds1 protein kinases. Mol Cell Biol. 1999;19:4262–4269. doi: 10.1128/mcb.19.6.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchsbaum RJ. Rho activation at a glance. J Cell Sci. 2007;120:1149–1152. doi: 10.1242/jcs.03428. [DOI] [PubMed] [Google Scholar]
- Chalamalasetty RB, Hümmer S, Nigg EA, Silljé HHW. Influence of human Ect2 depletion and overexpression on cleavage furrow formation and abscission. J Cell Sci. 2006;119:3008–3019. doi: 10.1242/jcs.03032. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubash AD, Guilluy C, Srougi MC, Boulter E, Burridge K, Garcia-Mata R. The small GTPase RhoA localizes to the nucleus and is activated by Net1 and DNA damage signals. PLoS One. 2011;6:e17380. doi: 10.1371/journal.pone.0017380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaway S, Liu H-Y, Walworth NC. Interaction of 14-3-3 protein with Chk1 affects localization and checkpoint function. J Cell Sci. 2005;118:39–50. doi: 10.1242/jcs.01570. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nat Rev Mol Cell Biol. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- Furuya K, Carr AM. DNA checkpoints in fission yeast. J Cell Sci. 2003;116:3847–3848. doi: 10.1242/jcs.00790. [DOI] [PubMed] [Google Scholar]
- García P, Tajadura V, García I, Sánchez Y. Rgf1p is a specific Rho1-GEF that coordinates cell polarization with cell wall biogenesis in fission yeast. Mol Biol Cell. 2006a;17:1620–1631. doi: 10.1091/mbc.E05-10-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García P, Tajadura V, García I, Sánchez Y. Role of Rho GTPases and Rho-GEFs in the regulation of cell shape and integrity in fission yeast. Yeast. 2006b;23:1031–1043. doi: 10.1002/yea.1409. [DOI] [PubMed] [Google Scholar]
- Garcia P, Tajadura V, Sanchez Y. The Rho1p exchange factor Rgf1p signals upstream from the Pmk1 mitogen-activated protein kinase pathway in fission yeast. Mol Biol Cell. 2009;20:721–731. doi: 10.1091/mbc.E08-07-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulli MP, Peter M. Temporal and spatial regulation of Rho-type guanine-nucleotide exchange factors: the yeast perspective. Genes Dev. 2001;4:365–379. doi: 10.1101/gad.876901. [DOI] [PubMed] [Google Scholar]
- Gupta S, Mana-Capelli S, McLean JR, Chen C-T, Ray S, Gould KL, McCollum D. Identification of SIN pathway targets reveals mechanisms of crosstalk between NDR kinase pathways. Curr Biol. 2013;23:1–6. doi: 10.1016/j.cub.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M, Wang TS-F. Checkpoint responses to replication stalling: inducing tolerance and preventing mutagenesis. Mutat Res. 2003;532:59–73. doi: 10.1016/j.mrfmmm.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Kono K, Nogami S, Abe M, Nishizawa M, Morishita S, Pellman D, Ohya Y. G1/S cyclin-dependent kinase regulates small GTPase Rho1p through phosphorylation of RhoGEF Tus1p in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19:1763–1771. doi: 10.1091/mbc.E07-09-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause SA, Cundell MJ, Poon PP, McGhie J, Johnston GC, Clive Price C, Gray JV. Functional specialisation of the yeast Rho1 GTP exchange factors. J Cell Sci. 2012;125:2721–2731. doi: 10.1242/jcs.100685. [DOI] [PubMed] [Google Scholar]
- Kutay U, Güttinger S. Leucine-rich nuclear-export signals: born to be weak. Trends Cell Biol. 2005;15:121–124. doi: 10.1016/j.tcb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Lagana A, Dorn JF, De Rop V, Ladouceur AM, Maddox AS, Maddox PS. A small GTPase molecular switch regulates epigenetic centromere maintenance by stabilizing newly incorporated CENP-A. Nat Cell Biol. 2010;12:1186–1193. doi: 10.1038/ncb2129. [DOI] [PubMed] [Google Scholar]
- Lambert S, Carr AM. Checkpoint responses to replication fork barriers. Biochimie. 2005;87:591–602. doi: 10.1016/j.biochi.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Lambert S, Watson A, Sheedy DM, Martin B, Carr AM. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005;121:689–702. doi: 10.1016/j.cell.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Langerak P, Russell P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos Trans R Soc Lond. 2011;366:3562–3571. doi: 10.1098/rstb.2011.0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay HD, Griffiths DJ, Edwards RJ, Christensen PU, Murray JM, Osman F, Walworth N, Carr AM. S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev. 1998;12:382–395. doi: 10.1101/gad.12.3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A, Furnari B, Mondesert O, Russell P. Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature. 1999;397:172–175. doi: 10.1038/16488. [DOI] [PubMed] [Google Scholar]
- Ma Y, Kuno T, Kita A, Asayama Y, Sugiura R. Rho2 is a target of the farnesiltransferase Cpp1 and acts upstream of Pmk1 mitogen-activated protein kinase signaling in fission yeast. Mol Biol Cell. 2006;17:5028–5037. doi: 10.1091/mbc.E06-08-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan CH, Russell P. The DNA damage response: sensing and signaling. Curr Opin Cell Biol. 2004;16:629–633. doi: 10.1016/j.ceb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Meister P, Poidevin M, Francesconi E, Tratner I, Zarzov P, Baldacci G. Nuclear factories for signalling and repairing DNA double strand breaks in living fission yeast. Nucleic Acids Res. 2003;31:5064–5073. doi: 10.1093/nar/gkg719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra M, Karagiannis J, Sevugan M, Singh P, Balasubramanian MK. The 14-3-3 protein Rad24p modulates function of the Cdc14p family phosphatase Clp1p/Flp1p in fission yeast. Curr Biol. 2005;15:1376–1383. doi: 10.1016/j.cub.2005.06.070. [DOI] [PubMed] [Google Scholar]
- Mohammad DH, Yaffe MB. 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair. 2009;8:1009–1017. doi: 10.1016/j.dnarep.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annu Rev Immunol. 2006;24:771–800. doi: 10.1146/annurev.immunol.24.021605.090723. [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Morrell-Falvey JL, Ren L, Feoktistova A, Haese GD, Gould KL. Cell wall remodeling at the fission yeast cell division site requires the Rho-GEF Rgf3p. J Cell Sci. 2005;118:5563–5573. doi: 10.1242/jcs.02664. [DOI] [PubMed] [Google Scholar]
- Murakami H, Okayama H. A kinase from fission yeast responsible for blocking mitosis in S phase. Nature. 1995;374:817–819. doi: 10.1038/374817a0. [DOI] [PubMed] [Google Scholar]
- Mutoh T, Nakano K, Mabuchi I. Rho1-GEFs Rgf1 and Rgf2 are involved in formation of cell wall and septum, while Rgf3 is involved in cytokinesis in fission yeast. Genes Cells. 2005;10:1189–1202. doi: 10.1111/j.1365-2443.2005.00908.x. [DOI] [PubMed] [Google Scholar]
- Nakano K, Arai R, Mabuchi I. The small GTP binding protein Rho1 is a multifunctional protein that regulates actin localization, cell polarity, and septum formation in the fission yeast Schizosaccharomyces pombe. Genes Cells. 1997;2:679–694. doi: 10.1046/j.1365-2443.1997.1540352.x. [DOI] [PubMed] [Google Scholar]
- Nern A, Arkowitz RA. Nucleocytoplasmic shuttling of the Cdc42p exchange factor Cdc24p. J Cell Biol. 2000;148:1115–1122. doi: 10.1083/jcb.148.6.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi E, Noguchi C, Du LL, Russell P. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol Cell Biol. 2003;23:7861–7874. doi: 10.1128/MCB.23.21.7861-7874.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. Toward maintaining the genome: DNA damage and replication checkpoints. Annu Rev Genet. 2002;36:617–656. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Bachelerie F, Dargemont C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science. 1997;278:141–144. doi: 10.1126/science.278.5335.141. [DOI] [PubMed] [Google Scholar]
- Park H-O, Bi E, Pringle JR, Herskowitz I. Two active states of the Ras-related Bud1/Rsr1 protein bind to different effectors to determine yeast cell polarity. Proc Natl Acad Sci USA. 1997;94:4463–4468. doi: 10.1073/pnas.94.9.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C-Y, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- Perez P, Rincón SA. Rho GTPases: regulation of cell polarity and growth in yeasts. Biochem J. 2010;426:243–253. doi: 10.1042/BJ20091823. [DOI] [PubMed] [Google Scholar]
- Rossman KL, Channing JD, Sondek J. GEF means go: turning on Rho GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- Russo C, Gao Y, Mancini P, Vanni C, Porotto M, Falasca M, Torrisi MR, Zheng Y, Eva A. Modulation of oncogenic DBL activity by phosphoinositol phosphate binding to pleckstrin homology domain. J Biol Chem. 2001;276:19524–19523. doi: 10.1074/jbc.M009742200. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002a;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Hall A. The Rho exchange factor Net1 is regulated by nuclear sequestration. J Biol Chem. 2002b;277:14581–14588. doi: 10.1074/jbc.M111108200. [DOI] [PubMed] [Google Scholar]
- Shimada Y, Gulli MP, Peter M. Nuclear sequestration of the exchange factor Cdc24 by Far1 regulates cell polarity during yeast mating. Nat Cell Biol. 2000;2:117–124. doi: 10.1038/35000073. [DOI] [PubMed] [Google Scholar]
- Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol. 1999;147:921–927. doi: 10.1083/jcb.147.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heusden PGH, Steensma HY. Yeast 14-3-3 proteins. Yeast. 2006;23:159–171. doi: 10.1002/yea.1338. [DOI] [PubMed] [Google Scholar]
- Viana RA, Pinar M, Soto T, Coll PM, Cansado J, Perez P. Negative functional interaction between cell integrity MAPK pathway and Rho1 GTPase in fission yeast. Genetics. 2013;195:421–432. doi: 10.1534/genetics.113.154807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth N, Davey S, Beach D. Fission yeast chk1 protein kinase links the rad checkpoint pathway to cdc2. Nature. 1993;363:368–371. doi: 10.1038/363368a0. [DOI] [PubMed] [Google Scholar]
- Weis K. Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell. 2003;112:441–451. doi: 10.1016/s0092-8674(03)00082-5. [DOI] [PubMed] [Google Scholar]
- Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell. 1995;82:463–473. doi: 10.1016/0092-8674(95)90435-2. [DOI] [PubMed] [Google Scholar]
- Wolfe BA, Glotzer M. Single cells (put a ring on it) Genes Dev. 2009;23:896–901. doi: 10.1101/gad.1801209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MB. How do 14-3-3 proteins work? Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett. 2002;513:53–57. doi: 10.1016/s0014-5793(01)03288-4. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Bartolini S, Pellman D. Mechanisms for concentrating Rho1 during cytokinesis. Genes Dev. 2009;23:810–823. doi: 10.1101/gad.1785209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Piwnica-Worms H. DNA damage and replication checkpoints in fission yeast require nuclear exclusion of the Cdc25 phosphatase via 14-3-3 binding. Mol Cell Biol. 1999;19:7410–7419. doi: 10.1128/mcb.19.11.7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.