

Abstract
Several subtypes of voltage-gated Na+ (NaV) channels are important targets for pain management. μ-Conotoxins isolated from venoms of cone snails are potent and specific blockers of different NaV channel isoforms. The inhibitory effect of μ-conotoxins on NaV channels has been examined extensively, but the mechanism of toxin specificity has not been understood in detail. Here the known structure of μ-conotoxin PIIIA and a model of the skeletal muscle channel NaV1.4 are used to elucidate elements that contribute to the structural basis of μ-conotoxin binding and specificity. The model of NaV1.4 is constructed based on the crystal structure of the bacterial NaV channel, NaVAb. Six different binding modes, in which the side chain of each of the basic residues carried by the toxin protrudes into the selectivity filter of NaV1.4, are examined in atomic detail using molecular dynamics simulations with explicit solvent. The dissociation constants (K d) computed for two selected binding modes in which Lys9 or Arg14 from the toxin protrudes into the filter of the channel are within 2 fold; both values in close proximity to those determined from dose response data for the block of NaV currents. To explore the mechanism of PIIIA specificity, a double mutant of NaV1.4 mimicking NaV channels resistant to μ-conotoxins and tetrodotoxin is constructed and the binding of PIIIA to this mutant channel examined. The double mutation causes the affinity of PIIIA to reduce by two orders of magnitude.
Introduction
Voltage-gated sodium (NaV) channels play a vital role in cell excitability. Several subtypes of NaV channels such as NaV1.3, NaV1.7, NaV1.8 and NaV1.9 are involved in the pain pathway [1], and thus are important targets for pain management. Many polypeptide toxins isolated from venomous animals such as scorpions, cone snails and spiders selectively interfere with the gating mechanisms and ion conduction properties of certain subtypes of NaV channels. These toxins are promising scaffolds for novel pesticides and analgesics [2], [3].
A family of peptide toxins isolated from venoms of cone snails, referred to as μ-conotoxins, are potent and selective blockers of NaV channels. μ-Conotoxins typically consist of 20–25 residues, six of which are cysteines forming three disulfide bridges, known as the inhibitor cystine knot [4], [5]. Some among the most well-characterized μ-conotoxins are GIIIA [6], PIIIA [7], SmIIIA [8] and KIIIA [9]. GIIIA and PIIIA selectively inhibit the skeletal muscle channel NaV1.4 with IC50 (half-maximal inhibitory concentration) values in the nanomolar range [7], [10], [11], and do not inhibit neuronal channels NaV1.7 and NaV1.8 at a concentration of 100 μM [12], [13]. On the other hand, SmIIIA and KIIIA inhibit NaV1.7 effectively at a concentration of 1 μM [12]. KIIIA was found to ease inflammatory pain in mice [14]. Various derivatives of KIIIA and other toxins have been designed [15]–[20], in an attempt to improve their analgesic effect and pharmaco-chemical properties. In particular, the R14A mutant of KIIIA was found to be 10-fold selective for NaV1.7 over NaV1.2 and NaV1.4 [18], suggesting that in principle selective inhibitors of NaV1.7 can be developed from μ-conotoxins.
μ-Conotoxins inhibit NaV channels via a pore-blocking mechanism [21], [22]. On binding to NaV channels, the side chain of a basic residue from a toxin protrudes into the selectivity filter of a channel, thereby inhibiting ion conduction. In the case of GIIIA, an arginine at position 13 has been proposed to be the residue that protrudes into the filter of the receptor channel [6], corresponding to the arginine at position 14 of PIIIA [7], [23]. Extensive mutagenesis experiments performed on PIIIA show that the mutation of each basic residue from the toxin to a neutral amino acid causes the affinity of the toxin for NaV1.4 to decrease by 10 to 50 fold, corresponding to a change of +2 to +4 kT in the free energy of binding [23], [24]. The change in binding free energy due to any of the charge-changing mutations is lower than the free energy of 6–7 kT for a typical salt bridge [25], [26], indicating that none of the mutations completely disrupts the salt bridge between the toxin and the channel pore.
Molecular dynamics (MD) simulations have been fruitfully used to construct model structures of peptide toxins in complex with ion channels such as K+ channels and the bacterial NaV channel NaVAb [27]–[29]. Such models, once validated against experimental data, would provide insight into the molecular determinants for the high-affinity and selective binding of a toxin to a channel, which can then be used to guide the design of novel toxins with improved potency and specificity. However, only few computational studies on the binding of μ-conotoxins to NaV channels have been reported [18], [23], [30], [31], largely due to the unavailability of atomic structures for mammalian NaV channels.
Here the binding of μ-conotoxin PIIIA to NaV1.4 is examined in atomic detail using MD simulations. Complex structures of all six possible binding modes of PIIIA-NaV1.4 are constructed using MD simulations with distance restraints. Similar pattern of interactions is observed in these complexes, suggesting that PIIIA can inhibit NaV1.4 with multiple alternative binding modes. The dissociation constant (K d) of PIIIA-NaV1.4 binding measured experimentally is reproduced within 5-fold using potential of mean force (PMF) calculations. A double mutant ([N181R, E172Q]) of NaV1.4 mimicking NaV channels resistant to tetrodotoxin (TTX) and μ-conotoxins is constructed. The K d value is found to increase by two orders of magnitude after the mutation, suggesting that NaV1.4 can be made resistant to PIIIA by this double mutation.
Methods
Homology model of NaV1.4
A homology model of the human muscle sodium channel NaV1.4 is generated using the 1836 amino acid sequence from the protein database of National Center for Biotechnology Information (Reference: NP_000325). Initially, seven partial models are generated from the full sequence on four different structural templates, including PDB IDs 2KBI [32], 2I53 [33], 3RW0 [34], and 3G43 [35], using the homology modeling server SWISS-MODEL [36]. Sequence identify of NaV1.4 segments with the different ion channels varies between 15% and 77%. The models are then visualized to determine how they correspond to the transmembrane pore forming domains of the channel. Those parts of NaV1.4 external to the pore forming domain are excluded from this study. The crystal structure of the bacterial NaV channel NaVAb [34], the filter of which is in the conductive state suitable for toxins to bind, is used as a template for the final model. NaVAb was used as a template in homology modeling of NaV1.4 previously [37]. Since the extracellular loops linking the S5, P1, P2 and S6 helices are significantly longer in NaV1.4 than in NaVAb, these loops cannot be modeled reliably on NaVAb. Therefore, the sequence of NaV1.4 is then modified to remove these loops (Fig. S1). The modified sequence is then used for homology modeling. As the sequence identity between the modified sequence and NaVAb is low (20–24%), structural similarity between NaV1.4 and NaVAb must be assumed. The four homologous domains of NaV1.4, referred to as domains I to IV (DI, DII, DIII and DIV), are modeled as discrete subunits. For DII, a second model is constructed on NaVRh, PDB ID 4DXW [38], and the resulting structure is found to be similar to that modeled on NaVAb (root mean square deviation 1.6 Å). This second model appears to align better with the other domains and thus is used in the final model. Each of the four domains generated comprises of the S5, P1, selectivity filter, P2 and S6 structural elements. Ramachandran plots show that more than 97% of the backbone dihedral angles are within favored or allowed regions. To configure the four-fold symmetry of the ion-channel, the four domains of NaV1.4 are aligned with NaVAb clockwise [39], [40]. The backbones of these two channels virtually superimpose (Fig. S2). The DEKA ring in the selectivity filter of NaV1.4 corresponds to the positions of the equivalent EEEE (NaVAb) and SSSS (NaVRh) rings in bacterial NaV channels. The resulting ion channel has a pore radius of 2.5 Å.
Molecular dynamics simulations
The NaV1.4 channel is embedded in a POPC (2-oleoyl-1-palmitoyl-sn-glycero-3-phosphocholine) bilayer built in VMD [41]. The simulation box contains the channel protein, 172 lipids, 68 Na+ ions, 59 Cl− ions and 15,743 water molecules. The concentration of NaCl is approximately 0.2 M. The system is equilibrated extensively for 50 ns, and the size of the simulation box evolves to about 85×85×100 Å3 at the end of the simulation (Fig. S2). In the first 20 ns of the equilibration during which the inner cavity of the channel becomes fully hydrated, a harmonic restraint is applied to maintain a rigid channel backbone. At 20 ns, a Na+ ion in the bulk is manually moved into the filter. Without the presence of this Na+ ion the side chain of Lys177 would spontaneously reorient and occlude the ion conduction pathway, consistent with the results of Xia et al. [42]. Subsequently the harmonic restraint is released and the system simulated for a further 30 ns without any restraints. The structure of the channel is stable, as evidenced by the fact that the root mean square deviation of the channel backbone with reference to the initial energy-minimized model fluctuates between 2 and 3 Å over the last 20 ns.
Toxin PIIIA is then added to the system, with its center of mass (COM) 40 Å above the COM of the channel, such that the toxin is not in direct contact with any part of the channel. The solution structure for PIIIA (PDB ID 1R9I) is used [43]. The toxin carries a net charge of +6 e, as histidine which adopts predominantly the deprotonated form at the physiological pH of 7.4 is assumed to be neutral. Six Na+ ions are removed to maintain charge neutrality. To accelerate the binding of the toxin to the channel, a flat-bottom harmonic distance restraint is applied to the side chain nitrogen atom of a basic residue of the toxin and the center of the carbonyl groups of residues 176 of the channel filter. This method has been successfully applied to several similar systems previously [44]–[46]. The upper boundary of the distance restraint is gradually decreased from 15 to 3 Å over a simulation period of 5 ns, such that the chosen basic residue of the toxin is drawn into the filter. The simulation is continued further for 15 ns with the harmonic restraints removed. To improve sampling each simulation is repeated a second time with a different orientation for the toxin.
MD simulations are performed at 1 atm and 300 K using NAMD 2.9 [47] with periodic boundary conditions applied. The CHARMM36 force field and the TIP3P model for water are used to describe the interatomic interactions [48]–[50]. The switch and cutoff distances for short-range non-bonded interactions are 8.0 Å and 12.0 Å, respectively. The particle mesh Ewald method is used to account for long-range electrostatic interactions, with a maximum grid spacing of 1.0 Å. The SHAKE [51] and SETTLE [52] algorithms are used to keep the bond lengths in the system rigid. A time step of 2 fs is used. Trajectories are saved every 20 ps for analysis. Molecular graphics are generated using VMD [41].
Umbrella sampling
The umbrella sampling method is used to construct the PMF profile for the unbinding of PIIIA from NaV1.4 along the channel axis. Based on the PMF profile the K d value for the formation of the toxin-channel complex can be calculated rigorously according to eq. 1 [27]. It is worth noting that K d and IC50 have been used interchangeably as it may be assumed that the experimentally measured IC50 and the true K d values are in the same order.
A constant force of 20 kcal mol−1 Å−1 is applied to pull the toxin out from the binding site, allowing the starting structures of the umbrella windows spaced at 0.5 Å intervals to be generated. The backbones of both the toxin and the channel are maintained rigid during the pulling. The COM of the toxin backbone is restrained to the center of each umbrella window using a harmonic force constant of 30 kcal mol−1 Å−2. The COM of the channel is at z = 0 Å. A flat-bottom harmonic restraint is applied to maintain the COM of the toxin backbone within a cylinder of 8 Å in radius centered on the channel axis. The toxin is allowed to move freely in the plane perpendicular to the channel axis within this cylinder. Each umbrella window is simulated for up to 8 ns until the PMF profile changes by less than 0.5 kT in depth over the last 1 ns. The first 1 ns of each window are removed from data analysis. The z coordinate of the toxin COM is saved every 1 ps for analysis.
Data analysis
A salt bridge is assumed to have formed if the distance is less than 4 Å between a side chain oxygen atom from an acidic residue and a side chain nitrogen atom from a basic residue [53]. A hydrogen bond is assumed to be formed if the donor and acceptor atoms (nitrogen or oxygen) are within 3.0 Å of each other and the donor-hydrogen-acceptor angle is ≥150° [54]. The length of a salt bridge is defined as the distance between the COM of the oxygen atoms in the side chain of the acidic residue and COM of the nitrogen atoms in the side chain of the basic residue. The weighted histogram analysis method is used to construct the PMF profile [55]. The K d value is derived using the following equation [27]:
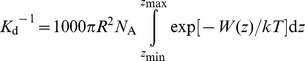 |
(1) |
where R is the radius of the cylinder (8 Å), N A is Avogadro's number, z min and z max are the boundaries of the binding site along the channel axis (z), and W(z) is the PMF.
Results and Discussion
Outer vestibule of NaV1.4
A homology model of the pore domain of NaV1.4 is constructed based on the sequence alignment displayed in Fig. 1A (details given in the Methods section). The model of NaV1.4 shows that there is an outer ring of acidic residues at positions 180 of DI, DII and DIV and 181 of DIII just outside the filter (Fig. 1B). In addition, two acidic residues at positions 184 and 187 of DII are also in close proximity to the filter. These negatively-charged residues may form strong electrostatic interactions with μ-conotoxins which typically carry several positively-charged basic residues. Thus, our model of NaV1.4 is consistent with the high-affinity inhibition of NaV1.4 by μ-conotoxins observed experimentally. We demonstrate below that our model of NaV1.4 allows both the binding affinity and the selectivity of PIIIA for NaV1.4 measured experimentally to be reproduced.
Figure 1. Sequence and structure of NaV1.4.
(A) Sequence alignment of NaVAb and the four domains (I–IV) of NaV1.4 in the pore domain. (B) The outer vestibule of NaV1.4 viewed from the extracellular side along the channel axis. For simplicity the numbering of NaVAb is used for NaV1.4.
Selectivity filter of NaV1.4
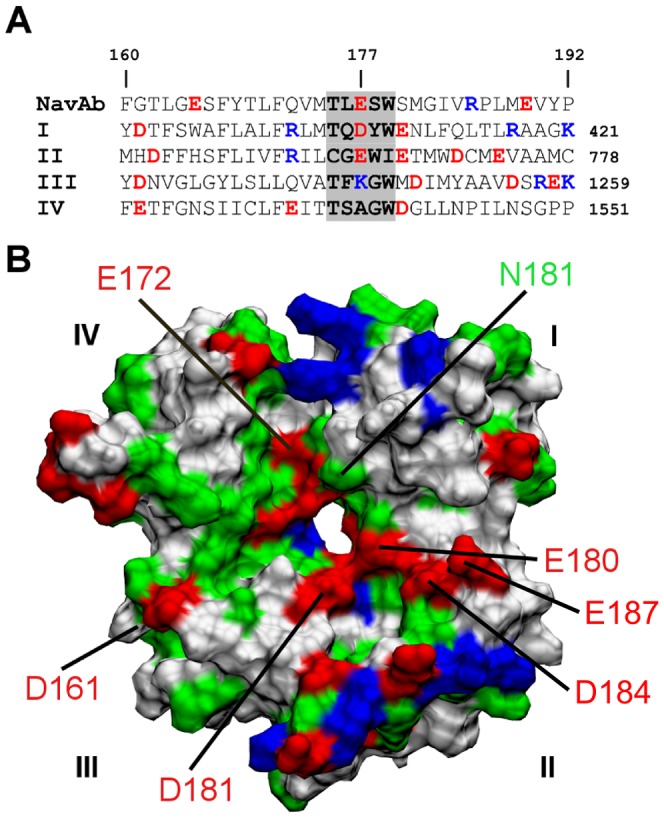
The selectivity filter of NaV1.4 carries a DEKA ring (Fig. 1A), as opposed to the EEEE ring found in NaVAb and voltage-gated Ca2+ (CaV) channels. This feature of NaV1.4 may have important implications for its ion conduction mechanism. After 50 ns of simulation, one Na+ ion is present in close proximity to the aspartate residue of the DEKA ring (Fig. 2A). The orientation of Lys177 side chain is stabilized by the two salt bridges it forms with the acidic residues at position 180 of DI and DIV, consistent with a recent study [42]. A second Na+ ion is found near the outer ring of acidic residues just outside the filter (Fig. 2A).
Figure 2. Ion binding site of NaV1.4 and structure of PIIIA.
(A) The selectivity filter of NaV1.4 with two Na+ ions (yellow spheres) bound. (B) Molecular structure of PIIIA with the side chains of six basic residues highlighted in blue.
Structure of PIIIA
PIIIA is a small peptide consisting of 22 residues. The primary structure of PIIIA is PcaRLCC-GFOKS-CRSRQ-CKOHR-CC, where Pca and O indicate pyroglutamate and hydroxyproline, respectively. The C-terminus of PIIIA is amidated and the overall charge of PIIIA is +6 e at neutral pH resulting from the six basic residues it carries. Fig. 2B shows that these six residues are approximately coplanar and symmetrically distributed around the globular surface of the toxin. This symmetry may be important for the high-affinity binding of PIIIA to NaV channels. For example, we show below that PIIIA can use each of the six basic residues it carries to occlude the selectivity filter of NaV1.4, similar to that observed previously for NaVAb [29].
Binding modes of PIIIA to NaV1.4
To predict the most favorable structures of PIIIA in complex with NaV1.4, we use MD simulations with distance restraints applied during the first 5 ns, as described in the Methods section. This method allows us to examine all the possible binding modes between the toxin and the channel with affordable computational cost. Two distinct binding modes, in which Lys9 and Arg14 of PIIIA occlude the selectivity filter of NaV1.4, respectively, are considered initially. For each binding mode, two simulations started from different initial configuration are performed to improve sampling. Based on the number of salt bridges and hydrogen bonds formed in the toxin-channel complex, the simulation predicting the more favorable structure for each binding mode is considered as representative and discussed in detail below.
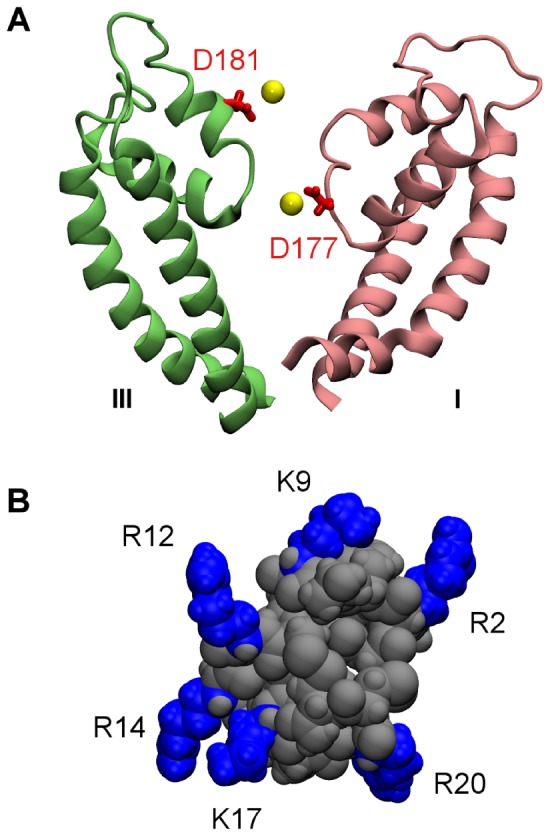
First we look at the simulation in which a distance restraint is applied to Lys9 of PIIIA and the selectivity filter of NaV1.4 over the first 5 ns. The distance restraint applied pulls the side chain of Lys9 rapidly into the filter, whereas other residues of the toxin interact favorably with the vestibular wall of the channel. Fig. 3A shows that three salt bridges are formed at 8 ns and remain intact for more than 10 ns during the remaining period of the simulation, suggesting that the complex structure is stable and well equilibrated. One of the three salt bridges (Lys9-Asp177) is located inside the filter, whereas the other two salt bridges are on the vestibular wall (Fig. 4 A and B). The Na+ ion inside the filter moves toward the inner cavity of the channel after the binding of PIIIA (Fig. 4A), possibly due to the repelling force from the positive charge carried by Lys9 of PIIIA.
Figure 3. Time evolution of the lengths of the salt bridges in two MD simulations.
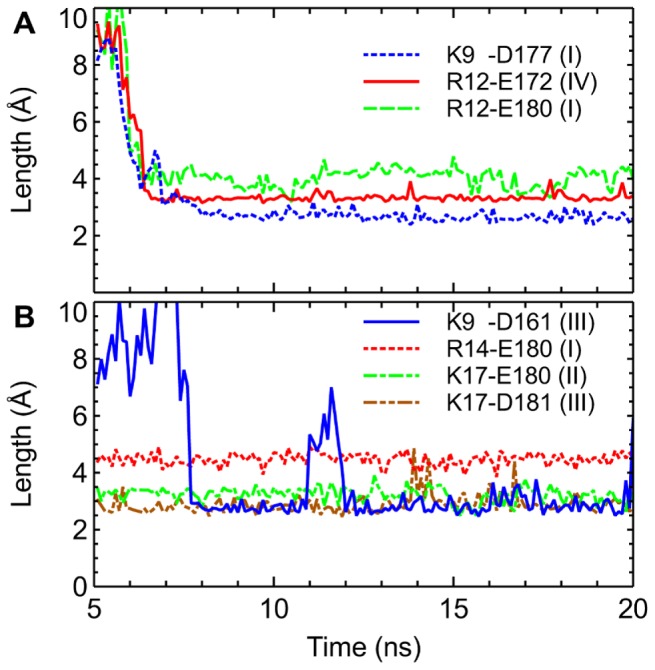
A distance restraint is applied to Lys9 (A) or Arg14 (B) of PIIIA and the filter of NaV1.4 over the first 5 ns of each simulation.
Figure 4. Structures of PIIIA in complex with NaV1.4 predicted from MD simulations biased with distance restraints.

In two binding modes of PIIIA-NaV1.4, Lys9 (A and B) and Arg14 (C and D) of the toxin protrudes into the filter of the channel, respectively. In (A) and (C), the Na+ ion inside the filter is shown as a yellow sphere. Toxin backbone is in yellow and channel backbone in grey.
Next we look at the simulation in which Arg14 of PIIIA is drawn into the filter of NaV1.4 by using a distance restraint. The position of PIIIA relative to the outer vestibule of NaV1.4 is displayed in Fig. 4 C and D. Four salt bridges are formed between the toxin and the channel, of which one is inside the filter and three on the vestibular wall. The salt bridge in the filter, Arg14-Glu180, is relatively weak, with its length longer than 4 Å most of the times (Fig. 3B). The three salt bridges on the vestibular wall, on the other hand, being less than 3.5 Å in length, are relatively strong. Although Arg14 of PIIIA does not form a strong salt bridge with the filter, the position of Arg14 with respect to the filter of the channel appears to be stable.
In the two binding modes discussed above, the toxin-channel complexes are stabilized by three or four salt bridges, whereas the filter residue (Lys9 or Arg14) protrudes deeply into the filter in both modes (Fig. 4). In these two binding modes the toxin interacts intimately with either DI-Glu180 or DII-Glu180, both of which have been shown experimentally to be important for the binding of a closely-related toxin, GIIIA [56]. Similar toxin-channel interactions are observed in all the other four possible binding modes, in which Arg2, Arg12, Lys17 or Arg20 of PIIIA protrudes into the filter of NaV1.4, predicted from MD simulations with distance restraints. In all the four modes, the toxin is able to form three or four salt bridges with the channel (Fig. S3), consistent with the Lys9 and Arg14 binding modes we observed (Fig. 4). Thus, the six binding modes of PIIIA-NaV1.4 may be of similar energetics. Subsequent PMF calculations are consistent with this proposal.
PMF profiles
To validate the binding modes of PIIIA-NaV1.4 predicted from MD simulations, we construct the PMF profile for the dissociation of the toxin from each complex along the channel axis (z). Based on the PMF profile, the corresponding K d value is calculated according to eq. 1 and compared to experiment. Due to the limited computational resource available, we derive PMF profiles for the selected two binding modes, in which Lys9 or Arg14 is the filter residue. The Arg14 mode is considered because it has been proposed as the predominant mode in previous experimental studies [23]. The PMF for the Lys9 mode is derived to illustrate that a binding mode equally favorable to the Arg14 mode exists.
For the Lys9 mode, extra umbrella windows are added at z = 23.8 Å and z = 25.2 Å to ensure good overlap between umbrella windows. Fig. S4 shows the convergence of the PMF profiles. The converged PMF profiles for the two binding modes in which Lys9 or Arg14 of PIIIA protrudes into the filter of NaV1.4 are displayed in Fig. 5. No significant differences in the two profiles are evident, indicating that the two binding modes are of similar energetics.
Figure 5. The PMF profiles for the dissociation of PIIIA from NaV1.4.
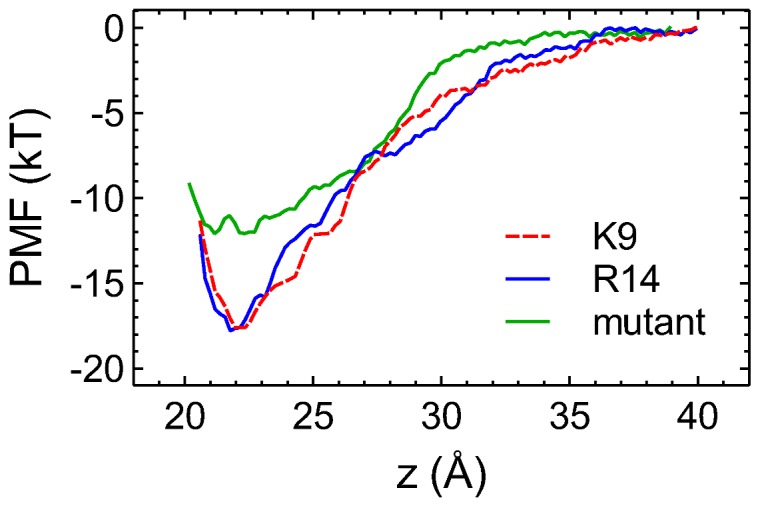
The profile for PIIIA binding to a [DI-N181R, DIV-E172Q] double mutant of NaV1.4 is also shown. The reaction coordinate z is the distance between the centers of mass of toxin and channel backbones along the channel axis. The random errors of the PMF profiles estimated from the bootstrapping method are less than 0.5 kT in all cases.
The depth of the PMF profile for the Lys9 mode is 17.6 kT, close to the value of 17.8 kT for the Arg14 mode. The K d values computed are 300 nM and 150 nM for the Lys9 and Arg14 modes, respectively. Experimentally the K d value for the binding of PIIIA to rat NaV1.4, which is identical to human NaV1.4 in the pore loops, has been estimated to be in the range of 36 nM to 200 nM [12], [23], [30]. Therefore, the K d values predicted from PMF calculations do not differ dramatically from those determined experimentally.
We note here that the two PMF profiles we derived are biased. In other words, the umbrella sampling simulations over a limited simulation time only explore a narrow configurational space. In real systems, the toxin would bind with different modes each contributing to an unbiased PMF. The unbiased PMF is reflected in the K d value measured experimentally. The fact that the K d values derived from the two biased PMF profiles are in good agreement with the experimental K d value suggests that the true unbiased PMF profile is similar to the biased PMF profiles we derived (Fig. 5).
Various factors can contribute to the uncertainty in the predicted K d values. The accuracy of the force field and molecular models, and the search of configurational space during umbrella sampling largely determine the systematic error, whereas approximating a PMF profile from umbrella sampling simulations can cause random errors. The random errors can be derived, e.g., from bootstrapping methods, while the systematic error can only be estimated by comparing the K d values predicted to that determined experimentally. Numerous studies performed on pore blockers of K+ channels showed that the systematic error was generally less than 3 kT [27], [28]. However, the systematic error for the PMF profiles in Fig. 5 could be higher, due to the uncertainty in the model of the channel. Also, the hetero-tetrameric nature of NaV1.4 poses difficulties in the sampling of the configurational space. Nevertheless, the overall error for the PMF profiles of PIIIA-NaV1.4 is found to be low, possibly because errors from different sources cancel out.
A NaV1.4 double mutant
Several isoforms of NaV channels (NaV1.5, NaV1.8 and NaV1.9) are resistant to TTX and μ-conotoxins [12], [57]. NaV1.5 is important for heart function [58], whereas NaV1.8 and NaV1.9 are involved in the perception of pain [1]. Sequence alignment shows that all these three channels differ from NaV1.4 at two positions, namely, position 181 of DI and position 172 of DIV (Fig. 6). At position 181 of DI, it is a neutral asparagine in NaV1.4, in contrast to a positively charged lysine or arginine in TTX-resistant channels. At position 172 of DIV, it is a negatively charged glutamate in NaV1.4, and a neutral glutamine in TTX-resistant channels. These two residues are in close proximity to each other, as illustrated in Fig. 1B. A double mutant of NaV1.4 (DI-N181R, DIV-E172Q) is constructed to ascertain the role of these two residues in the selectivity of PIIIA for NaV1.4. We show that the mutation causes the K d value for the binding of PIIIA to the channel to increase by two orders of magnitude, suggesting the importance of residues DI-Asn181 and DIV-Glu172 for the selectivity of PIIIA for NaV1.4 over TTX-resistant NaV channels.
Figure 6. Sequence alignment of NaV1.4 and TTX-resistant channels.
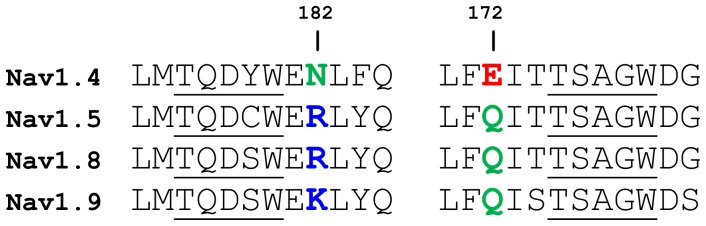
The selectivity filter region of DI (left) and DIV (right) is shown.
Computational mutagenesis is performed on the PIIIA-NaV1.4 complex in which toxin Arg14 protrudes into the filter of the channel. DI-Asn181 and DIV-Glu172 are replaced with an arginine and a glutamine, respectively, resulting in a net loss of -2 e in charge to the outer vestibule of the channel. Considering that PIIIA is positively charged, the mutant channel is expected to have a lower affinity for the toxin. Two Na+ ions are removed from the bulk to maintain charge neutrality. The complex is equilibrated for 10 ns in an unbiased MD simulation. The salt bridge Lys9-Asp161 is broken but an equivalent salt bridge, Arg20-Glu187, is formed after the equilibration. Although the same number of salt bridges is present in the structures of PIIIA in complex with the wild type and mutant NaV1.4, the binding affinity is found to decrease substantially after the mutation. Thus, the two residues mutated influence the binding affinity primarily via long-range electrostatic interactions with the toxin, and should have similar effects on different binding modes of the toxin and the channel. The PMF profile for the mutant channel, 12 kT in depth, predicts a K d value of 20 μM (Fig. 5). The mutant NaV1.4 binds PIIIA about 100 fold less effectively than the wild type, suggesting that the two residues (DI-Asn181 and DIV-Glu172) are important for toxin selectivity.
Conclusions
In this work the binding of μ-conotoxin PIIIA to a homology model of the voltage-gated sodium channel NaV1.4 is examined using MD simulations. Six binding modes, in which any of the six basic residues carried by PIIIA protrudes into the selectivity filter of NaV1.4, are considered. In all the binding modes the toxin-channel complex is stabilized by three or four salt bridges. PMF calculations predict K d values of 300 and 150 nM for the Lys9 and Arg14 modes, respectively, in close proximity to the experimental values of 36–200 nM [12], [23], [30]. Thus, our simulations suggest that PIIIA can inhibit NaV1.4 with multiple alternative binding modes of similar energetics. Further studies would be required to determine whether or not the multiple-binding-mode mechanism of PIIIA can be generalized to other μ-conotoxins such as GIIIA, for which one predominant binding mode to NaV1.4 has been suggested [59], [60].
The multiple binding modes of PIIIA to NaV1.4 uncovered here is consistent with the extensive mutagenesis experiments of McArthur et al. [24]. For example, it was found that the mutation of Arg14 to an alanine or glutamate reduces the affinity of PIIIA by only 11 fold, corresponding to a change of 2.4 kT in the binding free energy, substantially lower than the free energy of 6–7 kT for a typical salt bridge [25], [26]. In addition, the alanine mutation of several other basic residues, such as Arg2, Arg12 and Lys17, causes similar reduction in the toxin's binding affinity [23], [24]. The substitution of Lys8 in GIIIA, which corresponds to Lys9 in PIIIA, to a glutamine, also causes the toxin affinity to decrease by only 7 fold [10]. The similar effect of different mutations observed experimentally support a multiple-binding-mode mechanism by PIIIA. When a basic residue of the toxin is mutated to an alanine, only one of the several binding modes is disrupted. As such each charge-changing mutation causes a similar reduction in binding affinity by the removal of a positive charge. Different isomers of PIIIA having similar affinities for NaV1.4 may adopt distinct binding modes to the channel [30], suggesting that a unique binding mode is inadequate to describe PIIIA binding to NaV channels.
Experimental studies have found that several residues from DII and DIII of NaV channels are important for the specificity of μ-conotoxins [18], [61], [62]. Experimental data have also suggested that DI-Asn181 might be involved in μ-conotoxin specificity [63]. Here it is shown that residues DI-Asn181 and DIV-Glu172 are important for the selectivity of PIIIA for NaV1.4 over TTX-resistant NaV channels. As DI-Asn181 is replaced by a basic residue in TTX-resistant channels, one or more acidic residues may be introduced into the toxin to improve the affinity of the toxin for these channels. In fact, all known μ-conotoxins such as SmIIIA, KIIIA and BuIIIA with a K d value for NaV1.5 in the micromolar range carry one or more acidic residues [4], [12].
Supporting Information
Primary structure of human NaV1.4. The selectivity filter region is in purple and other regions of the pore domain included in homology modeling are highlighted in yellow. Regions of the pore domain not modeled are in grey.
(TIF)
(A) Structural alignment between NaVAb (red) and the four domains of our NaV1.4 model (blue). (B) The position of NaV1.4 model (purple ribbons) relative to the lipid bilayer viewed perpendicular to the bilayer normal is shown on the left. The view along the bilayer normal from the extracellular side is shown on the right (for channel protein color scheme is as follows: green, polar; white, hydrophobic; blue, basic; red, acidic). Yellow spheres indicate lipid phosphorus atoms. Blue lines indicate the boundary of the simulation box. Water molecules and ions are not shown for clarity.
(TIF)
Structures of toxin PIIIA (yellow ribbons) in complex with NaV1.4 (gray ribbons) in which the side chains of Arg2 (A), Arg12 (B), Lys17 (C) and Arg20 (D) protrude into the filter of the channel. In (D), Arg2 from PIIIA is in close contact with DIII-Asp161. The complex structures are predicted from MD simulations biased with distance restraints.
(TIF)
Block analysis of the PMF profiles for the Lys9 mode (A) and the Arg14 mode (B).
(TIF)
PDB coordinate files of NaV1.4 model and NaV1.4 in complex with PIIIA.
(ZIP)
Acknowledgments
This research was undertaken on the National Computational Infrastructure in Canberra, Australia, which is supported by the Australian Commonwealth Government.
Funding Statement
This research was undertaken on the National Computational Infrastructure in Canberra, Australia, which is supported by the Australian Commonwealth Government. This work was supported by the National Health and Medical Research Council of Australia and The Medical Advances Without Animals Trust (MAWA). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Dib-Hajj SD, Cummins TR, Black JA, Waxman SG (2010) Sodium channels in normal and pathological pain. Annu Rev Neurosci 33: 325–347. [DOI] [PubMed] [Google Scholar]
- 2. Saez NJ, Senff S, Jensen JE, Er SY, Herzig V, et al. (2010) Spider-venom peptides as therapeutics. Toxins 2: 2851–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith JJ, Herzig V, King GF, Alewood PF (2013) The insecticidal potential of venom peptides. Cell Mol Life Sci 70: 3665–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Norton RS (2010) μ-Conotoxins as leads in the development of new analgesics. Molecules 15: 2825–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Knapp O, McArthur JR, Adams DJ (2012) Conotoxins targeting neuronal voltage-gated sodium channel subtypes: potential analgesics? Toxins 4: 1236–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sato K, Ishida Y, Wakamatsu K, Kato R, Honda H, et al. (1991) Active site of μ-conotoxin GIIIA, a peptide blocker of muscle sodium channels. J Biol Chem 266: 16989–16991. [PubMed] [Google Scholar]
- 7. Shon KJ, Olivera BM, Watkins M, Jacobsen RB, Gray WR, et al. (1998) μ-Conotoxin PIIIA, a new peptide for discriminating among tetrodotoxin-sensitive Na channel subtypes. J Neurosci 18: 4473–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. West PJ, Bulaj G, Garrett JE, Olivera BM, Yoshikami D (2002) μ-Conotoxin SmIIIA, a potent inhibitor of tetrodotoxin-resistant sodium channels in amphibian sympathetic and sensory neurons. Biochemistry 41: 15388–15393. [DOI] [PubMed] [Google Scholar]
- 9. Bulaj G, West PJ, Garrett JE, Watkins M, Zhang MM, et al. (2005) Novel conotoxins from Conus striatus and Conus kinoshitai selectively block TTX-resistant sodium channels. Biochemistry 44: 7259–7265. [DOI] [PubMed] [Google Scholar]
- 10. Becker S, Prusak-Sochaczewski E, Zamponi G, Beck-Sickinger AG, Gordon RD, et al. (1992) Action of derivatives of μ-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry 31: 8229–8238. [DOI] [PubMed] [Google Scholar]
- 11. Wakamatsu K, Kohda D, Hatanaka H, Lancelin JM, Ishida Y, et al. (1992) Structure-activity relationships of μ-conotoxin GIIIA: structure determination of active and inactive sodium channel blocker peptides by NMR and simulated annealing calculations. Biochemistry 31: 12577–12584. [DOI] [PubMed] [Google Scholar]
- 12. Wilson MJ, Yoshikami D, Azam L, Gajewiak J, Olivera BM, et al. (2011) μ-Conotoxins that differentially block sodium channels NaV 1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc Natl Acad Sci U S A 108: 10302–10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lewis RJ, Dutertre S, Vetter I, Christie MJ (2012) Conus venom peptide pharmacology. Pharmacol Rev 64: 259–298. [DOI] [PubMed] [Google Scholar]
- 14. Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, et al. (2007) Structure/function characterization of μ-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J Biol Chem 282: 30699–30706. [DOI] [PubMed] [Google Scholar]
- 15. Khoo KK, Wilson MJ, Smith BJ, Zhang MM, Gulyas J, et al. (2011) Lactam-stabilized helical analogues of the analgesic μ-conotoxin KIIIA. J Med Chem 54: 7558–7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang MM, Gruszczynski P, Walewska A, Bulaj G, Olivera BM, et al. (2010) Cooccupancy of the outer vestibule of voltage-gated sodium channels by μ-conotoxin KIIIA and saxitoxin or tetrodotoxin. J Neurophysiol 104: 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khoo KK, Feng ZP, Smith BJ, Zhang MM, Yoshikami D, et al. (2009) Structure of the analgesic μ-conotoxin KIIIA and effects on the structure and function of disulfide deletion. Biochemistry 48: 1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McArthur JR, Singh G, McMaster D, Winkfein R, Tieleman DP, et al. (2011) Interactions of key charged residues contributing to selective block of neuronal sodium channels by μ-conotoxin KIIIA. Mol Pharmacol 80: 573–584. [DOI] [PubMed] [Google Scholar]
- 19. Zhang MM, Han TS, Olivera BM, Bulaj G, Yoshikami D (2010) μ-Conotoxin KIIIA derivatives with divergent affinities versus efficacies in blocking voltage-gated sodium channels. Biochemistry 49: 4804–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stevens M, Peigneur S, Dyubankova N, Lescrinier E, Herdewijn P, et al. (2012) Design of bioactive peptides from naturally occurring μ-conotoxin structures. J Biol Chem 287: 31382–31392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stevens M, Peigneur S, Tytgat J (2011) Neurotoxins and their binding areas on voltage-gated sodium channels. Front Pharmacol 2: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Catterall WA, Goldin AL, Waxman SG (2005) International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 57: 397–409. [DOI] [PubMed] [Google Scholar]
- 23. McArthur JR, Singh G, O'Mara ML, McMaster D, Ostroumov V, et al. (2011) Orientation of μ-Conotoxin PIIIA in a sodium channel vestibule, based on voltage dependence of its binding. Mol Pharmacol 80: 219–227. [DOI] [PubMed] [Google Scholar]
- 24. McArthur JR, Ostroumov V, Al-Sabi A, McMaster D, French RJ (2011) Multiple, distributed interactions of μ-conotoxin PIIIA associated with broad targeting among voltage-gated sodium channels. Biochemistry 50: 116–124. [DOI] [PubMed] [Google Scholar]
- 25. Wimley WC, Gawrisch K, Creamer TP, White SH (1996) Direct measurement of salt-bridge solvation energies using a peptide model system: Implications for protein stability. Proc Natl Acad Sci U S A 93: 2985–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anderson DE, Becktel WJ, Dahlquist FW (1990) pH-induced denaturation of proteins: a single salt bridge contributes 3–5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry 29: 2403–2408. [DOI] [PubMed] [Google Scholar]
- 27. Gordon D, Chen R, Chung SH (2013) Computational methods of studying the binding of toxins from venomous animals to biological ion channels: theory and applications. Physiol Rev 93: 767–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rashid MH, Mahdavi S, Kuyucak S (2013) Computational studies of marine toxins targeting ion channels. Mar Drugs 11: 848–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen R, Chung SH (2012) Binding modes of μ-conotoxin to the bacterial sodium channel (NaVAb). Biophys J 102: 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tietze AA, Tietze D, Ohlenschlager O, Leipold E, Ullrich F, et al. (2012) Structurally diverse μ-conotoxin PIIIA isomers block sodium channel NaV1.4. Angew Chem Int Edit 51: 4058–4061. [DOI] [PubMed] [Google Scholar]
- 31. Choudhary G, Aliste MP, Tieleman DP, French RJ, Dudley SC (2007) Docking of μ-conotoxin GIIIA in the sodium channel outer vestibule. Channels 1: 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chagot B, Potet F, Balser JR, Chazin WJ (2009) Solution NMR structure of the C-terminal EF-hand domain of human cardiac sodium channel NaV1.5. J Biol Chem 284: 6436–6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chagot B, Chazin WJ (2011) Solution NMR structure of Apo-calmodulin in complex with the IQ motif of human cardiac sodium channel NaV1.5. J Mol Biol 406: 106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Payandeh J, Scheuer T, Zheng N, Catterall WA (2011) The crystal structure of a voltage-gated sodium channel. Nature 475: 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fallon JL, Baker MR, Xiong LW, Loy RE, Yang GJ, et al. (2009) Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+•calmodulins. Proc Natl Acad Sci U S A 106: 5135–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31: 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tikhonov DB, Zhorov BS (2012) Architecture and pore block of eukaryotic voltage-gated sodium channels in view of NavAb bacterial sodium channel structure. Mol Pharmacol 82: 97–104. [DOI] [PubMed] [Google Scholar]
- 38. Zhang X, Ren W, DeCaen P, Yan C, Tao X, et al. (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486: 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dudley SC, Chang N, Hall J, Lipkind G, Fozzard HA, et al. (2000) μ-Conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J Gen Physiol 116: 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li RA, Ennis IL, French RJ, Dudley SC, Tomaselli GF, et al. (2001) Clockwise domain arrangement of the sodium channel revealed by μ-conotoxin (GIIIA) docking orientation. J Biol Chem 276: 11072–11077. [DOI] [PubMed] [Google Scholar]
- 41. Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14: 33–38. [DOI] [PubMed] [Google Scholar]
- 42. Xia MD, Liu H, Li Y, Yan N, Gong HP (2013) The mechanism of Na+/K+ selectivity in mammalian voltage-gated sodium channels based on molecular dynamics simulation. Biophys J 104: 2401–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nielsen KJ, Watson M, Adams DJ, Hammarstrom AK, Gage PW, et al. (2002) Solution structure of μ-conotoxin PIIIA, a preferential inhibitor of persistent tetrodotoxin-sensitive sodium channels. J Biol Chem 277: 27247–27255. [DOI] [PubMed] [Google Scholar]
- 44. Eriksson MA, Roux B (2002) Modeling the structure of agitoxin in complex with the Shaker K+ channel: a computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys J 83: 2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen R, Chung SH (2012) Structural basis of the selective block of Kv1.2 by maurotoxin from computer simulations. PLoS One 7: e47253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen R, Chung SH (2013) Complex structures between the N-type calcium channel (CaV2.2) and ω-conotoxin GVIA predicted via molecular dynamics. Biochemistry 52: 3765–3772. [DOI] [PubMed] [Google Scholar]
- 47. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, et al. (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26: 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102: 3586–3616. [DOI] [PubMed] [Google Scholar]
- 49. Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, et al. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114: 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1982) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79: 926–935. [Google Scholar]
- 51. Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23: 327–341. [Google Scholar]
- 52. Miyamoto S, Kollman PA (1992) SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem 13: 952–962. [Google Scholar]
- 53. Kumar S, Nussinov R (2002) Close-range electrostatic interactions in proteins. ChemBioChem 3: 604–617. [DOI] [PubMed] [Google Scholar]
- 54. Mills JE, Dean PM (1996) Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J Comput Aided Mol Des 10: 607–622. [DOI] [PubMed] [Google Scholar]
- 55. Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM (1992) The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comput Chem 13: 1011–1021. [Google Scholar]
- 56. Chang NS, French RJ, Lipkind GM, Fozzard HA, Dudley S (1998) Predominant interactions between μ-conotoxin Arg-13 and the skeletal muscle Na+ channel localized by mutant cycle analysis. Biochemistry 37: 4407–4419. [DOI] [PubMed] [Google Scholar]
- 57. Lee CH, Ruben PC (2008) Interaction between voltage-gated sodium channels and the neurotoxin, tetrodotoxin. Channels 2: 407–412. [DOI] [PubMed] [Google Scholar]
- 58. Abriel H (2007) Roles and regulation of the cardiac sodium channel NaV1.5: Recent insights from experimental studies. Cardiovasc Res 76: 381–389. [DOI] [PubMed] [Google Scholar]
- 59. Hui K, Lipkind G, Fozzard HA, French RJ (2002) Electrostatic and steric contributions to block of the skeletal muscle sodium channel by μ-conotoxin. J Gen Physiol 119: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hui KY, McIntyre D, French RJ (2003) Conotoxins as sensors of local pH and electrostatic potential in the outer vestibule of the sodium channel. J Gen Physiol 122: 63–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li RA, Ennis IL, Xue T, Nguyen HM, Tomaselli GF, et al. (2003) Molecular basis of isoform-specific μ-conotoxin block of cardiac, skeletal muscle, and brain Na+ channels. J Biol Chem 278: 8717–8724. [DOI] [PubMed] [Google Scholar]
- 62. Cummins TR, Aglieco F, Dib-Hajj SD (2002) Critical molecular determinants of voltage-gated sodium channel sensitivity to μ-conotoxins GIIIA/B. Mol Pharmacol 61: 1192–1201. [DOI] [PubMed] [Google Scholar]
- 63. Leipold E, Markgraf R, Miloslavina A, Kijas M, Schirmeyer J, et al. (2011) Molecular determinants for the subtype specificity of μ-conotoxin SIIIA targeting neuronal voltage-gated sodium channels. Neuropharmacology 61: 105–111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primary structure of human NaV1.4. The selectivity filter region is in purple and other regions of the pore domain included in homology modeling are highlighted in yellow. Regions of the pore domain not modeled are in grey.
(TIF)
(A) Structural alignment between NaVAb (red) and the four domains of our NaV1.4 model (blue). (B) The position of NaV1.4 model (purple ribbons) relative to the lipid bilayer viewed perpendicular to the bilayer normal is shown on the left. The view along the bilayer normal from the extracellular side is shown on the right (for channel protein color scheme is as follows: green, polar; white, hydrophobic; blue, basic; red, acidic). Yellow spheres indicate lipid phosphorus atoms. Blue lines indicate the boundary of the simulation box. Water molecules and ions are not shown for clarity.
(TIF)
Structures of toxin PIIIA (yellow ribbons) in complex with NaV1.4 (gray ribbons) in which the side chains of Arg2 (A), Arg12 (B), Lys17 (C) and Arg20 (D) protrude into the filter of the channel. In (D), Arg2 from PIIIA is in close contact with DIII-Asp161. The complex structures are predicted from MD simulations biased with distance restraints.
(TIF)
Block analysis of the PMF profiles for the Lys9 mode (A) and the Arg14 mode (B).
(TIF)
PDB coordinate files of NaV1.4 model and NaV1.4 in complex with PIIIA.
(ZIP)