

Abstract
Breast cancers classified as triple-negative (TNBC) and BRCA1-deficient, are particularly aggressive and difficult to treat. A major breakthrough was the finding that these tumors are exquisitely sensitive to inhibitors of poly(ADP-ribose) polymerase (PARPi). Phase II clinical trials have shown encouraging outcomes, with tolerable side effects. However, a significant fraction of these cancers acquire resistance. Elegant studies demonstrated that loss of the DNA repair protein 53BP1 contributes to the resistance of BRCA1-deficient cells and tumors to PARPi. Thus, raising the levels of 53BP1 in these aggressive tumors could potentially restore their sensitivity to PARPi and other genotoxic agents. We will review here our studies revealing that 1α,25(OH)2D3, an active form of vitamin D, stabilizes 53BP1 levels in tumor cells. Breast tumor cells that become BRCA1-deficient activate cathepsin L-mediated degradation of 53BP1 to ensure genome stability and proliferation. Importantly, 1α,25(OH)2D3 treatment restores the levels of 53BP1 as efficiently as cathepsin L inhibitors, which results in increased genomic instability in response to PARPi or radiation, and reduced proliferation. Furthermore, analysis of human breast tumors identified nuclear cathepsin L as a positive biomarker for TNBC, which correlates inversely with 53BP1 when vitamin D receptor (VDR) nuclear levels are low. The major findings of these studies are: 1) identification of a new pathway contributing to breast cancers with the poorest prognosis; 2) discovery of the ability of 1α,25(OH)2D3 to inhibit this pathway; and 3) discovery of a triple biomarker signature for identification of patients that could benefit from the treatment.
Keywords: BRCA1; 53BP1; cathepsin L; DNA repair; vitamin D; 1α,25(OH)2D3; breast cancer
Introduction
Breast cancer is the leading cause of cancer death in women worldwide (Jemal, Center et al. 2010). Women carrying germline mutations in BRCA1 have a 50–80% risk of developing breast cancer during their lifetime (King et al. 2003). The tumors that arise are highly invasive, tend to lack expression of estrogen and progesterone receptors, and do not show upregulation of HER2, being classified as “triple negative breast cancers” (TNBC) (Sorlie et al. 2003; Turner and Reis-Filho 2006). Interestingly, a subset of sporadic TNBC present with DNA repair defects and gene expression profiles that phenocopy BRCA1 related cancers and as such, are often responsive to therapeutic strategies that exploit DNA repair deficiencies (Foulkes, Smith et al. 2010).
A major breakthrough in the treatment of BRCA1-deficient and TNBC patients was the finding that these tumors are exquisitely sensitive to poly(ADP-ribose) polymerase inhibitors (PARPi) (Farmer, McCabe et al. 2005; Helleday, Bryant et al. 2005). Inhibition of PARP1 and the closely related PARP2 proteins (Satoh and Lindahl 1992; Ame, Rolli et al. 1999; Allinson, Dianova et al. 2003; Woodhouse and Dianov 2008) hinders single-strand breaks (SSBs) repair, which in turn leads to the stalling of the replication fork and the formation of double-strand breaks (DSBs) that need to be repaired primarily by homologous recombination (HR) (Helleday, Bryant et al. 2005; Ashworth 2008). BRCA1 plays a critical role in the repair of DNA DSBs by HR (Scully, Ganesan et al. 1996; Scully and Livingston 2000), a process that utilizes sister chromatids as templates for recombination resulting in error-free DNA DSBs repair. Therefore, BRCA1-deficient cells cannot deal with the amount of DSBs generated by PARPi, resulting in proliferation arrest and cell death. The demonstrated vulnerability of BRCA1-deficient cells and tumors to PARPi has expedited their use in the clinic (Rottenberg, Jaspers et al. 2008; Fong, Boss et al. 2009; Audeh, Carmichael et al. 2010). Phase II studies with PARPi have shown a significant response rate in women carrying BRCA1 mutations, with tolerable side effects (Tutt, Robson et al. 2010). Thus, the use of PARPi as single agents or in combination with radiation and chemotherapy represents a leading strategy for the management of breast cancers, especially BRCA1-deficient tumors. However, a significant fraction of these cancers acquire resistance to PARPi, stressing the importance of understanding the molecular mechanisms behind resistance, which will allow the design of novel therapeutic strategies.
Recent ground-breaking studies demonstrated that loss of 53BP1 is “synthetically viable” with BRCA1 loss (Aly and Ganesan 2011). Loss of 53BP1 promotes the viability of BRCA1-deficient cells by rescuing some of the phenotypes associated with the loss of BRCA1 function in HR (Cao, Xu et al. 2009; Bothmer, Robbiani et al. 2010; Bouwman, Aly et al. 2010; Bunting, Callen et al. 2010. Most importantly, loss of 53BP1 induces resistance of BRCA1-deficient cells to PARPi (Jaspers, Kersbergen et al. 2013). 53BP1 facilitates the repair of DNA DSBs by the error prone NHEJ mechanism (Schultz, Chehab et al. 2000; Fernandez-Capetillo, Chen et al. 2002; Wang, Matsuoka et al. 2002; Xie, Hartlerode et al. 2007; Difilippantonio, Gapud et al. 2008; Dimitrova, Chen et al. 2008). The current view is that loss of BRCA1 results in defective end-resection of DNA DSBs, an essential event in HR. Accumulation of 53BP1 at the breaks in this context promotes massive NHEJ with the consequent genomic instability that causes proliferation arrest. However, in cells double deficient in BRCA1 and 53BP1 end-resection is allowed, rescuing at least partially, the HR repair mechanism. As a consequence, these cells exhibit a much lesser degree of genomic instability resulting in increased survival and are less sensitive to PARPi. Altogether these studies indicate that loss of 53BP1 in the context of BRCA1 deficiency is critical for breast tumor progression. As such, loss of 53BP1 negatively correlates with a greater likelihood of metastases and significant decreased survival (Bouwman, Aly et al. 2010; Li, Xu et al. 2012). Thus, upregulation of 53BP1 levels represents a promising strategy to prevent progression of breast tumors with the poorest prognosis as well as to improve their response to PARPi and other DNA damaging strategies such as radiation. However, a limiting factor in the progress towards this goal is the lack of information about how the levels of 53BP1 are regulated in normal cells or downregulated in tumor cells. Here, we review the studies that led us to the identification of a new pathway regulating the levels of 53BP1 protein in mammalian cells, and how this pathway is activated in different disease states. Lastly, we will discuss the possible significance of this novel pathway for diagnosis and design of cancer therapies.
Studies in A-type lamins-deficient cells reveal a new pathway regulating 53BP1
Our initial studies in A-type lamins-deficient mouse embryonic fibroblasts (Lmna−/− MEFs) and lamins A/C-depleted human and mouse cells, revealed a marked decrease in the levels of 53BP1, which was accompanied by defects in DNA repair by NHEJ (Gonzalez-Suarez, Redwood et al. 2009; Gonzalez-Suarez, Redwood et al. 2009; Redwood, Perkins et al. 2011). In addition, we found that loss of A-type lamins results in upregulation of the cysteine protease cathepsin L (CTSL), and its accumulation in the nucleus (Gonzalez-Suarez, Redwood et al. 2011). We also demonstrated that the increase in CTSL, a hallmark of numerous cancers (Lankelma, Voorend et al. 2010), is responsible for the degradation of 53BP1 in lamins A/C-deficient cells. Accordingly, depletion of CTSL rescued normal levels of 53BP1 in these cells, as well as their defects in NHEJ (Gonzalez-Suarez, Redwood et al. 2011) (Figure 1).
Figure 1. Model of mechanisms contributing to genomic instability in lamins-deficient cells.
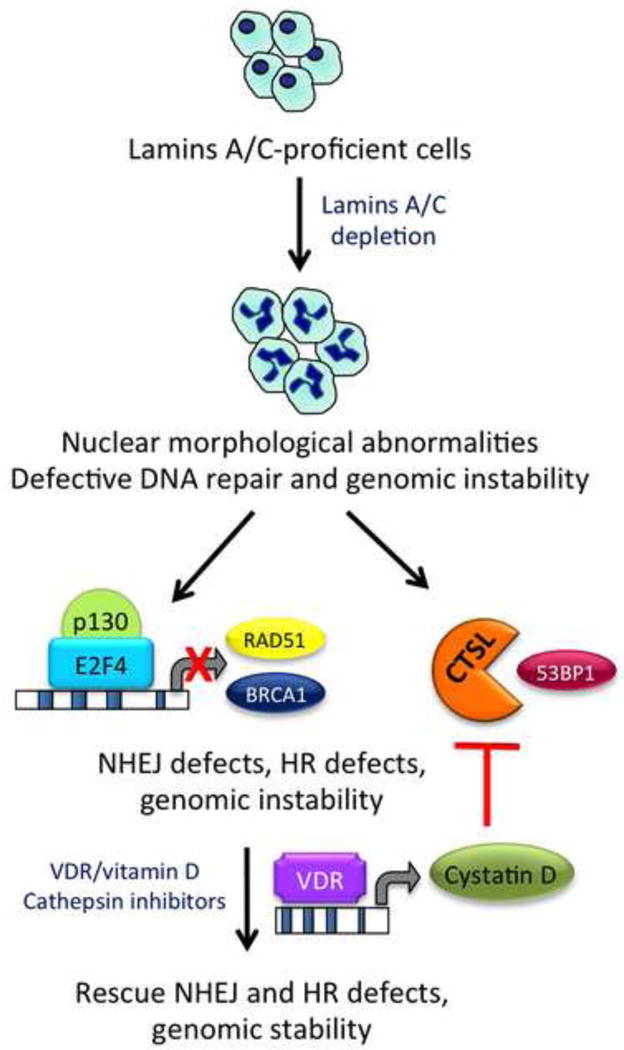
A-type lamins play a key role in the maintenance of nuclear architecture and genome integrity. Loss of A-type lamins results in profound nuclear morphological abnormalities, defects in DNA repair, and genomic instability. The figure illustrates some of the mechanisms behind DNA repair deficiencies, including activation of CTSL-mediated degradation of 53BP1 protein, and transcriptional downregulation of HR proteins BRCA1 and RAD51. A repressor complex formed by the Rb family member p130 and the transcription factor E2F4 participates in the repression. As a consequence, the two main mechanisms of DNA DSB repair -NHEJ and HR- are hindered, resulting in genomic instability. Importantly, inhibition of CTSL via vitamin D treatment restores 53BP1 protein levels as well as unrepaired DNA damage in lamins-deficient cells. Although the mechanism by which vitamin D inhibits CTSL is still under investigation, some data suggests a role for cystatin D mediating the effect.
Given the role of CTSL in regulating 53BP1 stability, we tested whether inhibition of CTSL activity could restore the levels of 53BP1 in A-type lamins-deficient cells, since such strategy could be relevant for therapy. We found that treatment with the specific CTSL inhibitor Z-FY-CHO restored the levels of 53BP1 in these cells (Gonzalez-Suarez, Redwood et al. 2011). Interestingly, we also found that 1α,25(OH)2D3 (the active form of vitamin D), via inhibition of CTSL activity, stabilized 53BP1 protein levels. Although the molecular mechanism by which 1α,25(OH)2D3 inhibits CTSL activity is poorly understood, some studies have indicated that the effect might be mediated by cystatins, endogenous inhibitors of cathepsins. In particular, a study in human colon cancer cells demonstrated that 1α,25(OH)2D3, via the vitamin D receptor (VDR), upregulates the levels of cystatin D, which in turn inhibits the activity of cathepsins, including CTSL (Alvarez-Diaz, Valle et al. 2009). Our gene promoter analysis identified mouse cystatin B as a gene containing at least two VDR/RXR heterodimer binding sites. In addition, we found that cystatin B is downregulated in lamins A/C-deficient cells, thus suggesting that the effect of 1α,25(OH)2D3 could be mediated by cystatins (Figure 1).
Overall, the studies in A-type lamins-deficient cells allowed us to identify a new pathway responsible for regulating the stability of 53BP1 protein in mammalian cells (CTSL-mediated degradation), and a way to inhibit this pathway with therapeutic purposes (CTSL inhibition via specific inhibitors or 1α,25(OH)2D3). Importantly, the effect of CTSL on 53BP1, and as a consequence on DNA repair, is not exclusive of A-type lamins-deficient cells, as overexpression of CTSL in wild-type MEFs results in degradation of 53BP1 and defective NHEJ (Gonzalez-Suarez, Redwood et al. 2011). This suggests that tumors in which CTSL is upregulated could present with genomic instability due to loss of 53BP1, in addition to the characteristic effects of CTSL overexpression on the degradation of the extracellular matrix (Lankelma, Voorend et al. 2010).
Additional studies revealed that loss of A-type lamins results in a marked decrease in the levels of BRCA1 and RAD51 transcripts and proteins (Redwood, Perkins et al. 2011) (Figure 1). As a consequence, these cells present with a 40% reduction in DNA repair by HR. Whether or not 1α,25(OH)2D3 or cathepsin inhibitors are able to rescue the levels of BRCA1 and RAD51 remains unanswered. However, the fact that 1α,25(OH)2D3 treatment reduced the basal levels of unrepaired DNA damage and the nuclear morphological abnormalities that characterize lamins A/C-deficient cells suggests that this is a possibility. Consistent with this possibility, a previous study showed that 1α,25(OH)2D3 treatment results in VDR-mediated induction of BRCA1 gene expression (Campbell, Gombart et al. 2000). Future experiments are needed to test the effect of 1α,25(OH)2D3 treatment on the expression of BRCA1 and RAD51 in lamins A/C-deficient cells, which could provide important clues for therapy.
Mechanisms behind downregulation of 53BP1 in BRCA1-deficient cells
Loss of BRCA1 results in profound genomic instability and proliferation arrest, in part due to inhibition of HR. Thus, an intriguing question in the field has been how BRCA1-deficient tumor cells are able to survive in the context of such profound genomic instability. Loss of 53BP1 seems to play a major role in the survival and proliferation of BRCA1-deficient cells (Cao, Xu et al. 2009; Bothmer, Robbiani et al. 2010; Bouwman, Aly et al. 2010; Bunting, Callen et al. 2010). Thus, understanding the mechanisms by which 53BP1 is lost in cancer cells could provide new therapeutic targets.
Based on the data obtained in A-type lamins-deficient cells, we hypothesized that BRCA1-deficient breast cancer cells might activate CTSL-mediated degradation of 53BP1 as a means to ensure viability and proliferation. If our hypothesis were correct, we could use 1α,25(OH)2D3 or cathepsin inhibitors to increase 53BP1 levels with therapeutic purposes.
To test our hypothesis, we depleted BRCA1 with a specific shRNAs in MCF7 cells, breast tumor cells that are proficient in both BRCA1 and 53BP1. As expected, depletion of BRCA1 leads to a proliferation arrest, which was accompanied by an increase in genomic instability (Grotsky, Gonzalez-Suarez et al. 2013). Growth arrested cells exhibited normal levels of 53BP1 and CTSL immediately after depletion of BRCA1. However, after approximately 2 weeks, BRCA1-deficient cells resumed proliferation, although at a lower rate than control cells. Interestingly, cells that overcome the growth arrest referred to as BOGA cells (BRCA1-deficient cells that Overcome Growth Arrest) exhibit decreased 53BP1 levels and increased CTSL levels, while still maintaining the depletion of BRCA1 (Figure 2). Monitoring transcripts levels revealed that CTSL is upregulated transcriptionally in these cells, and that 53BP1 transcripts levels are not decreased, indicating a decrease in 53BP1 protein stability, similarly to what we observed in A-type lamins-deficient cells.
Figure 2. Model of mechanisms contributing to the bypass of growth arrest in BRCA1-deficient cells.
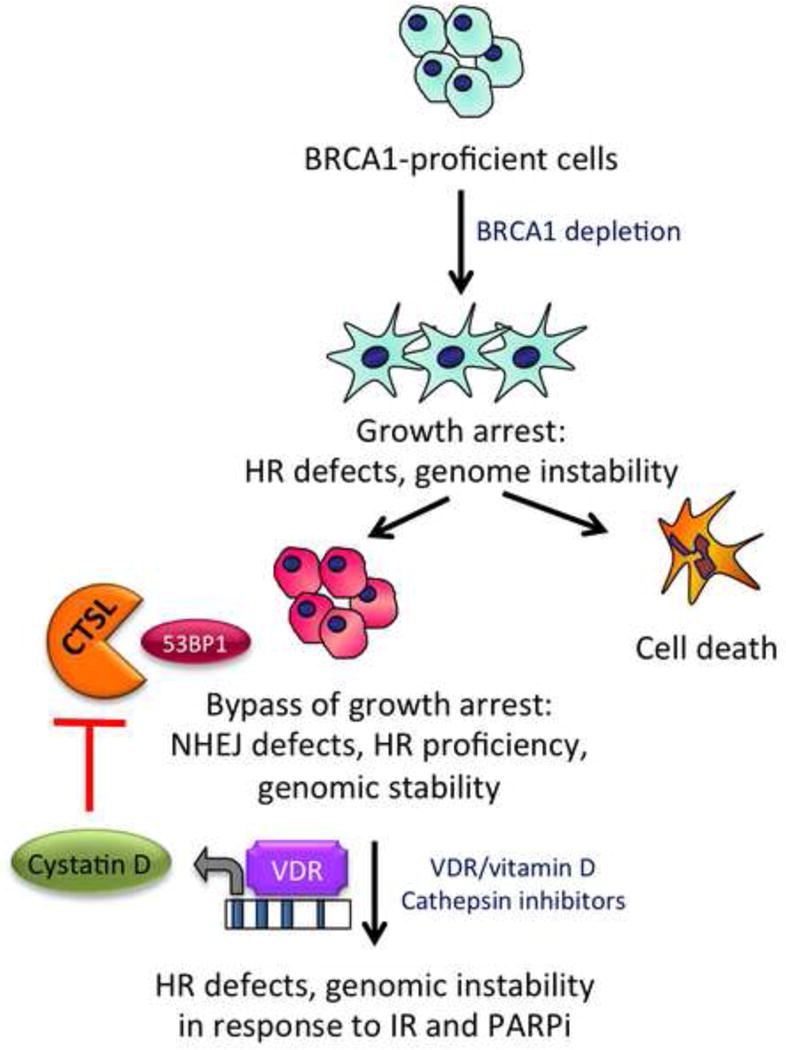
Loss of BRCA1 causes profound genomic instability due to defects in HR, and proliferation arrest. However, after some time in culture, BRCA1-deficient cells activate some mechanisms that allow them to overcome genomic instability and growth arrest. One of these mechanisms is CTSL-mediated degradation of 53BP1. Thus, inhibition of CTSL and stabilization of 53BP1 via treatment with vitamin D or cathepsin inhibitors causes genomic instability and decreased proliferation and survival in the context of BRCA1-deficiency. Furthermore, our model predicts that nuclear VDR regulates the identified mechanism. This provides a new possible therapeutic strategy for BRCA1-related cancers and TNBC.
To determine if the loss of 53BP1 is responsible for the bypass of growth arrest we depleted 53BP1 via specific shRNAs prior to depletion of BRCA1. Importantly, we found that while cells depleted only of BRCA1 undergo growth arrest, cells depleted of 53BP1 continue proliferating after depletion of BRCA1. These results demonstrate that 53BP1 plays a major role in the growth arrest induced by BRCA1 depletion (Grotsky, Gonzalez-Suarez et al. 2013).
Next, we determined if CTSL is responsible for the degradation of 53BP1 following depletion of BRCA1. We performed acute depletion of CTSL in control and BOGA cells. Importantly, depletion of CTSL stabilized 53BP1 protein levels in BOGA cells mirroring those of control cells. In addition, we inhibited CTSL activity in control and BOGA cells via treatment with 1α,25(OH)2D3 or the broad cathepsin inhibitor E64. In both cases, inhibition of CTSL activity leads to stabilization of 53BP1. These data demonstrate that cells growth arrested following depletion of BRCA1 activate CTSL-mediated degradation of 53BP1, allowing cells to bypass growth arrest (Figure 2). In addition, inhibition of CTSL could be used to increase 53BP1 levels in the context of BRCA1 deficiency.
Consequences of stabilization of 53BP1 in BRCA1-deficient cells
The generation of DNA DSBs by ionizing radiation (IR) leads to activation of the DNA damage response (DDR), a complex pathway that senses, signals, and ultimately repairs the DNA lesions generated (Zhou and Elledge 2000; Khanna and Jackson 2001; Hoeijmakers 2009; Jackson and Bartek 2009; Ciccia and Elledge 2010). Activation of DDR leads to the recruitment of a whole variety of factors to the site of damage, which can be easily visualized as nuclear foci by immunofluorescence with specific antibodies, and are referred to as ionizing radiation-induced foci (IRIF). 53BP1 and BRCA1 are recruited to DNA DSBs shortly after radiation (Figure 3). 53BP1 binding facilitates the recruitment of the NHEJ repair machinery (Wyman and Kanaar 2006; Shibata, Conrad et al. 2011), while inhibiting HR. In contrast, recruitment of BRCA1 is associated with repair by HR. BRCA1 facilitates recruitment of RAD51, a protein that is essential for homology search and strand invasion during recombination, and that is commonly used as a readout of HR (Scully, Chen et al. 1997; Scully, Chen et al. 1997; Moynahan, Chiu et al. 1999; Snouwaert, Gowen et al. 1999; Schlegel, Jodelka et al. 2006).
Figure 3. Relationship between 53BP1 and BRCA1 in DNA repair.
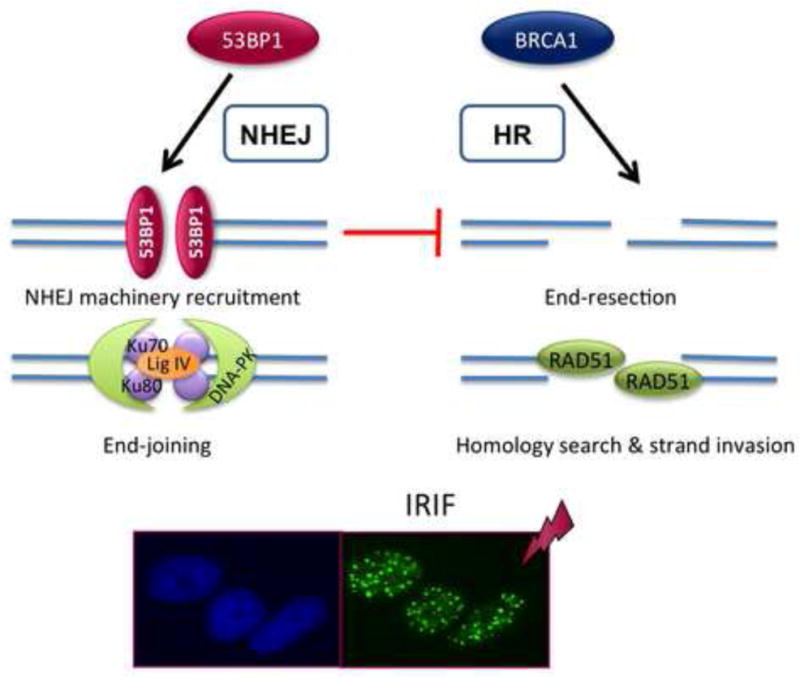
53BP1 and BRCA1 proteins that have been recently in the spotlight for their key role regulating the choice of mechanism of DNA DSB repair. BRCA1 protein promotes DNA end-resection at the break, a prerequisite for binding of RAD51, a protein that is essential for HR. In contrast, 53BP1 binding to the break facilitates the recruitment of the NHEJ machinery. In addition, binding of 53BP1 prevents BRCA1-dependent end-resection and thus functions as an inhibitor of HR. The recruitment of all these factors to DNA repair sites can be easily visualized as foci by IF with specific antibodies. The image shows an example of irradiated cells showing IRIF, and labeled with 53BP1 antibody.
To determine the ability of BOGA cells to repair IR-induced DSBs by NHEJ and HR, we monitored the recruitment of 53BP1 and RAD51 to sites of damage. As expected, BOGA cells were unable to form BRCA1 or 53BP1 IRIF, consistent with the decreased protein levels (Grotsky, Gonzalez-Suarez et al. 2013). In contrast, we found that RAD51 IRIF readily formed in BOGA cells, although their time of retention at the DSBs was reduced. This is consistent with previous studies showing that loss of both BRCA1 and 53BP1 restores at least partially the ability of cells to repair DSBs by HR (Bunting, Callen et al. 2010). Importantly, we found that formation of 53BP1 IRIF was rescued in BOGA cells by inhibiting CTSL activity via 1α,25(OH)2D3 treatment. These results indicate that inhibition of CTSL by 1α,25(OH)2D3 not only rescues the levels of 53BP1, but also its ability to be recruited to sites of DNA damage. Interestingly, treatment with 1α,25(OH)2D3 or depletion of CTSL partially reduced the formation of RAD51 IRIF in BOGA cells. Altogether, these studies reveal an unprecedented role for 1α,25(OH)2D3 in regulating the two main pathways of DNA DSBs repair, NHEJ and HR, especially in tumor cells that are deficient in BRCA1 and 53BP1, as is the case of some TNBC and BRCA1-related tumors. Stabilization of 53BP1 by 1α,25(OH)2D3 seems to promote NHEJ and inhibit HR. Given that HR-deficient cells are sensitivite to PARPi and other DNA damaging strategies, 1α,25(OH)2D3 treatment could potentially improve the sensitivity of BRCA1-deficient cells that become resistant to these compounds due to the loss of 53BP1, as discussed below.
Vitamin D as a new strategy to induce genomic instability and growth arrest
Based on the previous data, we hypothesized that stabilization of 53BP1 in the context of BRCA1 depletion could hinder the ability of these cells to deal with IR-induced damage, leading to genomic instability and cell death. To test this hypothesis, control and BOGA cells were treated with 1α,25(OH)2D3 in order to stabilize 53BP1 twenty-four hours prior to irradiation. Metaphase spreads analysis revealed that both, control cells and BOGA cells were able to repair breaks induced by IR, and did not present with much genomic instability. In contrast, treatment with 1α,25(OH)2D3 or with the cathepsin inhibitor E64 increased profoundly the extent of chromosomal aberrations in BOGA cells, but not in control cells. Both, 1α,25(OH)2D3 and cathepsin inhibitors are able to increase genomic instability in response to IR (Grotsky, Gonzalez-Suarez et al. 2013). In addition, these treatments increased radiosensitization, as shown by a decrease in proliferation with the combined treatment. These results provide a possible strategy to induce radiosensitization in BRCA1-deficient cells that activate CTSL-mediated degradation of 53BP1. Furthermore, stabilization of 53BP1 by 1α,25(OH)2D3 results in increased genomic instability in response to PARPi. These results indicate that 1α,25(OH)2D3 or cathepsin inhibitors could be used in combination with radiation or PARPi to reduce the growth of tumors that have activated CTSL-mediated degradation of 53BP1, if these tumors could be identified.
Activation of CTSL-mediated degradation of 53BP1 in TNBC
A key question raised by our findings was whether activation of CTSL-mediated degradation of 53BP1 is observed in human breast tumors. In collaboration with the groups of Adriana Dusso and Xavier Matias-Guiu in the University of Lleida, Spain, we performed immunohistochemical (IHC) analysis of a breast tumor tissue microarray constructed from biopsies of different molecular types to monitor the levels of CTSL and 53BP1 (Grotsky, Gonzalez-Suarez et al. 2013). A total of 249 sporadic tumors classified in four different types were analyzed: Luminal A, Luminal B, HER2 and TN. We found that all tumor types present with high levels of cytoplasmic CTSL. However, a subset of TNBC (60%) exhibits high levels of nuclear CTSL, in contrast to approximately 30% of all other molecular types. We also confirmed that a high percentage of TNBC (75%) have low levels of 53BP1, as previously reported (Bouwman, Aly et al. 2010). Thus, we identified nuclear CTSL as a novel biomarker for TNBC tumors, and a correlation between high nuclear CTSL and low 53BP1 levels in these tumors. These results suggest that CTSL-mediated degradation of 53BP1 could be one of the mechanisms responsible for the observed loss of this DNA repair factor in TNBC. However, not all TNBC exhibited increased CTSL and decreased 53BP1. In some tumors, we observed high levels of both CTSL and 53BP1, suggesting that additional factors could be regulating the ability of CTSL to degrade 53BP1 in these tumors. Identifying these factors could help to discriminate subsets of patients in which this pathway is activated.
Previous studies in human colon cancer cells found a correlation between levels of VDR and expression of cystatin D, an endogenous inhibitor of several cathepsins including CTSL (Alvarez-Diaz, Valle et al. 2009; Alvarez-Diaz, Larriba et al. 2010). They also showed upregulation of cystatin D by 1α,25(OH)2D3. In addition, our data in BOGA cells show that 1α,25(OH)2D3 inhibits CTSL-mediated degradation of 53BP1 (Grotsky, Gonzalez-Suarez et al. 2013), and most 1α,25(OH)2D3 actions require a functional nuclear VDR (Dusso, Brown et al. 2005). Thus, we hypothesized that an increase in nuclear VDR might lead to activation of cystatins and inhibition of CTSL-mediated degradation of 53BP1. High levels of nuclear VDR could explain the signature of tumors with high levels of both nuclear CTSL and 53BP1. To test this model, we performed IHC analysis of nuclear VDR in all 249 sporadic tumors. Then, we analyzed the linear relationship between 53BP1 and CTSL for those tumors with low nuclear VDR expression (below the median). We found that in 30% of all 249 tumors with low VDR expression, the decrease in 53BP1 can be account for by the increase in nuclear CTSL. This effect is even more striking if we consider only TNBC. In this case, 80% of the TNBC with low nuclear VDR, the decrease in 53BP1 can be accounted for by the increase in nuclear CTSL. This represents a strong correlation between levels of nuclear VDR and the ability of CTSL to decrease 53BP1 levels.
Thus, we have identified a novel triple nuclear biomarker signature to stratify TNBC patients. We find two signatures in these patients. One of the signatures shows high nuclear CTSL, high 53BP1 and high VDR. We hypothesize that the high levels of VDR are inhibiting CTSL-mediated degradation of 53BP1, thus the pathway is turned off. A second signature shows high CTSL, low 53BP1 and low VDR, thus suggesting that CTSL-mediated degradation of 53BP1 is turned on. We envision that these signatures could potentially have predictive value for drug response, such that patients with signature 1 might respond to PARPi, due to the high levels of 53BP1. In contrast, patients with signature 2 might be resistant to PARPi due to the loss of 53BP1. These patients might benefit from inhibition of CTSL-mediated degradation of 53BP1 via treatment with 1α,25(OH)2D3 or cathepsin inhibitors.
A new biomarker signature for BRCA1-related tumors
Given our findings that loss of BRCA1 activates CTSL-mediated degradation of 53BP1, we tested whether increased CTSL is observed in patients with breast cancer that carry germline mutations in BRCA1 or BRCA2. We found increased levels of nuclear CTSL in BRCA1-related tumors, but not in BRCA2 tumors. Similarly, we observed a marked decrease in 53BP1 levels in BRCA1 tumors when compared to BRCA2. Interestingly, BRCA1-related tumors exhibit lower levels of nuclear VDR than BRCA2-related tumors. Specifically, 85% of patients with BRCA1 mutations exhibit nuclear CTSL levels over the median, versus 53% of patients with BRCA2 mutations. Accordingly, 88% of patients with BRCA1 mutations present with 53BP1 levels below the median versus 15% of BRCA2 patients. Lastly, 83% of BRCA1 patients present with nuclear VDR levels under the median. These results in breast cancer patients support our model that loss of BRCA1 leads to activation of CTSL-mediated degradation of 53BP1, and that 1α,25(OH)2D3 via activation of VDR, can inactivate this pathway.
Summary
Our studies showed that depletion of BRCA1 leads to growth arrest due to defects in HR and increased genomic instability. BRCA1-deficient cells can activate compensatory mechanisms that allow them to overcome genomic instability and growth arrest. We found that one of these mechanisms is the activation of CTSL-mediated degradation of 53BP1. Importantly, inhibition of CTSL-mediated degradation of 53BP1 via increased levels of VDR or treatment with 1α,25(OH)2D3 or cathepsin inhibitors results in HR defects, genomic instability, and increased sensitivity to DNA damaging therapeutic strategies, such as IR or PARPi. In addition, we identified a strong inverse linear correlation between nuclear CTSL and 53BP1 levels in BRCA1-deficient tumors and TNBC that present with low levels of nuclear VDR. This novel triple biomarker signature could potentially have predictive value for drug response, as it could allow the identification of breast cancer patients that activate CTSL-mediated degradation of 53BP1. These patients could benefit from a therapeutic strategy such as 1α,25(OH)2D3 or cathepsin inhibitors to stabilize 53BP1 and restore sensitivity to PARPi or radiation. Although preclinical studies in mice are needed to evaluate the effect of inhibition of CTSL activity in the progression of specific types of breast cancers, 1α,25(OH)2D3 and cathepsin inhibitors have emerged in these studies as new possible therapeutic strategies for breast cancers with the poorest prognosis, such is the case of TNBC and BRCA1-related tumors.
The major findings of these studies are: 1) identification of a new pathway -CTSL-mediated degradation of 53BP1- contributing to breast cancers with the poorest prognosis; 2) discovery of the ability of 1α,25(OH)2D3 to inhibit this pathway; and 3) discovery of a triple biomarker signature -nuclear levels of VDR, CTSL and 53BP1- for the identification of patients that could benefit from the treatment.
Highlights.
Loss of 53BP1 in tumors leads to therapy resistance.
Identified a new pathway responsible for 53BP1 loss in breast cancers with poor prognosis.
Discovered a way to inhibit 53BP1 loss (vitamin D treatment).
Identified a triple nuclear biomarker signature to classify patients with pathway active that could benefit from treatment.
Abbreviations list
- TNBC
triple negative breast cancers
- BRCA1
breast cancer susceptibility gene 1
- PARPi
poly(ADP-ribose) polymerase inhibitors
- CTSL
cathepsin L
- DSBs
double strand breaks
- HR
homologous recombination
- NHEJ
non homologous end joining
- VDR
vitamin D receptor
- IRIF
ionizing radiation induced foci
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allinson SL, Dianova II, et al. Poly(ADP-ribose) polymerase in base excision repair: always engaged, but not essential for DNA damage processing. Acta Biochim Pol. 2003;50(1):169–179. [PubMed] [Google Scholar]
- Alvarez-Diaz S, Larriba MJ, et al. Vitamin D: Proteases, protease inhibitors and cancer. Cell Cycle. 2010;9(1):32–37. doi: 10.4161/cc.9.1.10266. [DOI] [PubMed] [Google Scholar]
- Alvarez-Diaz S, Valle N, et al. Cystatin D is a candidate tumor suppressor gene induced by vitamin D in human colon cancer cells. J Clin Invest. 2009;119(8):2343–2358. doi: 10.1172/JCI37205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aly A, Ganesan S. BRCA1, PARP, and 53BP1: conditional synthetic lethality and synthetic viability. J Mol Cell Biol. 2011;3(1):66–74. doi: 10.1093/jmcb/mjq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ame JC, Rolli V, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999;274(25):17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26(22):3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- Audeh MW, Carmichael J, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, et al. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207(4):855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ, Gombart AF, et al. The anti-proliferative effects of 1alpha,25(OH)2D3 on breast and prostate cancer cells are associated with induction of BRCA1 gene expression. Oncogene. 2000;19(44):5091–5097. doi: 10.1038/sj.onc.1203888. [DOI] [PubMed] [Google Scholar]
- Cao L, Xu X, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009;35(4):534–541. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S, Gapud E, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456(7221):529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, et al. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456(7221):524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusso AS, Brown AJ, et al. Vitamin D. Am J Physiol Renal Physiol. 2005;289(1):F8–28. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Chen HT, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol. 2002;4(12):993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- Fong PC, Boss DS, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Smith IE, et al. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Suarez I, Redwood AB, et al. Loss of A-type lamins and genomic instability. Cell Cycle. 2009;8(23):3860–3865. doi: 10.4161/cc.8.23.10092. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Suarez I, Redwood AB, et al. A new pathway that regulates 53BP1 stability implicates Cathepsin L and vitamin D in DNA repair. Embo J. 2011;30(16):3383–3396. doi: 10.1038/emboj.2011.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Suarez I, Redwood AB, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. Embo J. 2009;28(16):2414–2427. doi: 10.1038/emboj.2009.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotsky DA, Gonzalez-Suarez I, et al. BRCA1 loss activates cathepsin L-mediated degradation of 53BP1 in breast cancer cells. J Cell Biol. 2013;200(2):187–202. doi: 10.1083/jcb.201204053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Bryant HE, et al. Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle. 2005;4(9):1176–1178. doi: 10.4161/cc.4.9.2031. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers JE, Kersbergen A, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81. doi: 10.1158/2159-8290.CD-12-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Center MM, et al. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1893–1907. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27(3):247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- Lankelma JM, Voorend DM, et al. Cathepsin L, target in cancer treatment? Life Sci. 2010;86(7–8):225–233. doi: 10.1016/j.lfs.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Li X, Xu B, et al. 53BP1 functions as a tumor suppressor in breast cancer via the inhibition of NF-kappaB through miR-146a. Carcinogenesis. 2012;33(12):2593–2600. doi: 10.1093/carcin/bgs298. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, et al. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4(4):511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Redwood AB, Perkins SM, et al. A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle. 2011;10(15):2549–2560. doi: 10.4161/cc.10.15.16531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg S, Jaspers JE, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356(6367):356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- Schlegel BP, Jodelka FM, et al. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res. 2006;66(10):5181–5189. doi: 10.1158/0008-5472.CAN-05-3209. [DOI] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, et al. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol. 2000;151(7):1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Chen J, et al. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997;90(3):425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- Scully R, Chen J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88(2):265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- Scully R, Ganesan S, et al. Location of BRCA1 in human breast and ovarian cancer cells. Science. 1996;272(5258):123–126. doi: 10.1126/science.272.5258.123. [DOI] [PubMed] [Google Scholar]
- Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408(6811):429–432. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Conrad S, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. Embo J. 2011;30(6):1079–1092. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snouwaert JN, Gowen LC, et al. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene. 1999;18(55):7900–7907. doi: 10.1038/sj.onc.1203334. [DOI] [PubMed] [Google Scholar]
- Tutt A, Robson M, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, et al. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298(5597):1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- Woodhouse BC, Dianov GL. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair (Amst) 2008;7(7):1077–1086. doi: 10.1016/j.dnarep.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu Rev Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- Xie A, Hartlerode A, et al. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol Cell. 2007;28(6):1045–1057. doi: 10.1016/j.molcel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408(6811):433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]