

1. Summary
A traditional view of protein–DNA interaction regards the protein as an active element and the DNA as a passive element. This view is often implied across molecular biology where DNA is assumed to function as a relatively stable and uniform polymer probed by proteins with its sequence elements serving to define location. However, DNA is highly dynamic, enwrapping layers of complexity. In particular, DNA actively contributes to processes that dictate how protein–DNA interactions form. Furthermore, its structural features are often crucial to the formation of both transient and stable protein–DNA complexes. Such structure-dependent interaction is distinguishable from proteins that use DNA solely as a sequence-defined scaffold.
Here, we provide a general conceptual framework to explore the relationship between DNA and DNA-binding proteins. We consider how it is relevant in vivo and why it should be factored into experimental design and interpretation. We then review illustrative examples that have applied the single-molecule DNA curtain technique to study protein–DNA interactions and show how this framework enhances the understanding of single-molecule results.
2. Introduction
DNA stores information. Its function as a universal genetic material is among the most highly conserved qualities of living things. Its system of four bases, when overlaid with spatial and temporal controls, governs biology across the entire scale of life, from enzymatic reactions inside E. coli to embryogenesis in humans. Because the primary function of DNA is to store and propagate information, its sequence is often taken to be its most important biochemical property. However, during the ordinary course of cellular activity, DNA must be manipulated in many ways as a physical structure—a polymer in solution—regardless of whether a specific sequence is relevant to a given process. The dominant functional unit of this manipulation in the cell is the interaction between DNA and protein. Some interactions access or even extract the underlying sequence content of the DNA, whereas many others are largely oblivious to that information.
DNA’s primary function in storing genetic information requires a degree of stability. Indeed, the cell’s ability to correctly maintain and propagate DNA sequences through many cycles of duplication at low error rates is the source of both the continuity and the variability so fundamental to evolution.1 DNA is also constrained by the more immediate and practical concerns of the cell, which, in handling and passing on genetic content, must efficiently carry out duplication of chromosomes.2 The two intertwined factors of long-term stability and duplication efficiency are served by the fairly uniform global structure of DNA. The familiar double helix is generally not taken to vary significantly in structure along its length, and this is true on average—for example, across the span of a large genomic fragment. However, the physical properties of DNA are not uniform across regions of a size relevant to protein binding.3 Nor are they uniform across cellular environments, which are heterogeneous and fluctuating,2a,4 or even across experimental conditions.5
Appreciating the basic physical qualities of DNA as a function of physiologically and experimentally relevant variables is essential to understanding protein–DNA interactions, and the physical properties of DNA are particularly important when interpreting the results of single-molecule experiments. Important factors to consider are the polymer nature of DNA and the effect of various forces on this polymer, the effect of temperature and salt concentration, DNA length, and sequence-dependent variations in structure. Furthermore, DNA dynamics influence complex, subtle, and time-dependent protein behaviors on short spatial and temporal scales, a swath of which are only accessible through single-molecule methodologies. (Recent reviews that explore the nature of single-molecule experiments include refs (6a−6c)).
In this review, we focus on illustrative examples from our laboratory that have employed “DNA curtains”: parallel arrays of individual DNA molecules used to visualize the behavior of DNA-interacting proteins. However, we consider these concepts broadly applicable to single-molecule studies. Single-molecule bioscience is still coming of age, and given recent technical and conceptual advances, it is poised to tackle progressively more complex and physiologically relevant problems. However, designing and interpreting single-molecule experiments to study protein–DNA interactions often does not map directly onto comparable biochemical studies, and yet characterizing a particular system comprehensively requires data from as many approaches as possible. Constructing such cohesive understanding from disparate viewpoints, all directed toward an understanding of the same biological system, requires that each field possess a general framework to interpret results which can be universally applied to interpretation of phenomena as witnessed by that field. A successful framework should grant access to fundamental concepts about the biology underlying a given system, and it is these fundamental concepts that allow for a lucid dialogue among multiple fields. Here we present a framework that describes the relationship between DNA and DNA-binding proteins by considering the general factors that affect their interaction. Others have previously triangulated the relationships between DNA structure, DNA sequence, and protein–DNA interactions in various combinations, and the concepts presented here are informed by these earlier ideas.3a−3e
3. Recognition of DNA Sequences and Structures
DNA sequence and structure are inextricably linked, but we can conceptually separate the two with respect to protein binding. We assume that all DNA-binding proteins exhibit some degree of specificity in DNA binding; that is, all DNA-binding proteins exhibit preferences for certain stretches of DNA. We further assume that this specificity can emerge from DNA sequence recognition, DNA structure recognition, or some measured combination of the two. The core of this framework is a continuum of sequence–structure preferences anchored between a pure sequence-interacting protein and an opposing pure structure-interacting protein. All DNA-interacting proteins can be placed on this continuum, which is wholly defined if the opposing extreme cases are defined (Figure 1a).
Figure 1.
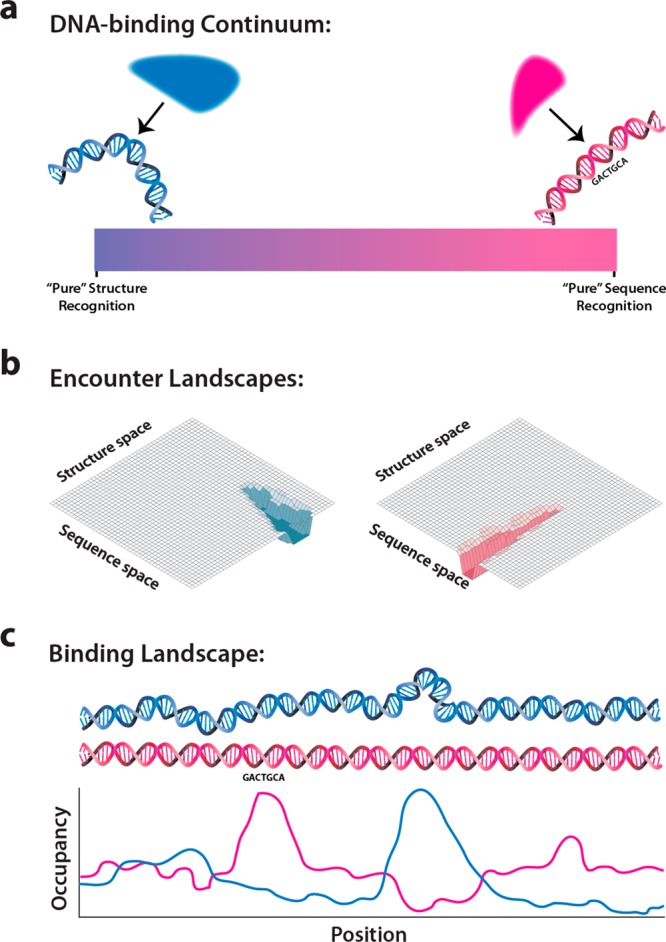
Principles contributing to DNA target recognition and binding by proteins. (a) Schematic illustration representing the DNA-binding continuum, the boundaries of which are defined by hypothetical examples of “structure interactor” or “sequence interactor”; real proteins are expected to fall somewhere in the continuum flanked by these two extremes. (b) Examples of hypothetical encounter landscapes: (left) protein whose binding behavior is dominated by DNA structure, and (right) protein whose target recognition properties are dominated by DNA sequence. (c) Binding landscape describes the stability of protein–DNA complexes along a defined DNA sequence.
First, consider a pure sequence interactor that recognizes only the sequence of its target DNA. This can be realized, in principle, by probing a pattern of hydrogen donors and acceptors that uniquely describes a DNA sequence. Our definition of a pure sequence interactor further precludes any residue on the protein from interacting with any chemical signature on the DNA other than what is necessary for target recognition and therefore an element of the sequence itself. Finally, a pure sequence interactor loses all specificity for its target if even a single base pair of that target is altered. We recognize that no such absolutely “pure” sequence interactors are expected to exist, and the extreme cases are considered only as a conceptual illustration. Second, on the opposite end of the continuum, a pure structure interactor recognizes only a specific DNA structure, without any reference to the underlying sequence. This definition precludes any residue on the protein from interacting with any chemical signature on the DNA that is directly defined by sequence. As all DNA structures are to some extent a function of sequence, this extreme case is also not expected to exist and is presented as an illustrative extreme case.
3.1. Classical View of Interactions
The standard view of site-specific binding typically addresses definitive interaction parameters such as kinetic rates or occupation probabilities. These parameters are determined wholly by the free energy change involved in a particular physical process—for example, a protein binding to DNA. A more nuanced view considers the identity of free energy changes: although there exists a global free energy minimum (commonly referred to as the thermodynamically stable state), there may also be local free energy minima (commonly referred to as kinetically stable intermediates). Sequence and structure preferences can be incorporated into this scheme: the change in free energy is due in part to structural cues and in part to sequence cues, and the relative contribution of each could place the interaction on the above continuum.
While this standard model of binding in terms of free energy change or stability is attractive due to its simplicity, it is also limited and can even lead to misinterpretation of experimental results. Specifically, the assumption that the free energy change of an interaction is fixed during a single binding event, or even similar across different physical environments, may cause results from disparate fields to seem contradictory despite identical physical origins. Indeed, the free energy associated with a given process is a dynamic variable and in vivo affords a remarkable level of cellular control, a concept that has only recently been recognized.7 To be clear, a description based only on the classical kinetically and thermodynamically stable states collapses an inherently dynamic process, shaped by many biologically important factors, into an energetically static picture. It is an incomplete description of the underlying physical processes. Furthermore, relying wholly on the free energy change to define a protein–DNA interaction obscures the multiple subcomponents of that interaction. To highlight the significance of this underlying complexity, we imagine the free energy change of DNA binding as an average over dynamical energy changes along the reaction coordinate. Specifically, we consider that the factors determining the probability of interaction between a protein and a particular sequence–structure component of DNA are related to but not necessarily identical to those that determine the stability of an interaction, as explored below.
3.2. Encounter Landscape
The first component of our deconvolution concerns how a protein encounters the DNA polymer. We define an encounter landscape as an instructive conceptualization reflecting the probability that a particular protein will interact with any DNA sequence or structure. The character of the encounter landscape contributes to placement of a protein at some point along the DNA-interaction continuum (i.e., more heavily sequence defined or more heavily structure defined), reflecting the extent to which DNA sequence or structure is important to the protein as it interacts with DNA. Importantly, the encounter landscape describes only the time-dependent likelihood that a protein will interact with a particular DNA site if it encounters that site. (The time dependence arises from the inherently dynamic qualities of all macromolecular interactions, including thermal fluctuations, steric occlusion, etc.) It does not account for possible downstream conformational changes or other factors that may significantly influence how an initial transient encounter matures or the stability of the resulting protein–DNA complex. One important implication is that the encounter landscape does not necessarily predict the likelihood that a binding event will result in a physiologically competent complex.
Our definition of an encounter landscape serving as a gateway to a binding landscape (see below) deconvolves the more traditional presentation in which the free energy landscape changes in response to protein binding.8 However, we feel that to highlight the significance of sequence and structure in the continuum and to better understand the dynamics of protein–DNA interactions it is instructive to distinguish between these two conceptually distinct landscapes, even though transitions between the two are smoothly connected in time.
3.3. Binding Landscape
The concepts presented above yield important corollaries for interpreting real biological systems and experimental data. Once a protein encounters and binds a segment of DNA, be it a specific sequence and/or a preferred structure, the resulting protein–DNA complex becomes a distinct entity. A protein cannot interact with DNA independently of mutual conformational changes because all interactions between biological molecules, and especially those between specifically interacting biological molecules, will alter each constituent’s structure.8d,9 Therefore, it is useful to consider a protein–DNA complex as distinct from its two constituent parts and as distinct from the initial encounter event. We define the binding landscape as describing the stability of protein–DNA complexes across sequence–structure space (Figure 1c), encompassing both kinetically and thermodynamically stable states. The lifetime of a protein–DNA complex at a specific location on a defined DNA substrate is a proxy for this landscape. As described below through illustrative examples, the binding distribution of a protein on a specific stretch of DNA under defined conditions is a direct readout of a portion of the binding landscape.
3.4. Significance of Distinguishing between the Landscapes
It is instructive to examine the nature of protein–DNA interactions for each of four cases defined by different contributions from encounter probability and binding stability. Let the first case, [↑↑], describe a protein–DNA interaction wherein the interaction is favored in both the encounter and the binding landscapes, the second case, [↓↓], describe an interaction that is disfavored in both landscapes, the third case, [↑↓], describe an interaction that is favored in the encounter landscape but disfavored in the binding landscape, and the fourth case, [↓↑], describe an interaction that is disfavored in the encounter landscape but favored in the binding landscape. Regardless of experimental technique, measurements of protein occupancy and kinetics in the first case, [↑↑], will yield the same result. That is, whether a single-molecule or bulk experiment is performed, investigators will find the DNA–protein complex highly enriched and the complex long lived. The second case, [↓↓], will yield a low occupancy and unstable signal, again irrespective of method.
The third and fourth cases have remarkable consequences that may be harnessed by the cell and affect experimental interpretation. Take the third case, [↑↓]. This interaction is favored in the encounter landscape, meaning that the protein–DNA complex is highly sampled but not stable. It can give contrasting results depending on the method of examination. For example, a chromatin immunoprecipitation (ChIP) experiment may conclude that this complex is enriched and therefore that a stable interaction is required for biological relevance. In contrast, a single-molecule assay may conclude the opposite: that the complex is rare because the frequency of complex formation may be too transient to capture. The actual picture is more complicated and more interesting. That is OR more interesting; that is, the complex may cycle in and out of existence, at times providing an authentic target for downstream steps. The transient nature of the interaction may, for example, allow competitor complexes to arise. This difference in apparent outcome is not exclusive to particular pairings of experimental methods; it is a direct consequence of the time scale accessible to a method—that is, any method that probes an interaction at a rate longer than the lifetime of the interaction will miss it, whereas other methods that can sample faster or artificially stabilize the interaction will observe it. This is also evident in the fourth case, [↓↑]. Here encounters are rare but long lived. Any method that probes events over short time scales may completely miss or only partially observe such interactions, whereas techniques adapted to longer time scales will have no problems in detection.
The cellular advantage of these two possible scenarios, [↓↑] and [↑↓], is control. The first two cases, [↑↑] and [↓↓], are fairly straightforward from the cellular perspective, a complex either exists or does not. The second two cases, [↓↑] and [↑↓], allow for a range of control over interactions that span the space between cases one and two, that is, the respective contributions from the encounter landscape and the binding landscape can be tuned to vary smoothly across countless combinations. This tunability allows for any state between completely “on” and completely “off”. Transitions between the cases are also possible—in response to allosteric signals, for example—allowing the cell to alternate how a DNA-binding event is controlled.
3.5. Building to Physiological Complexity
Many physiological processes require the cumulative actions of multiple protein–DNA interactions. For example, initiation of eukaryotic DNA replication requires at least 32 different polypeptides, many of which are known to contact DNA.10 Moreover, native DNA–protein interactions must be established within the context of highly crowded cellular environments where the DNA is not naked but rather covered with other proteins such as nucleosomes in eukaryotes or nucleoid associated proteins (NAPs) in prokaryotes.2b,11 Once a protein binds to DNA, the resulting complex can serve as a substrate for other proteins; this protein–DNA complex presents an entirely new encounter landscape for other DNA-binding proteins. It is possible to build any number of iterations of this pattern to accommodate highly complex systems. In principle, these concepts, as derived for a minimal in vitro system, can be stacked and arranged up to the in vivo state.
4. DNA Structure
Translating the conceptual framework described above to experimental settings requires a brief grounding in the basic physical properties of DNA. Our current understanding of DNA structure is based in significant part on X-ray crystallography data.12 Idealized B-form DNA, generally considered the physiologically prevalent and therefore relevant form of DNA, is 2 nm wide, with a 3.4 nm rise for each helical turn consisting of 10.5 bases. The two antiparallel strands of DNA twist around each other such that two distinct surfaces are generated: a minor groove that is 1.2 nm wide, and a major groove that is 2.2 nm wide. DNA-binding proteins can access sequence content through either the minor groove or the major groove, and this is an important distinction because these two interfaces present different information. Specifically, the pattern of atomic signatures presented by bases in the major groove specifies all four possible base pair combinations, whereas the pattern in the minor groove does not specify a difference between G–C and C–G or between A–T and T–A.
A simple model that is helpful in understanding the physical properties of DNA treats each strand as a negatively charged wire, the pair held together by hydrogen bonds across opposite bases and stabilized by stacking interactions between successive bases. The global shape and flexibility of such a structure at equilibrium is identical to an ideal polymer’s: it essentially behaves like a wadded up rubber hose. In fact, DNA is one of the best physical manifestations of an ideal polymer, and basic polymer physics offers the best picture of the physical behavior of DNA, especially for situations in which it is pushed out of equilibrium by external forces. DNA in vivo is expected to be almost continuously out of equilibrium because of the dynamic cellular environment, which includes the activity of DNA-manipulating proteins.
The most general effects are stretching and bending. For low forces, these are well described by the worm-like chain (WLC) model of a polymer.13 This model is effectively defined by three variables: (i) torsional rigidity describes the resistance of the polymer to twist about the long axis; (ii) persistence length describes the unit length over which it remains essentially straight (the persistence length of DNA is ∼50 nm or ∼150 base pairs (bp);5 and (iii) dynamics depend on the diameter and length of the polymer, and DNA can be treated as a uniform polymer if its sequence is sufficiently randomized.
4.1. Forces on DNA
The WLC model is applicable under many experimental conditions and perhaps physiological settings in which DNA experiences low to intermediate linear forces in the pico-Newton range. For example, DNA stretching may occur during bacterial division, when the daughter chromosome is extruded through the division septum by motor proteins,14 or during eukaryotic chromosome segregation.15 Mutant dicentric chromosomes in eukaryotes can rip apart during segregation; so, whatever the source of the forces involved, they are not trivial.15b However, the WLC model does not account for overstretching,5,16 and all polymer models of DNA break down at short length scales below the persistence length.17
Even small torsional forces can drastically change DNA structure. Underwinding (i.e., twisting in the direction opposite to the handedness of the helix) causes the DNA to relax into negative supercoils and destabilizes the hydrogen bonds between bases, increasing the probability of transient local melting. E. coli actively underwinds its chromosomal DNA, possibly to decrease the energetic barrier needed for transiently accessing sequence information (during transcription, for example).18 Conversely, overwinding energy is initially stored as by a spring, and continued overwinding causes the DNA to relax into positive supercoils. Transcription and replication both require the action of topoisomerases to relieve positive supercoils that form ahead of progressing polymerases.19 Note that generation of either negative or positive supercoils absolutely requires topologically constrained DNA so that the twist cannot be relieved by rotation of the DNA along its length. The constraint may be a pair of physical clamps, as in single-molecule studies of DNA supercoiling, motor proteins with multiple DNA-binding domains,20 circularization of DNA, or the effective mass of a chromosome in the viscous cellular environment.18a,19a
4.2. Effects of Ionic Strength
Two of the most easily manipulable factors that influence how DNA behaves in solution are temperature and salt concentration. At relatively low temperatures DNA is quite stable and rigid, but at higher temperatures, with energy on the order of two or three hydrogen bonds passing through each unit of the DNA, the two strands begin to breathe. Temperature is particularly interesting from the in vivo perspective because many organisms can withstand wide fluctuations in temperature, and many extremophile species are known to live at temperature extremes.21 Yet in all cases DNA must function in fundamentally the same way. Physiological conditions may shield DNA from the effects of temperature. For example, the hyperthermophile S. acidocaldarius actively pumps positive supercoils into its chromosomes, possibly to counter the destabilizing effects of high temperature.22
Ionic strength and the nature of the ions present can have major effects on DNA stability. There is no consensus about the exact physiological ionic strength in vivo, but it is almost certainly higher than commonly applied in vitro. Changes in salt concentration predominantly affect DNA structure through interactions with the negatively charged phosphates along the backbone.23 Monovalent ions shield the backbone charges, and at high concentrations of monovalent salt the DNA rigidity is mostly a function of the hydrogen bonds between bases.23 Multivalent salts impart rigidity and regularity to the backbone by coordinating multiple charged phosphates.24 When the total salt concentration drops below the millimolar range, the resulting charge–charge repulsion due to the phosphates of the backbone has a large but poorly understood influence on DNA structure.23b Initially this increases DNA rigidity as phosphates on the same strand repulse one another, but as the salt concentration drops further, the DNA denatures as the two strands are pushed apart. Protein–DNA interactions have evolved within a specific range of salt environments, and given the drastic effects of low salt on DNA structure it is important to treat this variable judiciously. The potential influence of nonphysiological salt concentrations may often be overlooked in bulk studies because these often employ exceptionally high concentrations of protein and measurements often take a relatively long time, so a given reaction is forced to an end despite unnatural ionic conditions. However, in single-molecule studies, perturbations to DNA structure due to ionic strength can so skew reaction kinetics on short spatial and temporal scales as to fundamentally alter measurements and thereby render a result difficult or impossible to interpret.
4.3. DNA Length Is Important
Another common experimental deviation is the use of short DNA oligonucleotides—sometimes only tens of base pairs long—to measure various aspects of protein–DNA interactions. Base stacking and long-range stabilization through the regularity of the backbone are both cooperative and contribute to the stability of the DNA helix and to the local stability of individual bases.25 Short DNA oligomers lack these qualities: the DNA helix is less globally stable, and the stability of individual bases is compromised, especially the set of bases at the ends of a fragment with no neighbors and one of their long-range-stabilizing arms missing.23a,26 The reduced overall surface area of a short oligonucleotide also results in a lower local concentration of condensing bodies; for DNA, these may be ions or DNA-binding proteins that form a local region of high concentration simply by their affinity for DNA (and in the case of proteins, irrespective of sequence). This phenomenon influences all protein–DNA interactions and probably has a major role in vivo where DNA tends to be confined and at a high local concentration.
4.4. Sequence Influences Structure
The specific nucleotide sequence of a DNA molecule also contributes to its structure.3 For example, A–T base pairs make two hydrogen bonds whereas G–C pairs make three, resulting in a relatively higher stabilization across the two strands of the double helix in G/C-rich sequences (experimentally manifested in the linear relationship between melting temperature and percent G/C content). G/C-rich regions also have relatively wide major grooves, whereas A/T-rich regions have relatively narrow minor grooves.3a−3c Proteins with G/C- or A/T-rich target sequences may therefore exhibit some preference for one or the other average groove width.3a−3c The narrower minor groove in A/T-rich regions also brings the backbone phosphates closer together, and the resulting increase in electrostatic interactions results in an overall higher stiffness. This effect is especially important for proteins that bend DNA on binding,27 as explored below with respect to nucleosomes.
4.5. Structural Perturbations at Protein–DNA Interfaces
The discussion above illustrates that DNA is readily perturbed away from the idealized structure of the B-form helix. Notably, the wealth of information generated by protein–DNA cocrystal structures over the past three decades reveals local deviations in helical structure are in fact commonly found at protein–DNA interfaces.28 The DNA within nucleoprotein complexes is often kinked, bent, unwound, and/or stretched, and in many cases, these structural perturbations can appear quite drastic.29 For example, the I-SceI homing endonuclease bends DNA by ∼50°,29k the eukaryotic transcription factor TBP (TATA-binding protein) bends DNA by ∼80°,29e and the prokaryotic IHF protein (integration host factor) induces a nearly 180° bend in DNA.29i In all three cases there are extensive local changes in both the base pair geometry as well as the dimensions of the major and minor grooves, which are required to accommodate the drastic bend angles. While it is not always clear whether the deformations at protein–DNA interfaces in cocrystal structures reflect an intrinsic property of the DNA or are induced entirely by the binding of the protein, it is clear that the pliability of DNA as a physical entity is crucial for association of many protein–DNA complexes.
In summary, the structural variability of DNA and the structural perturbations of DNA due to external conditions are very important factors with respect to protein–DNA interactions. A full understanding of protein–DNA interactions should reference the contribution of the DNA scaffold to the maturation of an interaction and account for how that contribution may change as a function of environmental and experimental conditions.
This overview of the role of DNA informs why we envision a continuum of sequence–structure preferences for DNA-binding proteins. The position of each protein on this continuum is determined by whether the minima across the protein’s encounter landscape are largely sequence defined or largely structure defined. Proteins bound to DNA have by definition already passed through the encounter landscape, and the resulting occupancy levels reflect the binding landscape. In addition, a protein–DNA complex can serve as a new substrate for other proteins, effectively establishing new encounter landscapes that will influence the association of any downstream proteins.
5. Single-Molecule Studies of Protein–DNA Interactions
Direct visualization of individual proteins as they interact with DNA has been crucial in unraveling many biological mechanisms previously obscured in bulk studies. In determining the nature of naked DNA, as above, atomic force microscopy (AFM) and optical and magnetic tweezers remain indispensible. These methods have translated well to the investigation of protein–DNA interactions, yielding insights into the physics of translocases,30 insights into nonspecific protein DNA interactions,31 and even complex processes such as transcriptional motion in the presence of nucleosomes.32 Other methods affix DNA to the surface of microscope slides, and buffer flow can be used to extend large DNA macromolecules. These surface association and flow stretched methods have been used to characterize the motion of transcription factors,33 the loading and dynamics of repair proteins,34 and the motion of viral replisomes.35 The diversity of the above approaches reflects that specific questions about the nature of proteins interacting with DNA are best suited to specific single-molecule techniques. Although here we outline the DNA curtain methodology of our laboratory, along with several examples, it is important to note that the concepts are general.
The DNA curtain technique for visualizing protein–DNA interactions has been reviewed elsewhere,36 and a series of technical articles describes the methodology and its applications in depth.36b,37 This technique allows for real-time and simultaneous observation of hundreds of individual DNA strands all aligned in a defined orientation. Briefly, the setup involves microfluidic flow cells constructed around nanofabricated microscope slides (Figure 2a). The fabrication process involves standard electron-beam lithography and metal deposition to generate defined nanoscale patterns on the slide surface.36b,37g These patterns define the arrangement of DNA molecules within a curtain. An experiment begins with deposition of a two-dimensionally fluid lipid bilayer on the slide surface. Individual lipid molecules are free to diffuse within the bilayer, but they cannot traverse the nanofabricated barriers. Some of the lipids are functionalized so that DNA strands can be linked to the bilayer via a biotin–streptavidin–biotin interaction. In the presence of buffer flow, DNA molecules are pushed up to the barriers and stretched out along the surface. To illuminate fluorescently labeled DNA or fluorescently labeled proteins bound to the DNA we use total internal reflection fluorescence microscopy (TIRFM). A laser beam is reflected off the interface between the glass slide and the buffer underneath. A shallow (hundreds of nanometers) evanescent field penetrates the buffer and illuminates only a small sample volume near the surface, where the DNA curtains have been established (Figure 2b). Importantly, the TIRF field does not excite fluorophores in bulk solution, thereby significantly increasing the signal-to-noise ratio. It is ideal for the study of double-stranded DNA-interacting proteins at the single-molecule level, and its versatility has recently been exploited to study single-stranded DNA-binding proteins38 as well as the formin-mediated assembly of actin filaments.39 As described in greater detail below, DNA curtain experiments have proven to be an efficient technique for determining binding distributions for a number of DNA-binding proteins.37a,37d,37i,40 In the following sections, we explore two recent studies from our laboratory that have harnessed the DNA curtain approach for measuring binding distributions, and we illustrate the usefulness of the conceptual framework presented above as a lens to interpreting these types of experiments.
Figure 2.
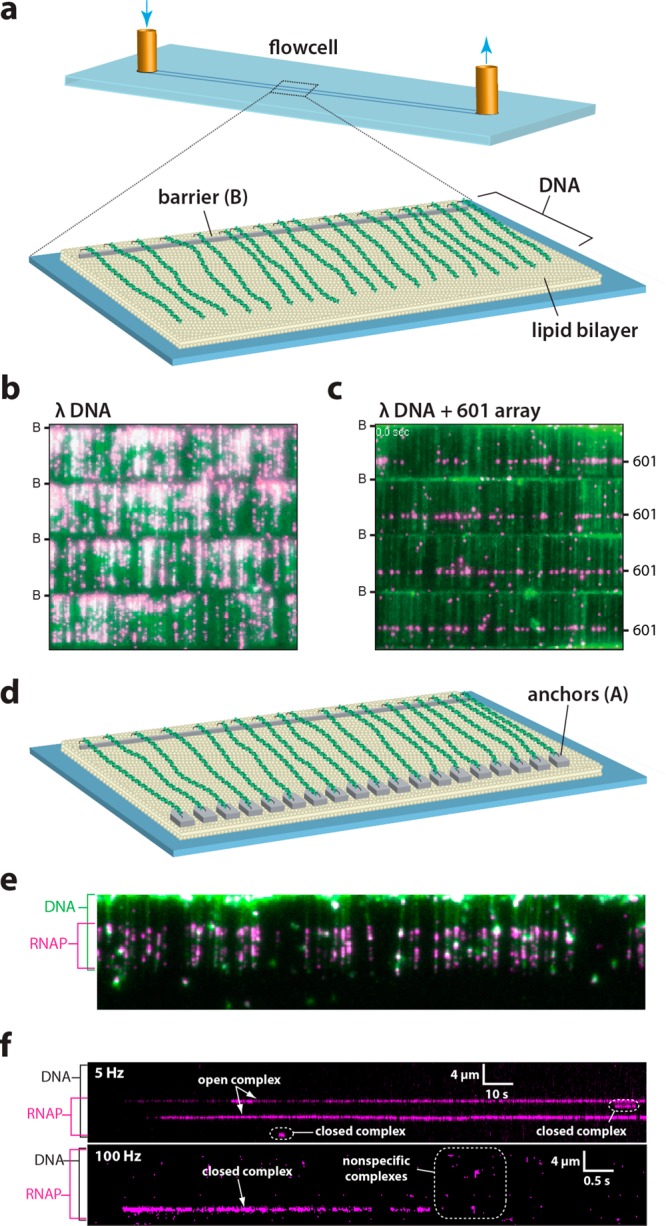
DNA curtains as a tool for studying protein–DNA interactions. (a) Schematic depiction a single-tethered DNA curtain in which one end of the DNA is anchored to a lipid bilayer and aligned along the leading edge of a nanofabricated barrier on the surface of the flow cell.37g (b) Example of a 4-tiered DNA curtain bound by recombinant nucleosomes each of which is tagged with a fluorescent quantum dot. Lambda phage DNA is stained with YOYO1 and shown in green, the nucleosomes are in magenta, and the location of the nanofabricated barriers (B) are indicated. Adapted with permission from ref (37i). (c) Example of a DNA curtain made using a lambda phage DNA substrate containing a 13× array of the Widom 601 nucleosome positioning sequence (unpublished). (d) Schematic depiction a double-tethered DNA curtain in which one end of the DNA is anchored to a lipid bilayer and aligned along the leading edge of a nanofabricated barrier and the second end of the DNA is anchored to an antibody-coated pedestal.37b (e) Wide-field TIRFM image showing quantum dot-tagged RNA polymerase (magenta) stably bound to the native promoters within the lambda phage genome. (f) Kymographs showing the association of RNA polymerase with an individual molecule of DNA (unlabeled) in real time. Adapted with permission from ref (37j).
6. RNA Polymerase
In all kingdoms of life, information stored in DNA is extracted by proteins called RNA polymerases, which read out genomic information, base-by-base, and transfer that information to single strands of RNA that can be translated into proteins or used for an evergrowing list of other biological functions (miRNA, siRNA, ncRNA, etc.).41 A reductionist view of biological regulation places RNA polymerase (RNAP) at the first step of regulatory pathways; all cellular processes require synthesis of RNA at some early stage. This fundamental role in gene regulation underlies the historically intense work around RNA polymerases,42 and its interactions with DNA have proven particularly interesting from the single-molecule perspective.43 RNA polymerase also provides an informative system to consider within the conceptual framework presented here.
In order for RNAP to express specific genes, it must first find and recognize promoter sequences embedded within the genome. E. coli has ∼3000 promoters, each of which contains a core sequence ∼35 base pairs in length comprised of hexameric consensus sites at the −35 (TTGACA) and −10 (TATAAT) regions relative to the transcription start site,42a,42c,44 although the spacing and sequence composition of these sites can vary significantly.45 Examining how polymerases locate promoters can show how the sequence and structural elements of DNA contribute to the regulation of expressed RNA levels. RNAP’s search for promoters is an example of a more general DNA-based target search problem that is common throughout biology.46 In particular, nontranscribing RNAP likely spends most of the promoter search exploring the cytoplasm by Brownian motion.37j,47 Once a polymerase encounters DNA, it must “determine” if it is on a promoter; if it has indeed bound a bona fide promoter then it can form a closed complex, which reflects the first in a series of structural intermediates on the path toward a transcriptionally active open complex that has separated the two strands of DNA and is capable of transcribing the encoded sequence information.42b−42f
6.1. Encounter and Binding Landscapes of RNA Polymerase
The process of open complex formation, the probability of which is tied up in the binding landscape of RNAP, represents the first major level of transcriptional regulation and funnels the entire biochemical pathway of gene expression toward its physiological end. A binding profile for RNAP (as well as any other protein) can be experimentally determined by measuring position- and species-dependent survival probability along a defined DNA substrate. This profile is a proxy for a portion of the aforementioned binding landscape determined by the substrate DNA and the experimental conditions, though, as discussed in section 3.4, one must take care in interpreting this profile. This is due to experimental constraints in determining the character of complexes that any one measurement accesses. In particular, for RNAP it is necessary to distinguish between initial, closed, and open complexes while being mindful of the influence transferred by the limits of resolution, both temporal or spatial.
Using DNA curtains, our laboratory has directly visualized E. coli RNAP in real time as it locates and binds physiological promoter sequences along the DNA of phage lambda, thus revealing the interactions of RNAP across the entire lambda phage genome (Figures 2e, 2f, and 3a).37j In contrast to experiments with nucleosomes described below, which were only able to probe the binding profile after the proteins had been deposited at their final positions on the DNA, experiments with RNAP could be conducted in real time, enabling us to assess the time-dependent evolution of the binding distributions. From these experiments, we obtained the survival probabilities and by extension a portion of the binding landscapes for each of the main intermediates along the biochemical pathway that leads to transcriptionally active complexes, including RNAP transiently bound to nonspecific sites (Figure 3a, top), and both closed (Figure 3a, middle) and open complexes (Figure 3a, bottom) at the native promoter sequences found in the lambda phage genome.37j
Figure 3.
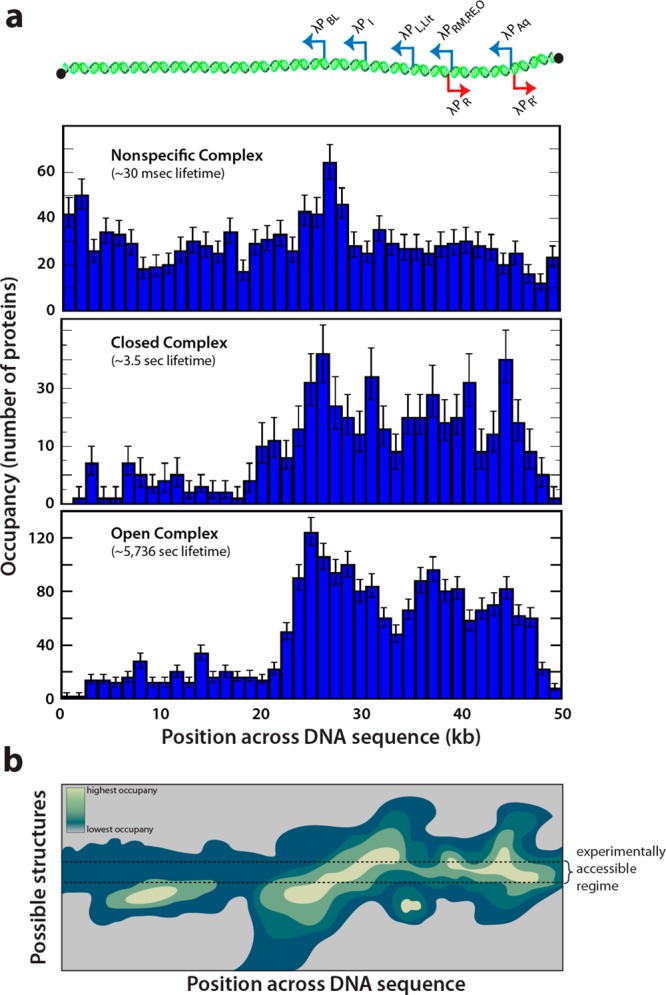
Promoter recognition by RNA polymerase. (a) Schematic overview of the promoter distribution in the lambda phage genome aligned with binding distributions of nonspecifically bound RNA polymerase (top), closed complexes (middle), and open complexes (bottom).37j Adapted with permission from ref (37j). (b) Schematic representation of a hypothetical 2D binding landscape across all particular DNA structural conformations available to a given DNA molecule including those brought about by different environmental settings and/or the presence of other DNA binding proteins. This schematic helps illustrate that in principle any given methodology used to probe a binding landscape can only access a relatively restricted region of potential structural space for a given DNA sequence.
Inspection of these binding distributions reveals several informative features. First, the nonspecific complex reflects a transiently bound intermediate that binds uniformly along the length of the DNA within our resolution limits (Figure 3a, top). This finding implies that RNAP can access the entirety of structure and sequence space available through the lambda phage genome within the context of this experiment and that for the nonspecific complex the binding landscape is likely defined predominantly by the sequence-independent qualities of DNA. This ability to nonspecifically associate with the DNA phosphate backbone regardless of underlying sequence is a characteristic of most DNA-binding proteins.
Importantly, the binding landscape of the nonspecifically bound complex does not represent the encounter landscape defined above. Recall, the encounter landscape describes the probability that a complex will form, not any characteristics of already-formed complexes, however transient the interaction. Further, it is important to recognize that the experimentally observed binding distribution of the nonspecific complex is not the same as that for the closed or open complexes, illustrating that this binding landscape is a function of reaction coordinate–nonspecific complexes that quickly dissociate from the DNA when they fail to encounter a promoter, whereas closed and open complexes remain stably bound for appreciably longer periods of time.42
Another significant result of these experiments is that the binding profiles of closed and open complexes map onto the locations of known phage promoters (Figure 3a). In addition, there are no sites within the profiles of the closed and open complexes that are completely devoid of polymerase, indicating that the protein is capable of transitioning to these latter stage intermediates at a low (but nonzero) efficiency regardless of the underlying sequence. Interestingly, this characteristic of RNA polymerase is not exclusive to the particular sigma subunit in the holoenzyme, which is to say not particular to the type of promoter that RNAP is primed to recognize.47 This ability of RNA polymerase to form open complexes at nonpromoter DNA may seem to contradict the fact that transcription initiates from specific promoter sequences. However, it is possible that these regions of the genome would be suppressed in vivo, through the cumulative action of other nonspecific DNA binding proteins such as HU or Fis, which are highly abundant in bacteria.11,31b Furthermore, the binding minima at promoters may be deeper in the presence of transcriptional activators, such as catabolite activator protein (CAP).42e,42f (Interestingly, recent work from the ENCODE project has suggested that most of the human genome is transcribed into RNA.48) It is also important to recognize that experiments for studying transcript production are typically designed to look at single, specific transcripts originating from known promoters and often require production of a full-length transcript leading to generation of a measurable reporter protein. These types of experiments would fail to detect low-level transcripts arising from nonpromoter sites. Given these considerations, it is possible that RNA polymerases may simply be much more promiscuous in initiating transcript production at nonpromoter DNA than generally appreciated.
These findings on the nature of DNA–RNAP interactions, when lensed through the presented conceptualization, yield an interesting take on how cellular control can be influenced by DNA. Consider the hypothetical scenario where all steps in transcription downstream of open complex formation are irreversible and homogeneous. In this case, the relative depths of the energetic minima revealed in the binding profile of the RNA polymerase open complex would in principle completely describe all control on gene expression levels. For example, in such an idealized system one would predict that the deepest minima in the open complex binding landscape would reflect the most highly expressed genes. However, as to be expected for a simplified in vitro measurement, the binding distribution of the RNA polymerase open complex clearly does not directly recapitulate either known expression levels or biological timing for each of the respective lambda phage promoters. As the most extreme example of this apparent incongruence, the most prominent peak found in the in vitro binding profile for the closed and open complexes coincides with the λPBL promoter (Figure 3a, middle and bottom).37a,37j DNA curtain measurements have revealed that RNAPs bound to the λPBL promoter are transcriptionally active in the presence of rNTPs37j and in fact display more efficient transcription than any of the other phage promoters in these single-molecule assays (S.R. and E.C.G., unpublished observations). Yet remarkably, the λPBL promoter is transcriptionally inactive in vivo.49 This discrepancy arises because the region of DNA encompassing the λPBL promoter harbors a total of 23 binding sites for the E. coli protein IHF, the presence of which prevents RNA polymerase from accessing the promoter in both the test tube and living cells49 (although it is formally possible that RNAP binds the λPBL promoter in the presence of IHF but fails to produce a transcript). In other words, the binding preferences that are revealed from the in vitro DNA curtain measurements would be otherwise obscured in an in vivo scenario due to the presence of other proteins. To reiterate, the encounter and binding landscapes for RNAP on naked extended λ-phage DNA, as in curtain experiments, reflect a baseline of interactions, though by inclusion of other interacting species (e.g., IHF), with each permutation defining a new encounter and binding landscape, the in vitro and in vivo results for the λPBL promoter are bridged. This observation provides a clear example of how experimental context defines the region of the binding landscape that can be accessed by any given technique and how the presence of extrinsic factors brings about distinct encounter and binding landscapes (Figure 3b).
6.2. Limits to Experimentally Accessing Structure Space
As alluded to above, in a DNA curtain experiment the binding landscape for a given DNA molecule is limited to a subset of structure space centered about a linear DNA conformation at relatively low tension (∼0.1–1 pico-Newton) under a given set of experimental buffer conditions, typically in the absence of other DNA-binding proteins. It would in principle be ideal to probe as much of the binding landscape as possible, but it is crucial to realize that most experimental methods query nonoverlapping regions of this field, with many regions remaining inaccessible due to experimental constraints (Figure 3b). For example, current DNA curtain experiments prevent access to any information regarding how the RNA polymerase binding landscape may be influenced by structural features of DNA such as those induced by supercoiling, higher order DNA conformations, and/or effects arising from the presence of transcriptional regulatory proteins. While curtain-assay-available DNA conformations define a limited region of this field that can be measured, it can be expanded upon or combined with other techniques to produce a more complete picture of DNA-based cellular regulation. At the other end of the experimental spectrum, genomics-based approaches used to study RNA polymerase can provide a population-averaged picture of global promoter occupancy in living cells,50 although it remains a challenge to interpret these types of experiments with respect to the influence of specific system parameters. Furthermore, information regarding protein dynamics is almost completely lacking from these approaches. However, the ideas presented here lobby for communication among techniques as the best way to holistically understand the nuances of cellular control.
6.3. Target Sizes Inherently Restrict Sequence Space
While determining the binding stability of proteins across all possible DNA structures is not feasible, it is in principle possible. However, there are absolute limitations placed on the extent of accessible sequence space arising from the physical restrictions of a protein–DNA interaction. This limitation in sequence space results from the finite size of the interface between a given protein and its DNA substrate. This concept is reflected in the “target size” of the protein–DNA interaction, which is a geometric constraint that describes the orientation and size of the binding surface of a protein as it samples DNA while searching for its target site.37j,51 The magnitude of a protein’s target size yields information regarding the potential scope of relevant sequence space. If the target size is small, then only a small number of contacts between the protein and the DNA can be utilized to read out genomic information and sequence space is consequently minimal. For example, if RNAP only “sees” one base pair at a time then the relevant sequence space consists of just four potential elements, A, T, C, and/or G, but would not require any combinations of two or more bases. Alternatively, if the target size were larger, then a proportionally larger amount of sequence space would be required to define both the binding and the encounter landscapes.
Using RNA polymerase as a model system, our laboratory has shown that it is possible to measure the apparent (or effective) target size of a DNA-binding protein using DNA curtains to visualize how RNAP searches for promoters in real time.37j The results revealed an E. coli RNAP target size of ∼7.5 Å,37j which corresponds to roughly 1/10th the length of the entire protein or just 1–1.5% of the protein’s surface;52 in other words, a relatively small fraction of the protein’s surface is required to discriminate its target sites from nonpromoter sequences. Interestingly, computational analysis has revealed that relatively few nucleotides within bacterial promoters are highly conserved.45 In addition, recent crystal structures of the RNA polymerase sigma subunit bound to promoter DNA fragments of the −10 element indicate that base-specific contacts with just two of these highly conserved nucleotides are sufficient to permit promoter binding and open complex formation, with the remaining contacts arising from electrostatic interactions with the phosphate backbone.53 While our measurements focused on the primary sigma subunit in E. coli, σ,70 promoter engagement by RNA polymerase containing the specialized subunit, σ,54 has recently been shown to behave remarkably similarly.47 Importantly, measurements of the association rate of RNAP to promoter DNA showed that the rate of promoter recognition and subsequent engagement was unperturbed by removal of flanking DNA up to 7 bp downstream or 78 bp upstream of the transcriptional start site.47 Given these findings, along with the relatively small target size revealed from our measurements, it is tempting to speculate that target discrimination by E. coli RNAP may involve initial recognition of as few as 2 or 3 base pairs within the −10 promoter region.
6.4. Beyond Promoter Binding
DNA curtain experiments with RNAP offer a satisfying representation of regulation, originating at the level of DNA, by highlighting the distinct binding landscapes of RNAP during the early stages of promoter binding and how the observed profiles can be influenced by experimental settings. While the above discussion is concerned with the possible extent of DNA-based control in gene expression, it is far from the in vivo picture. As indicated above, promoter binding and transcription must occur in a complex environment that contains many other proteins. This includes transcription factors, which provide crucial regulatory control over gene expression in response to cellular needs and environmental cues. Transcription factors act as activators or repressors and can alter the affinity between RNAP and promoter DNA.42e,42f When considered within the context of our arguments, activators can be said to function by creating new minima along the encounter and/or binding landscape or deepening existing ones, thereby increasing recruitment to and/or lifetime at particular locations. Alternatively, repressors may have the opposite effect by eliminating or attenuating existing minima. In addition to transcription factors, DNA in vivo is cluttered by many other proteins, each of which distinctly defines the frequency and/or stability of particular interactions, altering the baseline regulation patterns due solely to the DNA. The case of the λPBL promoter shows how the presence of other proteins can alter interactions. Although this may be a relatively extreme all-or-none example of how competition for the same region of DNA by two different proteins can affect binding, it is relatively easy to envision how more subtle effects, positive or negative, may be brought about under the influence of other DNA-binding proteins. Design and execution of experiments that can bring about an understanding of how the dynamic interplay in multicomponent systems can lead to physiological outcomes remains an exciting challenge in single-molecule bioscience.
7. Nucleosomes
The complex organization of genomic DNA is an important feature of eukaryotic cells. Highly conserved core histone proteins, their variants, and other associated nonhistone proteins complex with DNA to form nucleosomes, which in turn comprise the functional unit of chromatin.54 A canonical nucleosome consists of a histone octamer (two copies each of histone H2A, H2B, H3, and H4) bound to ∼147 bp of DNA.29a,55 The bound DNA wraps entirely around the outer surface of the histone octamer, making a total of ∼1.7 turns.29a,55 Nucleosomes serve in part to compact chromosomes in eukaryotes and to otherwise make DNA physically manageable, especially during cell division. Certain distinct features of chromosomes are defined by types of chromatin, including highly compact heterochromatin, which is generally associated with gene silencing, and the specialized chromatin found at centromeres which mediate the interaction between chromosomes and microtubules, required for chromosome segregation during mitosis and meiosis.
Chromatin creates a substrate that is decidedly distinct from the naked DNA contained within, which has profound implications for genome regulation. The basal encounter and binding landscapes associated with DNA-binding proteins in a eukaryotic cell will therefore be defined with respect to chromatin rather than naked DNA; the many ways in which cells use chromatin to regulate access to the underlying DNA is a major area of research in genetics and cell biology but remains largely unexplored by single-molecule biology. A major step in that direction is a basic single-molecule understanding of the relationship between DNA sequences and structures and nucleosome positioning.
7.1. Nucleosomes Must Access Vast Regions of Sequence Space
Nucleosomes do not specifically target defined DNA sequences, and nucleosomes do not extract detailed sequence information from DNA. Indeed, nucleosomes function as global DNA packaging elements; on average, the DNA within entire chromosomes needs to be accessible to nucleosomes, and so nucleosomes cannot possess high intrinsic sequence specificity.54b,55,56 However, each fully formed nucleosome drastically perturbs DNA structure,29a,55 and so nucleosomes are expected to exhibit some preference for sequences or sequence signatures that favor the extensive DNA bending needed to wrap DNA.55−57 This accords with our assumption that all DNA-binding proteins, regardless of whether or not they need to be sequence or structure specific, will exhibit some sequence or structure preferences (see Figure 1a); as a consequence, the binding landscape for any given DNA-binding protein is never completely flat. From this fundamental perspective the information content of DNA sequences is irrelevant to the nucleosome, and the significance of the sequences stems entirely from their capacity to form certain structures. Nucleosomes are therefore a good example of a structure interactor on the continuum.
It has been suggested that evolution has harnessed the sequence preferences of nucleosome formation to define a “genomic code” for nucleosome positioning,57d,58 but the idea remains highly controversial, and it seems clear that other facets of chromosome organization also impact nucleosome positioning (see below).54b,59 It would not be surprising if in certain cases sequences coevolved with nucleosome positioning, but there would remain a counter-evolutionary pressure to allow nucleosomes to package all DNA, and so the extent, or strength, of any such genomic code for nucleosome positioning cannot be absolute, that is, the wells of the binding landscape cannot vary too extensively across sequences. This is especially apparent given that the target size of a nucleosome is 147 bp, such that there are 4147 possible sequences that can be probed. This is a vast expanse of sequence space upon which to impose sequence-based regulation when a large portion of that space must be accessible to nucleosome binding.
7.2. Sequence and Structure Preferences of Nucleosomes
It is known that nucleosomes preferentially form on DNA with WW dinucleotides (W = A or T) that follow a 10 bp periodicity—approximately the number of bases in a single turn of the DNA double helix—with SS dinucleotides (S = G or C) 5 bp out of phase.57b This signature allows for helix distortions that favor bending of the DNA around the histones.29a,57b,57d Although there are no absolute sequence restrictions to nucleosome formation based on in vitro and in vivo nucleosome localization data, it has been suggested that the yeast genome has evolved a general 10 bp periodicity in WW dinucleotides to facilitate nucleosome deposition.59c In addition, A/T-rich sequences, which are relatively stiff and resistant to bending, do not favor nucleosome formation.58b,60 Despite this, nucleosomes still readily bind poly-(dA-dT) tracts, illustrating their ability to bind almost any DNA sequence.61 There has been tremendous interest in developing in silico models for predicting nucleosome distributions based on DNA sequence composition and nucleosome binding preferences and the exclusionary effects of stiff DNA sequences that resist bending, such as poly(dA-dT) tracts.58b,58d,60c,62 These modeling efforts together with in vivo mapping studies of nucleosome positions are helping to yield details about chromatin structure and its relationship to gene regulation.
7.3. Visualizing Nucleosome Positioning with DNA Curtains
Our laboratory has used the DNA curtain technique to measure nucleosome binding distributions as a function of DNA sequence.37i For this work, nucleosomes were assembled on DNA by salt dialysis in the absence of any remodelers or chaperones (Figure 2);37i this minimal in vitro approach allows nucleosomes to localize to their thermodynamically favored or nearly thermodynamically favored sites,55,58d that is, localization is determined strictly by the DNA-defined binding landscape, and an experimentally measured distribution using DNA curtains is therefore a directly proportional manifestation of that landscape.
The primary substrate in this study was the 48.5 kbp DNA genome of the bacteriophage lambda (as in the RNAP work described above), which infects E. coli and has not coevolved with nucleosome deposition. However, it does have the useful quality of being distinctly polar in base content; one-half of the DNA is G/C rich, while the other half is A/T rich, with a predominance of stiff poly(dA-dT) tracts in the A/T-rich half of the molecule (Figure 4a).37i This quality of the DNA substrate allowed the authors to examine the influence of intrinsic sequence content on the nucleosome binding landscape in the absence of any other factors. Binding distribution histograms built from data collected for either canonical nucleosomes or nucleosomes containing the histone variant H2AZ provided a course-grained profile of the thermodynamically favored intrinsic binding profile for nucleosome deposition (Figure 4b and 4c, top). The observed nucleosome binding distributions were anticorrelated with the distribution of poly(dA-dT) tracts and strongly correlated with the in silico predictions of Field et al.58b and Kaplan et al.,58d reinforcing the hypothesis that DNA contains intrinsic sequence and structure information capable of dictating nucleosomes binding. The distribution shows the same sequence-based “positioning rules” as observed for eukaryotic DNA, reflecting the capacity of nucleosomes to form on any DNA source while following the same physical principles to dictate binding site preferences.
Figure 4.
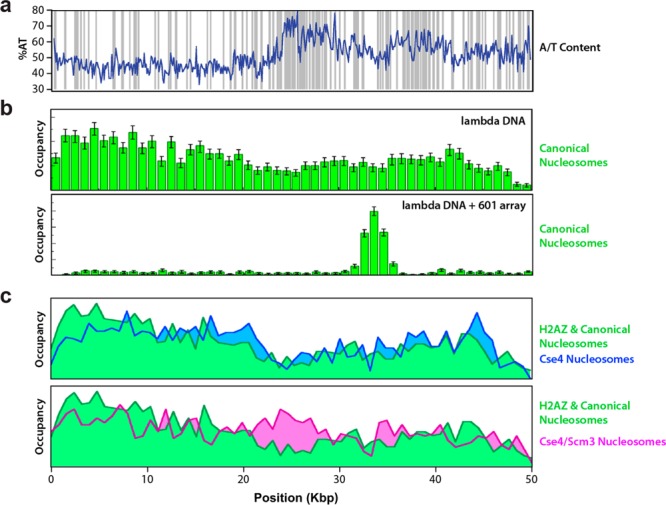
Nucleosome binding landscapes. (a) Graph depicting the unusual polar A/T-content distribution of the bacteriophage lambda genome along with the location of poly(dA-dT) tracts.37i (b) Binding distributions of S. cerevisiae nucleosomes on the lambda phage genome. (Top) Binding landscape for the wild-type phage DNA,37i and (bottom) distribution of nucleosomes on a lambda phage genome containing an engineered array of 13 tandem Widom 601 sequences (Unpublished results). (c) Comparison of canonical nucleosomes and H2AZ containing nucleosomes (combined data set) with nucleosomes containing the centromere specific histone H3 variant Cse4 (top) or nucleosomes assembled with both Cse4 and the nonhistone chaperone protein Scm3 (bottom).37i Adapted with permission from ref (37i).
Interestingly, the tightest known nucleosome binding sequence, called the “Widom 601” positioning sequence, was generated by in vitro selection.63 Remarkably, the Widom 601 sequence does not follow the same composition “rules” for nucleosome positioning sequences based on 10 bp nucleotide periodicity, and although this unusual DNA sequence is undoubtedly more “bendable” than a typical DNA sequence, it is still not entirely clear why it binds so well to nucleosomes.64
In DNA curtain experiments performed with an engineered lambda phage DNA containing either a single 601 sequence (not shown) or a 13× tandem array of 601 sequences (Figure 4b, bottom), the nucleosome binding profile becomes dominated by a single defined location. Notably, the peaks and valleys in the binding distributions measured on natural DNA, be it from phage (Figure 4b, top) or human,37i are much more subtle when compared to the stark binding profile induced by the presence of a 601 sequence (compare Figures 2b, 2c, and 4b). This result highlights the peculiarities of the Widom 601 sequence and its unique ability to bind nucleosomes much more tightly than any other sequence and also provides a clear example of DNA structure dominating an observed binding distribution. Although the Widom 601 positioning sequence has proven indispensible for many in vitro studies requiring positioned nucleosomes, note that sequences of similarly high nucleosome affinity are not found in nature; a nucleosome that cannot budge would constitute a major physical obstacle on DNA and could prove intransigent to regulation (i.e., positioning or repositioning by nucleosome remodelers or other factors). Interestingly, Gracey et al.65 and Perales et al.66 recently looked at the nucleosome occupancy of a 601 site in living cells. These studies revealed that the 601 site does not efficiently sustain positioned nucleosomes in vivo, demonstrating that extrinsic factors such as the local chromatin structure and/or the transcriptional status of DNA can supersede the potential capacity of the underlying sequence content to dominate the nucleosome binding landscape (see below).
DNA curtains were also used to determine whether the binding profile of centromeric nucleosomes was distinct from that of canonical histones. The binding landscape for S. cerevisiae centromeric nucleosomes, which contain the histone H3 variant Cse4, is particularly intriguing because centromeric nucleosomes must bind exclusively to centromeric DNA in vivo, a defining feature of which is high AT content (86–98%) and highly enriched homopolymeric tracts of poly(dA-dT).67 These A/T-rich sequences do not have the characteristic dinucleotide compositions that would favor nucleosome binding but instead would be expected to exclude histone octamers,37i,58b,58d suggesting that nucleosomes bearing centromere-specific histones might display binding profiles distinct from those observed for canonical nucleosomes. Surprisingly, nucleosomes assembled with the histone variant Cse4 exhibit the same distribution patterns as canonical nucleosomes (Figure 4d), indicating that the binding profiles for both types of nucleosomes are fundamentally similar; the Cse4 histone variant does not contribute to localizing nucleosomes to otherwise exclusionary sequences found at centromeres. However, centromeric nucleosomes can also form an unusual hexameric intermediate in which H2A/H2B is replaced with the nonhistone protein Scm3.68 Inclusion of this nonhistone protein, Scm3, to Cse4-containing nucleosomes does alter the binding distribution, allowing the nucleosomes to better bind regions of DNA enriched with poly(dA-dT) tracts (Figure 4d). Therefore, addition of a histone variant (H2AZ or Cse4) can leave a binding profile substantially unaltered, whereas a nonhistone protein (Scm3) can dramatically reconfigure the binding profile. This result demonstrates how complex and unexpected higher orders of binding regulation can be. The centromeric region of chromosomes must be highly occupied by nucleosomes for a very specific biological function (proper spindle formation and chromosome segregation), yet this region is rich in sequence features that specificaly antagonize nucleosome binding. Centromeric nucleosomes contain a noncanonical histone variant, Cse4, yet this histone does not increase binding to the exclusionary sequences of centromeres. Another nonhistone protein, Scm3, is needed to shift the binding landscape into its correct, physiological form.
7.4. Extrinsic Factors Can Affect Binding Landscapes
To determine the binding profile associated with a more complex eukaryotic substrate, the authors selected a 23 kb fragment derived from the human β-globin locus.37i Inspection of the β-globin nucleosome distribution indicates that the experimentally measured binding distribution is dictated by DNA sequence and also reflects the underlying organizational features of the DNA. Every peak within the binding distribution for the human DNA substrate coincides with regulatory sequences, including the promoter-proximal regions of the globin genes. An additional peak in the binding distribution occurs at a developmental stage-specific promoter located within a region necessary for silencing transcription of the fetal globin genes.69 In contrast, the valleys within the binding distribution correspond to nontranscribed and nonregulatory DNA. This data reveals that regulatory regions within the human β-globin DNA contain preferred nucleosome-binding sites as reflected by the in vitro binding profile. This organization has likely arisen not because more or more tightly bound nucleosomes are required at these regulatory sequences but rather because precise nucleosome positioning within these regions may be much more critical compared to other parts of the genome.70 This interpretation is consistent with the finding that eukaryotic transcriptional start sites are typically flanked by well-positioned nucleosomes.54b,59d
The locations of nucleosomes across the β-globin locus have been mapped in vivo,71 allowing for a comparison of the in vitro and in vivo DNA binding profiles. While some regions of the in vivo and in vitro landscapes overlap, other regions are clearly dissimilar.37i This comparison as well as the in vitro versus in vivo results with the Widom 601 sequence cited above illustrate that thermodynamically preferred sequences alone cannot predict nucleosome occupancy within living cells. This example illustrates that the intrinsic role of DNA in dictating protein–DNA interactions can in some cases pervade through to the in vivo state and in other cases become occluded by other factors. The precise relationship between the intrinsic nucleosome binding landscape and the extent to which it is exploited by the cell remains unknown, but it is clear that if the intrinsic binding profile associated with the β-globin locus plays a role during initial nucleosome deposition, then the nucleosomes must subsequently be shifted by unknown mechanisms to establish the in vivo localization pattern.
8. Concluding Perspectives
The role of DNA as both a carrier of sequence information and a structural scaffold hugely influences its interactions with DNA-binding proteins. It is crucial to understand how the respective effects of these qualities propagate through to the in vivo state. To examine and clarify this we presented a framework to interpret experimental results from across methodologies and have shown how it can lead to insightful interpretations. We discuss experiments from our laboratory to show how this framework functions in practice. We demonstrate that DNA curtains provide information about a protein’s DNA binding landscape and can be utilized to explore how the landscape may change over the course of a reaction trajectory. Our DNA curtain studies of RNA polymerase and nucleosomes both yield informative single-molecule binding distributions on phage lambda DNA substrates, highlighting how both sequence and structure bring about different but important consequences in each system. The protein distributions observed in these measurements reflect fundamental characteristics of the proteins and their DNA substrate. We find that while RNA polymerase localized to promoter sequences as expected, single-molecule studies of nucleosome positioning yielded counterintuitive results: a noncanonical histone variant unique to centromeres did not alter binding distributions, whereas addition of a nonhistone protein did. An appreciation of the dynamic and layered nature of binding landscapes helps place these results in a broader context. It becomes possible, for example, to develop hypotheses about the additional layers of control needed to reach the in vivo state. A significant amount of conceptual understanding can be layered onto the results by considering the framework presented in this review. We anticipate that the DNA curtain methodology can also be applied to increasingly complex biochemical questions involving multicomponent systems and/or higher order chromatin structures. We find that these prompts point to fundamental qualities about the biology underlying single-molecule experiments and hope that they can be applied to guide fruitful experimental design and data interpretation.
Acknowledgments
We thank members of the Greene laboratory for insightful discussions and Dr. P. Halálarc for incisive notes. We especially thank Myles Marshall for assistance with the figures and Luke Kaplan for sharing results on DNA curtain experiments using the 601 positioning sequence. Research in the Greene laboratory is funded by the National Institutes of Health (GM074739 and GM082848), the National Science Foundation (MCB1154511), and the Howard Hughes Medical Institute. D.D. is supported in part by an award from the Paul and Daisy Soros Fellowships for New Americans.
Biographies

Daniel Duzdevich received his B.A. degree in Biophysics from Columbia University and his M.Phil. from Churchill College, Cambridge, where he studied gross DNA structures with atomic force microscopy. He is currently pursuing his Ph.D. degree in the laboratory of Professor Eric C. Greene at Columbia University.

Sy E. Redding received his B.S. degree in Physics from Texas State University in 2009 and entered the graduate program in Chemical Physics at Columbia University that same year. He is currently completing his Ph.D. work using a combination of single-molecule imaging and theory to understand how proteins find binding targets within DNA.

Eric C. Greene received his B.S. degree in Biochemistry from the University of Illinois and Ph.D. degree from Texas A&M University, and he conducted postdoctoral studies at the National Institute of Health. He joined the Department of Biochemistry and Molecular Biophysics at Columbia University in 2004 and was appointed as an Early Career Scientist with the Howard Hughes Medical Institute in 2009. His laboratory has developed novel technologies for studying protein–DNA interactions at the single-molecule level.
Author Contributions
D.D. and S.E.R. contributed equally to this review.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- a Nishant K. T.; Singh N. D.; Alani E. Bioessays 2009, 31, 912. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Eyre-Walker A.; Keightley P. D. Nat. Rev. Genet. 2007, 8, 610. [DOI] [PubMed] [Google Scholar]; c Foster P. L. Methods Enzymol. 2006, 409, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Parker S. C.; Tullius T. D. Curr. Opin. Struct. Biol. 2011, 21, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Aparicio O. M. Genes Dev. 2013, 27, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Groth A.; Rocha W.; Verreault A.; Almouzni G. Cell 2007, 128, 721. [DOI] [PubMed] [Google Scholar]
- a Rohs R.; Jin X.; West S. M.; Joshi R.; Honig B.; Mann R. S. Annu. Rev. Biochem. 2010, 79, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rohs R.; West S. M.; Liu P.; Honig B. Curr. Opin. Struct. Biol. 2009, 19, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rohs R.; West S. M.; Sosinsky A.; Liu P.; Mann R. S.; Honig B. Nature 2009, 461, 1248. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Parker S. C.; Hansen L.; Abaan H. O.; Tullius T. D.; Margulies E. H. Science 2009, 324, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bishop E. P.; Rohs R.; Parker S. C.; West S. M.; Liu P.; Mann R. S.; Honig B.; Tullius T. D. ACS Chem. Biol. 2011, 6, 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Pabo C. O.; Sauer R. T. Annu. Rev. Biochem. 1984, 53, 293. [DOI] [PubMed] [Google Scholar]; g De Santis P.; Scipioni A. Phys. Life Rev. 2013, 10, 41. [DOI] [PubMed] [Google Scholar]
- Cavalli G.; Misteli T. Nat. Struct. Mol. Biol. 2013, 20, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante C.; Smith S. B.; Liphardt J.; Smith D. Curr. Opin. Struct. Biol. 2000, 10, 279. [DOI] [PubMed] [Google Scholar]
- a Tinoco I. Jr.; Gonzalez R. L. Jr. Genes Dev. 2011, 25, 1205. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wennmalm S.; Simon S. M. Annu. Rev. Biochem. 2007, 76, 419. [DOI] [PubMed] [Google Scholar]; c Duzdevich D.; Greene E. C. Philos. Trans. R. Soc. London, B: Biol. Sci. 2013, 368, 20120271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dion M. F.; Kaplan T.; Kim M.; Buratowski S.; Friedman N.; Rando O. J. Science 2007, 315, 1405. [DOI] [PubMed] [Google Scholar]; b Lickwar C. R.; Mueller F.; Hanlon S. E.; McNally J. G.; Lieb J. D. Nature 2012, 484, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kastritis P. L.; Bonvin A. M. J. R. Soc., Interface 2013, 10, 20120835. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kodera N.; Yamamoto D.; Ishikawa R.; Ando T. Nature 2010, 468, 72. [DOI] [PubMed] [Google Scholar]; c Phillips R.; Kondev J.; Theriot J.. Physical biology of the cell; Garland Science: New York, 2009. [Google Scholar]; d Spolar R. S.; Record M. T. Jr. Science 1994, 263, 777. [DOI] [PubMed] [Google Scholar]; e Van Holde K. E.; Johnson W. C.; Ho P. S.. Principles of physical biochemistry, 2nd ed.; Pearson/Prentice Hall: Upper Saddle River, N.J., 2006. [Google Scholar]; f Zhou H. X. Q. Rev. Biophys. 2010, 43, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. L.; Nikolov D. B.; Burley S. K. Nature 1993, 365, 520. [DOI] [PubMed] [Google Scholar]
- a Aves S. J.; Liu Y.; Richards T. A. Subcell Biochem. 2012, 62, 19. [DOI] [PubMed] [Google Scholar]; b Johnson A.; O’Donnell M. Annu. Rev. Biochem. 2005, 74, 283. [DOI] [PubMed] [Google Scholar]; c Bell S. P.; Dutta A. Annu. Rev. Biochem. 2002, 71, 333. [DOI] [PubMed] [Google Scholar]
- Dillon S. C.; Dorman C. J. Nat. Rev. Microbiol. 2010, 8, 185. [DOI] [PubMed] [Google Scholar]
- a Watson J. D.; Crick F. H. Nature 1953, 171, 737. [DOI] [PubMed] [Google Scholar]; b Wang A. H.; Fujii S.; van Boom J. H.; Rich A. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 3968. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang A. H.; Hakoshima T.; van der Marel G.; van Boom J. H.; Rich A. Cell 1984, 37, 321. [DOI] [PubMed] [Google Scholar]; d Larsen T. A.; Kopka M. L.; Dickerson R. E. Biochemistry 1991, 30, 4443. [DOI] [PubMed] [Google Scholar]; e Siggers T. W.; Silkov A.; Honig B. J. Mol. Biol. 2005, 345, 1027. [DOI] [PubMed] [Google Scholar]
- Bustamante C.; Bryant Z.; Smith S. B. Nature 2003, 421, 423. [DOI] [PubMed] [Google Scholar]
- Reyes-Lamothe R.; Nicolas E.; Sherratt D. J. Annu. Rev. Genet. 2012, 46, 121. [DOI] [PubMed] [Google Scholar]
- a Ferraro-Gideon J.; Sheykhani R.; Zhu Q.; Duquette M. L.; Berns M. W.; Forer A. Mol. Biol. Cell 2013, 24, 1375. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Brock J. A.; Bloom K. J Cell Sci. 1994, 107Pt 4891. [DOI] [PubMed] [Google Scholar]
- Smith S. B.; Cui Y.; Bustamante C. Science 1996, 271, 795. [DOI] [PubMed] [Google Scholar]
- a Vafabakhsh R.; Ha T. Science 2012, 337, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Peters J. P. 3rd; Maher L. J. Q. Rev. Biophys. 2010, 43, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cloutier T. E.; Widom J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Liu L. F.; Wang J. C. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 7024.2823250 [Google Scholar]; b Wang J. C.; Lynch A. S. Curr. Opin. Genet. Dev. 1993, 3, 764. [DOI] [PubMed] [Google Scholar]
- a Baranello L.; Levens D.; Gupta A.; Kouzine F. Biochim. Biophys. Acta 2012, 1819, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kouzine F.; Gupta A.; Baranello L.; Wojtowicz D.; Ben-Aissa K.; Liu J.; Przytycka T. M.; Levens D. Nat. Struct. Mol. Biol. 2013, 20, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kouzine F.; Sanford S.; Elisha-Feil Z.; Levens D. Nat. Struct. Mol. Biol. 2008, 15, 146. [DOI] [PubMed] [Google Scholar]; d Chen S. H.; Chan N. L.; Hsieh T. S. Annu. Rev. Biochem. 2013, 82, 139. [DOI] [PubMed] [Google Scholar]; e Wu H. Y.; Shyy S. H.; Wang J. C.; Liu L. F. Cell 1988, 53, 433. [DOI] [PubMed] [Google Scholar]
- a Koster D. A.; Crut A.; Shuman S.; Bjornsti M. A.; Dekker N. H. Cell 2010, 142, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ostrander E. A.; Benedetti P.; Wang J. C. Science 1990, 249, 1261. [DOI] [PubMed] [Google Scholar]; c Ristic D.; Wyman C.; Paulusma C.; Kanaar R. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesbah N. M.; Wiegel J. Ann. N.Y. Acad. Sci. 2008, 1125, 44. [DOI] [PubMed] [Google Scholar]
- Kikuchi A.; Asai K. Nature 1984, 309, 677. [DOI] [PubMed] [Google Scholar]
- a Yakovchuk P.; Protozanova E.; Frank-Kamenetskii M. D. Nucleic Acids Res. 2006, 34, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Podgornik R.; Hansen P. L.; Parsegian V. A. J. Chem. Phys. 2000, 113, 9343. [Google Scholar]
- Record M. T. Jr. Bioploymers 1975, 14, 2137. [DOI] [PubMed] [Google Scholar]
- SantaLucia J. Jr.; Hicks D. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 415. [DOI] [PubMed] [Google Scholar]
- Jose D.; Datta K.; Johnson N. P.; von Hippel P. H. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers A. A. Philos. Trans. A: Math. Phys. Eng. Sci. 2004, 362, 1423. [DOI] [PubMed] [Google Scholar]
- a Dickerson R. E. Nucleic Acids Res. 1998, 26, 1906. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jones S.; van Heyningen P.; Berman H. M.; Thornton J. M. J. Mol. Biol. 1999, 287, 877. [DOI] [PubMed] [Google Scholar]; c Locasale J. W.; Napoli A. A.; Chen S.; Berman H. M.; Lawson C. L. J. Mol. Biol. 2009, 386, 1054. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Olson W. K.; Gorin A. A.; Lu X. J.; Hock L. M.; Zhurkin V. B. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 11163. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Sunami T.; Kono H. PLoS One 2013, 8, e56080. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zhang Y.; Xi Z.; Hegde R. S.; Shakked Z.; Crothers D. M. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Luger K.; Mader A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Nature 1997, 389, 251. [DOI] [PubMed] [Google Scholar]; b Biswas T.; Aihara H.; Radman-Livaja M.; Filman D.; Landy A.; Ellenberger T. Nature 2005, 435, 1059. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chen Z.; Yang H.; Pavletich N. P. Nature 2008, 453, 489. [DOI] [PubMed] [Google Scholar]; d Gaudier M.; Schuwirth B. S.; Westcott S. L.; Wigley D. B. Science 2007, 317, 1213. [DOI] [PubMed] [Google Scholar]; e Kim Y.; Geiger J. H.; Hahn S.; Sigler P. B. Nature 1993, 365, 512. [DOI] [PubMed] [Google Scholar]; f Montano S. P.; Pigli Y. Z.; Rice P. A. Nature 2012, 491, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Murakami K. S.; Masuda S.; Campbell E. A.; Muzzin O.; Darst S. A. Science 2002, 296, 1285. [DOI] [PubMed] [Google Scholar]; h Obmolova G.; Ban C.; Hsieh P.; Yang W. Nature 2000, 407, 703. [DOI] [PubMed] [Google Scholar]; i Rice P. A.; Yang S.; Mizuuchi K.; Nash H. A. Cell 1996, 87, 1295. [DOI] [PubMed] [Google Scholar]; j Schultz S. C.; Shields G. C.; Steitz T. A. Science 1991, 253, 1001. [DOI] [PubMed] [Google Scholar]; k Moure C. M.; Gimble F. S.; Quiocho F. A. Nat. Struct. Biol. 2002, 9, 764. [DOI] [PubMed] [Google Scholar]
- Abbondanzieri E. A.; Greenleaf W. J.; Shaevitz J. W.; Landick R.; Block S. M. Nature 2005, 438, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Graham J. S.; Johnson R. C.; Marko J. F. Biochem. Biophys. Res. Commun. 2011, 415, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Graham J. S.; Johnson R. C.; Marko J. F. Nucleic Acids Res. 2011, 39, 2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bintu L.; Kopaczynska M.; Hodges C.; Lubkowska L.; Kashlev M.; Bustamante C. Nat. Struct. Mol. Biol. 2011, 18, 1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi B.; Blainey P. C.; Xie X. S. J. Phys. Chem. B 2008, 112, 6282. [DOI] [PubMed] [Google Scholar]
- Ragunathan K.; Liu C.; Ha T. eLife 2012, 1, e00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kulczyk A. W.; Tanner N. A.; Loparo J. J.; Richardson C. C.; van Oijen A. M.. J. Visualized Exp. 2010. [DOI] [PMC free article] [PubMed]; b Loparo J. J.; Kulczyk A. W.; Richardson C. C.; van Oijen A. M. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Finkelstein I. J.; Greene E. C. Methods Mol. Biol. 2011, 745, 447. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Greene E. C.; Wind S.; Fazio T.; Gorman J.; Visnapuu M. L. Methods Enzymol. 2010, 472, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Finkelstein I. J.; Visnapuu M. L.; Greene E. C. Nature 2010, 468, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gorman J.; Fazio T.; Wang F.; Wind S.; Greene E. C. Langmuir 2010, 26, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gorman J.; Plys A. J.; Visnapuu M. L.; Alani E.; Greene E. C. Nat. Struct. Mol. Biol. 2010, 17, 932. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gorman J.; Wang F.; Redding S.; Plys A. J.; Fazio T.; Wind S.; Alani E. E.; Greene E. C. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, E3074. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Graneli A.; Yeykal C. C.; Prasad T. K.; Greene E. C. Langmuir 2006, 22, 292. [DOI] [PubMed] [Google Scholar]; f Graneli A.; Yeykal C. C.; Robertson R. B.; Greene E. C. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Fazio T.; Visnapuu M. L.; Wind S.; Greene E. C. Langmuir 2008, 24, 10524. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Visnapuu M. L.; Fazio T.; Wind S.; Greene E. C. Langmuir 2008, 24, 11293. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Visnapuu M. L.; Greene E. C. Nat. Struct. Mol. Biol. 2009, 16, 1056. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Wang F.; Redding S.; Finkelstein I. J.; Gorman J.; Reichman D. R.; Greene E. C. Nat. Struct. Mol. Biol. 2013, 20, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb B.; Silverstein T. D.; Finkelstein I. J.; Greene E. C. Anal. Chem. 2012, 84, 7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtemanche N.; Lee J. Y.; Pollard T. D.; Greene E. C. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y.; Finkelstein I. J.; Crozat E.; Sherratt D. J.; Greene E. C. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Batista P. J.; Chang H. Y. Cell 2013, 152, 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lee J. T.; Bartolomei M. S. Cell 2013, 152, 1308. [DOI] [PubMed] [Google Scholar]; c Sabin L. R.; Delas M. J.; Hannon G. J. Mol. Cell 2013, 49, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Serganov A.; Nudler E. Cell 2013, 152, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wilson R. C.; Doudna J. A. Annu. Rev. Biophys. 2013, 42, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Yates L. A.; Norbury C. J.; Gilbert R. J. Cell 2013, 153, 516. [DOI] [PubMed] [Google Scholar]
- a Nudler E. Annu. Rev. Biochem. 2009, 78, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]; b DeHaseth P. L.; Zupancic M.; Record M. T. Jr. J. Bacteriol. 1998, 180, 3019. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Saecker R.; Record M.; Dehaseth P. J. Mol. Biol. 2011, 412, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]; d McClure W. R. Annu. Rev. Biochem. 1985, 54, 171. [DOI] [PubMed] [Google Scholar]; e Haugen S. P.; Ross W.; Gourse R. L. Nat. Rev. Microbiol. 2008, 6, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Browning D. F.; Busby S. J. Nat. Rev. Microbiol. 2004, 2, 57. [DOI] [PubMed] [Google Scholar]
- a Herbert K. M.; Greenleaf W. J.; Block S. M. Annu. Rev. Biochem. 2008, 77, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Larson M. H.; Landick R.; Block S. M. Mol. Cell 2011, 41, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mendoza-Vargas A.; Olvera L.; Olvera M.; Grande R.; Vega-Alvarado L.; Taboada B.; Jimenez-Jacinto V.; Salgado H.; Ju·rez K.; Contreras-Moreira B.; Huerta A.; Collado-Vides J.; Morett E. PLoS One 2009, 4, e7526. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cho B.; Zengler K.; Qiu Y.; Park Y.; Knight E.; Barrett C.; Gao Y.; Palsson B. Nat. Biotechnol. 2009, 27, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Browning D.; Busby S. Nat. Rev. Microbiol. 2004, 2, 57. [DOI] [PubMed] [Google Scholar]; d Haugen S.; Ross W.; Gourse R. Nat. Rev. Microbiol. 2008, 6, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultzaberger R. K.; Chen Z.; Lewis K. A.; Schneider T. D. Nucleic Acids Res. 2007, 35, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Berg O. G.; von Hippel P. H. Annu. Rev. Biophys. Biophys. Chem. 1985, 14, 131. [DOI] [PubMed] [Google Scholar]; b Halford S. E.; Marko J. F. Nucleic Acids Res. 2004, 32, 3040. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tafvizi A.; Mirny L. A.; van Oijen A. M. ChemPhysChem 2011, 12, 1481. [DOI] [PMC free article] [PubMed] [Google Scholar]; d von Hippel P. H.; Berg O. G. J. Biol. Chem. 1989, 264, 675. [PubMed] [Google Scholar]
- Friedman L. J.; Mumm J. P.; Gelles J. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 9740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E.; Stamatoyannopoulos J. A.; Dutta A.; Guigo R.; Gingeras T. R.; Margulies E. H.; Weng Z.; Snyder M.; Dermitzakis E. T.; Thurman R. E.; Kuehn M. S.; Taylor C. M.; Neph S.; Koch C. M.; Asthana S.; Malhotra A.; Adzhubei I.; Greenbaum J. A.; Andrews R. M.; Flicek P.; Boyle P. J.; Cao H.; Carter N. P.; Clelland G. K.; Davis S.; Day N.; Dhami P.; Dillon S. C.; Dorschner M. O.; Fiegler H.; Giresi P. G.; Goldy J.; Hawrylycz M.; Haydock A.; Humbert R.; James K. D.; Johnson B. E.; Johnson E. M.; Frum T. T.; Rosenzweig E. R.; Karnani N.; Lee K.; Lefebvre G. C.; Navas P. A.; Neri F.; Parker S. C.; Sabo P. J.; Sandstrom R.; Shafer A.; Vetrie D.; Weaver M.; Wilcox S.; Yu M.; Collins F. S.; Dekker J.; Lieb J. D.; Tullius T. D.; Crawford G. E.; Sunyaev S.; Noble W. S.; Dunham I.; Denoeud F.; Reymond A.; Kapranov P.; Rozowsky J.; Zheng D.; Castelo R.; Frankish A.; Harrow J.; Ghosh S.; Sandelin A.; Hofacker I. L.; Baertsch R.; Keefe D.; Dike S.; Cheng J.; Hirsch H. A.; Sekinger E. A.; Lagarde J.; Abril J. F.; Shahab A.; Flamm C.; Fried C.; Hackermuller J.; Hertel J.; Lindemeyer M.; Missal K.; Tanzer A.; Washietl S.; Korbel J.; Emanuelsson O.; Pedersen J. S.; Holroyd N.; Taylor R.; Swarbreck D.; Matthews N.; Dickson M. C.; Thomas D. J.; Weirauch M. T.; Gilbert J. Nature 2007, 447, 799.17571346 [Google Scholar]
- a Kur J.; Hasan N.; Szybalski W. Virology 1989, 168, 236. [DOI] [PubMed] [Google Scholar]; b Kur J.; Hasan N.; Szybalski W. Gene 1992, 111, 1. [DOI] [PubMed] [Google Scholar]
- a Cho B. K.; Zengler K.; Qiu Y.; Park Y. S.; Knight E. M.; Barrett C. L.; Gao Y.; Palsson B. O. Nat. Biotechnol. 2009, 27, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mendoza-Vargas A.; Olvera L.; Olvera M.; Grande R.; Vega-Alvarado L.; Taboada B.; Jimenez-Jacinto V.; Salgado H.; Juarez K.; Contreras-Moreira B.; Huerta A. M.; Collado-Vides J.; Morett E. PLoS One 2009, 4, e7526. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Reppas N. B.; Wade J. T.; Church G. M.; Struhl K. Mol. Cell 2006, 24, 747. [DOI] [PubMed] [Google Scholar]
- a Berg O.; Blomberg C. Biophys. Chem. 1976, 4, 367. [DOI] [PubMed] [Google Scholar]; b Berg O. Biophys. J. 1985, 47, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opalka N.; Brown J.; Lane W. J.; Twist K. A.; Landick R.; Asturias F. J.; Darst S. A. PLoS Biol 2010, 8, e1000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A.; Darst S. A. Cell 2011, 147, 1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Luger K.; Dechassa M. L.; Tremethick D. J. Nat. Rev. Mol. Cell. Biol. 2012, 13, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rando O. J.; Chang H. Y. Annu. Rev. Biochem. 2009, 78, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widom J. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 285. [DOI] [PubMed] [Google Scholar]
- Iyer V. R. Trends Cell Biol 2012, 22, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Drew H. R.; Travers A. A. J. Mol. Biol. 1985, 186, 773. [DOI] [PubMed] [Google Scholar]; b Satchwell S. C.; Drew H. R.; Travers A. A. J. Mol. Biol. 1986, 191, 659. [DOI] [PubMed] [Google Scholar]; c Travers A. A.; Klug A. Philos. Trans. R. Soc. London, B: Biol. Sci. 1987, 317, 537. [DOI] [PubMed] [Google Scholar]; d Segal E.; Fondufe-Mittendorf Y.; Chen L.; Thastrom A.; Field Y.; Moore I. K.; Wang J. P.; Widom J. Nature 2006, 442, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Morozov A. V.; Fortney K.; Gaykalova D. A.; Studitsky V. M.; Widom J.; Siggia E. D. Nucleic Acids Res. 2009, 37, 4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Segal E.; Widom J. Trends Genet. 2009, 25, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Field Y.; Kaplan N.; Fondufe-Mittendorf Y.; Moore I. K.; Sharon E.; Lubling Y.; Widom J.; Segal E. PLoS Comput. Biol. 2008, 4, e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kaplan N.; Moore I.; Fondufe-Mittendorf Y.; Gossett A. J.; Tillo D.; Field Y.; Hughes T. R.; Lieb J. D.; Widom J.; Segal E. Nat. Struct. Mol. Biol. 2010, 17, 918. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kaplan N.; Moore I. K.; Fondufe-Mittendorf Y.; Gossett A. J.; Tillo D.; Field Y.; LeProust E. M.; Hughes T. R.; Lieb J. D.; Widom J.; Segal E. Nature 2009, 458, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hughes A. L.; Jin Y.; Rando O. J.; Struhl K. Mol. Cell 2012, 48, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Struhl K.; Segal E. Nat. Struct. Mol. Biol. 2013, 20, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang Y.; Moqtaderi Z.; Rattner B. P.; Euskirchen G.; Snyder M.; Kadonaga J. T.; Liu X. S.; Struhl K. Nat. Struct. Mol. Biol. 2009, 16, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Radman-Livaja M.; Rando O. J. Dev. Biol. 2010, 339, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Vincent J. A.; Kwong T. J.; Tsukiyama T. Nat. Struct. Mol. Biol. 2008, 15, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zhang Y.; Moqtaderi Z.; Rattner B. P.; Euskirchen G.; Snyder M.; Kadonaga J. T.; Liu X. S.; Struhl K. Nat. Struct. Mol. Biol. 2010, 17, 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Iyer V.; Struhl K. EMBO J. 1995, 14, 2570. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Struhl K. Proc. Natl. Acad. Sci. U.S.A. 1985, 82, 8419. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Segal E.; Widom J. Curr. Opin. Struct. Biol. 2009, 19, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y.; White C. L.; Luger K. J. Mol. Biol. 2006, 361, 617. [DOI] [PubMed] [Google Scholar]
- a Yuan G. C.; Liu Y. J.; Dion M. F.; Slack M. D.; Wu L. F.; Altschuler S. J.; Rando O. J. Science 2005, 309, 626. [DOI] [PubMed] [Google Scholar]; b Lee W.; Tillo D.; Bray N.; Morse R. H.; Davis R. W.; Hughes T. R.; Nislow C. Nat. Genet. 2007, 39, 1235. [DOI] [PubMed] [Google Scholar]
- Lowary P. T.; Widom J. J. Mol. Biol. 1998, 276, 19. [DOI] [PubMed] [Google Scholar]
- a Makde R. D.; England J. R.; Yennawar H. P.; Tan S. Nature 2010, 467, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cloutier T. E.; Widom J. Mol. Cell 2004, 14, 355. [DOI] [PubMed] [Google Scholar]; c Du Q.; Smith C.; Shiffeldrim N.; Vologodskaia M.; Vologodskii A. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracey L. E.; Chen Z. Y.; Maniar J. M.; Valouev A.; Sidow A.; Kay M. A.; Fire A. Z. Epigenetics Chromatin 2010, 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales R.; Zhang L.; Bentley D. Mol. Cell. Biol. 2011, 31, 3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker R. E.; Rogers K. Genetics 2005, 171, 1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuguchi G.; Xiao H.; Wisniewski J.; Smith M. M.; Wu C. Cell 2007, 129, 1153. [DOI] [PubMed] [Google Scholar]
- a Gribnau J.; Diderich K.; Pruzina S.; Calzolari R.; Fraser P. Mol. Cell 2000, 5, 377. [DOI] [PubMed] [Google Scholar]; b Bank A. Blood 2006, 107, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sankaran V. G.; Menne T. F.; Xu J.; Akie T. E.; Lettre G.; Van Handel B.; Mikkola H. K.; Hirschhorn J. N.; Cantor A. B.; Orkin S. H. Science 2008, 322, 1839. [DOI] [PubMed] [Google Scholar]
- a Albert I.; Mavrich T. N.; Tomsho L. P.; Qi J.; Zanton S. J.; Schuster S. C.; Pugh B. F. Nature 2007, 446, 572. [DOI] [PubMed] [Google Scholar]; b Guenther M. G.; Levine S. S.; Boyer L. A.; Jaenisch R.; Young R. A. Cell 2007, 130, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Mavrich T. N.; Jiang C.; Ioshikhes I. P.; Li X.; Venters B. J.; Zanton S. J.; Tomsho L. P.; Qi J.; Glaser R. L.; Schuster S. C.; Gilmour D. S.; Albert I.; Pugh B. F. Nature 2008, 453, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mavrich T. N.; Ioshikhes I. P.; Venters B. J.; Jiang C.; Tomsho L. P.; Qi J.; Schuster S. C.; Albert I.; Pugh B. F. Genome Res. 2008, 18, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones D. E.; Cui K.; Cuddapah S.; Roh T. Y.; Barski A.; Wang Z.; Wei G.; Zhao K. Cell 2008, 132, 887. [DOI] [PMC free article] [PubMed] [Google Scholar]