

Abstract
L-type calcium channels (Cav1) represent one of the three major classes (Cav1–3) of voltage-gated calcium channels. They were identified as the target of clinically used calcium channel blockers (CCBs; so-called calcium antagonists) and were the first class accessible to biochemical characterization. Four of the 10 known α1 subunits (Cav1.1–Cav1.4) form the pore of L-type calcium channels (LTCCs) and contain the high-affinity drug-binding sites for dihydropyridines and other chemical classes of organic CCBs. In essentially all electrically excitable cells one or more of these LTCC isoforms is expressed, and therefore it is not surprising that many body functions including muscle, brain, endocrine, and sensory function depend on proper LTCC activity. Gene knockouts and inherited human diseases have allowed detailed insight into the physiological and pathophysiological role of these channels. Genome-wide association studies and analysis of human genomes are currently providing even more hints that even small changes of channel expression or activity may be associated with disease, such as psychiatric disease or cardiac arrhythmias. Therefore, it is important to understand the structure–function relationship of LTCC isoforms, their differential contribution to physiological function, as well as their fine-tuning by modulatory cellular processes.
INTRODUCTION
In experiments almost 50 years ago the German physiologist Albrecht Fleckenstein discovered that organic molecules, such as verapamil, closely mimicked the cardiodepressant actions of β-receptor antagonists but that their action could not be explained by binding to β-adrenergic receptors. They also did not alter sodium-dependent action potential (AP) parameters but their effects could be mimicked by withdrawal of extracellular Ca2+ and weakened (antagonized) by elevated extracellular Ca2+. Instead, they specifically inhibited Ca2+ ion influx into cardiomyocytes. Fleckenstein thus first coined the pharmacodynamic principle of ‘Ca2+ antagonism’. Later, it became evident that their pharmacological actions are fully explained by block of so-called voltage-gated Ca2+ channels (VGCCs) in the nanomolar concentration range. VGCCs open in response to membrane depolarizations and allow Ca2+ ions to enter cells along its 10,000-fold chemical gradient. This finding triggered a successful search for other Ca2+ channel blockers (CCBs) and their cardiodepressant and vasodilating properties are clinically used since then to treat hypertension, myocardial ischemia, and arrhythmias. This drug discovery process also led to the synthesis of radioactive and fluorescent CCBs. These pharmacological tools allowed purification of the first VGCC from skeletal muscle, which paved the way for the biochemical isolation and molecular cloning of its subunits. It enabled the assignment of pore-forming subunit genes to VGCC families previously classified on the basis of different pharmacological and biophysical properties. Four VGCC genes (see below) were found to mediate currents highly sensitive to CCBs, also termed L-type channels (LTCCs).
Although the pharmacological actions of CCBs at therapeutic doses are limited to the cardiovascular system, CCBs were successfully used as highly specific probes to unequivocally demonstrate the existence of LTCC proteins and currents in many other tissues, including the brain, sensory cells, pancreatic β-cells, adrenal chromaffin cells, and even neural progenitor cells. In this review, we summarize our current knowledge about the physiological and pathophysiological role of LTCCs in heart and brain. We will outline the differential contribution of different LTCC isoforms for organ function and the resulting implications for disease and novel therapies.
LTCC STRUCTURE AND REGULATION
The voltage-sensitive pores of all VGCC types are formed by so-called α1 subunits. Ten α1-subunit isoforms encoded by separate genes are the central building blocks of the different channel types1 (Table 1). They associate with other subunits to form hetero-oligomeric complexes. β Subunits are tightly associated at the cytoplasmic face of α1 (through the I–II linker), whereas α2δ subunits are GPI-anchored to the plasma membrane and interact with extracellular domains of α1 (Figure 1). In contrast to Cav1 and Cav2 channels, Cav3 channels appear not to form stable complexes with auxiliary subunits. γ Subunits also exist, but have so far only been found as part of muscle (Cav1.1 and Cav1.2) Ca2+ channels. More comprehensive reviews of LTCC subunits and topology have been published.2,3
TABLE 1.
Voltage-Gated Ca2+ Channel Types and Their Pore-Forming Subunits1
| Type | α1 Subunit (Old Nomenclature, Gene) | Predominant Tissue Expression | Pharmacology | |
|---|---|---|---|---|
| Cav1 | L | Cav1.1 (α1S; CACNA1S) | Skeletal muscle | Dihydropyridines (isradipine and nifedipine), phenylalkylamines (verapamil), and benzothiazepines (diltiazem) |
| Cav1.2 (α1C; CACNA1C) | Heart/smooth muscle, neurons (somatodendritic), and endocrine cells | |||
| Cav1.3 (α1D, CACNA1D) | Heart, neurons (somatodendritic), endocrine cells, and sensory cells | |||
| Cav1.4 (α1F, CACNA1F) | Retina and immune cells | |||
| Cav2 | P/Q | Cav2.1 (α1A, CACNA1A) | Neurons and endocrine cells | ω-Agatoxin IVA and ω-conotoxin MVIIC |
| N | Cav2.2 (α1B, CACNA1B) | Neurons and endocrine cells | ω-Conotoxin GVIA | |
| R | Cav2.3 (α1E, CACNA1E), Cav? | Cardiac/smooth muscle, endocrine cells, and neurons | SNX-482 | |
| Cav3 | T | Cav3.1 (α1G, CACNA1G) | Neurons and cardiac muscle | TTA-A2 and Z944 |
| Cav3.2 (α1H, CACNA1H) | Cardiac/vascular smooth muscle, kidney, and liver | |||
| Cav3.3 (α1I, CACNA1I) | Neurons | |||
FIGURE 1.
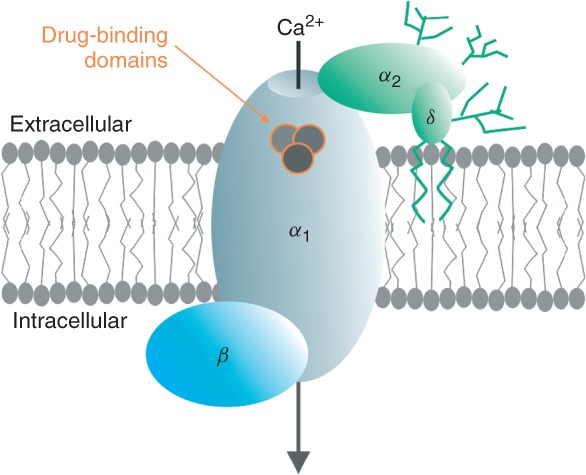
Voltage-gated Ca2+ channel (VGCC) complex. γ Subunits associate only with VGCC complexes in skeletal muscle and heart; drug-binding domains for Ca2+-channel blockers are located only on the α1 subunit; and their binding domains have been mapped.
Most of the pharmacological and gating properties are determined by α1 (for review see Refs 1 and 4) but auxiliary β and α2δ subunits support channel targeting to the plasma membrane and fine-tune channel function.1–3 As outlined below, calmodulin (CaM) can also be considered an important regulatory subunit of Cav1 and Cav2 channel complexes. In addition, as shown for Cav2 channels in brain,5 VGCCs engage in the formation of large protein signaling networks.
Structural Determinants of LTCC Function
Figure 2 highlights key functional domains within the proposed transmembrane topology of α1 subunits: the voltage sensor module comprised by transmembrane segments S1–S4 in each homologous repeat (I–IV) and the pore-forming region comprised by S5 and S6 segments together with their connecting linker, which contains helical regions contributing to the formation of the selectivity filter. From crystallographic data of related K+ and Na+ channels the S1–S3 helices of the voltage sensor appear to form a scaffold through which the positive charges of the S4 helices move outward upon depolarization. Their movement is transmitted through S4–S5 linkers to the cytoplasmic ends of the S5 and S6 helices. This opens the activation gate formed by the S6 helices on the inner side of the channel (not illustrated). Movement of the positively charged S4 segments is visible in patch-clamp recordings as nonlinear capacitive-like currents at the beginning (ON-gating charge) and end (OFF-gating charge) of a depolarizing pulse. As the magnitude of this charge movement is proportional to the number of channels in the plasma membrane, it can be used to quantify the surface expression of active channels in excitable cells.6 This is particularly useful to determine if mutations in channel subunits can alter the surface expression of the complex (see below). For more detailed information on voltage-dependent activation and inactivation gating mechanisms of VGCCs and related channels, see Refs 7–9.
FIGURE 2.

Pore-forming α1 subunits. Upper panel: Important functional domains discussed in this review are indicated. CaM, Ca2+-calmodulin (blue circles indicate EF-hands); IQ, PreIQ, and EF, CaM interaction domains in C-terminus; NSCaTE, CaM interaction domains in N-terminus (for N-lobe of CaM, Cav1.3 only); PDZ, PDZ-binding domain; DCRD and PCRD form the C-terminal modulatory domain (CTM); AKAP, A-kinase-anchoring protein interaction site; cAMP-PK and CaMKII, phosphorylation sites for kinases (Cav1.2: red dots; Cav1.3: blue dots); , proteolytic cleavage site in Cav1.1 and Cav1.2 α1; (sinoatrial node dysfunction and deafness) SANDD, in-frame glycine insertion in SANDD patients. Lower panel: Cartoon of voltage sensing and pore domains of Cav α1 subunits; only two domains (half of the channel) are shown for clarity. Movements of the positively charged S4 helices (which serve as voltage sensors) in response to membrane potential changes are transmitted to the pore domain through the cytoplasmic S4–S5 linkers. S4 movement within the membrane is guided by interactions with negative charges provided by the S1–S3 helices.
, proteolytic cleavage site in Cav1.1 and Cav1.2 α1; (sinoatrial node dysfunction and deafness) SANDD, in-frame glycine insertion in SANDD patients. Lower panel: Cartoon of voltage sensing and pore domains of Cav α1 subunits; only two domains (half of the channel) are shown for clarity. Movements of the positively charged S4 helices (which serve as voltage sensors) in response to membrane potential changes are transmitted to the pore domain through the cytoplasmic S4–S5 linkers. S4 movement within the membrane is guided by interactions with negative charges provided by the S1–S3 helices.
The long cytoplasmic C-terminal region of LTCC α1 subunits serves as an important modulatory domain and is a target of numerous protein–protein interactions (see Tables 2 and 3). In particular, the proximal C-terminus (IQ motif plus C-terminal portion linking it to IVS6) binds apo-CaM and contains effector domains for Ca2+–CaM modulation.36 The efficiency of this modulatory mechanism is further regulated by competing CaM-like Ca2+-binding proteins (CaBPs, Box 1, see also below) and, in the case of Cav1.3 channels, by alternative splicing. This allows generation of LTCC complexes with a wide range of gating properties to adjust the dynamics of Ca2+ entry to different physiological needs.
TABLE 2.
Protein Interactions of Cav1.2 and Cav1.3 in the Heart
| Protein | Interaction Partner1 | Tissue | Function | Notes/References |
|---|---|---|---|---|
| Actinin2 | Cav1.2 and Cav1.3 | Cardiomyocytes | Crosslinks SK2 K+-channels to both LTCCs | 10 |
| Ahnak | β2 Subunit | t-Tubules | May be involved in PKA-mediated upregulation of cardiac L-type currents | 11 |
| AKAP79/150 (AKAP5) | Cav1.2 | Cardiac myocytes | AKAPs are required to recruit PKA, leading to phosphorylation and current augmentation | Required to increase Ca2+ transients but not whole cell ICa by β-receptor activation12 |
| BIN-1 | Cav1.2 | t-Tubules | Targets Cav1.2 to t-tubules | 13 |
| CaN | Cav1.2 | Cardiac myocytes | Increases current density | Involved in development of cardiac hypertrophy14 |
| CamKII | Cav1.2 and auxiliary β subunit | Cardiac myocytes | Promotes CDF and VDF | As CamKII interacts with auxiliary β subunits of VGCC, interaction might also be present in neurons15–17 |
| Caveolin-3 | Cav1.2 | Ventricular myocytes | Targets channel to caveolae | 18 |
| KChIP2 | Cav1.2 | Cardiac myocytes | Enhances current density and current amplitude | 19 |
| Phospholemman/FXYD1 | Cav1.2 | t-Tubules and sarcolemma | Modulates gating kinetics: slows down activation and deactivation and voltage-dependent inactivation, large number of channels are inactivated owing to interaction | 20 |
| RGK-GTPases | β Subunits | Cardiac myocytes | Inhibits channel open probability and prevents PKA-mediated upregulation | 21 |
| Sorcin | Cav1.2 | Cardiac ventricular tissue | Enhances peak current magnitude and increases CDI | 22 |
LTCCs, L-type calcium channels; PKA, protein kinase A; AKAP, A-kinase-anchoring protein; CDF, calcium-dependent facilitation; CDI, calcium dependent inactivation; VDF, voltage dependent facilitation.
α1 Subunit if not further specified.
TABLE 3.
Protein Interactions of Cav1.2 and Cav1.3 in the Brain
| Protein | Interaction Partner1 | Tissue | Function | Notes/References |
|---|---|---|---|---|
| AKAP-MAP2B | Cav1.2 and Cav1.3 | Distal dendrites | Targets PKA, which is required for efficient phosphorylation and physiological regulation | 23 |
| AKAP-15 | Cav1.2 and Cav1.3 | Cell soma and proximal dendrites | Enhances channel activity by recruiting PKA | 23 |
| AKAP-79/150 | Cav1.2 | Postsynaptic densities of dendritic spines | Binds PKA and CaN, which both control channel activity | CaN enhances-calcium dependent gene regulation through NFAT and reduces peak calcium current owing to its binding to AKAP23,24 |
| CaBPs | Cav1.2 | Somatodendritic domains | Inhibit CDI and cause CDF | Interaction of CaBPs has also been shown for Cav1.3 in recombinant systems and cochlear inner hair cells25,26 |
| Densin | Cav1.3 | Dendritic spines | Recruits CaMKII, which enhances activity by inducing CDF | 27 |
| Erbin | Cav1.3 | Cell soma and proximal dendrites | Increases activity by enhancing VDF | Effect dependent on auxiliary β-subunit isoform28 |
| NIL-16 | Cav1.2 | Cerebellum and hippocampus | Scaffolding protein, links the channel to cytoskeletal and signaling proteins | May be involved in pCREB signaling29 |
| Rem2 | Auxiliary β subunit | Neuronal cells | Inhibits channel activity | 30 |
| RIM | Cav1.2 and auxiliary β-subunit | Presynaptic active zone | Involved in targeting and docking of secretory vesicles near calcium channels and slows down current inactivation | Owing to interaction with auxiliary β subunit, it might interact with Cav1.2 as well as with Cav1.3 complexes31,32 |
| Shank | Cav1.3 | Postsynaptic areas of hippocampal neurons | Mediates synaptic clustering of Cav1.3, and interaction plays an important role in pCREB signaling | 33 |
| STIM1 | Cav1.2 | Endoplasmatic reticulum | Inhibits Cav1.2 activity by physical interaction with the channel and by causing its internalization | 34,35 |
AKAP, A-kinase-anchoring protein; CaBPs, calcium-binding proteins; CaMKII, CaM-dependent kinase II; CaN, Ca2+/calmodulin-activated-phosphatase calcineurin; CDF, calcium-dependent facilitation; CDI, calcium-dependent inactivation; CIPP, channel-interacting PDZ domain protein; NFAT, nuclear factor of activated T-cells; NIL-16, neuronal interleukin-16; VGCCs, voltage-gated calcium channels; pCREB, phosphorylated cAMP response element-binding protein; PKA, protein kinase A; RIM, rab3-interacting molecule; STIM1, stromal interaction molecule 1.
α1 Subunit if not further specified.
BOX 1 CaM-LIKE CaBPs
CaBPs comprise a large family of Ca2+-sensing proteins that closely resemble CaM. They contain four potential Ca2+-binding EF-hand motifs like CaM, but one or two of the EF-hands are inactive in binding Ca2+. Moreover, these CaBPs are myristoylated allowing membrane anchoring. CaBPs, such as CaBP1, CaBP2, VILIP-2, and NCS-1, interact with different Ca2+ channel types and can modify the regulatory function of CaM. The crystal structure of CaBP1 in complex with the Cav1.2 α1-subunit C-terminus has been reported.37 The C-lobe of CaBP1 anchors it to the IQ domain, overlapping with the binding site for CaM. However, the N-lobe modulates channel function differently to CaM and by displacing CaM from the IQ domain prevents CDI.37
Properties of LTCC α1 Subunits
LTCC α1 subunits are all sensitive to the main chemical classes of CCBs [dihydropyridines (DHPs), phenylalkylamines, and benzothiazepines] but differ with respect to tissue expression and gating characteristics38 (Table 1). Cav1.1 channels are exclusively found in skeletal muscle where they trigger depolarization-induced Ca2+ release from ryanodine receptors (RYRs) of the sarcoplasmic reticulum (SR).39 Cav1.4 channel expression is also very restricted with its major site of expression in the retina.38 Therefore, Cav1.1 and Cav1.4 channels are not discussed here. In contrast, Cav1.2 and Cav1.3 show a highly overlapping expression pattern in many tissues and are even present in the same cells. However, their gating properties and protein interactions differ, which allows them to serve different physiological functions. As outlined below, a major distinguishing feature is the about 9–15 mV more negative activation range of Cav1.3 channels.40–42 As discussed below, they open at threshold potentials in sinoatrial node (SAN) cells and neurons and thereby contribute to pacemaking and stabilization of plateau potentials. They also differ with respect to their modulation by C-terminal alternative splicing.
Modulation by CaM, C-Terminal Domains, and Alternative Splicing
CaM can be considered an additional channel subunit because it is preassociated with the channel's C-terminus even at low intracellular Ca2+ concentrations.36 Upon Ca2+ binding CaM undergoes a conformational change that promotes inactivation (so-called Ca2+-dependent inactivation, CDI) by interaction with additional C-terminal (and in case of Cav1.3 also N-terminal; Figure 2) effector sites (for review see Ref 43). CDI occurs in addition to voltage-dependent inactivation (VDI) during depolarization. Both processes appear to involve conformational rearrangements of the intracellular channel mouth.9 CDI is an important autoinhibitory mechanism preventing excessive Ca2+ influx. The strength of CDI itself can be adjusted by regulating the strength of CaM binding. This is achieved by displacement of CaM from its C-terminal interaction sites either by competing CaM-like CaBPs that do not support CDI44 or by a modulatory domain within the C-terminus itself.
As illustrated in Figure 2 the C-terminus of Cav1.3 channels contains a modulatory domain that interferes with CaM modulation. This C-terminal modulator (CTM) consists of two putative α-helices: one on the C-terminal end (termed DCRD) and one immediately after the main CaM interaction site (PCRD), the so-called IQ motif.45,46 The positively charged PCRD and negatively charged DCRD bind to each other as shown by FRET studies46 and thereby form a structure that can compete with CaM binding (Figure 2). This causes a decrease in open probability and reduced CDI. In addition, this C-terminal modulatory domain (CTM) shifts the voltage dependence of channel activation to more positive voltages.45,46 Notice that these effects are very pronounced in Cav1.3 and Cav1.4 but much weaker in Cav1.2 channels.45–47
Interestingly, the CTM itself is also subject to modulation. In Cav1.2 there is evidence for partial proteolytic processing of the C-terminus with the site of cleavage located between PCRD and DCRD47,12 (Figure 2). The DCRD-containing Cav1.2 fragment remains associated to the channel through binding to PCRD and thereby still inhibits channel activity. Protein kinase A (PKA) phosphorylation of PCRD can relief this inhibition (see below), thereby increasing Ca2+ influx. In Cav1.3 the possibility of proteolytical processing has not yet been tested rigorously. Instead, several short splice variants have been identified, which lack DCRD and therefore prevent the C-terminus to inhibit CaM modulation.41 ‘Short’ channels exhibit much more pronounced CDI, and higher open probability,41 indicating higher CaM activity at the channel. Moreover, short channels also activate at more negative voltages (Figure 3). The latter effect seems to be independent of CaM.46 A similar modulation is also found in Cav1.4.45 Its C-terminus essentially prevents its own CDI and thereby stabilizes the long-lasting currents required for photoreceptor signaling.45
FIGURE 3.
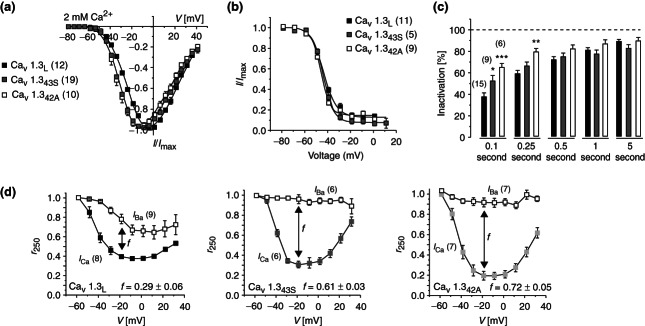
Cav1.3 splice variants have different biophysical properties. The C-terminal modulator (CTM) controls Cav1.3 gating leading to altered biophysical properties in naturally occurring splice variants lacking the CTM. (a) Current activation properties shown in representative normalized I–V curves recorded in tsA-201 cells expressing Cav1.3L (black), Cav1.343S (gray), and Cav1.342A (white) together with α2δ1 and β3 subunits; 2 mM Ca2+ was used as charge carrier. Half maximal activation voltage was significantly shifted by about 9 mV to more negative voltages and activation slope factor was significantly smaller. (b) Voltage dependence of inactivation elicited after 5-second conditioning prepulses using 20-millisecond test pulses to Vmax (no significant differences). (c) Percent ICa inactivation during 0.1-, 0.25-, 0.5-, 1-, and 5-second test pulses to Vmax revealing significantly faster inactivation time course of short variants. (d) Voltage dependence of CDI: r250 corresponds to the fraction of ICa or IBa remaining after 250 milliseconds; f is the difference in r250 of IBa and ICa at −19 mV. Number of experiments is given in parentheses. Error bars reflect SEM, *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA followed by Bonferroni post-test. (Reprinted with permission from Ref 41. Copyright 2011 American Society for Biochemistry and Molecular Biology)
Modulation of CTM activity is only one example of how alternative splicing can affect LTCC function. Extensive alternative splicing has been described for Cav1.2 α1, which has two important consequences: First, splice variants with preferential expression in arterial smooth muscle activate and inactivate at lower membrane potentials than ‘cardiac muscle’ splice variants.48 Because of the state-dependent block of LTCCs arterial smooth muscle variants are more sensitive to DHP CCBs. This contributes to their strong vasodilating but weak cardiodepressive properties.48,49 Second, alternative splicing can change in disease states, such as in vascular smooth muscle from patients with atherosclerosis and in hypertrophied rat and human failing hearts and in rat models of myocardial infarction (for an extensive review see Ref 50).
Modulation by Intracellular Signaling Pathways
Activation of GPCRs modulates VGCC activity either by direct channel interaction with activated G-proteins (Gα or Gβγ) or by G-protein-activated intracellular signaling pathways. Direct, membrane-delimited modulation is mainly observed for Cav2 VGCCs, such as Cav2.2 (N-type) and Cav2.1 (P/Q-type). The molecular interactions underlying this regulation are known and have been reviewed recently.51,52 The direct modulation of Cav2 channels by Gβγ is voltage dependent and relieved by strong depolarization of the channel.
Several important intracellular signaling pathways modulating Cav1.2 and Cav1.3 function are discussed below.
cAMP-Dependent Protein Kinase (PKA)
Cav1.2 LTCCs: During the ‘fight-or-flight’ response PKA-mediated phosphorylation of LTCC currents in cardiomyocytes (which are exclusively Cav1.2 mediated53) contributes to the increase of heart rate and contractile force. Although this could be unequivocally demonstrated 20 years ago in cardiomyocytes,54 the most likely molecular mechanism responsible for channel activation has been elucidated only recently.
Over the years many attempts to reconstitute the whole modulatory signaling cascade in recombinant systems failed because it requires a complicated interplay between intramolecular and intermolecular protein–protein interactions, posttranslational modification, and phosphorylation within Cav1.2 α1 subunits. As illustrated in Figure 2, posttranslational proteolytic processing occurs in a conserved region between DCRD and the PCRD putative α-helical C-terminal domains47,55 giving rise to a long (uncleaved) and short (cleaved) form. In heart and brain the cleaved fragment can either dissociate from the truncated α1 subunit and serve as a transcriptional modulator after entering the nucleus56,57 or remain noncovalently bound through the PCRD–DCRD interaction.12 When coexpressed in tsA-201 cells as a separate protein, binding of the C-terminal fragment (CT) strongly reduces ionic current through the short channel. Because the number of active channels does not change (as quantified by measuring ON-gating charge movement47) a lower probability of channel opening during depolarizations must explain reduced inward current. PKA exerts its activating effect by disinhibiting this inhibitory action of the noncovalently bound CT. This requires specific targeting of the kinase to the CT through an A-kinase-anchoring protein (AKAP, Figure 2), which directly associates with a leucine zipper-like motif within the CT. Once this signaling complex is formed, PKA activation phosphorylates Cav1.2 α1 residues serine-1700 and threonine-1704, which are located within the PCRD helix facing the interface with DCRD (Figure 2). The importance of serine-1700 and threonin-1704 for normal β-adrenergic phosphorylation and modulation of Cav1.2 channels in the heart has recently been confirmed in vivo in mutant mice.58 Although AKAP15 (AKAP7α) has been successfully used to reconstitute this physiological regulation, the identity of the AKAP responsible for upregulation of ICa in the intact heart is currently unknown [and is neither AKAP15 nor AKAP79/150 (AKAP5)].59,60
In heart and brain this phosphorylation machinery is complemented by C-terminally attached phosphatases to ensure rapid dynamics for regulation by phosphorylation/dephosphorylation. These include protein phosphatase 2A and 2B/calcineurin (CaN). Both are attracted to the channel through interaction sites at the distal C-terminus, although CaN can also interact through channel-bound AKAP79/150.61,62,24 When channel-bound, these phosphatases antagonize PKA-induced channel upregulation.61,62,24 Notably, CaN is activated by Ca2+–CaM which is also present in the channel's nanodomain (see above), thus providing a link between channel activity and phosphatase activity. A recent study indeed provided evidence that during increased depolarization frequency (i.e., enhanced Ca2+ influx) CaM associates with CaN and suppresses PKA enhancement of the channel in neurons.62,24 Moreover, this study also suggests that by opposing PKA phosphorylation CaN also accelerates inactivation of ICa during depolarizing steps. Because it acts in a Ca2+–CaM-dependent manner, CaN-mediated dephosphorylation provides another molecular explanation for the CDI of Cav1.2 channels.62 This hypothesis requires slowing of CDI by PKA phosphorylation, a phenomenon described for L-type currents in some neurons62,24 but not for Cav1.2-mediated currents in cardiomyocytes.63
The phosphatases ensure reversibility of PKA phosphorylation but do not terminate the cAMP signal. Therefore, phosphodiesterase activity is also closely associated with the Cav1.2 signaling complex in the heart.60,64 This limits β-adrenergic receptor-induced increases in ICa and limits Ca2+ transients, contraction and spontaneous Ca2+ release as shown in Pde4B-deficient mice.64
Serine-1928 (Figure 2) is another Cav1.2 C-terminal residue that gets reproducibly phosphorylated in heart and brain tissue and reports PKA activation in close proximity to Cav1.2. However, in contrast to some earlier findings in recombinant systems and neurons24 (for review see Ref 65), its phosphorylation is not involved in PKA-mediated enhancement of cardiac Cav1.2 channel currents.63,66,67 This was convincingly shown in cardiomyocytes from Ser-1928Ala knockin mice.67 In contrast to previous findings with HEK293 cell-expressed channels, phosphorylation of cardiac β2 subunits is also not required for adrenergic regulation of Cav1.2 channels.68
Cav1.3 LTCCs: Native Cav1.3 channels also form signaling complexes with AKAPs.23 They are also activated by PKA as demonstrated in adrenal chromaffin cells69 and Cav1.3-mediated current components in the SAN.42 In adrenal chromaffin cells both Cav1.2 and Cav1.3 current components are strongly dependent on PKA activity, suggesting an at least threefold stimulation from basal channel activity.69 The molecular details for this regulation of Cav1.3 are less clear than for Cav1.2. Site-directed mutations showed that phosphorylation of serines 1743,70,71 1817,70 and 1964,71 all located downstream the PCRD domain (Figure 2), mediates 8-Br-cAMP-70 or PKA-induced71 increase of Cav1.3 currents when expressed in HEK293 cells. Current stimulation was weak (20–40%) and the relative contribution of the individual serines was dependent on the coexpressed β-subunit isoform (β2a or β3). Effects of coexpressed AKAPs were not recorded.
CaM-Dependent Protein Kinase II (CamKII)
The CaM-dependent fine-tuning of LTCC activity does not only cause autoinhibition through CDI (see above) but can also enhance ICa. Ca2+-dependent facilitation is observed as an increase in ICa during repetitive stimuli, an effect partially masked by CDI. Consequently, it is very small in Cav1.2 channels but its effect in the heart can be largely amplified by Ca2+-induced SR Ca2+ release.72 CDF increases contractile force at faster heart rates and thus contributes to the positive inotropic response during exercise.73 In the brain, CDF may strengthen Ca2+ signals supporting the privileged role of LTCCs in excitation–transcription coupling (ETC).74 There is biochemical evidence that after CaM activation, CaMKII binds to the Cav1.2 α1 subunit's C-terminus where it phosphorylates two sites (Figure 2) and one on the β2 subunit.75 Once associated with α1, CaMKII no longer requires Ca2+–CaM for binding but only for reactivation after dephosphorylation.75 Therefore, both binding to the channel and enzymatic activity are required for this mechanism.75 Phosphorylation increases the channel's open probability and accelerates recovery from inactivation15 with both alterations contributing to the observed CDF at higher stimulation frequencies. In good agreement with these data largely derived from heterologous expression studies, this CaMKII-mediated regulation is reduced in mice in which the two phosphorylation sites in α1 (Ser-1512 and Ser-1570) were replaced by alanines.15
CaMKII also seems to contribute to the modulation of Cav1.3 channels. IGF-1 was found to enhance Cav1.3 activity in SH-SY5Y neuroblastoma cells and cortical neurons.76 This involves IGF-1-induced intracellular Ca2+ release with activation of CaMKII and phosphorylation of a serine residue within the C-terminal EF-hand motif of α1 (Figure 2). CaMKII also confers Ca2+-dependent facilitation to Cav1.3 channels. This requires CaMKII association with the postsynaptic scaffold protein densin as well as densin binding to the C-terminal PDZ-binding sequence of the Cav1.3 α1-subunit.27
Membrane Phospholipids
PLC-mediated breakdown of polyphosphoinositides also modulates LTCCs (as well as other VGCCs77). Two molecular mechanisms have been implied. One is stabilization of channels by phosphatidylinositol 4,5-bisphosphate (PIP2), suggesting that a cytoplasmic domain interacts with the negatively charged PIP2 headgroups. This well explains the reduction of inward current by PIP2 depletion. N-type channels (Cav2.2) are the most sensitive (50–60% inhibition), whereas L-type (Cav1.3 and Cav1.2) and P/Q-type (Cav2.1) currents are reduced by 20–30%. Another finding was that extracellular application of arachidonic acid (AA) mimicked inhibition seen by M1-receptor activation in superior cervical ganglion neurons.78 Therefore, it appeared possible that AA is released after PLC activation and activation of Ca2+-sensitive phospholipase A2. The latter liberates AA from the PIP2 backbone. Because experimental data appeared robust in supporting both the ‘PIP2’ and the ‘AA’ hypothesis a unifying hypothesis became likely.79 The crucial hints for this came from two observations: first the crystal structure of Kir2.2 K+ channels in which PIP2 tethers a cytoplasmic domain, which binds the inositol phosphate ring, to a transmembrane domain, which binds the fatty acid side chains.80 Second, the Ca2+ channel β2a subunit is palmitoylated at its N-terminus and (in contrast to nonpalmitoylated β subunits) was found to prevent the inhibitory effect of both PIP2 hydrolysis79 and AA, as if it could substitute for PIP2 (phospholipid mimic81). It is therefore likely that PIP2 and the β2a subunit stabilize active channel conformations by occupying a fatty acid-binding site on the channel and at the same time tether cytoplasmic domains to the membrane by either high-affinity interaction with the cytoplasmic I–II linker (β2a subunit) or through inositol phosphates attaching to cytoplasmic regions (PIP2). Instead, occupation of the fatty acid-binding site by AA alone (without ‘cross-linking’ function) would result in a less active form of the channel.
Protein Interactions with LTCCs
A large number of protein–protein interactions have been described for LTCCs in brain and heart. They serve as scaffold proteins, stabilize channel gating, recruit kinases (such as PKA and CaMKII, see above) to the channel, or target the channel to defined subcellular compartments. A summary of confirmed protein interaction partners is provided in Tables 2 and 3.
Regulation of LTCC Activity by Channel Trafficking
Availability of LTCCs on the cell surface is also an important determinant for their activity and therefore also subject to modulation by several intracellular processes. A central role for channel trafficking is known for β subunits. Impairment of their expression levels in native cells or heterologous expression systems reduces channel activity in the plasma membrane (for review see Ref 82). Heterologous expression in mammalian cells (such as HEK-293) revealed strong effects of β-subunit coexpression on current densities and kinetics of currents through Cav1.2,82 Cav1.3,40 and Cav1.483 LTCC α1 subunits. This is accomplished by a β-subunit-induced increase of the channels' open probability as well as enhanced trafficking to the plasma membrane.82,84,85 For Cav1.2 several molecular mechanisms have been proposed to explain this finding. One possibility is β-subunit binding to the α1 I–II loop causing a C-terminus-dependent conformational change of intracellular α1-subunit domains, promoting endoplasmic reticulum export through an acidic motif within the I–II loop.86 There is also increasing evidence that β subunits antagonize the ubiquitinylation of Cav1.2 α1 in HEK293 cells through an RFP2 (an E3 ubiquitin ligase)-dependent pathway and thus protect them from proteasomal degradation (for review see Ref 87). Because tonic ubiquitinylation can also be shown in rat brain88 it is likely that proper folding in the presence of β subunit also reduces ubiquitinylation and degradation of Cav1.2 in native cells. Regulation of channel expression by ubiquitinylation was also observed for Cav2 channels.87
As outlined above, AKAPs are required to anchor PKA to LTCCs. However, a second, phosphorylation-independent role for LTCC modulation has been postulated for AKAP79 by showing that it interacts with a proline-rich domain in the Cav1.2 α1 II–III cytoplasmic linker and thereby increases calcium current in oocytes or HEK-293 cells.89,90 Biochemical evidence suggests that AKAP79 binds to the channels' C-terminal tail and thereby competes for C-tail binding to the II–III loop. Activation is explained by relief of an autoinhibitory action of the C-terminus upon its binding to the II–III loop. The observed current stimulation by AKAP79 coexpression likely occurs through enhanced trafficking of channels to the plasma membrane. However, existing data do not rule out the possibility of direct channel activation, such as by inducing increased open probability by AKAP binding.
Once stably integrated in the membrane, β subunits may cause much smaller effects on LTCC activity than expected from heterologous expression systems.82 Inducing almost complete knockdown of β2-subunit protein in cardiomyocytes91 resulted only in minor changes of current density and current kinetics. It is important to note that most biochemical work concerning membrane trafficking has been performed in HEK-293 cells that are lacking most of the specific protein interaction partners for LTCCs present in their native environment in electrically excitable cells and are overexpressing the channel proteins after transfection. Therefore, more work is required to confirm the physiological relevance of biochemical mechanisms affecting channel trafficking in HEK-293 cells for the regulation of LTCC activity in native cells.
LTCCs IN THE HEART
LTCCs are expressed in all regions of the heart including pacemaker cells and the conduction system. β2 Subunits appear to be the predominant modulatory β-subunit isoform in cardiomyocytes.91,92 Several splice variants of this subunit exist. One of them, β2a, is palmitoylated and therefore not only tethered to the I–II linker of α1 subunits but also simultaneously anchored to the lipid bilayer as described above. In addition to altered regulation by lipids, this also stabilizes a prominent slowing of the inactivation kinetics. It is often coexpressed in heterologous expression systems with cardiac splice variants of Cav1.2. However, the β2a splice variant seems to be abundant in the brain but not in cardiac tissue93 where the not palmitoylated β2b seems to be the dominant cardiac β isoform.94
In cardiomyocytes Cav1.2 channels are found in signaling complexes together with β-adrenergic receptors and other molecules involved in cAMP and PKA signaling. Colocalization with caveolin-3 indicates their presence in caveolae and other caveolin-3-containing membrane compartments in the dyadic junctions (see Ref 95 for review).
LTCC Function in the SAN and AVN
Both Cav1.2 and Cav1.3 LTCCs are present in the SAN. Cav1.3−/− mice first allowed to demonstrate the importance of this sarcolemmal ion channel for SAN pacemaking.42,96 Resting heart rate of Cav1.3−/− mice was reduced and arrhythmic as was spontaneous beating frequency of isolated atria. Isolated SAN pacemaker cells displayed a longer and highly variable AP cycle length, slower diastolic depolarization, and prolonged APs. Maximum diastolic potential and AP threshold were unchanged.42 A very similar SAN dysfunction was found in human individuals carrying a loss of function mutation in the Cav1.3 (CACNA1D) gene (SANDD, Sinoatrial Node Dysfunction And Deafness, OMIM: 614896).6 Therefore, Cav1.3 channels are required for regular pacemaking in mouse and human and Cav1.2 cannot substitute for Cav1.3 in these cells.
Current models of SAN function are still controversial.97 However, numerous genetic models and human mutations clearly indicate that SAN pacemaking does not rely on a single pacemaker channel. Instead, there seem to be two separate but closely communicating pacemaker mechanisms (also termed ‘clocks’). A ‘membrane clock’ consists of a sarcolemmal ensemble of electrogenic molecules as its major components, including If (hyperpolarization-activated cyclic nucleotide-gated channels HCN4 and HCN2), LTCCs (Cav1.2 and Cav1.3), T-type VGCCs (Cav3.1), delayed rectifier K+ cannels and the Na/Ca exchanger (NCX). An intracellular ‘Ca2+ clock’ is characterized by rhythmic sarcoplasmic Ca2+ oscillations supported by SR Ca2+ release through RYRs and SR Ca2+ uptake through SERCA-2. Robust pacemaking results from a coupled action of the two clocks. During maximum diastolic depolarization after the SAN AP the K+ conductance decreases. Inward depolarizing currents, such as If channels long believed to be the single major pacemaker current97 and negatively activating ICa components, now induce spontaneous diastolic depolarization. At the same time local subsarcolemmal Ca2+ release from the SR occurs and, together with depolarization-induced Ca2+ entry, generates a Ca2+ signal that activates NCX. This results in NCX-mediated Na+ influx that contributes to late diastolic depolarization and accelerates reaching the SAN AP threshold. This coupled clock model agrees with the observation that single knockout or selective pharmacological inhibition of any of these clock components (Cav1.3, HCN2, HCN4, RYR, and NCX) does not completely prevent pacemaking but either reduces heart rate, induces arrhythmic SAN action,96,98,99 or causes SAN pauses.100
How do Cav1.3 and Cav1.2 fit into this complex scenario? Differences in their biophysical properties as well as subcellular localization provide us with some clues. Their relative contribution to ICa was determined in Cav1.3 knockout mice as well as in SAN cells from mice expressing DHP-insensitive Cav1.2 channels.101 In both models Cav1.3 carries more than 50% of ICa. As predicted from its biophysical properties in heterologous systems Cav1.3 is responsible for the ICa component activating at negative voltages.41,42,101,102 Both channels colocalize with sarcolemmal RYRs but Cav1.3 also strongly colocalizes with sarcomeric RYRs101 throughout the SAN cell. This may be relevant for the functional role of RYR-mediated Ca2+ release in pacemaking during the late phase of the diastolic depolarization as described above. Close apposition of Cav1.3 with RYRs may facilitate SR Ca2+ release because ICa stimulates RYR open probability (Ca2+-induced Ca2+ release). In contrast, Cav1.2 may contribute little to this close coupling because of its more positive activation threshold and less pronounced colocalization with sarcomeric RYRs. However, Cav1.2 is ideally suited to support the SAN AP.
Cav1.3 also conducts almost all of the L-type current in atrioventricular node (AVN) cells.103 Cav1.3−/− mice (as well as SANDD patients, see above) display AV-node conductance disturbances ranging from a longer PR interval to complete AV block.6,96 This emphasizes the role of this channel for normal AV-node function. Cav1.3 knockout or pharmacological channel block abolishes spontaneous AVN activity and results in more depolarized membrane potentials (Figure 4). Current injection restored negative membrane potentials and automaticity, although at a lower rate than in wild type and with reduced AP amplitude. The latter suggests a contribution of Cav1.3 also for the AP itself. Like in SAN, Cav1.3 is not the only ion channel responsible for pacemaking, because inhibition of voltage-gated Na+-channels, If channels, and Cav3.1 T-type Ca2+ channel also reduced automaticity.104
FIGURE 4.

Role of Cav1.3 channels for atrioventricular node (AVN) automaticity. Automaticity of wild-type (WT) AVN cells (AVNCs) is dependent on both INa and ICa,L. (a and b) Application of 20-μM tetrodotoxin (TTX) blocked action potential (AP) discharge. The membrane potential of AVNCs exposed to 20 μM TTX was stable at −59 ± 2 mV (n = 8). (c and d) Inhibition of ICa,L by 0.3 μM of the L-type channel blocker isradipine in WT mouse AVNCs stopped pacemaker activity of AVNCs and the cell membrane potential depolarized to −35 ± 3 mV (n = 6). Only low-amplitude oscillations of the membrane potential could be observed in isradipine-treated AVNCs. These results indicated that pacemaking of mouse AVNCs required both INa and ICa,L for AP discharge. (e) Cav1.3−/− AVNCs display positive membrane potential and low-amplitude oscillations without spontaneous APs very similar to isradipine-blocked WT AVNCs. (f) Tonic hyperpolarizing current injection (black arrow) induced spontaneous AP firing in Cav1.3−/− AVNCs but with slower pacemaker activity and smaller AP amplitude. This suggests contribution of Cav1.3 channels to both diastolic depolarization as well as to the AP itself. The positive resting membrane potential in Cav1.3−/− AVNCs likely is due to the loss of crosstalk between Cav1.3 channels and SK2 K+ channels. In the intact AVN, Cav1.3−/− myocytes must be sufficiently hyperpolarized (e.g., by electrical coupling with the right atrium) to enable INa-dependent APs and triggering by SAN impulses. (Reprinted with permission from Refs 103 and 104. Copyright 2011 Landes Bioscience)
Notice that in a second Cav1.3 knockout model, an apparent compensatory upregulation of Cav1.2 channels is observed in SAN,105 AVN,105 and also pancreatic β-cells.106 Therefore, the phenotype in SAN and AVN cells is less pronounced than in the studies described above without compensation by Cav1.2. At present the molecular basis for the differences between the two knockout models remains unknown.
LTCCs in the Working Myocardium
With the exception of low expression of Cav1.3 in atrial myocytes, Cav1.2 is the predominant LTCC in the working myocardium. Its activation supplies Ca2+ to trigger Ca2+-induced Ca2+ release from the SR RYRs and contraction. A significant contribution of Cav1.3 for cardiac contractility was ruled out in mice expressing mutant DHP-insensitive Cav1.2 LTCCs. In these mice DHPs no longer inhibited ventricular contractility in Langendorff hearts.53 Consequently, Cav1.2 channels are essential for heart function. Knockout of the Cav1.2 α1 subunit is embryonic lethal and causes death before day 14.5 p.c.107 Homozygous loss-of-function mutations in humans are therefore not expected to be compatible with life.
Cardiac-specific deletion of one or two Cav1.2 α1 wild-type alleles revealed that already less than 50% reduction of ICa is not tolerated and results in heart failure and enhanced lethality.108 Apparently, reduced Cav1.2 function leads to reduced SR Ca2+ load and a compensatory neuroendocrine activation with sensitized SR Ca2+ release (evident as increased Ca2+ spark activity) to preserve contractility.108–110 Heterozygous knockouts are also prone to development of cardiac hypertrophy and ventricular dilatation when subjected to pathological or physiological cardiovascular stress.108 In another cardiomyocyte-specific inducible Cav1.2 knockout mouse model mortality was not increased in heterozygous knockouts, because Cav1.2 α1 protein expression and ICa amplitude were almost at wild-type levels.110
Other mutant mouse models leading to diminished Cav1.2 ICa were also not viable or resulted in dilated cardiomyopathy. This included mice with the I1624E mutation,109 which disrupts interaction with CaM.
Cav1.2 α1 truncation mutants were also constructed to study the role of the distal C-terminal domain (DCRD). As explained above, DCRD exerts an inhibitory function on Cav1.2, seems to be noncovalently associated with a more proximal site (PCRD) even if proteolytically cleaved, and its inhibitory effect can be reversed by PKA phosphorylation. Truncations were introduced after N1904111 or G1796.112 In both mutants, this removed not only the DCRD but also the terminal PDZ-binding domain as well as a leucine zipper-like domain able to bind AKAPs (see above, Figure 2). Both mutant mice died in utero or immediately after birth. In both cases, Cav1.2 α1-subunit protein (but not mRNA) expression and ICa were severely reduced in cardiomyocytes from embryos or newborn pups. Another splice variant of Cav1.2 forms the major LTCC in vascular smooth muscle.50,111,113 In contrast to cardiac Cav1.2 channels their expression was not affected in the mutants. This suggests a splice-variant-specific protective role of the C-terminus against protein degradation.111 Because truncated Cav1.2 α1 subunits are expected to have higher open probabilities it is possible that cardiac dysfunction results from a combination of reduced cardiac contractility (owing to reduced ICa) with enhanced (pulmonary and peripheral) vascular resistance (owing to normal expression and enhanced activity in smooth muscle).
Cardiac dysfunction is not only induced by permanent loss of Cav1.2 activity but also by enhanced Cav1.2-mediated ICa in transgenic mice overexpressing accessory β subunits.114 The sustained increase in Ca2+ influx induces pathological cardiac hypertrophy with a larger ejection fraction, enhanced fractional shortening on the single myocyte and organ level, as well as increased peak Ca2+ transients in myocytes.114 Hypertrophy was associated with activation of the CaN/NFAT and CaMKII/HDAC pathways.
The fact that both increased as well as decreased Cav1.2 activity in the heart predispose for cardiac disease emphasizes the need for close control of its activity and expression. In addition to well-known direct or indirect regulators of cardiac Cav1.2 channels, including Ca2+/CaM, PKA, protein phosphatases, and phosphodiesterases, novel modulatory mechanisms have also been taken into account. Recently, microRNAs have been identified as potential regulators of Cav1.2. For example, the Cav1.2 α1-subunit gene (CACNA1C) is a known target of miR-1, which reduces its expression.115 Loss of miR-1 in patients with myotonic dystrophy (DM1 and DM2) could be responsible for the upregulation of Cav1.2 α1 protein in cardiac specimens and the observed cardiac pathology (including arrhythmias) in affected individuals.115
De novo mutations in Cav1.2 α1 or its associated subunits also cause cardiac disease. Timothy syndrome is an autosomal dominant condition with multiorgan dysfunction including prolonged QT, congenital heart disease, hand and foot abnormalities, mental retardation, and autism.116–118 The underlying mutations analyzed so far almost completely reduce VDI,116,117 which explains delayed cardiomyocyte repolarization with the risk of ventricular arrhythmias and death. Missense mutations in the genes of Cav1.2 α1 and the accessory subunits β2 and α2δ1 have also been associated with cardiac disease, resulting in Brugada Syndrome with and without short QT, idiopathic ventricular fibrillation, and early repolarization syndrome (for review see Refs 119 and 120).
LTCCs IN BRAIN
LTCC function in the brain is much more difficult to study than in the cardiovascular system. Reasons are the absence of specific pharmacological tools potently blocking LTCC activity in brain in vivo, the higher structural and functional heterogeneity of LTCCs in the brain, the additional presence of Cav2 and Cav3 channels (often in the same neuron), the larger complexity of accessory subunits, the heterogeneity of neuronal firing patterns, and the much more complex readout of changes in brain function due to altered LTCC activity.
The high LTCC complexity is not due to more LTCC isoforms being expressed. Like in the heart only Cav1.2 and Cav1.3 are present in the brain.121 However, their α1 subunits can combine with all four different β-subunit isoforms92 exist in different alternatively spliced variants (in particular Cav1.3) and are even subject to RNA editing.122 Unlike in the heart therapeutic plasma concentrations of organic CCBs (such as nifedipine or diltiazem) cause no obvious changes of brain function in hypertensive patients or animals. Higher (experimental) in vivo doses in animals are dominated by pronounced cardiovascular effects that hamper interpretation of any behavioral phenotype.123 Moreover, CCBs only weakly discriminate between Cav1.2 and Cav1.3 channels and can therefore not be considered isoform-selective tools. On the other hand, it is known that LTCCs are widely expressed in the brain (about 90% Cav1.2 and 10% Cav1.353,121) and in most cases a single neuron expresses both of them.124
Cav1.3 and Cav1.2 are postsynaptic channels localized predominantly in the spines and shafts of dendrites.27,125 There they shape neuronal firing or activate Ca2+ signaling pathways involved in ETC. ETC transforms synaptic activity patterns into neuronal remodeling associated with learning, memory, drug addiction, and neuronal development. Both channel isoforms seem to participate in this process126 in which channel-bound CaM and CaMKII are crucial in decoding voltage-gated changes in channel conformation and activity.127,128
Both Cav1.3 and Cav1.2 require additional protein interactions for proper function and targeting (Table 3). Examples are the PDZ domain-containing scaffolding protein densin for Cav1.3 channel fine-tuning by CaMKII (see above27), shank for attaching Cav1.3 to GPCRs-mediated signaling,129 and AKAPs for anchoring PKA to Cav1.2130 (Table 3).
Central nervous system (CNS) effects of LTCC antagonists in humans are subtle but have been detected in clinical experimental studies.131 Noninvasive continuous theta-burst stimulation (cTBS, a repetitive transcranial magnetic stimulation protocol) has been used to unequivocally demonstrate the in vivo effects of the CCB nimodipine on alterations of cTBS-induced changes of corticospinal excitability. This was interpreted with in vivo effects of nimodipine on LTP and LTD.131
In animals, reliable data regarding the physiological role of the two brain LTCC isoforms have been primarily revealed by using genetically modified mice.38 These included Cav1.3−/− mice, conditional Cav1.2−/− mice, and a mouse model expressing fully functional but DHP-resistant Cav1.2 channels (for review see Ref 38). Cav1.3 α1-deficient mice reproduce, have no gross anatomical defects, are deaf but have normal vision (Box 2). Unlike in Cav1.4−/− mice132 there is no evidence for retinal degeneration although they show minor changes in synaptic ribbon morphology in the outer plexiform layer and a reduced light peak in an otherwise normal electroretinogram.123,133
BOX 2L-TYPE CHANNELS IN SENSORY CELLS
Fast neurotransmitter release in neurons is tightly regulated by voltage-gated Cav2 channels. They conduct Ca2+ currents previously classified as P/Q-, N-, and R-type, respectively (Table 1), and initiate the fast release of important neurotransmitters such as glutamate, GABA, and acetylcholine. Specialized presynaptic structures tether these channels to the presynaptic vesicle release machinery by direct and indirect protein interactions with the channel complex.5 Specialized presynaptic structures providing highly localized Ca2+ signals for neurotransmitter release are also present in sensory cells, such as photoreceptors and cochlear inner hair cells. However, the Ca2+ channels involved there are members of the Cav1 family, which in neurons are mainly found postsynaptically in somatodendritic locations. Cav1.3 predominates in sensory cells of the inner ear (inner and outer cochlear hair cells and vestibular hair cells) and Cav1.4 in retinal photoreceptors.
Hippocampal Function
Hippocampal function depends much more on Cav1.2 than Cav1.3. Conditional deletion of Cav1.2 α1 in the cerebral cortex and hippocampus (NEX-driven cre deleter mice134) resulted in a selective loss of protein synthesis-dependent, N-methyl-d-aspartate (NMDA)-receptor-independent late-phase LTP (L-LTP) in CA3–CA1 synapses and a severe impairment of hippocampus-dependent spatial memory which became evident after 2 days. Activation of the mitogen-activated protein kinase (MAPK) pathway and cAMP response element (CRE)-dependent transcription in CA1 pyramidal neurons were reduced. This suggested that Cav1.2-mediated Ca2+ influx underlies hippocampal L-LTP, activation of the MAPK/CRE-binding protein signaling cascade, and hippocampal spatial memory formation. Another conditional brain knockout (CaMKIIα-driven cre deleter mice135) also affected remote spatial memory probed after 30 days. Interestingly, Cav1.2 deficiency did not alter the electrical properties, single and repetitive AP firing, or the post-burst afterhyperpolarization (AHP)136,137 in CA1 pyramidal neurons. However, the total area of the slow AHP was significantly diminished in Cav1.3−/− CA1 neurons.137 This indicates distinct function of these channel isoforms in the same cells: Cav1.2 preferentially couples to pathways initiating ETC and mediates LTP, whereas Cav1.3 couples to Ca2+-activated K+ channels and shapes the electrical properties of these cells. In contrast to Cav1.2, Cav1.3 is dispensable for CA3–CA1 hippocampal LTP and no deficit in spatial memory encoding in the Morris water maze was observed in Cav1.3−/− mice.138
Fear, Anxiety-, and Depression-Like Behaviors
Distinct roles of the two LTCCs were also found for fear memory. Cav1.3 is not essential for acquisition and extinction of conditioned contextual fear memory138,139 but is required for consolidation.140 Impaired consolidation was attributed to a significant decrease of the LTP in the basolateral amygdala (BLA) synapse receiving input from the entorhinal cortex.140 BLA neurons also showed signs of enhanced excitability. They fired more APs in response to depolarizing steps, exhibited reduced spike accommodation and a reduced post-burst AHP. The role of Cav1.2 for fear memory seems to be more complex. In brain-specific Cav1.2 knockout mice Cav1.2 carries essentially all the measurable L-type current in lateral amygdala (LA) neurons. Acute pharmacological block of these channels inhibited thalamo-LA LTP, which could explain the observed inhibition of auditory cued fear memory acquisition.141 However, this was not observed in Cav1.2−/− mice. This discrepancy was due to a shift of the dependence of LTP from Cav1.2 LTCCs to Ca2+-permeable AMPA receptors, apparently triggered by a homeostatic switch due to permanent Cav1.2 deficiency in these neurons.
Cav1.2 and Cav1.3 deficiency also lead to opposite effects on anxiety- and depression-like behaviors. Reduction of Cav1.2 expression in mouse forebrain by constitutive heterozygous knockout, CaMKII-driven cre deleter mice or adeno-associated viral (AAV) vector-expressing Cre recombinase induces anxiety-like behaviors in different experimental paradigms.142 This was not affected by shRNA-induced knockdown of Cav1.3 α1 subunits. In contrast, Cav1.3 deficiency induces anxiolytic-like behavior (which, however, may be explained by the deaf phenotype123) and an antidepressant-like behavior (not explained by deafness123). These data were well complemented by experiments in Cav1.2DHP−/− mice in which the application of the Ca2+-channel activator BAYK8644 selectively stimulates Cav1.3 channels and induces depression-like behavior.53,143 Notice that acute treatment of wild-type mice with BayK8644 induces a severe toxic dystonic neurobehavioral syndrome associated with massive neuronal activation in most brain regions and widespread neurotransmitter release.53,143 This syndrome is completely absent in Cav1.2DHP−/− mice indicating its dependence on Cav1.2 channels. Cav1.2DHP−/− mice can therefore be used to study specific behavioral effects of Cav1.3 activation in mouse brain.
As mentioned above, Timothy syndrome is associated with a Cav1.2 gain-of-function phenotype and also affects neuronal channels. Autism is one of the symptoms found in these patients. A recent in vitro study in rodent neurons and induced pluripotent stem cell-derived neurons from patients suggests that channels with the Timothy syndrome mutation cause activity-dependent dendrite retraction.144 This appears to be independent of Ca2+ entry through the mutant channel but involves activation of RhoA signaling known to affect dendrite retraction.
Taken together, these data indicate that Cav1.2 function in the brain must be tightly controlled within a narrow range of activity. Timothy syndrome shows that gain of function induces autism. However, if the rodent data also apply to human brain function, then SNPs causing significant loss of Cav1.2 activity should contribute to psychiatric disease risk as well. These observations support findings from genome-wide association studies linking (intronic) CACNA1C SNPs with enhanced risk for a range of psychiatric disorders with child and adult onset145 (see Ref 146 for review).
Drug-Taking Behaviors
LTCC activity controls signaling pathways that are involved in neuronal plasticity associated with drug dependence. Using locomotor sensitization as a model for psychostimulant-induced long-term plasticity in Cav1.3−/− and Cav1.2DHP−/− mice, a distinct role for Cav1.2 and Cav1.3 LTCCs was found.147 Although Cav1.3 mediates the development of sensitization, Cav1.2 is responsible for expression of the psychostimulant-induced sensitized response. Acute psychostimulant treatment in drug-naive mice was associated with activation of a D1/Cav1.3/CREB pathway in the NAc.146 During development of sensitization the ERK pathway was additionally recruited.146 Interestingly, expression of the psychostimulant-induced sensitized response after extended withdrawal from drug recruited activity of a D1/Cav1.2/ERK pathway that blunts CREB activation. It was therefore proposed that this molecular switch from Cav1.3 to Cav1.2 channels may determine the transition from the drug-naive to the drug-dependent state.147 Extended withdrawal from repeated cocaine administration increases phosphorylation of the AMPA-receptor GluA1 subunit at S845 in the NAc with a parallel increase in cell surface GluA1 that occurs independently of Cav1.2 or Cav1.3 channels (Figure 5).148 A challenge injection of cocaine that elicits expression of the sensitized response further increased surface GluA1 via both a D1/Cav1.2-mediated increase in GluA1 phosphorylation at S831 by CaMKII and by an ERK2-dependent mechanism. This long-term change in the NAc is dependent on the Cav1.3/ERK2 pathway in the ventral tegmental area (VTA) during the development of cocaine sensitization (Figure 5).148 In contrast, in the dorsal striatum repeated cocaine administration causes a non-VTA Cav1.3-dependent recruitment of the D2L signaling pathway following extended withdrawal, which decreases basal GluA1 phosphorylation at S845 and cell surface GluA1 levels.149 This highlights the complexity of LTCC-mediated activity in mediating cocaine-induced persistent behavioral changes and their utilization of different signaling pathways.
FIGURE 5.

Schematic representation of the role of Cav1.2 and Cav1.3 L-type calcium channels (LTCCs) in the persistent nucleus accumbens (NAc) molecular adaptations following extended withdrawal from repeated cocaine exposure. (a) The cocaine-naive dopamine D1-containing NAc neuron expresses AMPA receptors (GluA1/GluA2 tetramers) and Cav1.2 channels on the cell surface. (b) Twenty-one days following withdrawal from repeated cocaine treatment increased phosphorylation of GluA1 at S845 in the NAc (a PKA site) was paralleled by an increase in cell surface GluA1 and GluA2 levels (and higher levels of Cav1.2 mRNA). (c) A cocaine challenge that elicits expression of cocaine psychomotor sensitization involves dopamine D1 receptors and Cav1.2-activated CaMKII that increases GluA1 phosphorylation at S831 and Cav1.2-activated ERK2, which further increases cell surface GluA1 over that seen in b. This long-term adaptation is dependent on Cav1.3 channels and ERK2 in the ventral tegmental area (VTA) during the development of sensitization. (Reprinted with permission from Ref 148. Copyright 2011 Society for Neuroscience)
Dopamine Neuron Physiology and Pathophysiology
The negative activation voltage range enables Cav1.3 channels not only to serve as pacemaker channels in the heart (see above) but also to shape firing properties of neurons. In striatal medium spiny neurons (MSNs) glutamatergic synaptic input initiates a depolarized upstate during which neurons can spike. This upstate is sustained by Cav1.3 channel activity and absent in Cav1.3−/− mice.129 The progressive death of SNc neurons in Parkinson's disease (PD) leads to dopamine (DA) loss in the striatum, which disinhibits the suppression of Cav1.3 channel activity through D2 receptor activation. It causes a rapid and profound loss of spines and glutamatergic synapses on D2R-expressing striatopallidal MSNs but not on neighboring D1R-expressing striatonigral MSNs.129 This synaptic pruning requires Cav1.3 activity.129 The resulting disconnection of striatopallidal neurons from motor control structures could be a key mechanism in the emergence of pathological motor activity in PD and of therapy-associated l-DOPA-induced dyskinesias.150
Both Cav1.2 and Cav1.3 are also present in spontaneously firing substantia nigra pars compacta (SNc) DA neurons, which are vulnerable to degeneration in PD.151 Low doses of DHPs resulting in plasma levels corresponding to therapeutic levels in humans protect SNc neurons from degeneration in neurotoxin-based models of PD in rodents152–154 and non-human primates.155 Similarly, Cav1.3 deficiency also protected SNc DA neurons from rotenone toxicity in brain slices. Therefore, it is likely that inhibition of LTCC activity, including Cav1.3, in SNc DA neurons accounts for this neuroprotective effect. One molecular mechanism that can link reduced LTCC activity to neuroprotection is reduction of intracellular Ca2+ load and of oxidative stress.156 It has been proposed that LTCCs, and in particular Cav1.3,152,157 play a major role for the spontaneous pacemaking of these neurons. High micromolar concentrations of the DHP CCBs nimodipine or isradipine slowed spontaneous ‘tonic’ pacemaker frequency in acutely dissociated neurons or in brain slices, suggesting that LTCCs drive autonomous pacemaking in these cells.152,157,158 These data are in contrast to findings with lower concentrations of these drugs that should block LTCCs efficiently but have no major effect on pacemaking activity.159 These contradictory results most likely result from the fact that several cation channels contribute to subthreshold depolarizing currents158,159 and that their relative contribution varies depending on experimental conditions. DHPs may exert their neuroprotective action by inhibiting the LTCC-dependent dendritic Ca2+ transients that occur during spontaneous APs and cause a constant Ca2+ load. They may also affect Ca2+ entry into SNc DA neurons during burst firing, which occurs, e.g., in response to reward-predicting stimuli.151 In slices LTCCs appear to enhance NMDA-receptor activation-induced bursting, an effect that can be blocked by DHPs.157 LTCC-mediated Ca2+ influx also promotes DA synthesis and α-synuclein-dependent l-DOPA-induced degeneration of SNc DA neurons160 providing yet another link between LTCC activity and oxidative stress.
Taken together, animal models strongly support a role of LTCCs for the selective vulnerability of SNc DA neurons in PD. At present it is unclear if Cav1.3 alone or both LTCC isoforms contribute to this pathology. Case–control and cohort studies from Denmark and the United Kingdom found a significant association between long-term use of CCBs as antihypertensives and reduced risk for a first-time diagnosis of PD (odds ratios of 0.71–0.78).161–163 This effect was consistently seen for brain-permeable DHPs but not for amlodipine (a DHP with poor penetration into the brain), non-DHP CCBs (like verapamil and diltiazem), β-blockers, and ACE inhibitors. One prospective cohort study (with data from the Nurses Health Study and the Health Professionals Follow-up Study)164 did not confirm these findings but was clearly underpowered with only 18 cases (PD patients) receiving CCBs. Based on this available preclinical and clinical evidence LTCCs are currently pursued as an attractive target for drug discovery in pharmaceutical industry worldwide. Cav1.3-selective drugs appear most promising because peripheral side effects arising from vascular Cav1.2 LTCC block (vasodilatation and edema) would be minimized. Moreover, Cav1.3 block may have other beneficial CNS effects, such as antidepressant actions (see above). However, at present it is unknown if Cav1.3-selective inhibitors would miss a neuroprotective component mediated by Cav1.2 channels.
Neuronal Development and Synaptic Refinement
In addition to the above observations in MSNs on synaptic pruning, a role for Cav1.3 in synaptic refinement has also been described in the auditory pathway. In general, neuronal circuits underlie a developmental process in which initially imprecisely formed synapses become refined by selective elimination of redundant immature synapses and strengthening of remaining ones. In the auditory brainstem Cav1.3 crucially controls the refinement of inhibitory synapses in projections from the medial nucleus of the trapezoid body (MNTB) to the lateral superior olive (LSO).165 Normally, during the first 2 postnatal weeks the number of axons of MNTB neurons projecting to one LSO neuron in mice declines sharply, whereas the remaining ones consolidate. In Cav1.3−/− mice projections were not eliminated up to hearing onset and synaptic strengthening was strongly impaired. Moreover, the mediolateral topography was less precise and the shift from a mixed GABA/glycinergic to a purely glycinergic transmission normally seen in wild-type mice before hearing onset did not occur.165
Cav1.3 deficiency is also associated with a drastically reduced volume in all auditory brainstem centers (but not other brain regions) already before hearing onset.166 The LSO contains fewer neurons and is abnormally shaped. The remaining LSO neurons receive functional glutamatergic input through normal dendritic trees but show an abnormal firing pattern upon depolarization, which is attributed to reduced K+-channel function. Notably, this effect was not due to inner ear dysfunction because it was also present in mice in which Cav1.3 channels were conditionally knocked out only in the auditory brainstem, but with preserved expression in the cochlea.167
CONCLUSION
Gene knockout animal models have allowed to dissect the distinct physiological roles of LTCC isoforms for different physiological functions. This furnished an essential basis for understanding how their loss or gain of function can cause human disease. However, 30 years after the discovery that 3H-dihydropyridine-binding sites in rodent brain represent functional LTCCs168 an important pharmacological question still remains unexplained: given the essential role of LTCCs for synaptic plasticity,169 why do brain-permeable LTCC blockers used for treating hypertension not cause memory impairment even at higher therapeutic doses? The low sensitivity of brain LTCCs for DHPs could be one explanation. It is most likely due to the short AP duration and negative resting membrane potential of most neurons. This disfavors inactivated channel states to which DHPs preferentially bind. Alternative splicing also affects DHP sensitivity. Cardiac splice variants of Cav1.2 are less sensitive to DHPs than their smooth muscle counterparts,113 which partially explains why arterial vasodilation occurs at much lower plasma concentrations than cardiodepressant effects. The low DHP sensitivity of neuronal Cav1.2 LTCCs may therefore also be due to alternative splicing. Cav1.3 LTCCs are slightly less sensitive to DHPs than Cav1.2 although their binding pockets can bind DHPs with similar affinities.40 This emphasizes the important role of state-dependent (i.e., voltage-induced) differences in channel architecture. At present big efforts are made to discover Cav1.3-selective LTCC blockers with the aim to inhibit potentially toxic Cav1.3-mediated Ca2+ load in SNc DA neurons without causing cardiovascular depression through Cav1.2 channel block. Once drug candidates become available it will also be interesting to see if they also exert other pharmacological effects of potential therapeutic value as predicted by mouse models (antidepressant effects, reduced development of drug addiction, and heart rate lowering without negative inotropy).
Acknowledgments
This work is supported by the Austrian Science Fund (SFB-F44020).
REFERENCES
- 1.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- 2.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3:a003947. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dolphin AC. Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat Rev Neurosci. 2012;13:542–555. doi: 10.1038/nrn3311. [DOI] [PubMed] [Google Scholar]
- 4.Striessnig J, Grabner M, Mitterdorfer J, Hering S, Sinnegger MJ, Glossmann H. Structural basis of drug binding to L calcium channels. Trends Pharmacol Sci. 1998;19:108–115. doi: 10.1016/s0165-6147(98)01171-7. [DOI] [PubMed] [Google Scholar]
- 5.Muller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B, et al. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A. 2010;107:14950–14957. doi: 10.1073/pnas.1005940107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, Ali A, Ahmad I, Sinnegger-Brauns MJ, Brandt N, et al. Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14:77–84. doi: 10.1038/nn.2694. [DOI] [PubMed] [Google Scholar]
- 7.Catterall WA. Ion channel voltage sensors: structure, function, and pathophysiology. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stotz SC, Jarvis SE, Zamponi GW. Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J Physiol. 2004;554:263–273. doi: 10.1113/jphysiol.2003.047068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tadross MR, Ben Johny M, Yue DT. Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels. J Gen Physiol. 2010;135:197–215. doi: 10.1085/jgp.200910308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N. Molecular coupling of a Ca2+-activated K+ channel to L-type Ca2+ channels via α-actinin2. Circ Res. 2007;100:112–120. doi: 10.1161/01.RES.0000253095.44186.72. [DOI] [PubMed] [Google Scholar]
- 11.Pankonien I, Alvarez JL, Doller A, Kohncke C, Rotte D, Regitz-Zagrosek V, Morano I, Haase H. Ahnak1 is a tuneable modulator of cardiac Cav1.2 calcium channel activity. J Muscle Res Cell Motil. 2011;32:281–290. doi: 10.1007/s10974-011-9269-2. [DOI] [PubMed] [Google Scholar]
- 12.Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal. 2010;3:ra70. doi: 10.1126/scisignal.2001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong TT, Smyth JW, Gao D, Chu KY, Vogan JM, Fong TS, Jensen BC, Colecraft HM, Shaw RM. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS Biol. 2010;8:e1000312. doi: 10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tandan S, Wang Y, Wang TT, Jiang N, Hall DD, Hell JW, Luo X, Rothermel BA, Hill JA. Physical and functional interaction between calcineurin and the cardiac L-type Ca2+ channel. Circ Res. 2009;105:51–60. doi: 10.1161/CIRCRESAHA.109.199828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blaich A, Welling A, Fischer S, Wegener JW, Kostner K, Hofmann F, Moosmang S. Facilitation of murine cardiac L-type Cav1.2 channel is modulated by calmodulin kinase II-dependent phosphorylation of S1512 and S1570. Proc Natl Acad Sci U S A. 2010;107:10285–10289. doi: 10.1073/pnas.0914287107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type calcium channels, establishing a local and dedicated integrator of calcium signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the β subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Balijepalli RC, Foell JD, Hall DD, Hell JW, Kamp TJ. Localization of cardiac L-type calcium channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomsen MB, Wang C, Ozgen N, Wang HG, Rosen MR, Pitt GS. Accessory subunit KChIP2 modulates the cardiac L-type calcium current. Circ Res. 2009;104:1382–1389. doi: 10.1161/CIRCRESAHA.109.196972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Gao G, Guo K, Yarotskyy V, Huang C, Elmslie KS, Peterson BZ. Phospholemman modulates the gating of cardiac L-type calcium channels. Biophys J. 2010;98:1149–1159. doi: 10.1016/j.bpj.2009.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X, Marx SO, Colecraft HM. Molecular mechanisms, and selective pharmacological rescue, of Rem-inhibited Cav1.2 channels in heart. Circ Res. 2010;107:620–630. doi: 10.1161/CIRCRESAHA.110.224717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fowler MR, Colotti G, Chiancone E, Smith GL, Fearon IM. Sorcin modulates cardiac L-type Ca2+ current by functional interaction with the α1C subunit in rabbits. Exp Physiol. 2008;93:1233–1238. doi: 10.1113/expphysiol.2008.043497. [DOI] [PubMed] [Google Scholar]
- 23.Marshall MR, Clark JP, III, Westenbroek R, Yu FH, Scheuer T, Catterall WA. Functional roles of a C-terminal signaling complex of Cav1 channels and A-kinase anchoring protein 15 in brain neurons. J Biol Chem. 2011;286:12627–12639. doi: 10.1074/jbc.M110.175257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliveria SF, Dell'Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-typeCa2+ channel activity and nuclear signaling. Neuron. 2007;55:261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou H, Kim SA, Kirk EA, Tippens AL, Sun H, Haeseleer F, Lee A. Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J Neurosci. 2004;24:4698–4708. doi: 10.1523/JNEUROSCI.5523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tippens AL, Lee A. Caldendrin, a neuron-specific modulator of Cav1.2 (L-type) Ca2+ channels. J Biol Chem. 2007;282:8464–8473. doi: 10.1074/jbc.M611384200. [DOI] [PubMed] [Google Scholar]
- 27.Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, Usachev YM, Obermair GJ, Colbran RJ, Lee A. Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J Neurosci. 2010;30:5125–5135. doi: 10.1523/JNEUROSCI.4367-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calin-Jageman I, Yu K, Hall RA, Mei L, Lee A. Erbin enhances voltage-dependent facilitation of Cav1.3 calcium channels through relief of an autoinhibitory domain in the Cav1.3 α1 subunit. J Neurosci. 2007;27:1374–1385. doi: 10.1523/JNEUROSCI.5191-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weick JP, Groth RD, Isaksen AL, Mermelstein PG. Interactions with PDZ proteins are required for L-type calcium channels to activate cAMP response element-binding protein-dependent gene expression. J Neurosci. 2003;23:3446–3456. doi: 10.1523/JNEUROSCI.23-08-03446.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Correll RN, Botzet GJ, Satin J, Andres DA, Finlin BS. Analysis of the Rem2—voltage dependant calcium channel β subunit interaction and Rem2 interaction with phosphorylated phosphatidylinositide lipids. Cell Signal. 2008;20:400–408. doi: 10.1016/j.cellsig.2007.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coppola T, Magnin-Luthi S, Perret-Menoud V, Gattesco S, Schiavo G, Regazzi R. Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and synaptotagmin. J Biol Chem. 2001;276:32756–32762. doi: 10.1074/jbc.M100929200. [DOI] [PubMed] [Google Scholar]
- 32.Gandini MA, Felix R. Functional interactions between voltage-gated Ca2+ channels and Rab3-interacting molecules (RIMs): new insights into stimulus-secretion coupling. Biochim Biophys Acta. 1818;2012:551–558. doi: 10.1016/j.bbamem.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I. Association of Cav1.3 L-type calcium channels with Shank. J Neurosci. 2005;25:1037–1049. doi: 10.1523/JNEUROSCI.4554-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J, Tang XD, Gill DL. The calcium store sensor, STIM1, reciprocally controls Orai and Cav1.2 channels. Science. 2010;330:105–109. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park CY, Shcheglovitov A, Dolmetsch R. The CRAC channel activator STIM1 binds and inhibits L-type voltage-gated calcium channels. Science. 2010;330:101–105. doi: 10.1126/science.1191027. [DOI] [PubMed] [Google Scholar]
- 36.Johny MB, Yang PS, Bazzazi H, Yue DT. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of Cav1.3 channels. Nat Commun. 2013;4:1717. doi: 10.1038/ncomms2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Findeisen F, Minor DL., Jr Structural basis for the differential effects of CaBP1 and calmodulin on Cav1.2 calcium-dependent inactivation. Structure. 2010;18:1617–1631. doi: 10.1016/j.str.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Striessnig J, Koschak A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin) 2008;2:233–251. doi: 10.4161/chan.2.4.5847. [DOI] [PubMed] [Google Scholar]
- 39.Tuluc P, Molenda N, Schlick B, Obermair GJ, Flucher BE, Jurkat-Rott K. A Cav1.1 Ca2+ channel splice variant with high conductance and voltage-sensitivity alters EC coupling in developing skeletal muscle. Biophys J. 2009;96:35–44. doi: 10.1016/j.bpj.2008.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. α1D (Cav1.3) subunits can form L-type calcium channels activating at negative voltages. J Biol Chem. 2001;276:22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- 41.Bock G, Gebhart M, Scharinger A, Jangsangthong W, Busquet P, Poggiani C, Sartori S, Mangoni ME, Sinnegger-Brauns MJ, Herzig S, et al. Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J Biol Chem. 2011;286:42736–42748. doi: 10.1074/jbc.M111.269951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 calcium channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003;100:5543–5548. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christel C, Lee A. Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim Biophys Acta. 1820;2012:1243–1252. doi: 10.1016/j.bbagen.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang PS, Alseikhan BA, Hiel H, Grant L, Mori MX, Yang W, Fuchs PA, Yue DT. Switching of calcium-dependent inactivation of Cav1.3 channels by calcium binding proteins of auditory hair cells. J Neurosci. 2006;26:10677–10689. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh A, Hamedinger D, Hoda JC, Gebhart M, Koschak A, Romanin C, Striessnig J. C-terminal modulator controls calcium-dependent gating of Cav1.4 L-type calcium channels. Nat Neurosci. 2006;9:1108–1116. doi: 10.1038/nn1751. [DOI] [PubMed] [Google Scholar]
- 46.Singh A, Gebhart M, Fritsch R, Sinnegger-Brauns MJ, Poggiani C, Hoda JC, Engel J, Romanin C, Striessnig J, Koschak A. Modulation of voltage- and Ca2+-dependent gating of Cav1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J Biol Chem. 2008;283:20733–20744. doi: 10.1074/jbc.M802254200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. Autoinhibitory control of the Cav1.2 Channel by its proteolytically processed distal C-terminal domain. J Physiol. 2006;576:87–102. doi: 10.1113/jphysiol.2006.111799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao P, Zhang HY, Soong TW. Alternative splicing of voltage-gated calcium channels: from molecular biology to disease. Pflugers Arch. 2009;458:481–487. doi: 10.1007/s00424-009-0635-5. [DOI] [PubMed] [Google Scholar]
- 49.Striessnig J. Pharmacology, structure and function of cardiac L-type calcium channels. Cell Physiol Biochem. 1999;9:242–269. doi: 10.1159/000016320. [DOI] [PubMed] [Google Scholar]
- 50.Liao P, Soong TW. Understanding alternative splicing of Cav1.2 calcium channels for a new approach towards individualized medicine. J Biomed Res. 2010;24:181–186. doi: 10.1016/S1674-8301(10)60027-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zamponi GW, Currie KP. Regulation of Cav2 calcium channels by G protein coupled receptors. Biochim Biophys Acta. 1828;2013:1629–1643. doi: 10.1016/j.bbamem.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lipscombe D, Raingo J. Alternative splicing matters: N-type calcium channels in nociceptors. Channels (Austin) 2007;1:225–227. doi: 10.4161/chan.4809. [DOI] [PubMed] [Google Scholar]
- 53.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renstrom E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschutz R, et al. Isoform-specific regulation of mood behavior and pancreatic β cell and cardiovascular function by L-type calcium channels. J Clin Invest. 2004;113:1430–1439. doi: 10.1172/JCI20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Osterrieder W, Brum G, Hescheler J, Trautwein W, Flockerzi V, Hofmann F. Injection of subunits of cyclic AMP-dependent protein kinase into cardiac myocytes modulates calcium current. Nature. 1982;298:576–578. doi: 10.1038/298576a0. [DOI] [PubMed] [Google Scholar]
- 55.Hulme JT, Konoki K, Lin TW, Gritsenko MA, Camp DG, II, Bigelow DJ, Catterall WA. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of Cav1.1 channels in skeletal muscle. Proc Natl Acad Sci U S A. 2005;102:5274–5279. doi: 10.1073/pnas.0409885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Cav1.2 encodes a transcription factor. Cell. 2006;127:591–606. doi: 10.1016/j.cell.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schroder E, Byse M, Satin J. L-type calcium channel C terminus autoregulates transcription. Circ Res. 2009;104:1373–1381. doi: 10.1161/CIRCRESAHA.108.191387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu Y, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation sites required for regulation of cardiac calcium channels in the fight-or-flight response. Proc Natl Acad Sci U S A. 2013;110:19621–19626. doi: 10.1073/pnas.1319421110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones BW, Brunet S, Gilbert ML, Nichols CB, Su T, Westenbroek RE, Scott JD, Catterall WA, McKnight GS. Cardiomyocytes from AKAP7 knockout mice respond normally to adrenergic stimulation. Proc Natl Acad Sci U S A. 2012;109:17099–17104. doi: 10.1073/pnas.1215219109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF, McKnight GS. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res. 2010;107:747–756. doi: 10.1161/CIRCRESAHA.109.216127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu H, Ginsburg KS, Hall DD, Zimmermann M, Stein IS, Zhang M, Tandan S, Hill JA, Horne MC, Bers D, et al. Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel Cav1.2. Biochemistry. 2010;49:10298–10307. doi: 10.1021/bi101018c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oliveria SF, Dittmer PJ, Youn DH, Dell'Acqua ML, Sather WA. Localized calcineurin confers Ca2+-dependent inactivation on neuronal L-type Ca2+ channels. J Neurosci. 2012;32:15328–15337. doi: 10.1523/JNEUROSCI.2302-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac Cav1.2 channels during β1-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:16574–16579. doi: 10.1073/pnas.0607294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leroy J, Richter W, Mika D, Castro LR, Abi-Gerges A, Xie M, Scheitrum C, Lefebvre F, Schittl J, Mateo P, Westenbroek R, et al. Phosphodiesterase 4B in the cardiac L-type calcium channel complex regulates calcium current and protects against ventricular arrhythmias in mice. J Clin Invest. 2011;121:2651–2661. doi: 10.1172/JCI44747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dai S, Hall DD, Hell JW. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol Rev. 2009;89:411–452. doi: 10.1152/physrev.00029.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ganesan AN, Maack C, Johns DC, Sidor A, O'Rourke B. β-Adrenergic stimulation of L-type calcium channels in cardiac myocytes requires the distal carboxyl terminus of α1C but not serine 1928. Circ Res. 2006;98:e11–e18. doi: 10.1161/01.RES.0000202692.23001.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, Lenhardt P, Hofmann F, Moosmang S. Unchanged β-adrenergic stimulation of cardiac L-type calcium channels in Cav1.2 phosphorylation site S1928A mutant mice. J Biol Chem. 2008;283:34738–34744. doi: 10.1074/jbc.M804981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brandmayr J, Poomvanicha M, Domes K, Ding J, Blaich A, Wegener JW, Moosmang S, Hofmann F. Deletion of the C-terminal phosphorylation sites in the cardiac β-subunit does not affect the basic β-adrenergic response of the heart and the Cav1.2 channel. J Biol Chem. 2012;287:22584–22592. doi: 10.1074/jbc.M112.366484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mahapatra S, Marcantoni A, Zuccotti A, Carabelli V, Carbone E. Equal sensitivity of Cav1.2 and Cav1.3 channels to the opposing modulations of PKA and PKG in mouse chromaffin cells. J Physiol. 2012;590:5053–5073. doi: 10.1113/jphysiol.2012.236729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramadan O, Qu Y, Wadgaonkar R, Baroudi G, Karnabi E, Chahine M, Boutjdir M. Phosphorylation of the consensus sites of protein kinase A on α1D L-type calcium channel. J Biol Chem. 2009;284:5042–5049. doi: 10.1074/jbc.M809132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liang Y, Tavalin SJ. Auxiliary β subunits differentially determine pka utilization of distinct regulatory sites on Cav1.3 L type Ca2+ channels. Channels (Austin) 2007;1:102–112. doi: 10.4161/chan.4284. [DOI] [PubMed] [Google Scholar]
- 72.Maier LS, Bers DM. Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol. 2002;34:919–939. doi: 10.1006/jmcc.2002.2038. [DOI] [PubMed] [Google Scholar]
- 73.Ross J, Jr, Miura T, Kambayashi M, Eising GP, Ryu KH. Adrenergic control of the force-frequency relation. Circulation. 1995;92:2327–2332. doi: 10.1161/01.cir.92.8.2327. [DOI] [PubMed] [Google Scholar]
- 74.Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/s0959-4388(03)00076-x. [DOI] [PubMed] [Google Scholar]
- 75.Pitt GS. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc Res. 2007;73:641–647. doi: 10.1016/j.cardiores.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 76.Gao L, Blair LA, Salinas GD, Needleman LA, Marshall J. Insulin-like growth factor-1 modulation of Cav1.3 calcium channels depends on calcium release from IP3-sensitive stores and calcium/calmodulin kinase II phosphorylation of the α1 subunit EF hand. J Neurosci. 2006;26:6259–6268. doi: 10.1523/JNEUROSCI.0481-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suh BC, Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol. 2005;15:370–378. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 78.Heneghan JF, Mitra-Ganguli T, Stanish LF, Liu L, Zhao R, Rittenhouse AR. The Ca2+ channel β subunit determines whether stimulation of Gq-coupled receptors enhances or inhibits N current. J Gen Physiol. 2009;134:369–384. doi: 10.1085/jgp.200910203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Suh BC, Kim DI, Falkenburger BH, Hille B. Membrane-localized β-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc Natl Acad Sci U S A. 2012;109:3161–3166. doi: 10.1073/pnas.1121434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. 2011;477:495–498. doi: 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roberts-Crowley ML, Mitra-Ganguli T, Liu L, Rittenhouse AR. Regulation of voltage-gated Ca2+ channels by lipids. Cell Calcium. 2009;45:589–601. doi: 10.1016/j.ceca.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Buraei Z, Yang J. The β-subunit of voltage-gated Ca2+ channels. Physiol Rev. 2010;90:1461–1506. doi: 10.1152/physrev.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koschak A, Reimer D, Walter D, Hoda JC, Heinzle T, Grabner M, Striessnig J. Cav1.4α1 subunits can form slowly inactivating dihydropyridine-sensitive L-type calcium channels lacking calcium-dependent inactivation. J Neurosci. 2003;23:6041–6049. doi: 10.1523/JNEUROSCI.23-14-06041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Herzig S, Khan IF, Grundemann D, Matthes J, Ludwig A, Michels G, Hoppe UC, Chaudhuri D, Schwartz A, Yue DT, et al. Mechanism of Ca(v)1.2 channel modulation by the amino terminus of cardiac β2-subunits. FASEB J. 2007;21:1527–1538. doi: 10.1096/fj.06-7377com. [DOI] [PubMed] [Google Scholar]
- 85.Dalton S, Takahashi SX, Miriyala J, Colecraft HM. A single CaVβ can reconstitute both trafficking and macroscopic conductance of voltage-dependent calcium channels. J Physiol. 2005;567:757–769. doi: 10.1113/jphysiol.2005.093195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fang K, Colecraft HM. Mechanism of auxiliary β-subunit-mediated membrane targeting of L-type (Ca(V)1.2) channels. J Physiol. 2011;589:4437–4455. doi: 10.1113/jphysiol.2011.214247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lipscombe D, Allen SE, Toro CP. Control of neuronal voltage-gated calcium ion channels from RNA to protein. Trends Neurosci. 2013;36:598–609. doi: 10.1016/j.tins.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Altier C, Garcia-Caballero A, Simms B, You H, Chen L, Walcher J, Tedford HW, Hermosilla T, Zamponi GW. The Cavβ subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat Neurosci. 2011;14:173–180. doi: 10.1038/nn.2712. [DOI] [PubMed] [Google Scholar]
- 89.Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Scott JD, Nargeot J, Zamponi GW, Bourinet E. AKAP79 modulation of L-type channels involves disruption of intramolecular interactions in the CaV1.2 subunit. Channels (Austin) 2012;6:157–165. doi: 10.4161/chan.20865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW, et al. Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem. 2002;277:33598–33603. doi: 10.1074/jbc.M202476200. [DOI] [PubMed] [Google Scholar]
- 91.Meissner M, Weissgerber P, Londono JE, Prenen J, Link S, Ruppenthal S, Molkentin JD, Lipp P, Nilius B, Freichel M, et al. Moderate calcium channel dysfunction in adult mice with inducible cardiomyocyte-specific excision of the cacnb2 gene. J Biol Chem. 2011;286:15875–15882. doi: 10.1074/jbc.M111.227819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pichler M, Cassidy TN, Reimer D, Haase H, Kraus R, Ostler D, Striessnig J. β Subunit heterogeneity in neuronal L-type calcium channels. J Biol Chem. 1997;272:13877–13882. doi: 10.1074/jbc.272.21.13877. [DOI] [PubMed] [Google Scholar]
- 93.Link S, Meissner M, Held B, Beck A, Weissgerber P, Freichel M, Flockerzi V. Diversity and developmental expression of L-type calcium channel β2 proteins and their influence on calcium current in murine heart. J Biol Chem. 2009;284:30129–30137. doi: 10.1074/jbc.M109.045583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takahashi SX, Mittman S, Colecraft HM. Distinctive modulatory effects of five human auxiliary β2 subunit splice variants on L-type calcium channel gating. Biophys J. 2003;84:3007–3021. doi: 10.1016/S0006-3495(03)70027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harvey RD, Hell JW. CaV1.2 signaling complexes in the heart. J Mol Cell Cardiol. 2013;58:143–152. doi: 10.1016/j.yjmcc.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type calcium channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 97.Lakatta EG, DiFrancesco D. What keeps us ticking: a funny current, a calcium clock, or both? J Mol Cell Cardiol. 2009;47:157–170. doi: 10.1016/j.yjmcc.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bround MJ, Asghari P, Wambolt RB, Bohunek L, Smits C, Philit M, Kieffer TJ, Lakatta EG, Boheler KR, Moore ED, et al. Cardiac ryanodine receptors control heart rate and rhythmicity in adult mice. Cardiovasc Res. 2012;96:372–380. doi: 10.1093/cvr/cvs260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Herrmann S, Stieber J, Stockl G, Hofmann F, Ludwig A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 2007;26:4423–4432. doi: 10.1038/sj.emboj.7601868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Christel CJ, Cardona N, Mesirca P, Herrmann S, Hofmann F, Striessnig J, Ludwig A, Mangoni ME, Lee A. Distinct localization and modulation of Cav1.2 and Cav1.3 L-type Ca2+ channels in mouse sinoatrial node. J Physiol. 2012;590:6327–6342. doi: 10.1113/jphysiol.2012.239954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mangoni ME, Traboulsie A, Leoni AL, Couette B, Marger L, Le Quang K, Kupfer E, Cohen-Solal A, Vilar J, Shin HS, et al. Bradycardia and slowing of the atrioventricular conduction in mice lacking Cav3.1/α1G T-type calcium channels. Circ Res. 2006;98:1422–1430. doi: 10.1161/01.RES.0000225862.14314.49. [DOI] [PubMed] [Google Scholar]
- 103.Marger L, Mesirca P, Alig J, Torrente A, Dubel S, Engeland B, Kanani S, Fontanaud P, Striessnig J, Shin HS, et al. Functional roles of Cav1.3, Cav3.1 and HCN channels in automaticity of mouse atrioventricular cells: insights into the atrioventricular pacemaker mechanism. Channels (Austin) 2011;5:251–261. doi: 10.4161/chan.5.3.15266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Marger L, Mesirca P, Alig J, Torrente A, Dubel S, Engeland B, Kanani S, Fontanaud P, Striessnig J, Shin HS, et al. Pacemaker activity and ionic currents in mouse atrioventricular node cells. Channels (Austin) 2011;5:241–250. doi: 10.4161/chan.5.3.15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS, Chiamvimonvat N. Functional roles of Cav1.3 (α1D) calcium channel in sinoatrial nodes: insight gained using gene-targeted null mutant mice. Circ Res. 2002;90:981–987. doi: 10.1161/01.res.0000018003.14304.e2. [DOI] [PubMed] [Google Scholar]
- 106.Namkung Y, Skrypnyk N, Jeong MJ, Lee T, Lee MS, Kim HL, Chin H, Suh PG, Kim SS, Shin HS. Requirement for the L-type calcium channel α1D subunit in postnatal pancreatic β cell generation. J Clin Invest. 2001;108:1015–1022. doi: 10.1172/JCI13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Seisenberger C, Specht V, Welling A, Platzer J, Pfeifer A, Kuhbandner S, Striessnig J, Klugbauer N, Feil R, Hofmann F. Functional embryonic cardiomyocytes after disruption of the L-type α1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem. 2000;275:39193–39199. doi: 10.1074/jbc.M006467200. [DOI] [PubMed] [Google Scholar]
- 108.Goonasekera SA, Hammer K, Auger-Messier M, Bodi I, Chen X, Zhang H, Reiken S, Elrod JW, Correll RN, York AJ, et al. Decreased cardiac L-type Ca2+ channel activity induces hypertrophy and heart failure in mice. J Clin Invest. 2012;122:280–290. doi: 10.1172/JCI58227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Blaich A, Pahlavan S, Tian Q, Oberhofer M, Poomvanicha M, Lenhardt P, Domes K, Wegener JW, Moosmang S, Ruppenthal S, et al. Mutation of the calmodulin binding motif IQ of the L-type Cav1.2 Ca2+ channel to EQ induces dilated cardiomyopathy and death. J Biol Chem. 2012;287:22616–22625. doi: 10.1074/jbc.M112.357921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rosati B, Yan Q, Lee MS, Liou SR, Ingalls B, Foell J, Kamp TJ, McKinnon D. Robust L-type calcium current expression following heterozygous knockout of the Cav1.2 gene in adult mouse heart. J Physiol. 2011;589:3275–3288. doi: 10.1113/jphysiol.2011.210237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Domes K, Ding J, Lemke T, Blaich A, Wegener JW, Brandmayr J, Moosmang S, Hofmann F. Truncation of murine Cav1.2 at Asp-1904 results in heart failure after birth. J Biol Chem. 2011;286:33863–33871. doi: 10.1074/jbc.M111.252312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fu Y, Westenbroek RE, Yu FH, Clark JP, III, Marshall MR, Scheuer T, Catterall WA. Deletion of the distal C terminus of Cav1.2 channels leads to loss of β-adrenergic regulation and heart failure in vivo. J Biol Chem. 2011;286:12617–12626. doi: 10.1074/jbc.M110.175307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liao P, Yong TF, Liang MC, Yue DT, Soong TW. Splicing for alternative structures of Cav1.2 calcium channels in cardiac and smooth muscles. Cardiovasc Res. 2005;68:197–203. doi: 10.1016/j.cardiores.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 114.Chen X, Nakayama H, Zhang X, Ai X, Harris DM, Tang M, Zhang H, Szeto C, Stockbower K, Berretta RM, et al. Calcium influx through Cav1.2 is a proximal signal for pathological cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2011;50:460–470. doi: 10.1016/j.yjmcc.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol. 2011;18:840–845. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- 116.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. discussion 8086–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barrett CF, Tsien RW. The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of Cav1.2 L-type calcium channels. Proc Natl Acad Sci U S A. 2008;105:2157–2162. doi: 10.1073/pnas.0710501105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gillis J, Burashnikov E, Antzelevitch C, Blaser S, Gross G, Turner L, Babul-Hirji R, Chitayat D. Long QT, syndactyly, joint contractures, stroke and novel CACNA1C mutation: expanding the spectrum of Timothy syndrome. Am J Med Genet A. 2012;158A:182–187. doi: 10.1002/ajmg.a.34355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fukuyama M, Ohno S, Wang Q, Kimura H, Makiyama T, Itoh H, Ito M, Horie M. L-type calcium channel mutations in Japanese patients with inherited arrhythmias. Circ J. 2013;77:1799–1806. doi: 10.1253/circj.cj-12-1457. [DOI] [PubMed] [Google Scholar]
- 120.Napolitano C, Antzelevitch C. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-type calcium channel. Circ Res. 2011;108:607–618. doi: 10.1161/CIRCRESAHA.110.224279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda JC, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol. 2009;75:407–414. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- 122.Huang H, Tan BZ, Shen Y, Tao J, Jiang F, Sung YY, Ng CK, Raida M, Kohr G, Higuchi M, et al. RNA editing of the IQ domain in Cav1.3 channels modulates their Ca2+-dependent inactivation. Neuron. 2012;73:304–316. doi: 10.1016/j.neuron.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Busquet P, Nguyen NK, Schmid E, Tanimoto N, Seeliger MW, Ben-Yosef T, Mizuno F, Akopian A, Striessnig J, Singewald N. Cav1.3 L-type calcium channels modulate depression-like behaviour in mice independent of deaf phenotype. Int J Neuropsychopharmacol. 2010;13:499–513. doi: 10.1017/S1461145709990368. [DOI] [PubMed] [Google Scholar]
- 124.Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α 1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Di Biase V, Obermair GJ, Szabo Z, Altier C, Sanguesa J, Bourinet E, Flucher BE. Stable membrane expression of postsynaptic Cav1.2 calcium channel clusters is independent of interactions with AKAP79/150 and PDZ proteins. J Neurosci. 2008;28:13845–13855. doi: 10.1523/JNEUROSCI.3213-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang H, Fu Y, Altier C, Platzer J, Surmeier DJ, Bezprozvanny I. Cav1.2 and Cav1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur J Neurosci. 2006;23:2297–2310. doi: 10.1111/j.1460-9568.2006.04734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wheeler DG, Barrett CF, Groth RD, Safa P, Tsien RW. CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J Cell Biol. 2008;183:849–863. doi: 10.1083/jcb.200805048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ma H, Cohen S, Li B, Tsien RW. Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci Rep. 2013;33:97–101. doi: 10.1042/BSR20120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal Cav1.3 L-type calcium channels is dependent on a Shank-binding domain. J Neurosci. 2005;25:1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry. 2007;46:1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- 131.Wankerl K, Weise D, Gentner R, Rumpf JJ, Classen J. L-type voltage-gated calcium channels: a single molecular switch for long-term potentiation/long-term depression-like plasticity and activity-dependent metaplasticity in humans. J Neurosci. 2010;30:6197–6204. doi: 10.1523/JNEUROSCI.4673-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mansergh F, Orton NC, Vessey JP, Lalonde MR, Stell WK, Tremblay F, Barnes S, Rancourt DE, Bech-Hansen NT. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet. 2005;14:3035–3046. doi: 10.1093/hmg/ddi336. [DOI] [PubMed] [Google Scholar]
- 133.Wu J, Marmorstein AD, Striessnig J, Peachey NS. Voltage-dependent calcium channel Cav1.3 subunits regulate the light peak of the electroretinogram. J Neurophysiol. 2007;97:3731–3735. doi: 10.1152/jn.00146.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, Stiess M, Marais E, Schulla V, Lacinova L, et al. Role of hippocampal Cav1.2 calcium channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.White JA, McKinney BC, John MC, Powers PA, Kamp TJ, Murphy GG. Conditional forebrain deletion of the L-type calcium channel Cav1.2 disrupts remote spatial memories in mice. Learn Mem. 2008;15:1–5. doi: 10.1101/lm.773208. [DOI] [PubMed] [Google Scholar]
- 136.Lacinova L, Moosmang S, Langwieser N, Hofmann F, Kleppisch T. Cav1.2 calcium channels modulate the spiking pattern of hippocampal pyramidal cells. Life Sci. 2008;82:41–49. doi: 10.1016/j.lfs.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 137.Gamelli AE, McKinney BC, White JA, Murphy GG. Deletion of the L-type calcium channel Cav 1.3 but not Cav1.2 results in a diminished sAHP in mouse CA1 pyramidal neurons. Hippocampus. 2011;21:133–141. doi: 10.1002/hipo.20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.McKinney BC, Murphy GG. The L-type voltage-gated calcium channel Cav1.3 mediates consolidation, but not extinction, of contextually conditioned fear in mice. Learn Mem. 2006;13:584–589. doi: 10.1101/lm.279006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Busquet P, Hetzenauer A, Sinnegger-Brauns MJ, Striessnig J, Singewald N. Role of L-type calcium channel isoforms in the extinction of conditioned fear. Learn Mem. 2008;15:378–386. doi: 10.1101/lm.886208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McKinney BC, Sze W, Lee B, Murphy GG. Impaired long-term potentiation and enhanced neuronal excitability in the amygdala of Cav1.3 knockout mice. Neurobiol Learn Mem. 2009;92:519–528. doi: 10.1016/j.nlm.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Langwieser N, Christel CJ, Kleppisch T, Hofmann F, Wotjak CT, Moosmang S. Homeostatic switch in hebbian plasticity and fear learning after sustained loss of Cav1.2 calcium channels. J Neurosci. 2010;30:8367–8375. doi: 10.1523/JNEUROSCI.4164-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lee AS, Ra S, Rajadhyaksha AM, Britt JK, De Jesus-Cortes H, Gonzales KL, Lee A, Moosmang S, Hofmann F, Pieper AA, et al. Forebrain elimination of cacna1c mediates anxiety-like behavior in mice. Mol Psychiatry. 2012;17:1054–1055. doi: 10.1038/mp.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hetzenauer A, Sinnegger-Brauns MJ, Striessnig J, Singewald N. Brain activation pattern induced by stimulation of L-type Ca2+-channels: contribution of Cav1.3 and Cav1.2 isoforms. Neuroscience. 2006;139:1005–1015. doi: 10.1016/j.neuroscience.2006.01.059. [DOI] [PubMed] [Google Scholar]
- 144.Krey JF, Pasca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, Dolmetsch RE. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16:201–209. doi: 10.1038/nn.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cross-Disorder Group of the Psychiatric Genomics Consortium. Smoller JW, Craddock N, Kendler K, Lee PH, Neale BM, Nurnberger JI, Ripke S, Santangelo S, Sullivan PF. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM, Gould TD. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol. 2012;99:1–14. doi: 10.1016/j.pneurobio.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Giordano TP, Tropea TF, Satpute SS, Sinnegger-Brauns MJ, Striessnig J, Kosofsky BE, Rajadhyaksha AM. Molecular switch from L-type Cav1.3 to Cav1.2 Ca2+ channel signaling underlies long-term psychostimulant-induced behavioral and molecular plasticity. J Neurosci. 2010;30:17051–17062. doi: 10.1523/JNEUROSCI.2255-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Schierberl K, Hao J, Tropea TF, Ra S, Giordano TP, Xu Q, Garraway SM, Hofmann F, Moosmang S, Striessnig J, et al. Cav1.2 L-type Ca2+-channels mediate cocaine-induced GluA1 trafficking in the nucleus accumbens, a long-term adaptation dependent on ventral tegmental area Cav1.3 channels. J Neurosci. 2011;31:13562–13575. doi: 10.1523/JNEUROSCI.2315-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schierberl K, Giordano T, Satpute S, Hao J, Kaur G, Hofmann F, Moosmang S, Striessnig J, Rajadhyaksha A. Cav 1.3 L-type Ca2+ channels mediate long-term adaptation in dopamine D2L-mediated GluA1 trafficking in the dorsal striatum following cocaine exposure. Channels (Austin) 2012;6:11–17. doi: 10.4161/chan.19324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schuster S, Doudnikoff E, Rylander D, Berthet A, Aubert I, Ittrich C, Bloch B, Cenci MA, Surmeier DJ, Hengerer B, et al. Antagonizing L-type Ca2+ channel reduces development of abnormal involuntary movement in the rat model of L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol Psychiatry. 2009;65:518–526. doi: 10.1016/j.biopsych.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 151.Liss B, Roeper J. Individual dopamine midbrain neurons: functional diversity and flexibility in health and disease. Brain Res Rev. 2008;58:314–321. doi: 10.1016/j.brainresrev.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 152.Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. 'Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 153.Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis. 2011;43:364–371. doi: 10.1016/j.nbd.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kupsch A, Gerlach M, Pupeter SC, Sautter J, Dirr A, Arnold G, Opitz W, Przuntek H, Riederer P, Oertel WH. Pretreatment with nimodipine prevents MPTP-induced neurotoxicity at the nigral, but not at the striatal level in mice. Neuroreport. 1995;6:621–625. doi: 10.1097/00001756-199503000-00009. [DOI] [PubMed] [Google Scholar]
- 155.Kupsch A, Sautter J, Schwarz J, Riederer P, Gerlach M, Oertel WH. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in non-human primates is antagonized by pretreatment with nimodipine at the nigral, but not at the striatal level. Brain Res. 1996;741:185–196. doi: 10.1016/s0006-8993(96)00917-1. [DOI] [PubMed] [Google Scholar]
- 156.Surmeier DJ, Guzman JN, Sanchez-Padilla J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson's disease. Cell Calcium. 2010;47:175–182. doi: 10.1016/j.ceca.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Putzier I, Kullmann PH, Horn JP, Levitan ES. Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J Neurosci. 2009;29:15414–15419. doi: 10.1523/JNEUROSCI.4742-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci. 2007;27:645–656. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH, Sulzer D. Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol. 2010;67:600–606. doi: 10.1002/ana.21937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Becker C, Jick SS, Meier CR. Use of antihypertensives and the risk of Parkinson disease. Neurology. 2008;70:1438–1444. doi: 10.1212/01.wnl.0000303818.38960.44. [DOI] [PubMed] [Google Scholar]
- 163.Pasternak B, Svanstrom H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson's disease. Am J Epidemiol. 2012;175:627–635. doi: 10.1093/aje/kwr362. [DOI] [PubMed] [Google Scholar]
- 164.Simon KC, Gao X, Chen H, Schwarzschild MA, Ascherio A. Calcium channel blocker use and risk of Parkinson's disease. Mov Disord. 2010;25:1818–1822. doi: 10.1002/mds.23191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hirtz JJ, Braun N, Griesemer D, Hannes C, Janz K, Lohrke S, Muller B, Friauf E. Synaptic refinement of an inhibitory topographic map in the auditory brainstem requires functional Cav1.3 calcium channels. J Neurosci. 2012;32:14602–14616. doi: 10.1523/JNEUROSCI.0765-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hirtz JJ, Boesen M, Braun N, Deitmer JW, Kramer F, Lohr C, Muller B, Nothwang HG, Striessnig J, Lohrke S, et al. Cav1.3 calcium channels are required for normal development of the auditory brainstem. J Neurosci. 2011;31:8280–8294. doi: 10.1523/JNEUROSCI.5098-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Satheesh SV, Kunert K, Ruttiger L, Zuccotti A, Schonig K, Friauf E, Knipper M, Bartsch D, Nothwang HG. Retrocochlear function of the peripheral deafness gene Cacna1d. Hum Mol Genet. 2012;21:3896–3909. doi: 10.1093/hmg/dds217. [DOI] [PubMed] [Google Scholar]
- 168.Middlemiss DN, Spedding M. A functional correlate for the dihydropyridine binding site in rat brain. Nature. 1985;314:94–96. doi: 10.1038/314094a0. [DOI] [PubMed] [Google Scholar]
- 169.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]