

Abstract
Quantitative analysis of administered drugs in biological tissues is essential for understanding the mechanisms underlying their efficacy or toxicity. Imaging mass spectrometry (IMS) may allow the quantification of targeted drugs in tissue sections along with the visualization of their spatial distribution. In this study, surrogate tissue-based calibration standards were prepared to quantify a small molecule drug (S-777469 or raclopride) in tissue sections of mice administered with the drug, followed by analysis with a linear ion trap mass spectrometer equipped with a matrix-assisted laser desorption/ionization (MALDI) source. The distribution of the drugs in the dissected organs was clearly visualized by MALDI-IMS. The drug concentration determined using the calibration standards prepared for MALDI-IMS analysis was highly consistent with that determined by liquid chromatography-tandem mass spectrometry, and the quantification in multiple organs was enabled. The results of this study show that MALDI-IMS can be used to quantify small molecule drugs in biological tissue sections using surrogate tissue-based calibration standards.
Keywords: imaging mass spectrometry, matrix-assisted laser desorption/ionization, quantification
INTRODUCTION
Quantitative analysis of the in vivo distribution of a drug candidate after its administration is essential in drug discovery and development as it facilitates the understanding of the mechanisms underlying the pharmacological or toxicological effects of the drug.1–3) Autoradiography (ARG) is currently the standard tool for examining drug distribution in animals in the pharmaceutical industry, because it allows quantification of drug concentration as well as visualization of drug distribution in tissue sections.4–7) However, ARG has some technical limitations as it is based on the detection of radioactivity. A large number of drug candidates are assessed during the discovery stage of pharmaceutical research and it is not time- or cost-effective to synthesize a radiolabeled form of each candidate. Further, the quantitative and spatial information obtained from ARG may be derived from both a drug and its metabolites, which complicates the interpretation of the pharmacokinetic properties of the drug. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) with electrospray ionization (ESI) is another method used for drug distribution studies. LC-MS/MS has advantages in quantification, sensitivity, and selectivity, and it has been applied to measure drug concentrations in various organs.8,9) However, LC-MS/MS cannot provide spatial information on the drug distribution within an organ, because samples have to be homogenized prior to analysis.
Imaging mass spectrometry (IMS) has been developed to visualize the distribution of drugs or biological molecules in tissue sections without radiolabeling,10,11) and the application of this technique has been increasing in recent years.12–14) IMS, due to its MS-based detection, can distinguish between the distribution of a drug and that of its metabolites in tissue sections. Therefore, it has the potential to be an effective imaging technique for drug distribution studies.15,16)
Matrix-assisted laser desorption/ionization (MALDI), a soft ionization technique that allows both small molecules (drugs, lipids, and endogenous metabolites)17–19) and large molecules (peptides and proteins) to be analyzed,20,21) has been used frequently to detect target molecules in biological tissue sections in IMS studies. However, there are some limitations on the quantitative analysis with MALDI technique. Ion suppression can be a major limitation because endogenous biomolecules in tissue samples may be ionized simultaneously with the analytes in the MALDI source. Specifically in MALDI-IMS, the heterogeneous distribution of endogenous biomolecules within a single tissue section could complicate the quantitative analysis of the analyte. In addition, the incorrect application of matrix compounds could cause the inhomogeneous formation of co-crystals within a tissue section, resulting in reduced quantitative information. Therefore, normalizing the ionization efficiency of a target analyte is necessary for quantitative analysis by MALDI-IMS.22–24) The preparation of analyte calibration standards is also a key factor for successful quantification because analyte extraction from tissue sections into the matrix solution should be considered.16) Several studies on MALDI-IMS method development for the quantification of small molecule drugs have been reported in the past few years.25) In most cases, analyte standard solution spotted onto control tissue sections was used as the calibration standard, but the methodology can still be improved by preparing calibration standards which mimic the incurred tissue samples well.
In this study, we developed a novel method for quantification of small molecule drugs by MALDI-IMS, which involves the combined use of homogenized tissue-based calibration standards and normalization of analyte ionization efficiency with a structural analog. The method was applied to quantify S-777469 (1-((6-ethyl-1-(4-fluorobenzyl)-5-methyl-2-oxo-1,2-dihydropyridine-3-carbonyl)amino)-cyclohexanecarboxylic acid), a synthetic cannabinoid receptor 2 selective agonist,26) or raclopride (RCP), a dopamine D2 receptor selective antagonist,27) in tissue sections from different mouse organs, and evaluated.
EXPERIMENTAL
Chemicals and reagents
S-777469 and deuterated S-777469 (S-777469-d10) were synthesized at Shionogi & Co., Ltd. (Osaka, Japan). RCP tartrate salt, sulpiride (SLP) and 2,5-dihydroxybenzoic acid (DHB) were purchased from Sigma-Aldrich (St. Louis, MO, USA). High-performance LC-grade methanol, acetonitrile, isopropanol, and special-grade formic acid, were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan). Dimethyl sulfoxide (DMSO) was obtained from Nacalai Tesque Inc. (Kyoto, Japan). N,N-Dimethylacetamide (DMA), propylene glycol (PG), and trifluoroacetic acid (TFA) were obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Isoflurane was purchased from Abbott Japan Co., Ltd. (Tokyo, Japan).
Animal experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee of Shionogi & Co., Ltd. Male 8-week old ddY mice were purchased from Japan SLC (Hamamatsu, Japan) and housed under normal conditions until the experiments. Mice, weighing 35.8–40.3 g (n=2), were administered S-777469 in DMA/PG (1 : 1) or RCP in saline (10 mg/10 mL/kg, each) by tail vein injection, and were then euthanized by isoflurane anesthesia and exsanguinated via the inferior vena cava at 10 min post-dose administration. The liver, brain, lungs, and kidneys of each mouse were dissected, and then the liver was divided in two. For LC-MS/MS analysis, half of the liver, the right brain, the right lung, and the right kidney were weighed, and 4 mL of distilled water/g of tissue were added before the organs were homogenized using an Ultra-Turrax T10 (IKA Works Inc., Wilmington, NC, USA). The obtained tissue homogenates were stored at −80°C until analysis. For the MALDI-IMS analysis, the other half of the liver, the left brain, the left lung, and the left kidney were snap-frozen in liquid nitrogen and stored at −80°C until sectioning.
LC-MS/MS analysis
An Acquity ultra-performance LC (UPLC) system (Waters Corp., Milford, MA, USA) with a triple quadrupole mass spectrometer (API 5000™, AB Sciex, Foster City, CA, USA) was used for LC-MS/MS analysis. The LC and MS/MS systems were controlled by Masslynx 4.1 (Waters Corp.) and Analyst 1.4.2 (AB Sciex) software, respectively. To determine the concentration of S-777469 or RCP in the samples, 500 μL of acetonitrile was added to 40 μL of the tissue samples. The mixture was stirred for a few minutes and centrifuged at 5,000 rpm for 5 min at 10°C, and the supernatant was diluted 10-fold with acetonitrile. Then, 2 μL of the diluted supernatant was injected into the LC-MS/MS system. Chromatographic separation was achieved using an Acquity UPLC™ BEH C18 column (50×2.1 mm i.d., 1.7 mm; Waters Corp.) at 40°C. A temperature of 10°C was maintained in the autosampler compartment throughout the analysis. Isopropanol and water containing 10% acetonitrile were used in the autosampler as strong- and weak-wash solvents, respectively. The analyte was eluted with a 10–95% gradient of acetonitrile containing 0.1% formic acid at a flow rate of 750 μL/min, and was electrosprayed into the mass spectrometer. ESI was performed in the positive-ion mode, and transitions of m/z 415→109 at a collision energy of 35 eV and m/z 347→112 at a collision energy of 25 eV were used for selected reaction monitoring of S-777469 and RCP, respectively. The concentration of S-777469 or RCP was calculated from a calibration curve generated by analyzing a series of tissue samples from non-treated mice, containing known quantities of S-777469 or RCP, using the Analyst software.
In vitro MALDI analysis of S-777469
A standard solution of S-777469 (2 μg/mL in methanol–water [1 : 1, v/v] with 0.2% TFA) was mixed with an equal volume of DHB solution (60 mg/mL in methanol–water [1 : 1, v/v] with 0.2% TFA), spotted onto a MALDI target plate (Thermo Fisher Scientific Inc., Waltham, MA, USA), and analyzed to obtain the MS or MS/MS spectra of S-777469 using a linear ion trap mass spectrometer with a MALDI source (MALDI LTQ XL, Thermo Fisher Scientific Inc.) equipped with a nitrogen laser (337 nm; 60 Hz). The laser energy was set at 15 μJ. Auto gain control (where the ion trap is filled with the optimum number of ions) was not used in any of the experiments. For the MS/MS analysis, S-777469 was detected in the positive-ion mode by using a product ion scan of the [M+H]+ ion of S-777469 (m/z 415) with a precursor ion isolation width of 3.0 Da.
Sample preparation for calibration standards in MALDI-IMS
A stock solution of 150 mg/mL S-777469 or 20 mg/mL RCP in DMSO was prepared and diluted stepwise with methanol–water (1 : 1, v/v) to prepare S-777469 and RCP standard solutions at concentrations from 0.1 to 15 mg/mL and 0.1 to 2 mg/mL, respectively. The liver dissected from non-treated mice was homogenized in 0.333 mL distilled water/g of liver tissue using an Ultra-Turrax T10, spiked with the S-777469 or RCP standard solutions to prepare calibration standards with final concentrations from 1 to 150 μg/g or 1 to 20 μg/g, respectively, and immediately frozen.
Sample preparation for MALDI-IMS
Frozen 20-μm thick sections were prepared from mouse tissues or the calibration standards with a cryostat (Leica CM3050 S; Leica Microsystems Inc., Wetzlar, Germany) at −20°C. The sections were placed onto glass microscope slides (Superfrost; Thermo Fisher Scientific Inc.) and stored at −80°C until analysis. Prior to matrix coating and mass spectrometric analysis, the sections were placed in a vacuum desiccator for 15 min at room temperature, and optical images were acquired using a scanner (Scanjet G4050; Hewlett-Packard Co., Palo Alto, CA, USA) to identify the location of each tissue. S-777469-d10 or SLP (10 μg/mL in 1 : 1, v/v methanol–water) were used as the internal standard solution for S-777469 or RCP, respectively, and were uniformly coated over the sections (3 mL/glass slide) using a 0.5 mm nozzle caliber airbrush (HP-TH, Anest Iwata Corp., Yokohama, Japan) held at 40 cm from the sections. The sections were then coated with the matrix solution (30 mg/mL DHB dissolved in 1 : 1, v/v methanol–water containing 0.2% TFA) using an ImagePrep™ automated device utilizing vibrational vaporization technology (Bruker Daltonics Inc., Billerica, MA, USA).
MALDI-IMS
The matrix-coated sections were analyzed with a MALDI LTQ XL. The laser energy and the raster step size were set at 32 μJ and 250 μm, respectively. S-777469, S-777469-d10, RCP, and SLP were detected in the positive-ion mode using product ion scans of their [M+H]+ ions, with a precursor ion isolation width of 3.0 Da. The collision energy was 35% and 40% of the maximum available energy required for the complete fragmentation of the precursor ion derived from the Met-Arg-Phe-Ala peptide for S-777469/S-777469-d10 and RCP/SLP, respectively.28) The spectral data were then transformed to image data using ImageQuest 1.0.1 software (Thermo Fisher Scientific Inc.). The entire visible tissue area in each section was selected as the region of interest (ROI) for the quantitative image data analysis, and the signal intensities of S-777469 (m/z 415→369), S-777469-d10 (m/z 425→379), RCP (m/z 347→129), and SLP (m/z 342→112) within each ROI were averaged respectively, using ImageQuest.
RESULTS AND DISCUSSION
Optimization of the analytical conditions for MALDI-IMS detection of S-777469 and RCP
Chemical structures of S-777469 and RCP are shown in Fig. 1. The MS spectrum from a standard solution of S-777469 mixed with DHB was obtained in the positive-ion mode and the [M+H]+ ion of S-777469 (m/z 415) was detected (Fig. 2a). The MS/MS spectrum of S-777469 was also obtained using a product ion scan of the [M+H]+ ion (m/z 415) with an optimized collision energy of 35%. As shown in Fig. 2b, a product ion at m/z 369, resulting from the loss of the carboxyl group, had a relatively high intensity. Thus, the m/z 415→369 transition was used to obtain the S-777469-specific ion images in subsequent MALDI-IMS studies. For RCP detection, the m/z 347→129 transition was used as previously described.29) TFA was added to the matrix solution as is the case in recent reports.22,23)
Fig. 1. Chemical structures of (a) S-777469 and (b) RCP. The cleavages by MS/MS analysis related with the detection for MALDI-IMS were also illustrated.
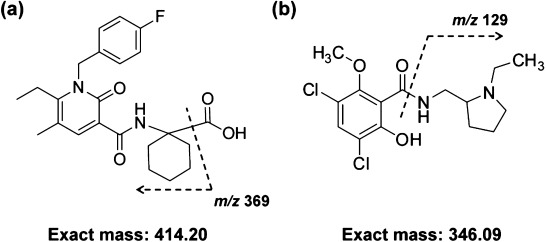
Fig. 2. (a) MALDI-MS spectrum from a standard solution of S-777469 mixed with DHB in the positive-ion mode. The [M+H]+ ion of S-777469 (m/z 415) was detected. (b) MALDI-MS/MS spectrum of m/z 415 from a standard solution of S-777469 mixed with DHB. m/z 369 had relatively high intensities.
![Fig. 2. (a) MALDI-MS spectrum from a standard solution of S-777469 mixed with DHB in the positive-ion mode. The [M+H]+ ion of S-777469 (m/z 415) was detected. (b) MALDI-MS/MS spectrum of m/z 415 from a standard solution of S-777469 mixed with DHB. m/z 369 had relatively high intensities.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=3968287_massspectrometry-3-1-A0025-g002.jpg)
Quantification of S-777469 in tissue sections by MALDI-IMS
The distribution of S-777469 (m/z 415→369) in mouse tissue sections was clearly visualized by MALDI-IMS (Fig. 3e). S-777469 was present in both the kidney and liver at 10 min post-dose administration.
Fig. 3. Representative optical images (a and d) and ion images of m/z 415→369 (b and e) and m/z 425→379 (c and f) on the sections of the calibration standards (a–c) or the kidney and liver (d–f). Bar=5 mm.

To determine S-777469 concentration in the tissue sections by MALDI-IMS, homogenized liver spiked with known quantities of S-777469 was used as a calibration standard. The analysis of the calibration standards by MALDI-IMS was performed with relative standard deviation (RSD) of 7.3–21.2% (Table 1). For quantification of S-777469 in mouse tissue sections, duplicate sections of the calibration standard with 6 different concentrations of S-777469 were coated with S-777469-d10 solution followed by matrix solution application and MALDI-IMS analysis, in the same manner as the test tissue sections (Fig. 3b). The signal intensity ratio of S-777469/S-777469-d10 versus S-777469 concentration (within the 1–150 μg/g range) was fit to a linear regression curve, as shown in Fig. 4. The correlation coefficient was 0.996 (p<0.01). Using the calibration curve, the concentration of S-777469 in the tissue sections was calculated from the signal intensity ratio of S-777469/S-777469-d10 measured by MALDI-IMS. The calculated concentration of S-777469 was divided by the concentration determined by LC-MS/MS to calculate the accuracy (%) for the validation of the MALDI-IMS-based quantification method. The S-777469 concentrations in the tissue sections determined by MALDI-IMS were similar to those quantified by LC-MS/MS with accuracy of 82–160% (Table 2). In agreement with this, the plots of the S-777469 concentration quantified by LC-MS/MS versus the signal intensity ratio of S-777469/S-777469-d10 obtained by MALDI-IMS were very close to the calibration curve (Fig. 4). These results indicate that the concentrations of S-777469 in tissue sections were successfully quantified with the MALDI-IMS-based method. For more precise quantification, the inhomogeneity of the distribution of a targeted drug across tissue sections should be considered. To overcome this, the 3D IMS technique might be useful.30,31)
Table 1. Reproducibility evaluation of the MALDI-IMS-based quantification method using the surrogate tissue-based calibration standards.
| Analyte | Analyte concentration (μg/g) | Signal intensity ratio of analyte/internal standard† | RSD (%) |
|---|---|---|---|
| S-777469 | 1 | 0.271±0.057 | 21.2 |
| 20 | 1.26±0.15 | 12.1 | |
| 150 | 7.65±0.56 | 7.3 | |
| RCP | 1 | 0.0499±0.0084 | 16.8 |
| 5 | 0.306±0.031 | 10.1 | |
| 20 | 1.24±0.05 | 4.4 |
† Mean±S.D., n=3.
Fig. 4. Correlation between the concentrations of S-777469 in the calibration standards and the signal intensity ratios of S-777469/S-777469-d10 measured by MALDI-IMS, indicated by open circles. The filled symbols indicate the tissue samples of the kidney (blue) or liver (green) of the mouse No. 1 (squares) or No. 2 (circles), plotted with the concentration of S-777469 determined by LC-MS/MS on the horizontal axis and the signal intensity ratio of S-777469/S-777469-d10 measured by MALDI-IMS on the vertical axis.
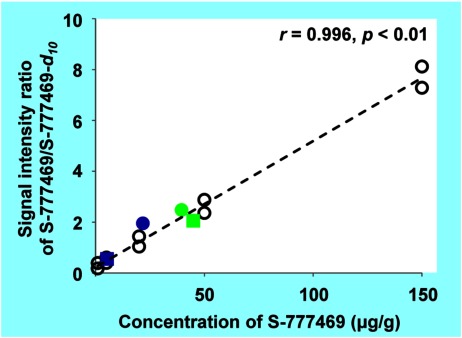
Table 2. Comparison between the drug concentrations quantified by LC-MS/MS and those by MALDI-IMS in each tissue of mice after intravenous administration of S-777469 or RCP.
| LC-MS/MS quantification (μg/g) | MALDI-IMS quantification (μg/g) | Accuracy (%) | |||
|---|---|---|---|---|---|
| S-777469-dosed | Kidney | Mouse | |||
| #1 | 5.31 | 6.51 | 122 | ||
| #2 | 21.7 | 34.8 | 160 | ||
| Liver | Mouse | ||||
| #1 | 45.0 | 36.9 | 82 | ||
| #2 | 39.5 | 45.4 | 115 | ||
| RCP-dosed | Brain | Mouse | |||
| #1 | 2.25 | 2.33 | 104 | ||
| #2 | 1.59 | 2.50 | 157 | ||
| Lung | Mouse | ||||
| #1 | 1.60 | 1.97 | 123 | ||
| #2 | 1.36 | 2.14 | 157 | ||
| Kidney | Mouse | ||||
| #1 | 2.23 | 2.92 | 131 | ||
| #2 | 1.69 | 2.06 | 122 | ||
| Liver | Mouse | ||||
| #1 | 13.4 | 16.1 | 120 | ||
| #2 | 9.04 | 10.3 | 113 | ||
MALDI-IMS analysis often requires a normalization method for precise imaging and quantification because it is necessary to control for inhomogeneous sample-matrix co-crystallization within a single tissue section and for tissue-specific ion suppression. We previously reported a normalization method that used the signal intensity of a matrix compound, uniformly coated on tissue sections, as an internal standard.29,32) This method has the advantages that it can be applied to different types of drugs and it can compensate for the variability of sample-matrix co-crystallization. On the other hand, a stable isotope-labeled version of the target drug is considered ideal for the normalization of ion suppression.24) In this study, we used S-777469-d10, a deuterium-labeled version of S-777469, as an internal standard to maximize the quantitative capability of MALDI-IMS, and uniformly coated S-777469-d10 solution over the sections to allow pixel-by-pixel normalization (Figs. 3c and f).
The calibration standard preparation method is also important for MALDI-IMS quantification. Although several groups have successfully used analyte solutions with different concentrations spotted onto control tissue sections as calibration standards,25) an improved method for calibration standard preparation could allow more precise MALDI-IMS quantification. MS-based quantification generally requires calibration standards which mimic the incurred samples as closely as possible.33) Therefore, we used blank liver homogenates spiked with S-777469 as surrogate tissue samples for calibration standards, in which the volume of distilled water added prior to homogenization was reduced to 1/3 equivalents to avoid excessive dilution of endogenous salts and proteins. The successful quantification of S-777469 concentration in tissue sections suggests that the surrogate tissue-based calibration standards are useful for MALDI-IMS quantification considering the extraction process of the analyte from tissue sections. In addition, our method enabled the simultaneous quantification of S-777469 in multiple organs.
Quantification of RCP in tissue sections by MALDI-IMS
The distribution of RCP (m/z 347→129) in mouse tissue sections was clearly visualized by MALDI-IMS (Fig. 5e). At 10 min post-dose administration, RCP was present in the analyzed organs, and a higher signal was observed in the liver than in the brain, lung, and kidney.
Fig. 5. Representative optical images (a and d) and ion images of m/z 347→129 (b and e) and m/z 342→112 (c and f) on the sections of the calibration standards (a–c) or the brain, lung, kidney, and liver (d–f). Bar=5 mm.

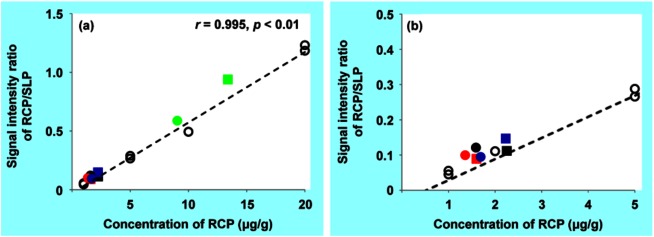
Quantification of the RCP concentration in tissue sections by MALDI-IMS was conducted in the same manner as for S-777469. In this case, SLP was used as an internal standard for normalization of the ionization efficiency of RCP, as shown in Figs. 5c and f. SLP is not a stable isotope-labeled version of RCP, but a structural analog with dopamine D2 receptor selective antagonism function.34) The analysis of the calibration standards by MALDI-IMS was performed with RSD of 4.4–16.8% (Table 1). The signal intensity ratio of RCP/SLP in the calibration standard sections versus RCP concentration (within the 1–20 μg/g range) was fit to a linear regression curve, as shown in Fig. 6. The correlation coefficient was 0.995 (p<0.01). Using the calibration curve, the concentration of RCP in the tissue sections was calculated from the signal intensity ratio of RCP/SLP measured by MALDI-IMS. Table 2 shows the calculated concentrations of RCP in the tissue sections, which are similar to those measured by LC-MS/MS with accuracy of 104–157%. Consistent with this, the plots of the RCP concentration measured by LC-MS/MS versus the signal intensity ratio of RCP/SLP, determined by MALDI-IMS, were very close to the calibration curve (Fig. 6). These results suggest that the RCP concentrations in the tissue sections were successfully measured with the MALDI-IMS-based method. The successful quantification of RCP by using SLP as an internal standard indicates that a structural analog of the target drug can be used to normalize for ionization efficiency if a stable isotope-labeled compound is not available due to time- or cost-constraints.
Fig. 6. (a) Correlation between the concentrations of RCP in the calibration standards and the signal intensity ratios of RCP/SLP measured by MALDI-IMS, indicated by open circles. The filled symbols indicate the tissue samples of the brain (black), lung (red), kidney (blue), or liver (green) of the mouse No. 1 (squares) or No. 2 (circles), plotted with the concentration of RCP determined by LC-MS/MS on the horizontal axis and the signal intensity ratio of RCP/SLP measured by MALDI-IMS on the vertical axis. (b) shows the correlation for lower concentrations.
The surrogate tissue-based calibration standards were also used to quantify RCP in tissue sections of multiple organs (liver, brain, lung, and kidney). The liver homogenate calibration standards quantified RCP concentration not only in the liver, but also in the brain, lung, and kidney tissue sections. This suggests that the extraction process of RCP from tissue sections might not widely vary between the four organs that were examined. To extend the use of these calibration standards, they would need to be tested for the analysis of different types of tissue, such as adipose and hard bone.
CONCLUSION
Our study demonstrated that MALDI-IMS could achieve the quantification of small molecule drugs in biological tissue sections using the surrogate tissue-based calibration standards as well as the visualization of drug distribution. Furthermore, the calibration standards could work for the simultaneous quantification in multiple organs. These findings suggest that MALDI-IMS can be applied at the drug discovery stage for the selection of drug candidates with an ideal distribution profile in target tissues.
Acknowledgments
The authors would like to thank Shuhei Shigaki, Yukiko Masaki, and Tomoyuki Ohkawa (Drug Developmental Research Laboratories, Shionogi & Co., Ltd.) for their helpful discussions.
Please cite this article as: N. Takai, et al., Quantification of Small Molecule Drugs in Biological Tissue Sections by Imaging Mass Spectrometry Using Surrogate Tissue-Based Calibration Standards, Mass Spectrom (Tokyo) 2014; 3(1): A0025; DOI: 10.5702/massspectrometry.A0025
References
- 1) A. M. Monro. Are routine tissue distribution studies justifiable for approval of human drugs? Drug Metab. Dispos. 22: 341–342, 1994 [PubMed] [Google Scholar]
- 2) D. B. Campbell. Are we doing too many animal biodisposition investigations before phase I studies in man? A re-evaluation of the timing and extent of ADME studies. Eur. J. Drug Metab. Pharmacokinet. 19: 283–293, 1994 [DOI] [PubMed] [Google Scholar]
- 3) K. Mizojiri, H. Shindo, Y. Ohno. The possibility of predicting tissue accumulation after repeated dosing using a single-dose tissue distribution study. J. Toxicol. Sci. 21: 523–527, 1996 [DOI] [PubMed] [Google Scholar]
- 4) W. Steinke, Y. Archimbaud, M. Becka, R. Binder, U. Busch, P. Dupont, J. Maas. Quantitative distribution studies in animals: Cross-validation of radioluminography versus liquid-scintillation measurement. Regul. Toxicol. Pharmacol. 31: S33–S43, 2000 [DOI] [PubMed] [Google Scholar]
- 5) R. d’Argy, A. Sundwall. Quantitative whole-body radioluminography—Future strategy for balance and tissue distribution studies. Regul. Toxicol. Pharmacol. 31: S57–S62, 2000 [DOI] [PubMed] [Google Scholar]
- 6) E. G. Solon, L. Kraus. Quantitative whole-body autoradiography in the pharmaceutical industry: Survey results on study design, methods, and regulatory compliance. J. Pharmacol. Toxicol. Methods 46: 73–81, 2002 [DOI] [PubMed] [Google Scholar]
- 7) K. Inazawa, M. Koike, T. Yamaguchi. Study on the practical use of quantitative whole-body auto-radioluminography. Exp. Mol. Pathol. 76: 153–165, 2004 [DOI] [PubMed] [Google Scholar]
- 8) H. Jiang, J. Zeng, N. Zheng, H. Kandoussi, Q. Peng, J. L. Valentine, R. W. Lange, M. E. Arnold. A convenient strategy for quantitative determination of drug concentrations in tissue homogenates using a liquid chromatography/tandem mass spectrometry assay for plasma samples. Anal. Chem. 83: 6237–6244, 2011 [DOI] [PubMed] [Google Scholar]
- 9) Y. J. Xue, H. Gao, Q. C. Ji, Z. Lam, X. Fang, Z. J. Lin, M. Hoffman, D. Schulz-Jander, N. Weng. Bioanalysis of drug in tissue: Current status and challenges. Bioanalysis 4: 2637–2653, 2012 [DOI] [PubMed] [Google Scholar]
- 10) R. M. Caprioli, T. B. Farmer, J. Gile. Molecular imaging of biological samples: Localization of peptides and proteins using MALDI-TOF MS. Anal. Chem. 69: 4751–4760, 1997 [DOI] [PubMed] [Google Scholar]
- 11) M. L. Reyzer, Y. Hsieh, K. Ng, W. A. Korfmacher, R. M. Caprioli. Direct analysis of drug candidates in tissue by matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 38: 1081–1092, 2003 [DOI] [PubMed] [Google Scholar]
- 12) M. T. M. Blaze, A. Akhmetov, B. Aydin, P. D. Edirisinghe, G. Uygur, L. Hanley. Quantification of antibiotic in biofilm-inhibiting multilayers by 7.87 eV laser desorption postionization MS imaging. Anal. Chem. 84: 9410–9415, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13) L. Fornai, A. Angelini, I. Klinkert, F. Giskes, A. Kiss, G. Eijkel, E. A. Amstalden-van Hove, L. A. Klerk, M. Fedrigo, G. Pieraccini, G. Moneti, M. Valente, G. Thiene, R. M. Heeren. Three-dimensional molecular reconstruction of rat heart with mass spectrometry imaging. Anal. Bioanal. Chem. 404: 2927–2938, 2012 [DOI] [PubMed] [Google Scholar]
- 14) X. Liu, E. M. Weaver, A. B. Hummon. Evaluation of therapeutics in three-dimensional cell culture systems by MALDI imaging mass spectrometry. Anal. Chem. 85: 6295–6302, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15) S. Khatib-Shahidi, M. Andersson, J. L. Herman, T. A. Gillespie, R. M. Caprioli. Direct molecular analysis of whole-body animal tissue sections by imaging MALDI mass spectrometry. Anal. Chem. 78: 6448–6456, 2006 [DOI] [PubMed] [Google Scholar]
- 16) S. Castellino, M. R. Groseclose, D. Wagner. MALDI imaging mass spectrometry: Bridging biology and chemistry in drug development. Bioanalysis 3: 2427–2441, 2011 [DOI] [PubMed] [Google Scholar]
- 17) J. Gobey, M. Cole, J. Janiszewski, T. Covey, T. Chau, P. Kovarik, J. Corr. Characterization and performance of MALDI on a triple quadrupole mass spectrometer for analysis and quantification of small molecules. Anal. Chem. 77: 5643–5654, 2005 [DOI] [PubMed] [Google Scholar]
- 18) J. A. Marto, F. M. White, S. Seldomridge, A. G. Marshall. Structural characterization of phospholipids by matrix-assisted laser desorption/ionization Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 67: 3979–3984, 1995 [DOI] [PubMed] [Google Scholar]
- 19) S. Vaidyanathan, S. Gaskell, R. Goodacre. Matrix-suppressed laser desorption/ionisation mass spectrometry and its suitability for metabolome analyses. Rapid Commun. Mass Spectrom. 20: 1192–1198, 2006 [DOI] [PubMed] [Google Scholar]
- 20) M. Karas, F. Hillenkamp. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 60: 2299–2301, 1988 [DOI] [PubMed] [Google Scholar]
- 21) F. Hillenkamp, M. Karas. Mass spectrometry of peptides and proteins by matrix-assisted ultraviolet laser desorption/ionization. Methods Enzymol. 193: 280–295, 1990 [DOI] [PubMed] [Google Scholar]
- 22) B. Prideaux, V. Dartois, D. Staab, D. M. Weiner, A. Goh, L. E. Via, C. E. Barry 3rd, M. Stoeckli. High-sensitivity MALDI-MRM-MS imaging of moxifloxacin distribution in tuberculosis-infected rabbit lungs and granulomatous lesions. Anal. Chem. 83: 2112–2118, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23) G. Hamm, D. Bonnel, R. Legouffe, F. Pamelard, J. M. Delbos, F. Bouzom, J. Stauber. Quantitative mass spectrometry imaging of propranolol and olanzapine using tissue extinction calculation as normalization factor. J. Proteomics 75: 4952–4961, 2012 [DOI] [PubMed] [Google Scholar]
- 24) D. A. Pirman, R. F. Reich, A. Kiss, R. M. Heeren, R. A. Yost. Quantitative MALDI tandem mass spectrometric imaging of cocaine from brain tissue with a deuterated internal standard. Anal. Chem. 85: 1081–1089, 2013 [DOI] [PubMed] [Google Scholar]
- 25) N. Sun, A. Walch. Qualitative and quantitative mass spectrometry imaging of drugs and metabolites in tissue at therapeutic levels. Histochem. Cell Biol. 140: 93–104, 2013 [DOI] [PubMed] [Google Scholar]
- 26) M. Odan, N. Ishizuka, Y. Hiramatsu, M. Inagaki, H. Hashizume, Y. Fujii, S. Mitsumori, Y. Morioka, M. Soga, M. Deguchi, K. Yasui, A. Arimura. Discovery of S-777469: An orally available CB2 agonist as an antipruritic agent. Bioorg. Med. Chem. Lett. 22: 2803–2806, 2012 [DOI] [PubMed] [Google Scholar]
- 27) C. Köhler, H. Hall, S. O. Ogren, L. Gawell. Specific in vitro and in vivo binding of 3H-raclopride. A potent substituted benzamide drug with high affinity for dopamine D-2 receptors in the rat brain. Biochem. Pharmacol. 34: 2251–2259, 1985 [DOI] [PubMed] [Google Scholar]
- 28) L. L. Lopez, P. R. Tiller, M. W. Senko, J. C. Schwartz. Automated strategies for obtaining standardized collisionally induced dissociation spectra on a benchtop ion trap mass spectrometer. Rapid Commun. Mass Spectrom. 13: 663–668, 1999 [Google Scholar]
- 29) N. Takai, Y. Tanaka, K. Inazawa, H. Saji. Quantitative analysis of pharmaceutical drug distribution in multiple organs by imaging mass spectrometry. Rapid Commun. Mass Spectrom. 26: 1549–1556, 2012 [DOI] [PubMed] [Google Scholar]
- 30) E. H. Seeley, R. M. Caprioli. 3D imaging by mass spectrometry: A new frontier. Anal. Chem. 84: 2105–2110, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31) D. Trede, S. Schiffler, M. Becker, S. Wirtz, K. Steinhorst, J. Strehlow, M. Aichler, J. H. Kobarg, J. Oetjen, A. Dyatlov, S. Heldmann, A. Walch, H. Thiele, P. Maass, T. Alexandrov. Exploring three-dimensional matrix-assisted laser desorption/ionization imaging mass spectrometry data: Three-dimensional spatial segmentation of mouse kidney. Anal. Chem. 84: 6079–6087, 2012 [DOI] [PubMed] [Google Scholar]
- 32) N. Takai, Y. Tanaka, A. Watanabe, H. Saji. Quantitative imaging of a therapeutic peptide in biological tissue sections by MALDI MS. Bioanalysis 5: 603–612, 2013 [DOI] [PubMed] [Google Scholar]
- 33) M. T. Reimer, X. Wan, J. Zhang. A case study of validation and sample analysis for the determination of zotarolimus in various tissues. Bioanalysis 5: 811–817, 2013 [DOI] [PubMed] [Google Scholar]
- 34) M. Memo, L. Lucchi, P. F. Spano, M. Trabucchi. Aging process affects a single class of dopamine receptors. Brain Res. 202: 488–492, 1980 [DOI] [PubMed] [Google Scholar]