

Abstract
Traumatic brain injury (TBI) is a leading cause of death and disability among children and young adults in the United States. In this study, we explored whether changes in the gene expression profile of peripheral blood mononuclear cells (PBMC) may provide a clinically assessable “window” into the brain, reflecting molecular alterations following TBI that might contribute to the onset and progression of TBI clinical complications. We identified three olfactory receptor (OR) TBI biomarkers that are aberrantly down-regulated in PBMC specimens from TBI subjects. Down-regulation of these OR biomarkers in PBMC was correlated with the severity of brain injury and TBI-specific symptoms. A two- biomarker panel comprised of OR11H1 and OR4M1 provided the best criterion for segregating the TBI and control cases with 90% accuracy, 83.3% sensitivity, and 100% specificity. We found that the OR biomarkers are ectopically expressed in multiple brain regions, including the entorhinal-hippocampus system known to play an important role in memory formation and consolidation. Activation of OR4M1 led to attenuation of abnormal tau phosphorylation, possibly through JNK signaling pathway. Our results suggested that addition of the two-OR biomarker model to current diagnostic criteria may lead to improved TBI detection for clinical trials, and decreased expression of OR TBI biomarkers might be associated with TBI-induced tauopathy. Future studies exploring the physiological relevance of OR TBI biomarkers in the normal brain and in the brain following TBI will provide a better understanding of the biological mechanisms underlying TBI and insights into novel therapeutic targets for TBI.
Keywords: Biomarker, olfactory receptor, peripheral blood mononuclear cell, tauopathy, traumatic brain injury
INTRODUCTION
Traumatic brain injury (TBI) is a leading cause of death and disability among children and young adults in the United States [1]. TBI is an acquired injury caused by a sudden trauma to the head that disrupts normal brain functioning, which leads to either transient or chronic impairments in physical, cognitive, emotional, and/or behavioral functions. In the civilian population, TBI is typically associated with direct, closed impact mechanical trauma to the brain due to falls, motor vehicle accidents, sports, etc. [2]. In contrast, TBI among military personnel, particularly among veterans returning from the Persian Gulf region, is primarily due to exposure to blast pressure waves stemming from blast-producing weaponry, leading to prototypical cognitive deficits including impairments in attention, memory, processing speed, and executive functioning [2–4]. Both civilians and veterans who suffer from TBI exhibit symptoms that range in severity from mild to very severe, with a minimal to profound impact on daily functioning. The reasons why TBI induces different clinical symptoms among affected individuals are not yet known.
New evidence has highlighted defects in neural circuit and synapses, and the plastic processes controlling these functions, in TBI [5–11]. While genes relevant to these processes are expressed in the brain, some of these genes are also expressed in circulating blood cells, such as peripheral blood mononuclear cells (PBMCs) [12–15]. Consistent with this, recent studies have illustrated that PBMC-associated biomarkers may provide insights into the pathogenesis of neurological disorders such as Alzheimer's disease and can be used to monitor disease diagnosis and progression [16, 17]. Thus, PBMC may also provide an ideal, clinically assessable “window” into the brain, reflecting molecular alterations following TBI which might contribute to the onset and progression of clinical TBI phenotypes.
Evidence suggests that appropriate interventions can reduce functional impairment after TBI [18–20]. In order to demonstrate the efficacy of clinical interventions, research must identify the biological, clinical, and neurological indexes that are sensitive to the detection of functional impairments after TBI. Therefore, in this study we explored the feasibility of identifying clinically assessable TBI biomarkers and the potential function of identified TBI biomarkers in TBI neuropathology.
MATERIALS AND METHODS
Civilian TBI and control subjects
Eleven individuals with TBI (five male and six female), with TBI severity ranging from mild to severe, and nine control participants (four male and five female) were recruited from the Brain Injury Research Center at the Mount Sinai School of Medicine (MSSM). Medical documentation of TBI was reviewed for each participant. The age- and education-matched controls were recruited using advertisements placed in local media and flyers, and through word of mouth. Following a capacity screening to assess the individual's ability to comprehend the purpose and procedures of the study and provide informed consent, the informed consent process was performed in accordance with MSSM IRB policies and procedures.
All participants were screened using the Brain Injury Screening Questionnaire (BISQ) [21, 22] to assess the kinds of situations in which a brain injury might have occurred, and the number and severity of hits to the head sustained throughout the lifespan. Using information gained from the BISQ and from individual interviews conducted by the authors, injury severity was classified using a 7-point scale ranging from 1 (no loss of consciousness, no confusion (i.e., no TBI)) to 7 (loss of consciousness greater than 4 weeks in duration) [21, 22]. For those who have been injured, questionnaires in the BISQ solicit participants’ self-reported measures of functional difficulties and symptoms associated with brain injury and events, conditions other than brain injury that might lead to symptoms similar to those seen in brain injury, as well as the occurrence of persistent, highly disruptive symptoms (functional changes) that do not “go away”, which TBI participants may continue to experience following the blow to the head.
Veteran TBI and control subjects
For this biomarker validation study, we obtained banked PBMCs from a cohort of five OEF/OIF veterans and a control cohort of seven age- and gender-matched veterans without TBI (non-TBI veterans). TBI diagnosis is based on confirmation according to Defense and Veterans Brain Injury Center (DVBIC) criteria of sustained injury to the head plus subsequent alteration of consciousness, and Repeatable Battery for Neuropsychological Testing (RBANS) scores of one standard deviation below the norms established for age and education of individuals in question. Non-TBI control classification is based on DVBIC confirmation of no sustained injury to the head and RBANS scores less than one standard deviation below the norms established. Most of the veteran TBI cases exhibit co-morbid post-traumatic stress disorder (PTSD). Thus, our non-TBI control cases are also matched for PTSD diagnosis. In this study, diagnosis of PSTD is based on a score of 50 or more in the PTSD Checklist – Civilian Version.
Postmortem brain specimens
Human postmortem brain samples from 4 neurologically normal cases (characterized by a Clinical Dementia Rating (CDR) of 0) were obtained from the Alzheimer's Disease Brain Bank of the Mount Sinai School of Medicine. A multistep approach based on cognitive and functional status during the last 6 months of life was applied to the assignment of CDR [23] as previously reported [24, 25].
PBMC and RNA isolation
Blood specimens were collected into BD Vacutaineer CPT Cell Preparation Tubes. PBMC were isolated following manufacturer's instructions (Becton, Dickinson and Company) and were stored at –80°C until use. Total RNA was isolated from approximately 10–50 mg of PBMC using RNA STAT-60 (Tel-Test) according to the manufacturer's instructions. The purity and concentration of RNA samples were determined from OD260/280 readings using a dual beam UV spectrophotometer and RNA integrity was determined by capillary electrophoresis using the RNA 6000 Nano Lab-on-a-Chip kit and the Bioanalyzer 2100 (Agilent Technologies).
Microarray study and analysis
Total RNA was directly labeled using the FlashTag™ HSR Biotin RNA Labeling Kit according to the manufacturer's instructions (Genisphere). Verification of biotin labeling was obtained by an enzyme-linked oligoabsorbant assay (ELOSA) using Immobilizer™ Amino – 8 well strips (Nunc/Thermo Fisher Scientific) according to instructions supplied by Genisphere. Labeled RNA (1.0 μg) was hybridized for 16 h at 48°C to Affymetrix HuGene 1.0 ST arrays containing probe sets for 28,869 genes. Arrays were washed and stained on a Fluidics Station 450 (Affymetrix) according to the manufacturer's recommended procedures. The arrays were stained with phycoerythrein-conjugated streptavidin (Life Technologies) and the fluorescence intensities were determined using a GCS 3000 7G high-resolution confocal laser scanner and AGCC software (Affymetrix). The scanned images were analyzed with the RNA QC tool (Affymetrix) using RMA global background correction, quantile normalization and median polish summarization to generate quantified data (as recommended by Genisphere). Quality control metrics for arrays included normalized signal values >1000 for five spike-in control oligo probe sets (Genisphere).
RNA probe sets exhibiting significant differential expression (SDE) were identified using the following steps in GeneMaths XT (Applied Maths): 1) Probe sets with array detection p-values ≤0.05 for all samples in at least one experimental group were selected for further analysis; 2) Performed Discriminant Analysis (DA) and determined the largest percentage of remaining probe sets that permitted correct group assignment of samples in unsupervised hierarchical clustering by the Unweighted Pair-Group Method using Arithmetic averages (UPGMA) based on cosine correlation of row mean centered log2 signal values; this was the top 50 percentile; and 3) in the DA top 50 percentile, selected probe sets with absolute signal log2 fold changes ≥1.0 and independent t-test p-values ≤0.05 adjusted for multiple testing error by the Benjamini-Hochberg FDR correction method. Unsupervised hierarchical clustering of probe sets and heat map generation were performed in GeneMaths XT following row mean centering of log2 transformed MAS5.0 signal values; probe set clustering was performed by the UPGMA method using Cosine correlation as the similarity metric. For comparative purposes, clustered heat maps included probe sets for spike-in controls (Genisphere), or endogenous small RNAs exhibiting: 1) Array detection p-values ≤0.05, and 2) either a) a log2 signal value standard deviation ≤0.025 for all samples or b) in the DA top 50 percentile with a FC >1.3 in the opposite direction of the selected SDE profile.
Confirmatory quantitative PCR
First strand cDNA was synthesized from 1 μg of total RNA using Superscript III Super-mix for qRT-PCR (Invitrogen). Quantitative RT-PCR was performed using Maxima SYBR Green master mix (Fermentas) in ABI Prism 7900HT in four replicates with primers amplifying the seven olfactory receptor candidates: OR4Q3 (Forward: CACCTGCTCCAATCTCCTATG, Reverse: TCCCCTAACATCTTTGGCAC); OR51L1 (Forward: TTCCCACACCTTTGCTACTG, Reverse: AATACTGTTGGTCCTGGCATC); OR4D10 (Forward: CCATCTCTGTCACCTTCACTG, Reverse: ATGGCTGACTTCATCTCATGG); OR4M1 (Forward: TCTGTTAATGTCCTATGCCTTCC, Reverse: AATGTGGGAATAGCAGGTGG); OR52N5 (Forward: ATGCTACCACACTCACCAAC, Reverse: CATCAGCAATACACCCCTCAG); OR2J3 (Forward: CCTCTCATCCTCATTCTCACTTC, Reverse: CAAACACTTTCTGAAGCCCAG); OR11H1 (Forward: AACTGGTCATACTGTGCTGG, Reverse: GGGCAGAAACACAATCCAATG). Human TATA-binding protein (TBP, Forward: TGCACAGGAGCCAAGAGTGAA, Reverse: CTGGAACGGTGAAGGTGACA) expression level was used as an internal control. Data were normalized using the 2–ΔΔCt method [26]. Levels of olfactory receptor mRNAs were expressed relative to those in control groups and plotted in GraphPad Prism.
Lentiviral plasmid construction and lentivirus packaging
We obtained pCMV6-XL5-OR4M1 cDNA clone from Origne and subcloned the OR4M1 ORF into lentivial plasmid pLVX-IRES-ZsGreen (Clontech). The inserted OR4M1 sequence was verified by sequencing. For lentivirus packaging, we transfected Lenti-X 293T cells with either pLVX-OR4M1-IRES-ZsGreen or pLVX-IRES-ZsGreen using Lentiviral Packaging System (Clontech). Medium was collected 48 h after transfection and a ten-fold concentration step was performed using Lenti-X Concentrator (Clontech).
Primary neuron culture, lentiviral transduction, and cAMP assay
Embryonic day 15 cortico-hippocampal neuronal cultures were prepared from C57BL6 mouse (Jackson Laboratory) as previously described [27]. Cells were seeded onto poly-D-lysine-coated 12-well plates at 5 × 105 cells per well and cultured in Neurobasal medium supplemented with 2% B27, 0.5 mM L-glutamine, and 1% penicillin-streptomycin (Life Technologies). On Day 5 of culture, primary cortico-hippocampal neuron cultures were transduced with lentiviral particles overexpressing OR4M1 or control lentiviral particles by spin infection (800 g × 90 min at 30°C). Transduction efficiency was monitored by ZsGreen expression. 72 h after lentiviral transduction, cells were pretreated with 3-isobutyl-l-methylxanthine (IBMX), an inhibitor of cAMP phosphodiesterase, for 10 min followed by ligand treatment (10 μM) for 10 min. cAMP assay was performed using a colorimetric cAMP ELISA assay kit (Cell Biolabs).
Luminex multiplex assay and western blot analysis
Primary cortico-hippocampal neurons were infected with OR4M1 lentiviral particles and stimulated with acetophenone (10 μM) for 1 h. Multiplex luminex assay was performed using the Milliplex xMAP 8-plex multipathway signaling-phosphoprotein kit (Millipore) as previously described [28]. Protein concentrations were determined using the BCA protein assay kit (Thermo Scientific). 30 μg of total protein were loaded onto 12% SDS-PAGE and subjected to western blot analysis with antibody recognizing phospho-JNK (T183/Y185) (Cell Signaling), phospho-ERK (T185/Y187) (Cell Signaling), and PHF-1 (phospho-tau (S396/S404), a generous gift from Peter Davies at Albert Einstein College of Medicine). Blots were quantified in ImageJ (NIH), normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnologies) and then plotted in GraphPad Prism.
RESULTS
Identification of select olfactory receptors as clinically accessible candidate TBI biomarkers
We analyzed the gene expression profile in PBMC derived from subjects with TBI and age-, gender-, and education-matched healthy controls. Our data showed that female subjects had a more robust profile than males probably due to a rather smaller number of male subjects in the study cohort. Taking this into consideration, further analysis and confirmatory studies were all performed in a subset of the study cohort which only contains female subjects. Demographic information for the female TBI and control participants is summarized in Table 1. Using microarray gene expression profile analysis, we identified a panel of 102 candidate biomarker genes (36 annotated and 66 non-annotated genes) that are differentially regulated (t-test p < 0.05) following Benjamini-Hochberg false discovery rate correction [29] by >1.5-fold in PBMC specimens of TBI cases compared to control cases (Fig. 1A). More importantly, we were able to correctly separate all cases into TBI and non-TBI groupings using this panel of 102 genes in an unsupervised hierarchical clustering analysis (Fig. 1A). Interestingly, among the panel of 102 candidate TBI biomarkers that we identified from microarray data, 7 were olfactory receptors (Fig. 1B). Our microarray studies revealed that each of the 7 ORs were down-regulated in PBMC of TBI cases compared to those of control cases in this study cohort (Figs. 1B, 2A).
Table 1.
Demographic information for the civilian study cohort. TBI severity was classified using a 7-point scale ranging from 1 (no loss of consciousness, no confusion (i.e., no TBI)) to 7 (loss of consciousness greater than 4 weeks in duration) [23]. Time post injury is the time frame between the occurrence of brain injury and volunteer's participation in this biomarker study
| Control | TBI | |
|---|---|---|
| Number of cases | 5 | 6 |
| Gender | 100% female | 100% female |
| TBI severity | ||
| Mean ± SD | 1 ± 0 | 4 ± 1.67 |
| Range | 1 | 2–6 |
| Age (mean y ± SD) at study participation | 34.80 ± 14.48 | 35.33 ± 13.32 |
| Post injury interval (mean y ± SD) | — | 5.42 ± 5.30 |
| Education (mean y ± SD) | 16.80 ± 1.10 | 14.50 ± 2.35 |
| Ethnicity composition | ||
| White-Caucasian | 80% | 66.7% |
| Black-African American | — | 33.3% |
| Hispanic | 20% | — |
Fig. 1.

Development of blood biological indices (biomarkers) capable of correctly segregating TBI and control cases. Gene expression profile analysis of PBMC specimens from TBI and 5 age-, gender-, and education-matched control cases using a microarray platform (Affymetrix) led to the identification of a panel of 102 candidate biomarker genes. A) The 108 differentially-regulated genes identified were subjected to unsupervised hierarchical clustering analysis using the UPGMA algorithm with cosine correlation as the similarity metric. Results are presented as a heat map (left panel) demonstrating that the content of the 108 biomarker panel is able to correctly segregate TBI from control cases. B) 7 ORs and one OR pseudogene that are down-regulated in PBMC of TBI cases.
Fig. 2.

Down-regulation of OR genes in PBMC of TBI cases provide a sensitive and specific criterion for distinguishing TBI from control cases. mRNA contents for each of the 7 candidate OR biomarker genes in TBI and control cases were analyzed by microarray (A) or independent qPCR (B). Bar graphs represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by student t-test, TBI versus control. The efficacy of using biomarker contents from PBMC as a criterion to correctly segregate TBI and control cases was tested by unsupervised clustering analysis using the UPGMA algorithm with cosine correlation as the similarity metric. C) The accuracy, sensitivity, and specificity of OR11H1, OR4M1, OR52N5, or panels of ORs to distinguish TBI from control cases. D) A heat map graphically depicting the efficacy of using the two biomarker panel to distinguish TBI cases and control cases by unsupervised clustering analysis.
Confirmatory qPCR studies
Using independent qPCR assays, we confirmed that three of the ORs (OR11H1, OR4M1, and OR52N5) were significantly down-regulated in PBMC of TBI cases (Fig. 2B). For the remaining four ORs (OR4D10, OR2J3, OR4Q3, OR51L1), our qPCR evidence showed lower levels of these ORs in PBMC of TBI compared to control cases, but these differences did not reach statistical significance (Fig. 2B). This particular result is conceivably due to a high degree of gene expression variability for these 4 ORs among healthy control cases and the relatively small sample sizes that we used in our exploratory study.
We continued to explore the sensitivity and specificity of an individual or combined role of OR11H1, OR4M1, and OR52N5 in distinguishing TBI cases from normal healthy controls in our study cohort (Fig. 2C). Using unsupervised clustering analyses, we found that a two- biomarker panel comprised of OR11H1 and OR4M1 provides the best criterion for segregating the TBI and control cases with 90% accuracy, 83.3% sensitivity, and 100% specificity (Fig. 2C, D).
TBI biomarker validation studies: Testing the validity of OR11H1, OR4M1, and OR52N5 to distinguish TBI from non-TBI control cases from a study cohort of OEF/OIF veterans
The pathophysiological mechanisms underlying mechanical and blast-related TBI may differ in some ways, but they share important pathophysiological features. Similarities between the pathophysiologies of mechanical and blast-related TBI [2] suggested that information gathered from TBI cases in the civilian population may also be relevant to combat-related TBI. Based on this, we tested the ability of OR11H1, OR4M1, and OR52N5 to distinguish TBI cases from non-TBI controls from a study cohort of OEF/OIF veterans. For this biomarker validation study, we obtained banked PBMCs from a cohort of five OEF/OIF veterans and a control cohort of seven ageand gender-matched veterans without TBI (non-TBI veterans) (Table 2). Similar to our above-mentioned observations regarding OR biomarkers from the civilian study cohort, we found that OR4M1 (Fig. 3A), OR11H1 (Fig. 3B), and OR52N5 (Fig. 3C) are also significantly down-regulated in PBMC specimens of veteran TBI cases compared to that of non-TBI control cases. The two biomarker panel of OR11H1 and OR4M1 we identified from the civilian TBI study cohort is also capable of distinguishing veteran TBI from control veteran cases with 83% accuracy, 80% sensitivity, and 86% specificity (Fig. 3D & E).
Table 2.
Demographic information for the veteran study cohort. TBI diagnosis is based on confirmation according to Defense and Veterans Brain Injury Center (DVBIC) criteria of sustained injury to the head plus subsequent alteration of consciousness, and Repeatable Battery for Neuropsychological Testing (RBANS) scores of one standard deviation below the norms established for age and education of individuals in question. Non-TBI control classification is based on DVBIC confirmation of no sustained injury to the head and RBANS scores less than one standard deviation below the norms established. Most of the veteran TBI cases exhibit co-morbid post-traumatic stress disorder (PTSD). Thus, our non-TBI control cases are also matched for PTSD diagnosis. In this study, diagnosis of PSTD is based on a score of 50 or more in the PTSD Checklist – Civilian Version
| Control | TBI | |
|---|---|---|
| Number of cases | 7 | 5 |
| Gender | 57% male | 80% male |
| Age (mean y ± SD) | 30.1 ± 9.4 | 31.0 ± 7.1 |
| Interval since last deployment (mean y ± SD) | 2.8 ± 2.3 | 4.0 ± 3.8 |
| Education (mean y ± SD) | 13.3 ± 2.8 | 13.8 ± 1.5 |
| Percentage of cases with co-morbid PTSD | 85.7% | 100% |
| Ethnicity composition | ||
| Black-African American | 14.3% | 40% |
| Hispanic | 85.7% | 60% |
Fig. 3.

Validation of OR TBI biomarkers in a veteran study cohort. The mRNA contents for (A) OR4M1, (B) OR11H1, and (C) OR52N5 in TBI and control cases from a veteran study cohort were analyzed by qPCR. Bar graphs represent mean + SEM. *p < 0.05 by student t-test, TBI versus control. D) The accuracy, sensitivity, and specificity of OR11H1, OR4M1, OR52N5, or panels of ORs to distinguish veteran TBI from control veteran cases. E) A heat map graphically depicting the efficacy of using the two biomarker panel to distinguish veteran TBI cases and control veteran cases by unsupervised clustering analysis.
OR biomarker contents in PBMCs are correlated with TBI severity & long-term clinical neuropsychological complications
Continuing to explore the potential relevance of OR11H1, OR4M1, and OR52N5 to TBI clinical symptoms, we found that the contents of the three OR biomarkers in PBMC was significantly and inversely correlated with TBI severity: lower OR mRNA contents in PBMCs are associated with exposure to more severe head injury (Fig. 4A–C). We also found that OR TBI biomarker contents in PBMC were significantly correlated with select aspects of self-reported clinical TBI symptoms. In particular, PBMC contents of OR11H1 or OR4M1 were significantly associated in an inverse fashion with TBI-specific symptoms, a summation of 25 cognitive symptoms that are sensitive and specific to TBI [22]. OR11H1 or OR4M1 content within PBMC were not associated with self-assessments of mood (Fig. 4D). Expression level of the third OR biomarker, OR52N5, was not correlated with TBI-specific symptoms or self-assessment of mood (Fig. 4D).
Fig. 4.

Olfactory receptor TBI biomarker contents in PBMCs are inversely correlated with TBI severity and long-term neuropsychological complications. Correlation analysis was used to test the potential association between PBMC OR11H1, OR4M1, and OR52N5 mRNA content and the severity of TBI injury or self-reported measures of TBI complications. OR11H1 (A), OR4M1 (B), and OR52N5 (C) content in PBMCs are inversely correlated with TBI severity. D) Correlation coefficients and p-values of associations between individual OR biomarker content in PBMCs and TBI severity, TBI-specific symptoms (a summation of 25 cognitive symptoms that are sensitive and specific to TBI) and self-assessment of mood.
Ectopic expression of OR TBI biomarkers in multiple brain regions outside of the olfactory bulb
The brain represents a key target tissue for understanding TBI clinical complications and for the development of clinical interventions. In order to explore the potential physiological relevance of OR11H1, OR4M1, and OR52N5 in the brain, we assessed the RNA contents of OR biomarkers in the brain. We found that the three TBI biomarker ORs were expressed in multiple regions of the brain from postmortem brain specimens, including temporal gyrus (BM22), entorhinal cortex (BM36), occipital cortex (BM17), and the hippocampal formation (Fig. 5A & B). In agreement with our RT-PCR data, we also found the expression of OR TBI biomarkers in the hippocampal formation region from the human brain genome-wide microarray database [30] (Fig. 5C–E). Our observation that the three OR biomarkers were ectopically expressed in multiple brain regions outside of the olfactory bulb suggested potential function(s) of these ORs in the brain unrelated to the detection and processing of olfactory information.
Fig 5.

Olfactory receptor TBI biomarkers expression in the brain. OR11H1, OR4M1, and OR52N5 mRNA expression by (A) RT-PCR and (B) relative expression level in postmortem superior temporal gyrus (BM22), hippocampal formation (HF), occipital cortex (BM17), and entorhinal cortex (BM36) specimens from neurologically normal cases. Expression pattern of (C) OR4M1, (D) OR11H1, and (E) OR52N5 in the brain by genome-wide microarray from the Allen Human Brain Atlas [30].
Activation of OR TBI biomarker modulates tau neuropathology-related phenotypes in vitro
Olfactory receptors are members of the class A rhodopsin-like family of G protein-coupled receptors (GPCRs). Once the odorant has bound to the odor receptor, the receptor undergoes structural changes and it binds and activates the olfactory-type G protein on the inside of the olfactory receptor neuron. The G protein (Golf and/or Gs) in turn activates adenylate cyclase, which converts ATP into cyclic AMP (cAMP). So we first examined the expression level of Golf and β-arrestin in the PBMC of TBI and control subject and no change was detected (Supplementary Figure 1; available online: http://www.j-alz.com/issues/34/vol34-2.html#supplementarydata04).
Considering that tau neuropathology is a main pathological feature following TBI, we continued to explore whether OR4M1 activation might influence tau processing mechanisms. We transduced primary cortical-hippocampal neuron culture with OR4M1 lentiviral particles and screened several odorants for their ability to activate OR4M1 using cAMP ELISA assay. In Fig. 6A, we presented some representative odorants that were capable or not capable of activating OR4M1. Positive ligands were used to stimulate OR4M1-overexpressing primary cortico-hippocampal neurons and the potential impact of activation of OR4M1 on signaling pathways and aberrant tau phosphorylation were assessed. Using luminex multiplex technology, we found that upon acetophenone stimulation, phosphorylation of c-Jun N-terminal kinase (JNK) at Thr183/Tyr185 was significantly reduced in neurons overexpressing OR4M1 (Fig. 6B), indicating the activation of OR4M1 might influence JNK signaling pathway. The phosphorylation of ERK1/2 (Fig. 6B), among others (STAT3, MEK, p70S6, IKBa, and CREB, see Supplemental Figure 2), did not change upon acetophenone stimulation, suggesting that this inhibition of JNK signaling is rather specific. The stress-activated kinase JNK belongs to the mitogen-activated protein kinase family and takes part in signaling cascades initiated by various forms of stress. Its targets include the microtubule-associated protein tau [31]. Hyperphosphorylation of tau at Ser396/Ser404 is a known feature associated with tau neuropathology in neurodegenerative disorders such as Alzheimer's disease. Encouragingly, we observed that overexpression and activation of OR4M1 in primary neurons significantly reduced cellular content of abnormally phosphorylated tau at Ser396/Ser404 (PHF1 epitope) (Fig. 6C), suggesting that activation of OR4M1 might result in protection against abnormal tau processing.
Fig. 6.

Activation of OR4M1 resulted in reduced tau phosphorylation via JNK signaling pathway. Primary cortico-hippocampal neuron culture was transduced with lentiviral particles overexpressin OR4M1 or control lentiviral particles by spin infection (250 g × 90 min at 30°C). A) Transduced cells were treated by (1) hedione, (2) (+) citronellal, (3) acetophenone, (4) pyrazine, and (5) 2-isobutyl-3-methopyrazine (all at 10 μM, Sigma-Aldrich) for 10 min and cAMP assay was performed. Transduced neurons were also treated with acetophenone for 1 h, and multiplex luminex assay was performed using the Milliplex xMAP 8-plex multipathway signaling-phosphoprotein kit (Millipore) according to manufacturer's protocol: (B) Change of JNK phosphorylation on Thr183/Tyr185 and (C) ERK1/2 phosphorylation on Thr185/Tyr187. JNK and tau phosphorylation were also measured by (D) western blot analysis using antibodies recognizing phospho-JNK, phospho-ERK, or phospho-tau (PHF-1 epitope) and quantified (E, F) using GAPDH as a loading control. *p<0.05, **p < 0.01, ***p < 0.001 by two-tailed student t-test.
Collectively, our data demonstrated that select olfac-tory receptors (e.g., OR11H1, OR4M1, and OR52N5) were down-regulated in the PBMC of TBI cases and could serve as TBI biomarkers. Activation of OR4M1 resulted in protection against tau neuropathological features possibly through the JNK signaling pathway (Fig. 7).
Fig. 7.
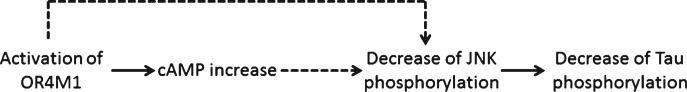
Scheme of working hypothesis.
DISCUSSION
Consistent with accumulation evidence suggesting that PBMC-associated biomarkers may provide insights into the pathogenesis of neurological disorders, results from our studies revealed that expression of select ORs in PBMC may serve as clinically assessable surrogate biological indices of TBI. ORs are G protein-coupled receptors known to be expressed in nasal epithelium olfactory neurons, where they are responsible for the detection of odorants [32]. However, ectopic expression of select ORs, defined as a biological event or process that occurs in an abnormal location or position in the brain or other tissues, has also been described [33]. Our high throughput microarray evidence, followed by data from independent qPCR confirmatory studies, led to the identification of three OR TBI biomarkers (OR11H1, OR4M1, and OR52N5) that are ectopically expressed in PBMC and are aberrantly down-regulated in PBMC specimens from subjects with a history of TBI. Among the TBI cases in our study cohort, there was a significant time lag between the occurrence of brain injury and the volunteers’ participation in this biomarker study; the average post-injury interval among the TBI cases was 5.4 ± 5.3 years (Table 1). Thus, down-regulation of the three ORs in PBMC among TBI cases was reflections of long-term physiological consequences of TBI.
We found that down-regulation of the three OR biomarkers in PBMC were directly correlated with the severity of brain injury in our TBI participants. Among the three OR biomarkers, we found that a two-biomarker panel, comprised of OR11H1 and OR4M1, provides the best criterion for segregating the TBI and control cases with 90% accuracy, 83.3% sensitivity, and 100% specificity. Interestingly, we found that PBMC contents of OR11H1 and OR4M1 were inversely correlated with cognitive-related symptoms. While additional studies will be required to clarify the mechanisms underlying the inter-relationships between changes in OR biomarker contents in PBMC, the initial severity of brain injury, and long-term clinical consequences of TBI, outcomes from our studies suggest that additional applications of this two-OR biomarker panel to current diagnostic criteria may lead to improved TBI detection and more sensitive outcome measures for clinical trials.
Initial and persistent cognitive deficits are the most common complaints after TBI [34, 35]. The entorhinal-hippocampus system is known to play an important role in the formation and consolidation of memories, particularly spatial memories [36–38]. Among the brain regions surveyed, we found the highest content of OR TBI biomarkers in the hippocampal formation, with relatively lower levels of OR TBI expression in the entorhinal cortex. Based on this and on our observation that the down-regulation of OR11H1 and OR4M1 in the PBMC are inversely correlated with self-reported indexes of cognitive functions, it is likely that ectopic expression of OR biomarkers in the entorhinal-hippocampus circuitry might be relevant to long-term deficits in cognitive functions following TBI.
It is well known that TBI may be a risk factor for dementia [39], but recent evidence suggests that Alzheimer's disease-related neuropathological mechanisms may contribute to cognitive dysfunction in TBI. The two characteristic neuropathologies of Alzheimer's disease are the abnormal accumulation and deposition of amyloid-β peptides and tau proteins in the brain. Evidence from humans [40–42] and experimental animal models [43] has also revealed abnormal accumulations of amyloid-β peptides and tau proteins in the brain and in cerebrospinal fluid following TBI. Interestingly, elevation of plasma tau levels has been associated with increasingly severe outcomes of TBI [44]. Multiple signaling pathways have been demonstrated to be involved in TBI pathology, including the AKT signaling pathway [45], glycogen synthase kinase-3 signaling pathway [46], STAT3 signaling pathway [47], ERK signaling pathway [48], and JNK signaling pathway [49]. We found that activation of OR4M1 could lead to attenuation of abnormal tau phosphorylation on S396/404 via JNK signaling pathway, suggesting a possible link between OR4M1 and TBI-related tauopathy. Further studies will be needed to investigate abnormal tau phosphorylation level in the serum/cerebrospinal fluid of TBI subjects, especially the phosphorylation of Thr181, which has been implicated as a fluid-based marker in dementia [50]. Studies will also be needed to dissect in detail the signaling pathways that are associated with the attenuation of tau neuropathology through OR activation.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by discretionary funding to GMP and in part from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) (UL1RR029887).
Footnotes
Supplementary data available online: http://dx.doi.org/10.3233/JAD-121894
Authors’ disclosures available online (http://www.jalz.com/disclosures/view.php?id=1571).
REFERENCES
- 1.Centers for Disease Control and Prevention Traumatic brain injury in the United States: A report to Congress. 1999 http://www.cdc.gov/ncipc/pub-res/tbi_congress/TBI in the US.PDF.
- 2.Elder GA, Cristian A. Blast-related mild traumatic brain injury: Mechanisms of injury and impact on clinical care. Mt Sinai J Med. 2009;76:111–118. doi: 10.1002/msj.20098. [DOI] [PubMed] [Google Scholar]
- 3.Lezak M, Howieson DB, Loring DW. Neuropsycho-logical Assessment. Oxford University Press; 2004. [Google Scholar]
- 4.Vasterling JJ, Proctor SP, Amoroso P, Kane R, Heeren T, White RF. Neuropsychological outcomes of army personnel following deployment to the Iraq war. JAMA. 2006;296:519–529. doi: 10.1001/jama.296.5.519. [DOI] [PubMed] [Google Scholar]
- 5.Redell JB, Zhao J, Dash PK. Altered expression of miRNA-21 and its targets in the hippocampus after traumatic brain injury. J Neurosci Res. 2011;89:212–221. doi: 10.1002/jnr.22539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reeves TM, Lyeth BG, Povlishock JT. Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp Brain Res. 1995;106:248–256. doi: 10.1007/BF00241120. [DOI] [PubMed] [Google Scholar]
- 7.Cohen AS, Pfister BJ, Schwarzbach E, Grady MS, Goforth PB, Satin LS. Injury-induced alterations in CNS electrophysiology. Prog Brain Res. 2007;161:143–169. doi: 10.1016/S0079-6123(06)61010-8. [DOI] [PubMed] [Google Scholar]
- 8.Gao X, Deng P, Xu ZC, Chen J. Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PLoS One. 2011;6:e24566. doi: 10.1371/journal.pone.0024566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gobbel GT, Bonfield C, Carson-Walter EB, Adelson PD. Diffuse alterations in synaptic protein expression following focal traumatic brain injury in the immature rat. Childs Nerv Syst. 2007;23:1171–1179. doi: 10.1007/s00381-007-0345-2. [DOI] [PubMed] [Google Scholar]
- 10.Witgen BM, Lifshitz J, Smith ML, Schwarzbach E, Liang SL, Grady MS, Cohen AS. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: A systems, network and cellular evaluation. Neuroscience. 2005;133:1–15. doi: 10.1016/j.neuroscience.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 11.Wu A, Molteni R, Ying Z, Gomez-Pinilla F. A saturated-fat diet aggravates the outcome of traumatic brain injury on hippocampal plasticity and cognitive function by reducing brain-derived neurotrophic factor. Neuroscience. 2003;119:365–375. doi: 10.1016/s0306-4522(03)00154-4. [DOI] [PubMed] [Google Scholar]
- 12.Patanella AK, Zinno M, Quaranta D, Nociti V, Frisullo G, Gainotti G, Tonali PA, Batocchi AP, Marra C. Correlations between peripheral blood mononuclear cell production of BDNF, TNF-alpha, IL-6, IL-10 and cognitive performances in multiple sclerosis patients. J Neurosci Res. 2010;88:1106–1112. doi: 10.1002/jnr.22276. [DOI] [PubMed] [Google Scholar]
- 13.van Heerden JH, Conesa A, Stein DJ, Montaner D, Russell V, Illing N. Parallel changes in gene expression in peripheral blood mononuclear cells and the brain after maternal separation in the mouse. BMC Res Notes. 2009;2:195. doi: 10.1186/1756-0500-2-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardiner E, Beveridge NJ, Wu JQ, Carr V, Scott RJ, Tooney PA, Cairns MJ. Imprinted DLK1-DIO3 region of 14q32 defines a schizophrenia-associated miRNA signature in peripheral blood mononuclear cells. Mol Psychiatry. 2011;17:827–840. doi: 10.1038/mp.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maes OC, Schipper HM, Chertkow HM, Wang E. Methodology for discovery of Alzheimer's disease blood-based biomarkers. J Gerontol A Biol Sci Med Sci. 2009;64:636–645. doi: 10.1093/gerona/glp045. [DOI] [PubMed] [Google Scholar]
- 17.Speciale L, Calabrese E, Saresella M, Tinelli C, Mariani C, Sanvito L, Longhi R, Ferrante P. Lymphocyte subset patterns and cytokine production in Alzheimer's disease patients. Neurobiol Aging. 2007;28:1163–1169. doi: 10.1016/j.neurobiolaging.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 18.Archer T, Svensson K, Alricsson M. Physical exercise ameliorates deficits induced by traumatic brain injury. Acta Neurol Scand. 2012;125:293–302. doi: 10.1111/j.1600-0404.2011.01638.x. [DOI] [PubMed] [Google Scholar]
- 19.Qu C, Mahmood A, Ning R, Xiong Y, Zhang L, Chen J, Jiang H, Chopp M. The treatment of traumatic brain injury with velcade. J Neurotrauma. 2010;27:1625–1634. doi: 10.1089/neu.2010.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dams-O'Connor K, Gordon WA. Role and impact of cognitive rehabilitation. Psychiatr Clin North Am. 2010;33:893–904. doi: 10.1016/j.psc.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Cantor JB, Gordon WA, Schwartz ME, Charatz HJ, Ashman TA, Abramowitz S. Child and parent responses to a brain injury screening questionnaire. Arch Phys Med Rehabil. 2004;85:S54–S60. doi: 10.1016/j.apmr.2003.08.113. [DOI] [PubMed] [Google Scholar]
- 22.Gordon WA, Haddad L, Brown M, Hibbard MR, Sliwinski M. The sensitivity and specificity of self-reported symptoms in individuals with traumatic brain injury. Brain Inj. 2000;14:21–33. [PubMed] [Google Scholar]
- 23.Berg L. Clinical dementia rating (CDR). Psychopharmacol Bull. 1988;24:637–639. [PubMed] [Google Scholar]
- 24.Haroutunian V, Purohit DP, Perl DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Neurofibrillary tangles in non-demented elderly subjects and mild Alzheimer disease. Arch Neurol. 1999;56:713–718. doi: 10.1001/archneur.56.6.713. [DOI] [PubMed] [Google Scholar]
- 25.Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, Humala N, Seror I, Bartholomew S, Rosendorff C, Pasinetti GM. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest. 2007;117:3393–3402. doi: 10.1172/JCI31547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao W, Varghese M, Yemul S, Pan Y, Cheng A, Marano P, Hassan S, Vempati P, Chen F, Qian X, Pasinetti GM. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1alpha) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener. 2011;6:51. doi: 10.1186/1750-1326-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 30.Allen Human Cortex Study [Internet] Allen Institute for Brain Science ©2009; Seattle, WA: 2012. http://humancortex.alleninstitute.org. [Google Scholar]
- 31.Reynolds CH, Betts JC, Blackstock WP, Nebreda AR, Anderton BH. Phosphorylation sites on tau identified by nanoelectrospray mass spectrometry: Differences in vitro between the mitogen-activated protein kinases ERK2, c-Jun N-terminal kinase and P38, and glycogen synthase kinase-3beta. J Neurochem. 2000;74:1587–1595. doi: 10.1046/j.1471-4159.2000.0741587.x. [DOI] [PubMed] [Google Scholar]
- 32.DeMaria S, Ngai J. The cell biology of smell. J Cell Biol. 2010;191:443–452. doi: 10.1083/jcb.201008163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feldmesser E, Olender T, Khen M, Yanai I, Ophir R, Lancet D. Widespread ectopic expression of olfactory receptor genes. BMC Genomic. 2006;7:121. doi: 10.1186/1471-2164-7-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lovell M, Franzen M. Neuropsychological assessment. In: Silver JM, Yudofsky SC, Hales RE, editors. Neuropsychiatry of Traumatic Brain Injury. American Psychiatric Press, Inc; Washington, DC: 2012. [Google Scholar]
- 35.Whyte J, Polansky M, Cavallucci C, Fleming M, Lhulier J, Coslett HB. Inattentive behavior after traumatic brain injury. J Int Neuropsychol Soc. 1996;2:274–281. doi: 10.1017/s1355617700001284. [DOI] [PubMed] [Google Scholar]
- 36.Derdikman D, Moser EI. A manifold of spatial maps in the brain. Trends Cogn Sci. 2010;14:561–569. doi: 10.1016/j.tics.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 37.Eichenbaum H, Lipton PA. Towards a functional organization of the medial temporal lobe memory system: Role of the parahippocampal and medial entorhinal cortical areas. Hippocampus. 2008;18:1314–1324. doi: 10.1002/hipo.20500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Squire LR, Zola-Morgan S. The medial temporal lobe memory system. Science. 1991;253:1380–1386. doi: 10.1126/science.1896849. [DOI] [PubMed] [Google Scholar]
- 39.van Duijn CM, Tanja TA, Haaxma R, Schulte W, Saan RJ, Lameris AJ, ntonides-Hendriks G, Hofman A. Head trauma and the risk of Alzheimer's disease. Am J Epidemiol. 1992;135:775–782. doi: 10.1093/oxfordjournals.aje.a116364. [DOI] [PubMed] [Google Scholar]
- 40.Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 41.Marklund N, Blennow K, Zetterberg H, Ronne-Engstrom E, Enblad P, Hillered L. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg. 2009;110:1227–1237. doi: 10.3171/2008.9.JNS08584. [DOI] [PubMed] [Google Scholar]
- 42.Olsson A, Csajbok L, Ost M, Hoglund K, Nylen K, Rosen-gren L, Nellgard B, Blennow K. Marked increase of beta-amyloid(1-42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J Neurol. 2004;251:870–876. doi: 10.1007/s00415-004-0451-y. [DOI] [PubMed] [Google Scholar]
- 43.Szczygielski J, Mautes A, Steudel WI, Falkai P, Bayer TA, Wirths O. Traumatic brain injury: Cause or risk of Alzheimer's disease? A review of experimental studies. J Neural Transm. 2005;112:1547–1564. doi: 10.1007/s00702-005-0326-0. [DOI] [PubMed] [Google Scholar]
- 44.Liliang PC, Liang CL, Weng HC, Lu K, Wang KW, Chen HJ, Chuang JH. Tau proteins in serum predict outcome after severe traumatic brain injury. J Surg Res. 2010;160:302–307. doi: 10.1016/j.jss.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 45.Chen T, Zhang L, Qu Y, Huo K, Jiang X, Fei Z. The selective mGluR5 agonist CHPG protects against traumatic brain injury in vitro and in vivo via ERK and Akt pathway. Int J Mol Med. 2012;29:630–636. doi: 10.3892/ijmm.2011.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dash PK, Johnson D, Clark J, Orsi SA, Zhang M, Zhao J, Grill RJ, Moore AN, Pati S. Involvement of the glycogen synthase kinase-3 signaling pathway in TBI pathology and neurocognitive outcome. PLoS One. 2011;6:e24648. doi: 10.1371/journal.pone.0024648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oliva AA, Kang Y, Sanchez-Molano J, Furones C, Atkins CM. STAT3 signaling after traumatic brain injury. J Neurochem. 2012;120:710–720. doi: 10.1111/j.1471-4159.2011.07610.x. [DOI] [PubMed] [Google Scholar]
- 48.Chen T, Cao L, Dong W, Luo P, Liu W, Qu Y, Fei Z. Protective effects of mGluR5 positive modulators against traumatic neuronal injury through PKC-dependent activation of MEK/ERK Pathway. Neurochem Res. 2012;37:983–990. doi: 10.1007/s11064-011-0691-z. [DOI] [PubMed] [Google Scholar]
- 49.Tran HT, Sanchez L, Brody DL. Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J Neuropathol Exp Neurol. 2012;71:116–129. doi: 10.1097/NEN.0b013e3182456aed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sjogren M, Davidsson P, Tullberg M, Minthon L, Wallin A, Wikkelso C, Granerus AK, Vanderstichele H, Vanmechelen E, Blennow K. Both total and phosphorylated tau are increased in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2001;70:624–630. doi: 10.1136/jnnp.70.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.