

Key Points
Human cGVHD B cells have increased proximal BCR signaling protein expression and are more BCR responsive than non-cGVHD B cells.
Inhibiting Syk kinase activity abrogates the BCR-driven ex vivo proliferative and survival advantage of human chronic GVHD B cells.
Abstract
Although B cells have emerged as important contributors to chronic graft-versus-host-disease (cGVHD) pathogenesis, the mechanisms responsible for their sustained activation remain unknown. We previously showed that patients with cGVHD have significantly increased B cell–activating factor (BAFF) levels and that their B cells are activated and resistant to apoptosis. Exogenous BAFF confers a state of immediate responsiveness to antigen stimulation in normal murine B cells. To address this in cGVHD, we studied B-cell receptor (BCR) responsiveness in 48 patients who were >1 year out from allogeneic hematopoietic stem cell transplantation (HSCT). We found that B cells from cGVHD patients had significantly increased proliferative responses to BCR stimulation along with elevated basal levels of the proximal BCR signaling components B cell linker protein (BLNK) and Syk. After initiation of BCR signaling, cGVHD B cells exhibited increased BLNK and Syk phosphorylation compared with B cells from patients without cGVHD. Blocking Syk kinase activity prevented relative post-HSCT BCR hyper-responsiveness of cGVHD B cells. These data suggest that a lowered BCR signaling threshold in cGVHD associates with increased B-cell proliferation and activation in response to antigen. We reveal a mechanism underpinning aberrant B-cell activation in cGVHD and suggest that therapeutic inhibition of the involved kinases may benefit these patients.
Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) is a potentially curative treatment of many hematologic diseases. Unfortunately, high mortality rates limit widespread use of this therapy. The leading cause of nonrelapse mortality in patients who survive >100 days after HSCT is chronic graft-versus-host disease (cGVHD), which affects 30% to 70% of patients.1 Currently, death rates from cGVHD remain high (30-50%),2 and established therapies for prevention and/or treatment of cGVHD remain inadequate.
B cells have emerged in recent years as key players in cGVHD pathogenesis.3 In murine models of cGVHD, depletion of donor B cells reduced disease incidence.4 The fibrosis associated with target organ pathology was additionally shown to be dependent on B-cell infiltration and alloantibody deposition.5 In humans, the presence of alloantibodies directed against host minor histocompatibility antigens were found to be associated with disease,6,7 and several phase 1-2 trials of B cell–directed therapy demonstrated efficacy.8-13
B-cell homeostasis is altered in cGVHD patients14-18 and is associated with excessive levels of B cell–activating factor (BAFF) per B cell.15 Our previous findings suggested a mechanistic link between elevated BAFF levels and B-cell activation.19 We found that peripheral B cells directly isolated from cGVHD patients signal through protein kinase B and extracellular signal-regulated kinase and have decreased expression of the proapoptotic molecule Bim. These findings are consistent with the heightened metabolic state and resistance to apoptosis of such B cells.19 Of note, BAFF-mediated signaling has been shown to maintain murine B cells in a state of immediate responsiveness to antigen stimulation, and B cells treated with BAFF have increased proliferative responses to BCR stimulation.20
Taken together, these data led to the hypothesis that B cells in patients with cGVHD respond more readily to the allo- and neo-autoantigens present after transplant. To examine this, we determined whether B cells from cGVHD patients had elevated responses to BCR stimulation. Our data show that peripheral B cells purified from patients with cGVHD have increased BCR-specific proliferation. We find that cGVHD B cells have elevated basal expression of the proximal signaling components B cell linker protein (BLNK) and Syk, which may contribute to increased responsiveness on BCR stimulation. When signaling through this pathway is blocked using a small molecule Syk inhibitor, we find that aberrant B-cell proliferation is attenuated. These data suggest a mechanistic link between proximal BCR signaling and increased BCR responsiveness in cGVHD patients after HSCT.
Methods
Patients
Samples were obtained from patients following written informed consent in accordance with the Declaration of Helsinki. The Institutional Review Boards at the University of North Carolina Chapel Hill (UNC), Duke University Medical Center (DUMC), Fred Hutchinson Cancer Research Center (FHCRC), and the Dana-Farber Cancer Institute (DFCI) approved all studies. Included were patient samples obtained from the UNC (n = 24), DUMC (n = 11), FHCRC (n = 10), and DFCI (n = 3). Clinical characteristics of the 48 patients included in our functional studies are shown in Table 1. Study criteria were as follows: (1) >12 months from time of allogeneic HSCT; (2) not receiving high-dose prednisone (≥0.3 mg/kg per day or ≥30 mg/day); and (3) never received rituximab (α-CD20 mAb). Importantly, cGVHD status on the day of sample collection, not necessarily on first diagnosis, was documented by clinical examination and laboratory testing (in accordance with the National Institutes of Health cGVHD consensus criteria). Twenty patients were classified as having active cGVHD (+cGVHD), defined as requiring addition of high-dose prednisone or continued multiagent immunosuppression after sample collection. Control groups included 28 patients that did not have cGVHD (−cGVHD) at the time of sample collection. Patients without cGVHD included those who never developed cGVHD (n = 22) and those with previous disease (n = 6) that had resolved, or became asymptomatic, by the time of sample collection. There were no significant differences in age, gender, time after transplant, conditioning regimen, source or type of graft, grade of acute GVHD, or underlying hematologic malignancy between patients with and without cGVHD in our study (Table 1). As expected, there was a significant difference in prednisone treatment (≥0.3 mg/kg per day or ≥30 mg/day) on the date of sample collection between patients with active cGVHD compared with those without cGVHD (P = .002).
Table 1.
Clinical characteristics of 48 allogeneic HSCT patients
| Characteristic | −cGVHD* (N = 28) | +cGVHD (N = 20) | P value |
|---|---|---|---|
| Median age, year (range) | 58 (21-73) | 51 (28-67) | .39 |
| Sex, no. (%) of females | 8 (40) | 14 (50) | .57 |
| Median time after transplant, mo (range) | 23 (10-110) | 25 (12-116) | .96 |
| Conditioning regimen (%) | |||
| Myeloablative | 15 (54) | 6 (30) | .14 |
| Nonmyeloablative | 13 (46) | 14 (70) | |
| Source of graft (%) | |||
| Peripheral blood | 26 (93) | 18 (90) | 1.00 |
| Bone marrow | 2 (7) | 2 (10) | |
| HLA matching (%) | |||
| Matched, unrelated | 8 (29) | 10 (50) | .15 |
| Matched, related | 18 (64) | 7 (35) | |
| Mismatched | 2 (7) | 3 (15) | |
| Immunosuppressive treatment (%)† | |||
| Prednisone ≤30 mg (2.5-30 mg)/day | 4 (14) | 12 (60) | .002 |
| MMF | 0 (0) | 1 (5) | .42 |
| Tacrolimus | 5 (18) | 6 (30) | .49 |
| Rapamycin | 3 (15) | 0 (0) | .07 |
| ATG | 5 (18) | 4 (20) | 1.00 |
| Alemtuzumab | 2 (7) | 0 (0) | .50 |
| aGVHD | |||
| Grade I, no. (%) | 7 (25) | 8 (40) | .74 |
| Grade II, no. (%) | 2 (7) | 1 (5) | |
| Grade III, no. (%) | 1 (4) | 0 (0) | |
| Disease (%) | |||
| AML / AML from MDS | 14 (50) | 9 (45) | |
| ALL | 2 (7) | 2 (10) | |
| CML | 1 (4) | 0 (0) | |
| CLL | 2 (7) | 4 (20) | .81 |
| MDS | 4 (14) | 2 (10) | |
| NHL | 1 (4) | 2 (10) | |
| MM | 1 (4) | 0 (0) | |
| AA | 2 (7) | 0 (0) | |
| HL | 1 (4) | 1 (5) |
AA, aplastic anemia; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; ATG, anti-thymocyte globulin; CLL, chronic lymphoblastic leukemia; CML, chronic myeloid leukemia; HL, Hodgkin lymphoma; MDS, myelodysplastic syndrome; MM, multiple myeloma; MMF, mycophenolate mofetil; NHL, non-Hodgkin lymphoma.
Included 22 patients who had never developed cGVHD and 6 patients with resolved or inactive cGVHD on date of sample collection.
On date of sample collection.
Peripheral blood mononuclear cell collection and B-cell purification
Blood was collected from patients in EDTA-containing tubes. Peripheral blood mononuclear cells (PBMCs) were isolated immediately after blood draws by Ficoll gradients (Ficoll-Paque PLUS; GE Healthcare) and cryopreserved (10% dimethylsulfoxide [DMSO]). Healthy donor PBMCs were isolated by Ficoll gradients from Gulf Coast Regional Blood Center products (Houston, TX) and cryopreserved (10% DMSO). Unless indicated, B cells were purified to ≥95% by positive selection with CD19 microbeads according to the manufacturer’s instructions (Miltenyi Biotec).
B-cell proliferation
B cells (1 × 106) were incubated with 2.5 μM of carboxyfluorescein succinimidyl ester (CFSE; Invitrogen) for 10 minutes at 37°C, plated (0.4 × 106), stimulated with 10 or 50 μg/mL of the F(ab′)2 fragment of IgM (Jackson Immunoresearch) or 100 ng/mL of α-CD40 (R&D Systems) plus 5 ng/mL of interleukin (IL)-4 (R&D Systems), and then incubated for 6 days at 37°C. For fluorescence-activated cell sorter analysis, B cells were acquired on a MACSQuant Analyzer (MACSQuantify software; Miltenyi Biotech) or on a Canto II (FACSDIVA software; BD Biosciences). Analysis and proliferation indices were calculated using FlowJo Version 8.8.7 (TreeStar).
RNA expression analysis
Large-volume leukapheresis samples were obtained from 3 patients with cGVHD and 3 healthy donors and cryopreserved before use. B cells were purified using the human B-cell isolation kit II (Miltenyi Biotech) to ≥98.4% (CD3− CD19+). CD27− and CD27+ B-cell fractions were subsequently purified by positive selection using CD27 microbeads (Miltenyi Biotech). Purity was confirmed; the CD27− B-cell fraction contained ≤1.8% of CD27+ B cells. RNA was extracted using the RNeasy kit (Qiagen), and cDNA was prepared using RT2 First Strand Kit (SABiosciences). Gene expression profiling was determined using the RT2 Profiler PCR Array System for Human T-cell and B-cell Activation (SABiosciences) containing a panel of 84 pathway-focused genes, 3 RNA and polymerase chain reaction (PCR) quality controls, and 5 housekeeping genes. Fold changes were determined using the web-based PCR Array Data Analysis specific for the kit, which used the ΔΔCt method (SABiosciences). Fold-increase of BLNK was validated by performing reverse transcriptase-PCR on B cells from patients with and without cGVHD using the ΔΔCt method.
Flow cytometric analysis of surface Ig
PBMCs were thawed, and 1 × 106 cells were stained using the following antibodies: CD19 (clone J3119; Beckman), CD20 (clone 2H7; ebioscience), CD27 (clone O323; ebioscience), IgM (clone SA-DA4; ebioscience), IgG (clone G18-145; BD Biosciences), IgD (clone IA6-2; BD Biosciences), and 7AAD (ebioscience).
Intracellular flow cytometry
B cells were isolated from cryopreserved PBMCs and allowed to rest overnight in complete media (2.5 × 106 cells/mL) to reduce background signaling.21 Intracellular staining was performed according to the manufacturer’s instructions (BD Biosciences). Cells were fixed (BD cytofix buffer), permeabilized (BD Perm Buffer III), and stained using the following antibodies from BD Biosciences: BLNK (clone 2B11) or Syk (clone 4D10).
For analysis of BLNK and Syk phosphorylation, cryopreserved PBMCs were thawed and allowed to rest overnight (5 × 106 cells/mL), as previously described; 1 × 106 cells were stimulated with 5 μg/mL of the F(ab′)2 fragment of IgM (Jackson Immunoresearch) for 5 minutes at 37°C. Following stimulation, cells were immediately fixed (BD cytofix buffer), permeabilized (BD Perm Buffer III), and stained using antibodies from BD Biosciences: CD20 (clone H1), CD27 (clone L128), and either BLNK (pY84, clone J117-1278) or Syk (pY348, clone 1120-722).
Syk inhibition
The Syk inhibitor, R406, was kindly provided by Rigel Pharmaceuticals. CFSE-stained B cells (as previously described) were plated (0.4 × 106 cells/mL) and treated with R406 (0.01 or 0.1 μM) or DMSO control (0.1%) for 30 minutes at 37°C. Cells were stimulated with 50 μg/mL of the F(ab′)2 fragment of IgM (Jackson Immunoresearch) or 100 ng/mL of anti-CD40 (R&D Systems) plus 5 ng/mL of IL-4 (R&D Systems) and incubated for 6 days at 37°C.
Statistical methods
For analysis of continuous variables among multiple groups, the analysis of variance (ANOVA) test was used. When the overall test was significant, pairwise comparisons using Tukey’s multiple comparison test were used to control the family-wise error rate (D’Agostino). For analysis of categorical variables, the Fisher's exact test was used. All tests were generated using SAS software, version 9.3 (SAS Institute) and considered statistically significant if P < .05.
Results
B cells from patients with cGVHD are hyper-responsive to BCR stimulation
To address the hypothesis that B cells in cGVHD patients respond more readily to antigenic stimulation, we purified B cells from patients with and without cGVHD and assessed proliferation in response to BCR stimulation. Initial dose titration experiments revealed that B cells from patients with cGVHD proliferated in response to a limiting dose of BCR ligand (10 μg/mL of α-IgM or 10 μg/mL of α-IgM + IgG) that did not elicit a response in B cells from healthy individuals or patients without cGVHD (supplemental Figure 1 on the Blood Web site). The concentration of α-IgM used for BCR stimulation in subsequent experiments was at a level that reproducibly induced proliferation in B cells from healthy donors or control B cells from patients without cGVHD (Figure 1A). We found that B cells from patients with active cGVHD had significantly increased proliferation in response to BCR stimulation with α-IgM compared with control patients without cGVHD or to healthy donors (Figure 1A). The proliferation index following stimulation with α-IgM was also significantly increased in cGVHD B cells (Figure 1A), suggesting an increase in both the rate of cellular division and the number of dividing cells. These findings were BCR specific as proliferation with α-CD40 plus IL-4 was similar in patients with and without cGVHD (Figure 1B). Taken together, these data suggest that B cells in patients with cGVHD proliferate more readily in response to antigenic stimulation than B cells in patients without cGVHD.
Figure 1.

B cells from patients with cGVHD have increased BCR-driven proliferation. (A) B cells isolated from healthy donors and patients with and without cGVHD, stained with CFSE and stimulated with α-IgM (50 μg/mL) for 6 days. Unstimulated controls are shown as dashed lines. (Left) Representative histograms of B-cell proliferation from patients without cGVHD (gray area, thin line) and with cGVHD (white area, bold line). (Center) Frequency of B-cell proliferation quantified in healthy donors (HD, filled triangles), patients without cGVHD (−cGVHD, open circles), and patients with cGVHD (+cGVHD, filled squares). (Right) Proliferation index quantified in healthy donors (HD, filled triangles), patients without cGVHD (−cGVHD, open circles), and patients with cGVHD (+cGVHD, filled squares). Data are median ± range pooled from 4 independent experiments. *P < .05. (B) B cells isolated from patients with and without cGVHD, stained with CFSE, and stimulated with α-CD40 (100 ng/mL) + IL-4 (5 ng/mL) for 6 days. Unstimulated controls are shown as dashed lines. (Left) Representative histograms of B-cell proliferation from patients without cGVHD (gray area, thin line) and with cGVHD (white area, bold line). (Center) Frequency of B-cell proliferation quantified in patients without cGVHD (−cGVHD, open circles) and with cGVHD (+cGVHD, filled squares). (Right) Proliferation index quantified in patients without cGVHD (−cGVHD, open circles) and with cGVHD (+cGVHD, filled squares). Data are median ± range pooled from 3 independent experiments. *P < .05. Proliferation index = total number of divisions divided by the number of cells that went into division calculated using FlowJo Version 8.8.7.
Proximal BCR signaling components are elevated in B cells from patients with cGVHD
Previous work found that the CD27+ B cells in patients with cGVHD are activated and can constitutively secrete IgG ex vivo.15,19 To define the mechanisms underlying the increased activation and elevated BCR-driven proliferation of B cells from patients with cGVHD, we performed mRNA expression profiling of highly purified CD27+ and CD27− B cells. As purification of these subsets requires large-volume leukapheresis samples, this analysis was limited to healthy donors and the 3 patients with cGVHD who consented to collection. RNA expression of 84 genes associated with lymphocyte activation was determined using a commercially available reverse transcriptase-PCR array. The fold regulation of each gene in CD27+ or CD27− B-cell subsets from patients with cGVHD was compared with healthy donors. Twenty of 84 genes in CD27+ B cells (Figure 2A) and 10 of 84 genes in CD27− B cells (Figure 2B) met statistical criteria and were increased or decreased by greater than threefold compared with healthy donors. Four genes, IL-12A, CD40, IFNGR2, and IRF4, were decreased in both B-cell subsets in cGVHD, and a role for these genes in disease requires further study. Importantly, these data suggest that CD27+ and CD27− B-cell subsets from patients with cGVHD have patterns of gene expression that are distinct from healthy memory B-cell counterparts.
Figure 2.

Expression of BLNK and Syk is increased in cGVHD B cells ex vivo. Relative mRNA expression of 84 genes known to be involved in lymphocyte activation were analyzed in CD27+ and CD27− B cells from patients with cGVHD (n = 3) and healthy donors (n = 3). Data indicate the fold regulation of each gene that met statistical criteria, depicted in (A) CD27+ or (B) CD27− B cells from patients with cGVHD compared with healthy donors. Dashed lines indicate a greater than threefold increase (above line) or decrease (below line) in mRNA expression. Gray bars represent genes that are altered in both CD27+ and CD27− B-cell subsets. The P value for all genes is <.05; Student t test of replicate 2^(−ΔCt) values for each gene in the cGVHD group compared with healthy as calculated by SAbioscience Web-Based Array Analysis. Protein expression by mean fluorescence intensity (MFI) of (C) BLNK and (D) Syk in total peripheral B cells isolated from patients without cGVHD (−cGVHD, open circles) and with cGVHD (+cGVHD, filled squares). Data are median ± range from 2 independent experiments. *P <.05. (E) Protein expression by MFI of BLNK and Syk in B cells isolated from healthy donors (filled triangles), plated (1.5 × 106 cell/mL) and stimulated with α-IgM (50 μg/mL) or phosphate-buffered saline (PBS) as a control for 36 hours. Fold increase of α-IgM divided by PBS is depicted. *P = .003 (1-sample Student t test, theoretical mean = 1). Data are median ± range pooled from 2 independent experiments. *P < .05.
To determine whether the altered B-cell gene expression was associated with cGVHD, we performed semiquantitative PCR on small numbers of total B cells from patients with cGVHD and our control group: patients without cGVHD. We began with the gene with the greatest fold increase in both B-cell subsets, BLNK (also known as SLP65 or BASH). BLNK was found to be increased 5.86-fold in B cells from patients with cGVHD (n = 5) compared with B cells from patients without cGVHD (n = 15). Work in murine B cells has shown that BLNK is required for BCR-driven proliferation.24-27 The elevated BLNK mRNA expression by B cells from patients with cGVHD, coupled with the role of BLNK in BCR-driven proliferation and its increase after BCR stimulation, made this a compelling signaling pathway to examine further. We examined BLNK protein levels in purified B cells from 10 patients, either with or without cGVHD. As shown in Figure 2B-C, cells from patients with cGVHD had significantly increased protein levels of BLNK compared with B cells from patients without cGVHD. Finally, because BLNK is an important scaffold protein bridging the gap between antigen, BCR activation, and proliferation,28 we quantified the expression of the upstream signaling molecule Syk in B cells from these 10 patients. Syk was significantly increased in cGVHD B cells compared with B cells from patients without cGVHD (Figure 2D). Because overexpression of proximal BCR signaling molecules BLNK and Syk had not previously been described, we tested whether BCR stimulation directly resulted in increased protein levels by using B cells from healthy donors. Although expression of Syk was not affected, expression of BLNK was significantly increased by BCR stimulation of healthy donor B cells with α-IgM (Figure 2E). Taken together, these data suggested that (1) elevated levels of BLNK in B cells from patients with cGVHD may be the result of BCR engagement and (2) increased expression of proximal BCR signaling components may render cGVHD B cells more responsive to activation by antigen compared with B cells from patients without cGVHD.
B cells from patients with cGVHD have increased BCR-driven signaling compared with patients without disease
Activation and signal transduction through the BCR pathway begins with phosphorylation of the tyrosine-based activation motifs by Src family kinases. This leads to recruitment of Syk to the plasma membrane, where it becomes phosphorylated and, in turn, phosphorylates BLNK. Activation of this pathway has been shown in murine B cells to be required for proliferation.24-27 Because we found that B cells from patients with cGVHD have increased levels of Syk and BLNK expression, we examined activation of this proliferation-inducing pathway in B cells from patients with and without cGVHD.
To determine whether cGVHD B cells were more readily activated after BCR stimulation, Syk and BLNK phosphorylation were quantified in CD27+ and CD27− B-cell subsets by flow cytometry. Antigen-experienced CD27+ B cells from patients with cGVHD had significantly increased BCR-driven phosphorylation of BLNK (Figure 3A) and Syk (Figure 3C) compared with B cells from patients without cGVHD. Phosphorylation of BLNK in response to BCR stimulation was also significantly increased in the CD27− B-cell subset from patients with cGVHD (Figure 3B). BCR-driven phosphorylation of Syk was, however, similar in CD27− B cells from patients with and without disease (Figure 3D). Similar to these findings, BCR stimulation appears to impact BLNK, but not Syk, protein expression in healthy donors (Figure 2E). Thus, we show an increased signaling capacity via BLNK and Syk in cGVHD B cells, likely resulting in aberrant BCR activation.
Figure 3.
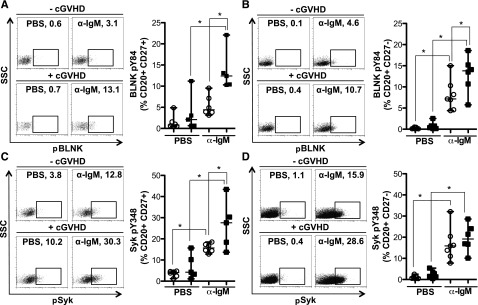
BCR-driven phosphorylation of BLNK is increased in CD27+ and CD27− B cells from patients with cGVHD. Phosphorylation of BLNK (tyrosine 84) and Syk (tyrosine 348) in CD27+ and CD27− B cells (CD20+) stimulated with α-IgM (5 μg/mL) or PBS as a control as indicated for 5 minutes. (A) (Left) representative dot plots of pBLNK in CD27+ B cells from a patient (upper) without cGVHD (−cGVHD) and (lower) with cGVHD (+cGVHD). Numbers indicate frequency of CD20+ CD27+ B cells that are pBLNK+. (Right) Frequency of BLNK phosphorylation in CD27+ B cells quantified in patients without cGVHD (n = 6, open circles) and with cGVHD (n = 5, filled squares). Data are median ± range pooled from 2 independent experiments, *P < .05. (B) (Left) Representative dot plots of pBLNK in CD27− B cells from a patient (upper) without cGVHD (−cGVHD) and (lower) with cGVHD (+cGVHD). Numbers indicate frequency of CD20+ CD27− B cells that are pBLNK+. (Right) Frequency of BLNK phosphorylation in CD27− B cells quantified in patients without cGVHD (n = 7, open circles) and with cGVHD (n = 6, filled squares). Data are median ± range pooled from 2 independent experiments, *P < .05. (C) (Left) Representative dot plots of pSyk in CD27+ B cells from a patient (upper) without cGVHD (−cGVHD) and (lower) with cGVHD (+cGVHD). Numbers indicate frequency of CD20+ CD27+ B cells that are pSyk+. (Right) Frequency of Syk phosphorylation in CD27+ B cells quantified in patients without cGVHD (n = 6, open circles) and with cGVHD (n = 5, filled squares). Data are median ± range pooled from 2 independent experiments, *P < .05. (D) (Left) Representative dot plots of pSyk in CD27− B cells from a patient (upper) without cGVHD (−cGVHD) and (lower) with cGVHD (+cGVHD). Numbers indicate frequency of CD20+ CD27− B cells that are pSyk+. (Right) Frequency of Syk phosphorylation in CD27− B cells quantified in patients without cGVHD (n = 7, open circles) and with cGVHD (n = 6, filled squares). Data are median ± range pooled from 2 independent experiments, *P < .05.
Interestingly, antigen-experienced CD27+ B cells from patients with cGVHD were activated to the same extent as healthy donors. Notably, this was not the case for B cells from patients without cGVHD as these cells had diminished signaling compared with healthy donors (supplemental Figure 2). Whether this is a mechanism of B-cell tolerance that prevents cGVHD warrants additional study.
BCR signaling was initiated in our studies by stimulating PBMCs with α-IgM. We therefore examined surface IgM expression to ascertain whether differences in signaling were the result of altered BCR expression. This was not the case; CD27− and CD27+ B-cell subsets from patients with and without cGVHD had similar surface expression of IgM (Figure 4), IgD, and IgG (supplemental Figure 3). Surface expression of BCR is variable between the CD27− and CD27+ B-cell subsets in healthy donors.21 We found similar results in our patients. Although not significantly increased in our study, patients with cGVHD appeared to have an increased frequency of IgM-expressing CD27+ B cells. It is tempting to speculate that these CD27+ IgM+ B cells in patients with cGVHD represent an infrequent, but uniquely activated, subset. Thus, although IgM is predominately expressed on the surface of CD27− B cells (85-90%), antigen-experienced CD27+ B cells also express IgM (44-49%) with no significant differences between patient phenotypes found (Figure 4).
Figure 4.

Surface expression of IgM is similar in CD27− and CD27+ B cells. Flow cytometric analysis of PBMCs for surface IgM expression. (A) representative histogram of CD27− B cells (CD19+ CD20+) from a patient without cGVHD (gray area, thin line) and with cGVHD (white area, bold line). Isotype is depicted as think gray line. (B) Representative histogram of CD27+ B cells (CD19+ CD20+) from a patient without cGVHD (gray area, thin line) and with cGVHD (white area, bold line). Gates indicate the frequency of B cells positive for IgM surface expression compared with isotype (thin gray line). (C) Frequency of IgM surface expression on CD27− and CD27+ B cells (CD19+ CD20+) quantified in patients without cGVHD (−cGVHD, n = 5, open circles) and with cGVHD (+cGVHD, n = 7 filled squares). Data are median ± range pooled from 2 independent experiments. *P < .05.
Increased BCR responsiveness of cGVHD B cells is abrogated by Syk inhibition
B cells from patients with cGVHD have increased proliferation in response to BCR stimulation (Figure 1). These cells also have elevated protein levels of BLNK and Syk (Figure 2) and increased pBLNK and pSyk in response to BCR stimulation (Figure 3). We wanted to determine whether the increased proliferation was a result of the elevated protein levels or of the kinase activity of these proteins. We used a small molecule inhibitor of Syk, R406 (the active metabolite of fostamatinib), to prevent signal transduction through the BCR pathway. R406 has been used in human B-cell lines to inhibit BCR signaling, including phosphorylation of Syk and BLNK, and proliferation.29,30 We first expanded these published findings to primary B cells and found that R406 inhibited α-IgM–driven phosphorylation of Syk (supplemental Figure 4A) and BLNK (supplemental Figure 4B) in purified B cells from a healthy donor. Proliferation of these cells was also inhibited in a dose-dependent manner (supplemental Figure 4C).
Next, we isolated B cells from 4 patients with cGVHD, known to have increased BCR-specific proliferation (Figure 1), and 4 patients without cGVHD. We treated these B cells ex vivo with R406 and initiated BCR signaling by stimulating with α-IgM. Consistent with our earlier findings, B cells from patients with cGVHD had increased BCR-specific proliferation (Figure 5A). As R406 inhibits the kinase activity of Syk,29 we expected BCR-driven proliferation (downstream of Syk activation) to be inhibited in both patient phenotypes. This was indeed the case, as B cells purified from both patient phenotypes had a dose-dependent inhibition of BCR-driven proliferation (Figure 5A). Importantly, these data suggest that the kinase activity initiated by Syk is important for the hyperproliferation of B cells from patients with cGVHD.
Figure 5.

Kinase activity of Syk is important for BCR hyper-responsiveness in cGVHD B cells ex vivo. B cells isolated from patients without cGVHD (n = 4, open circles) and with cGVHD (n = 4, filled squares) stained with CFSE, treated with R406 (0, 0.01, and 0.1 μM) as indicated, and stimulated with (A-B) α-IgM (50 μg/mL) or (C-D) α-CD40 (100 ng/mL) + IL-4 (5 ng/mL) for 6 days. (A) Frequency of proliferation in response to α-IgM in B cells from patients (left) without cGVHD (open circles) and (right) with cGVHD (filled squares). (Left) ANOVA, P < .0001; *P < .05. (Right) ANOVA, P < .0067; *P < .05. (B) Percent change in number of B cells from A: [Percent change = (V2 – V1)/V1 × 100]. ANOVA, P = .0002; *P < .05. (C) Frequency of proliferation in response to α-CD40 + IL-4 in B cells from patients (left) without cGVHD (open circles) and (right) with cGVHD (filled squares). (D) Percent change in number of B cells from C: [Percent change = (V2 – V1)/V1 × 100]. Data are median ± range pooled from 3 independent experiments.
In addition, although the higher dose of R406 (0.1 μM) almost completely abrogated B-cell proliferation in response to α-IgM, it also appeared to mildly reduce the proliferation in response to anti-CD40 plus IL-4 (Figure 5C). These data suggested that R406 treatment might also impact B-cell survival. We previously found a survival advantage in B cells from patients with cGVHD compared with B cells from patients without disease. Specifically, unmanipulated B cells from patients with cGVHD have decreased apoptosis 48 hours ex vivo.19 We find similar results here after stimulation through the BCR for 6 days: the number of B cells surviving from patients with cGVHD is significantly increased compared with B cells from patients without cGVHD (Figure 5B). Importantly, low-dose R406 (0.01 μM) abrogated this BCR-driven survival advantage of B cells from patients with cGVHD (Figure 5B). Together, these data suggest that BCR signaling, mediated by the kinase activity of Syk, is required for the hyper-responsiveness of B cells from patients with cGVHD.
Discussion
The role of B cells in cGVHD pathogenesis is not completely understood. We find that B cells isolated from patients with cGVHD have the capacity to respond more readily to BCR stimulation compared with B cells from patients without disease. Patients with cGVHD have significantly increased proliferation in response to anti-IgM (Figure 1). Such proliferation is unlikely due to residual homeostatic proliferation as there were no significant differences in the number of B cells present in patients with and without cGVHD included in our proliferative analysis (data not shown). Additionally, massive B-cell proliferation is not a characteristic of cGVHD, suggesting that additional signals resulting in a lowered B-cell activation threshold are necessary. Indeed, our data suggest a number of genes that are decreased in B cells from patients with cGVHD (Figure 2), such as the negative regulator of cell cycle progression and proliferation interferon regulatory factor-4.31,32 These data suggest the decrease of interferon regulatory factor-4 in cGVHD B cells may additionally increase cell cycle progression and their responsiveness.
In murine BCR-driven proliferation, BLNK-deficient mice have impaired cell cycle progression and nuclear factor-κB translocation.24-27 These data demonstrated the essential role of BLNK in BCR-driven proliferation in mice. Our data show that in addition to increased BCR-driven proliferation in cGVHD B cells, these cells have elevated basal protein levels of BLNK and Syk (Figure 2C-D). In healthy donors, signaling initiated through the BCR leads to elevated protein levels of BLNK (Figure 2E), suggesting that increased basal levels of BLNK in cGVHD B cells may be due to the continuous exposure to BCR stimulation after HSCT.
Previous data show that the CD27+ B-cell subset in patients with cGVHD are uniquely activated.15,19 Consistent with these data, CD27+ B cells from patients with cGVHD have significantly increased BCR-driven BLNK and Syk phosphorylation compared with patients without cGVHD (Figure 3A,C). A comparison with B-cell subsets from healthy donors suggested that (1) CD27+ B cells are more activated compared with their CD27− counterparts (supplemental Figure 2) and (2) CD27+ B cells from patients without cGVHD appear to have attenuated signaling compared with both healthy donors and patients with cGVHD (Figure 3; supplemental Figure 2). Unlike healthy donors, B cells from HSCT patients are under constant exposure to allo- and auto-antigens. Whether attenuated signaling in patients without cGVHD is involved in maintenance of B-cell tolerance, and thus lack of disease, requires further study.
Although patients with cGVHD have increased basal levels of Syk (Figure 2D), BCR stimulation does not appear to result in the up-regulation of Syk protein levels (Figure 2E). Thus, the mechanism by which Syk is increased in cGVHD B cells remains unknown. One possibility is increased BAFF-associated signaling. Recent work in murine B cells has revealed that BAFF-dependent B-cell survival requires signaling through Syk.36 Whether cross-talk between BAFF, known to be increased in patients with cGVHD, and BCR signaling occurs in cGVHD, warrants further investigation.
Using R406 to block Syk kinase activity suggested an integral role for BCR signaling in the hyper-responsiveness of cGVHD B cells. R406-mediated inhibition abrogated proliferation and the BCR-driven survival advantage of cGVHD B cells (Figure 5). Taken together, our data suggest a mechanistic link between antigen-BCR signaling, proliferation, and disease pathogenesis in patients with cGVHD. Our data also suggest that therapeutic inhibition of Syk kinase activity may prove beneficial in attenuating increased B-cell responsiveness in cGVHD.
Supplementary Material
Acknowledgments
The authors thank the patients, their families, and Drs Paul Armistead, Jay Coghill, and William Wood for clinical care provided at UNC, Dr Jerome Ritz at the DFCI, and Megan Baker and Krista Rowe at Duke University for providing patient samples. Jessica A. Lavery and Dr Lisong Yang provided additional statistical and technical help, respectively, and Dr Sally Hunsucker maintained the flow cytometer at UNC.
This work was supported by the National Marrow Donor Program through funding from the Amy Strelzer Manasevit Scholars Program funded through The Be The Match Foundation and the National Marrow Donor Program in addition to National Institutes of Health (NIH), National Heart, Lung and Blood Institute grant K08HL107756, National Cancer Institute (NCI) grants 5P01-CA047741-20 (PI, N.J.C.), CA118953 and CA163438 (PI, S.J.L.). The Chronic GVHD Consortium (NCI; U54 CA163438) is a part of the NIH Rare Diseases Clinical Research Network, supported through collaboration between the NIH Office of Rare Diseases Research at the National Center for Advancing Translational Science, and the NCI.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.L.A. designed and performed the research, analyzed and interpreted the data, and wrote the paper; P.V.T performed surface Ig expression; M.S.F., J.W., and S.R. provided technical assistance; A.M.D. calculated statistics; A.S., T.H., P.A.R., T.C.S., J.S.S., K.L.R., M.J., S.J.L., D.R., M.E.H., and N.J.C. collected and provided patient samples, clinical information, and helpful discussions; and S.S. supervised design, analysis, and interpretation of the study and edited the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Stefanie Sarantopoulos, Department of Medicine, Duke University Medical Center, CB# 3961, Durham, NC 27710; e-mail: stefanie.sarantopoulos@duke.edu.
References
- 1.Lee SJ, Flowers ME. Recognizing and managing chronic graft-versus-host disease. Hematology Am Soc Hematol Educ Program. 2008;2008(1):134–141. doi: 10.1182/asheducation-2008.1.134. [DOI] [PubMed] [Google Scholar]
- 2.Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. 2012;12(6):443–458. doi: 10.1038/nri3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimabukuro-Vornhagen A, Hallek MJ, Storb RF, von Bergwelt-Baildon MS. The role of B cells in the pathogenesis of graft-versus-host disease. Blood. 2009;114(24):4919–4927. doi: 10.1182/blood-2008-10-161638. [DOI] [PubMed] [Google Scholar]
- 4.Schultz KR, Paquet J, Bader S, HayGlass KT. Requirement for B cells in T cell priming to minor histocompatibility antigens and development of graft-versus-host disease. Bone Marrow Transplant. 1995;16(2):289–295. [PubMed] [Google Scholar]
- 5.Srinivasan M, Flynn R, Price A, et al. Donor B-cell alloantibody deposition and germinal center formation are required for the development of murine chronic GVHD and bronchiolitis obliterans. Blood. 2012;119(6):1570–1580. doi: 10.1182/blood-2011-07-364414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miklos DB, Kim HT, Miller KH, et al. Antibody responses to H-Y minor histocompatibility antigens correlate with chronic graft-versus-host disease and disease remission. Blood. 2005;105(7):2973–2978. doi: 10.1182/blood-2004-09-3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miklos DB, Kim HT, Zorn E, et al. Antibody response to DBY minor histocompatibility antigen is induced after allogeneic stem cell transplantation and in healthy female donors. Blood. 2004;103(1):353–359. doi: 10.1182/blood-2003-03-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arai S, Sahaf B, Narasimhan B, et al. Prophylactic rituximab after allogeneic transplantation decreases B-cell alloimmunity with low chronic GVHD incidence. Blood. 2012;119(25):6145–6154. doi: 10.1182/blood-2011-12-395970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SJ, Lee JW, Jung CW, et al. Weekly rituximab followed by monthly rituximab treatment for steroid-refractory chronic graft-versus-host disease: results from a prospective, multicenter, phase II study. Haematologica. 2010;95(11):1935–1942. doi: 10.3324/haematol.2010.026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ratanatharathorn V, Ayash L, Reynolds C, et al. Treatment of chronic graft-versus-host disease with anti-CD20 chimeric monoclonal antibody. Biol Blood Marrow Transplant. 2003;9(8):505–511. doi: 10.1016/s1083-8791(03)00216-7. [DOI] [PubMed] [Google Scholar]
- 11.Cutler C, Miklos D, Kim HT, et al. Rituximab for steroid-refractory chronic graft-versus-host disease. Blood. 2006;108(2):756–762. doi: 10.1182/blood-2006-01-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaja F, Bacigalupo A, Patriarca F, et al. GITMO (Gruppo Italiano Trapianto Midollo Osseo) Treatment of refractory chronic GVHD with rituximab: a GITMO study. Bone Marrow Transplant. 2007;40(3):273–277. doi: 10.1038/sj.bmt.1705725. [DOI] [PubMed] [Google Scholar]
- 13.Mohty M, Marchetti N, El-Cheikh J, Faucher C, Fürst S, Blaise D. Rituximab as salvage therapy for refractory chronic GVHD. Bone Marrow Transplant. 2008;41(10):909–911. doi: 10.1038/bmt.2008.12. [DOI] [PubMed] [Google Scholar]
- 14.Fedoriw Y, Samulski TD, Deal AM, et al. Bone marrow B cell precursor number after allogeneic stem cell transplantation and GVHD development. Biol Blood Marrow Transplant. 2012;18(6):968–973. doi: 10.1016/j.bbmt.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarantopoulos S, Stevenson KE, Kim HT, et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood. 2009;113(16):3865–3874. doi: 10.1182/blood-2008-09-177840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarantopoulos S, Stevenson KE, Kim HT, et al. Recovery of B-cell homeostasis after rituximab in chronic graft-versus-host disease. Blood. 2011;117(7):2275–2283. doi: 10.1182/blood-2010-10-307819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greinix HT, Pohlreich D, Kouba M, et al. Elevated numbers of immature/transitional CD21- B lymphocytes and deficiency of memory CD27+ B cells identify patients with active chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2008;14(2):208–219. doi: 10.1016/j.bbmt.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Kuzmina Z, Greinix HT, Weigl R, et al. Significant differences in B-cell subpopulations characterize patients with chronic graft-versus-host disease-associated dysgammaglobulinemia. Blood. 2011;117(7):2265–2274. doi: 10.1182/blood-2010-07-295766. [DOI] [PubMed] [Google Scholar]
- 19.Allen JL, Fore MS, Wooten J, et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood. 2012;120(12):2529–2536. doi: 10.1182/blood-2012-06-438911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patke A, Mecklenbräuker I, Erdjument-Bromage H, Tempst P, Tarakhovsky A. BAFF controls B cell metabolic fitness through a PKC beta- and Akt-dependent mechanism. J Exp Med. 2006;203(11):2551–2562. doi: 10.1084/jem.20060990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toapanta FR, Bernal PJ, Sztein MB. Diverse phosphorylation patterns of B cell receptor-associated signaling in naïve and memory human B cells revealed by phosphoflow, a powerful technique to study signaling at the single cell level. Front Cell Infect Microbiol. 2012;2:128. doi: 10.3389/fcimb.2012.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu JM, Thoburn CJ, Wisell J, Farmer ER, Hess AD. CD20, AIF-1, and TGF-beta in graft-versus-host disease: a study of mRNA expression in histologically matched skin biopsies. Mod Pathol. 2010;23(5):720–728. doi: 10.1038/modpathol.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawakami Y, Ohtsuka M, Kikuta A, Yamamoto T. Multiple morphea-like lesions associated with chronic graft-versus-host disease after cord blood transplantation. Acta Derm Venereol. 2009;89(1):86–87. doi: 10.2340/00015555-0527. [DOI] [PubMed] [Google Scholar]
- 24.Xu S, Tan JE, Wong EP, Manickam A, Ponniah S, Lam KP. B cell development and activation defects resulting in xid-like immunodeficiency in BLNK/SLP-65-deficient mice. Int Immunol. 2000;12(3):397–404. doi: 10.1093/intimm/12.3.397. [DOI] [PubMed] [Google Scholar]
- 25.Jumaa H, Wollscheid B, Mitterer M, Wienands J, Reth M, Nielsen PJ. Abnormal development and function of B lymphocytes in mice deficient for the signaling adaptor protein SLP-65. Immunity. 1999;11(5):547–554. doi: 10.1016/s1074-7613(00)80130-2. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi K, Nittono R, Okamoto N, et al. The B cell-restricted adaptor BASH is required for normal development and antigen receptor-mediated activation of B cells. Proc Natl Acad Sci U S A. 2000;97(6):2755–2760. doi: 10.1073/pnas.040575697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan JE, Wong SC, Gan SK, Xu S, Lam KP. The adaptor protein BLNK is required for b cell antigen receptor-induced activation of nuclear factor-kappa B and cell cycle entry and survival of B lymphocytes. J Biol Chem. 2001;276(23):20055–20063. doi: 10.1074/jbc.M010800200. [DOI] [PubMed] [Google Scholar]
- 28.Fu C, Turck CW, Kurosaki T, Chan AC. BLNK: a central linker protein in B cell activation. Immunity. 1998;9(1):93–103. doi: 10.1016/s1074-7613(00)80591-9. [DOI] [PubMed] [Google Scholar]
- 29.Chen L, Monti S, Juszczynski P, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. 2008;111(4):2230–2237. doi: 10.1182/blood-2007-07-100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quiroga MP, Balakrishnan K, Kurtova AV, et al. B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel spleen tyrosine kinase inhibitor, R406. Blood. 2009;114(5):1029–1037. doi: 10.1182/blood-2009-03-212837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein U, Casola S, Cattoretti G, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006;7(7):773–782. doi: 10.1038/ni1357. [DOI] [PubMed] [Google Scholar]
- 32.Ma S, Pathak S, Trinh L, Lu R. Interferon regulatory factors 4 and 8 induce the expression of Ikaros and Aiolos to down-regulate pre-B-cell receptor and promote cell-cycle withdrawal in pre-B-cell development. Blood. 2008;111(3):1396–1403. doi: 10.1182/blood-2007-08-110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackay F, Figgett WA, Saulep D, Lepage M, Hibbs ML. B-cell stage and context-dependent requirements for survival signals from BAFF and the B-cell receptor. Immunol Rev. 2010;237(1):205–225. doi: 10.1111/j.1600-065X.2010.00944.x. [DOI] [PubMed] [Google Scholar]
- 34.Hase H, Kanno Y, Kojima M, et al. BAFF/BLyS can potentiate B-cell selection with the B-cell coreceptor complex. Blood. 2004;103(6):2257–2265. doi: 10.1182/blood-2003-08-2694. [DOI] [PubMed] [Google Scholar]
- 35.Schebesta M, Pfeffer PL, Busslinger M. Control of pre-BCR signaling by Pax5-dependent activation of the BLNK gene. Immunity. 2002;17(4):473–485. doi: 10.1016/s1074-7613(02)00418-1. [DOI] [PubMed] [Google Scholar]
- 36.Schweighoffer E, Vanes L, Nys J, et al. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity. 2013;38(3):475–488. doi: 10.1016/j.immuni.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.