

Abstract
Somatostatin signals through somatostatin receptor subtypes (SSTR) 2 and 5 to attenuate GH secretion. Although expressed in normal pituitary glands and in GH-secreting pituitary tumors, SSTR3 function was unclear, and we have now determined the role of SSTR3 in somatotroph function. Stable rat pituitary tumor cell (GC) transfectants of human SSTR3 (GpSSTR3WT) showed suppression of rat (r) GH promoter activity, GH mRNA expression, and secreted GH concordant with suppressed cAMP/protein kinase A (PKA) signaling. In contrast, cAMP levels and GH expression were unchanged in cells expressing a mutant SSTR3 DRY motif (GpSSTR3R141A). GH expression was rescued by treatment of GpSSTR3WT with forskolin and 8-bromo-cAMP. GpSSTR3WT exhibited activation of glycogen synthase kinase3-β (GSK3-β), a PKA substrate, which was also reversed by 8-Bromo-cAMP treatment. Moreover, SSTR3-dependent GH transcriptional inhibition was rescued by inhibition of GSK3-β. GpSSTR3WT exhibited elevated Pit-1 serine phosphorylation and decreased Pit-1 occupancy of the rGH promoter with sustained Pit-1 expression. GSK3-β and Pit-1 physically interacted with each other, indicating that Pit-1 may be a GSK3-β phosphorylation substrate. In conclusion, constitutive SSTR3 activity mediates transcriptional repression of GH through cAMP/PKA, leading to subsequent activation of GSK3-β and increased Pit-1 phosphorylation and ultimately attenuating Pit-1 binding to the rGH promoter.
Somatostatin receptor subtype (SSTR) 3, a class A Gαi protein–coupled receptor (GPCR), is expressed in normal pituitary tissue and in ∼44% of GH-secreting pituitary tumors (1). Somatostatin (SRIF) and SRIF analogs suppress GH secretion from normal and tumoral somatotroph cells through inhibition of adenylate cyclase as well as decreased calcium influx (2–8). However, because SRIF-null mice do not exhibit a phenotype consistent with GH excess (9–11), compensatory SRIF-signaling mechanisms controlling basal GH regulation are probably present.
Ligand-independent constitutive GPCR activity occurs with class A GPCRs (2–8, 12, 13). Ligand-mediated pituitary SSTR activation is associated with inhibition of cAMP levels and regulation of protein kinase A (PKA) substrates including cAMP response element-binding protein (CREB) and glycogen synthase kinase 3-β (GSK3-β) (4, 5, 9–11). Knockdown of endogenous Sstr3 in mouse corticotroph AtT-20 cells results in elevated intracellular cAMP production and lower ACTH secretion, ie, evidence of constitutive SSTR3 activity (14). However, the role of ligand-independent SSTR3 signaling in regulation of GH synthesis remains unclear. Although several studies indicated that SRIF signaling does not regulate GH synthesis (15, 16), transient SSTR2 overexpression in primary somatotroph tumor cultures resulted in suppressed GH mRNA levels (17). In addition, we recently showed that stable overexpression of human (h) SSTR2 in rat somatotroph GC cells suppressed GH synthesis (18) and now sought to investigate the contribution of constitutive SSTR3 activity to somatotroph function.
Homeostatic control of GH and IGF-I synthesis and, thus, regulation of insulin availability, glucose homeostasis, and growth are maintained through several feedback loops (6, 19). Rat (r) GH transcriptional regulation is under sustained tissue-specific regulation by Pit-1 (Pou1f1) transcription factor (20). GHRH-induced GH transcriptional activation is mediated by a cAMP/protein kinase A (PKA)–dependent mechanism leading to increased CREB phosphorylation and Pit-1 activation (21–25). Pit-1 is a phosphorylation substrate for multiple kinases including PKA and protein kinase C (PKC), primarily at residues Ser115 and Thr220, mediating Pit-1 interaction with the GH promoter (26, 27). As of yet, Pit-1 has not been shown to be a direct GSK3-β substrate.
GSK3-β, a serine/threonine protein kinase, is a key component in regulating glycogen synthesis (28). GSK3-β is phosphorylated by kinases including PKA, PKC, and AKT (29). GSK3-β activity is enhanced by phosphorylation at tyrosine-216 (30), whereas phosphorylation at serine-9 inactivates the kinase activity (31, 32). Substrate phosphorylation by GSK3-β may result in proteolysis as well as inhibition of transcription factor DNA binding (28, 33).
We hypothesized that in the absence of ligand, SSTR3 exhibits constitutive receptor activity, which regulates GH synthesis. The ability to rigorously assess constitutive receptor activity requires the identification of naturally occurring mutants as well as the availability of inverse agonists, neither of which exist for SSTR3. Mutation of the DRY (aspartate-arginine-tyrosine) motif located within the third transmembrane domain and second intracellular loop transition, conserved in all 5 SSTRs, interferes with an “ionic lock” salt bridge stabilizing the GPCR in an inactive conformation, thereby attenuating constitutive receptor activity (34). Using rat pituitary GC cells, devoid of both endogenous rSstr3 and somatostatin, but expressing low levels of endogenous rSstr2, we generated stable wild-type (WT) and DRY mutant (R141A) SSTR3 transfectants. We observed that ligand-independent SSTR3 activity inhibits basal cAMP/PKA signaling and suppresses GH transcription through GSK3-β activation.
Materials and Methods
Plasmid generation
Human SSTR3 expression plasmids
SSTR3WT was amplified from pCMV-hSSTR3 (kindly provided by Dr Graeme Bell, University of Chicago, Chicago, Illinois) with primers SSTR3-F-EcoRI and SSTR3-R-BamHI then ligated into pIRES2-ZsGreen (Clontech). SSTR3R141A was generated by PCR assembly. A 389-bp fragment was amplified with Out-F-EcoRI and DRY-R, and an 820-bp fragment was amplified with DRY-F and Out-R-BamHI from the human (h) SSTR3WT template. DRY-F and DRY-R primers incorporate a missense R141A mutation upon translation of SSTR3. Assembly PCR was performed with F-Out-EcoRI and R-Out-BamHI using the 389- and 820-bp fragments as a template followed by ligation into pIRES2-ZsGreen. Primer sequences are listed in Supplemental Table 1.
rGH promoter plasmids
Generation of a 4.2-kb promoter construct (rGH4.2) was described previously (18). A 1.7-kb rGH promoter (rGH1.7) was generated by digesting an internal EcoRI site at −1.7 kb within rGH4.2 followed by ligation of a SacI linker. A 2.4-kb fragment was excised by SacI digestion and ligated into pGL4.10 (Promega). Promotor fragments (160- and 220-bp) were amplified with forward primers rGHP-160F and rGHP-220F, respectively, and reverse primer rGHP+167R, using rGH1.7 as a template. PCR products were ligated into pGL4.10 at KpnI and HindIII sites.
Pit-1 expression plasmid
Human Pit-1 cDNA was kindly provided by Dr Ronald Cohen (University of Chicago), and PCR amplified with primers Pit-1-F-BamHI and Pit-1-R-XhoI to generate a C-terminal Myc-tag upon ligation into BamHI and XhoI sites of pCDNA3.1-Myc/His (Invitrogen). All plasmids were sequence confirmed (Sequetech).
Stable transfected cell lines
GC cells from American Type Culture Collection were cultured in DMEM high glucose with 10% fetal bovine serum with antibiotic-antimycotic solution (Invitrogen). Cells were seeded at 3 × 105/well and transfected with 800 ng of pCMV-ZsGreen (empty vector), hSSTR3WT, or hSSTR3R141A with Lipofectamine 2000 according to the manufacturer (Invitrogen) to generate 3 stable transfectants, GpCon, GpSSTR3WT, and GpSSTR3R141A, respectively. After 48 hours, cells were transferred to T75 flasks and selected in medium supplemented with 450 μg/mL G418 sulfate (Santa Cruz Biotechnology) until cell death was no longer apparent. Stable transfectants were maintained in G418-containing medium and expanded as polyclonal lines, and early passage stocks were stored in liquid nitrogen. After 15 to 18 passages postsorting, cells were disposed of and new aliquots were thawed.
Promoter luciferase assays
Stable transfectants were seeded overnight at a density of 2.5 × 104/well of a 48-well plate. Cells were cotransfected with 2.5 ng of Renilla luciferase plasmid as an internal control and 200 ng of rGH promoter constructs or pGL4.10 (empty vector) with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. At 24 hours posttransfection, fresh medium was supplemented with the indicated treatments for an additional 24 hours. Cells were lysed at room temperature for 15 minutes and then were assayed with a Dual-Luciferase Reporter Assay kit (Promega).
cAMP assays
Cells seeded overnight at a density of 5 × 104/well in a 48-well plate were stimulated for 30 minutes at 37°C in medium containing 0.3% BSA and 1 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich) supplemented with indicated treatments. Quadruplicate samples were assayed for cAMP levels with the LANCE cAMP Kit (PerkinElmer) and modified for intracellular cAMP measurement (35). Results were extrapolated from standard curves measured by the Victor3 spectrophotometer (PerkinElmer) and corrected for cell viability.
WST-1 assay for cell viability
An experimental cell suspension was seeded at 100 μL/well of a 96-well plate in 6 replicate wells. Then 10 μL of WST-1 cell proliferation assay solution (Clontech) was added to each well and incubated with WST-1 substrate at 37°C for 4 hours and absorbance measured at 490 nM with the Victor3 spectrophotometer.
Quantitative TaqMan real-time PCR
Cells were grown in 12-well plates, treated accordingly, and total mRNA isolated with RNeasy (QIAGEN). RNA concentration and purity were quantified with the NanoPhotometer P-Class (Implen). Then 2 μg of RNA was reverse transcribed in a 20-μL reaction using QuantiTect Reverse Transcription Kit (QIAGEN). Gene expression was assayed by TaqMan quantitative RT-PCR on the IQ5 real-time PCR thermocycler (Bio-Rad Laboratories). Gene expression was quantified using comparative threshold cycles (CT) in triplicate samples at 50 ng of RNA/well. Then 20-μL reactions were prepared using 2× universal TaqMan Master Mix and 1 μL of a gene-specific TaqMan assay (Applied Biosystems): rGH (Rn01495894_g1), human SSTR3 (Hs00265633_s1), rat SSTR2 (Rn01464950_g1), or rat Pit-1 (Rn00564562_m1). Amplicons were detected using probes tagged with MGB quencher and FAM dye. A rat β-actin control gene expression assay with MGB and VIC dye (4352340E; Applied Biosystems) was used as a reference control. Thermal cycling conditions were 95°C for 10 minutes; 95°C for 15 seconds, and 60°C for 1 minute for 40 cycles. Standard curves were generated for each probe ranging from 0.02 to 200 ng of cDNA.
PKA enzymatic activity assay
Stable transfectants were grown for 72 hours, trypsinized, and washed with PBS, 1 × 106 cells were pelleted, and a PKA enzymatic activity assay was performed with a PepTag Assay for Non-Radioactive Detection of cAMP-Dependent Protein Kinase (Promega). Reactions were run in an 0.8% agarose Tris-HCl gel at 100 V for 20 minutes. Images were taken with Bio-Rad Molecular Imager ChemiDoc and Image Lab software.
Protein isolation and Western blotting
Total cell lysates were isolated with radioimmunoprecipitation assay buffer (Sigma-Aldrich) supplemented with protease and phosphatase inhibitor cocktail (Sigma-Aldrich). Nuclear lysates were prepared using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific). Protein concentrations were quantified by the Bradford assay with Bio-Rad protein assay dye. Absorbances were measured with the Victor3 spectrophotometer at 590 nM (PerkinElmer). Then 20 μg/sample was separated in 4% to 20% PROTEAN TGX gels (Bio-Rad Laboratories) and semidry transferred onto polyvinylidene difluoride membranes. Membranes were probed for total GSK-3β and p-GSK-3β-Ser9 (9832 and 9336, 1:1000; Cell Signaling Technology), β-catenin (610154, 1:1000; BD Transduction Laboratories), p-β-catenin-Ser33/37/Thr41 (9561, 1:1000; Cell Signaling Technology), pan-actin (1:20 000; Millipore), c-Jun and p-c-Jun-Ser243 (9165 and 2994, 1:1000; Cell Signaling Technology), Pit-1 (sc442, 1:200; Santa Cruz Biotechnology), and lamin A/C (sc7293, 1:500; Santa Cruz Biotechnology).
Hormone measurements
Cells were seeded overnight in quadruplicate wells at a density of 5 × 104/well of a 48-well plate in 300 μL of culture medium and simultaneously seeded for the WST-1 assay from the same cell suspension. After 48 hours, 250 μL of conditioned medium was collected. An RIA for rGH was performed with reagents provided by Dr A. F. Parlow from the National Hormone and Peptide Program, Harbor-UCLA Medical Center (Torrance, California) (18). Values were normalized to WST-1 readings for cell viability.
Chromatin immunoprecipitation (ChIP)
Stable transfectants were seeded at 5 × 106/15-cm plate for 72 hours and chromatin isolated with a ChIP-IT Express Enzymatic kit (Active Motif). Chromatin concentration was established after phenol chloroform extraction, and 25 μg of total chromatin added to each ChIP reaction. Immunoprecipitated chromatin was purified for quantitative PCR with the Chromatin IP DNA Purification Kit (Active Motif). For quantification of chromatin recovery, each ChIP reaction was compared with a 10-fold dilution of input chromatin. Quantitative real-time PCR was performed with SYBR Premix Ex Taq II (Takara) on the IQ5 real-time PCR thermocycler (Bio-Rad Laboratories). Values are reported as percent recovery of total input chromatin. rGHP-F and rGHP-R primers were used to detect the first kilobase of the rGH promoter.
Immunoprecipitation and Coimmunoprecipitation
Immunoprecipitation
Stable transfectants were seeded at a density of 1 × 106/per 10-cm plate for 72 hours. Immunoprecipitation was performed with the Universal Magnetic Co-IP Kit (Active Motif) with 750 μg of nuclear lysate and 2 μg of Pit-1 antibody. Beads were washed and then were boiled in 2× Laemmli loading buffer supplemented with β-mercaptoethanol. Samples were separated in 10% PROTEAN TGX gels (Bio-Rad Laboratories) and semidry transferred to polyvinylidene difluoride membrane. Membranes were blotted for total phosphoserine (612546, 1:1000; BD Transduction Laboratories) or total phosphothreonine (sc5267, 1:200; Santa Cruz Technology) and Pit-1 (sc442, 1:200; Santa Cruz Biotechnology).
Coimmunoprecipitation
HEK293T cells (293T) were seeded overnight at a density of 10 × 106/per 15-cm plate in DMEM supplemented with 10% fetal bovine serum. Cells were cotransfected with a human GSK3βWT-HA–tagged plasmid (14753, Addgene; provided by Dr Jim Woodgett, University of Toronto, Toronto, Ontario, Canada) together with either pcDNA3.1-Myc/His (Invitrogen)– or human Pit-1-Myc–tagged plasmid. At 36 hours posttransfection, nuclear lysates were collected, and coimmunoprecipitation was performed with the Universal Magnetic Co-IP Kit with 500 μg of nuclear lysate per condition either with 2 μg of Myc-Tag Antibody (2276; Cell Signaling Technology) or with the IgG isotype control (Cell Signaling Technology). Samples were prepared as mentioned previously, and the interaction between GSK3-β and Pit-1 was determined by sequential blotting with HA-tag antibody (3724, 1:1000; Cell Signaling) and Myc-tag antibody (2276, 1:1000; Cell Signaling Technology). Input protein equal to 1% of protein used in the immunoprecipitation reactions was loaded as a control in a separate gel similarly blotted with HA- and Myc-tag antibodies.
Statistical analysis
Dose-response curves and statistical analysis were generated using GraphPad Prism 5.0 (GraphPad Software). In figures, unless otherwise indicated, column bars represent the means ± SEM. Statistical significance was determined using the Student t test. Significance is indicated on each figure as follows: *, P < .05; **, P < .01; ***, P < .001.
Results
Stable transfectant characterization
Stable polyclonal GC transfectants expressing empty vector (GpCon) or WT hSSTR3 (GpSSTR3WT) were matched for equivalent ZsGreen protein tag expression intensity by flow cytometry. GpSSTR3WT exhibited enhanced dose-dependent SRIF-14 efficacy and potency compared with that of GpCon (Figure 1, A and B, respectively). Although SSTR3 expression is undetectable in GpCon cells, hSSTR3 transgene expression is ∼10-fold less than endogenous rSstr2 of GpCon. rSstr2 expression was suppressed in GpSSTR3WT compared with that in GpCon (Figure 1C). GpSSTR3WT exhibited 3-fold suppression of GH mRNA expression (P < .01) (Figure 1D) and 4-fold attenuation of GH secretion (P < .01) (Figure 1E) compared with those of GpCon.
Figure 1.

Characterization of a ligand-dependent and -independent SSTR3 phenotype in GC transfectants. A and B, SRIF-14 efficacy (A) and potency (B) in GpSSTR3WT and GpCon cotreated with 3-isobutyl-1-methylxanthine and 1 μM forskolin (FOR, Sigma-Aldrich) in DMEM supplemented with 0.3% BSA for 30 minutes, representative of 3 repeated experiments. C, Relative expression of endogenous rSstr2 and hSSTR3 transgene in untreated GpSSTR3WT and GpCon (n = 4). N.D., not detected. D, baseline rGH mRNA expression normalized to β-actin measured at 48 hours after plating (n = 5). E, GH levels measured in the medium of GpSSTR3WT and GpCon at 48 hours after plating normalized to WST-1 absorbance (n = 4). **, P < .01.
GpSSTR3WT exhibited 3-fold suppression of cAMP levels compared with that of GpCon (P < .01) (Figure 2A). GpSSTR3WT exhibited decreased cAMP-dependent PKA activity (Figure 2B) compared with that of GpCon but responded similarly to GpCon upon forskolin stimulation (Supplemental Figure 1). Basal rGH transcriptional activity was significantly inhibited in GpSSTR3WT compared with that in GpCon for 2 different rGH promoter-luciferase constructs at 48 hours posttransfection: a 55% reduction for a 4.3-kb rGH promoter fragment (rGH4.3, P < .01) and a 45% reduction for a 1.7-kb rGH promoter fragment (rGH1.7, P < .05) (Figure 2C). GpSSTR3WT treated with forskolin resulted in rebound of the rGH promoter activity (P < .01) (Figure 2D). GH mRNA suppression was rescued by forskolin (Figure 2E) and by 8-Br-cAMP in GpSSTR3WT (P < .05 (Figure 2F), whereas GpCon exhibited only a modest response to either treatment. With high doses of forskolin, GH secretion in GpSSTR3WT transfectants was increased at 48-hours posttreatment (Supplemental Figure 2).
Figure 2.

Effects of constitutive SSTR3 activity on basal GH synthesis. A, Relative intracellular cAMP in untreated GpSSTR3WT and GpCon (n = 4). Samples were measured with the LANCE cAMP assay kit. B, Basal PKA activity assayed with a PepTag nonradioactive PKA assay. Phosphorylated peptide migrates toward the negative charge and nonphosphorylated peptide migrates toward the positive charge, representative of 3 repeated experiments. Ctrl, control. C, Baseline rGH promoter activity in untreated GpSSTR3WT and GpCon at 48 hours post-transfection (n = 6) (normalized to the internal Renilla luciferase control). D, Effect of 24 hours of 1 μM forskolin on rGH promoter activity expressed as fold change from nontreated cells (n = 3). E and F, Effect of 24-hour cAMP stimulation by 1 μM forskolin (n = 4) and 3 mM 8-Br-cAMP (n = 3) on rGH expression, respectively. *, P < .05; **, P < .01.
R141A mutation of the SSTR3 DRY motif supports a constitutive phenotype
GpCon, GpSSTR3WT, and GpSSTR3R141A stable transfectants were reselected and sorted by flow cytometry for equivalent ZsGreen protein tag signal intensity. Equivalent hSSTR3 transgene expression in GpSSTR3WT and GpSSTR3R141A was further validated by TaqMan real-time PCR (Figure 3A). Both GpSSTR3WT and GpSSTR3R141A exhibited increased dose-dependent SRIF-14 efficacy and potency compared with that of GpCon, confirming that SSTR3R141A is capable of classic SRIF-14 ligand signaling (Figure 3, B and C, respectively). Both GpSSTR3WT and GpSSTR3R141A exhibited down-regulation of endogenous rSstr2 compared with that for GpCon (Supplemental Figure 3). GpSSTR3WT exhibited inhibition of cAMP levels (P < .01) (Figure 3D) and GH mRNA expression (P < .05) (Figure 3E), whereas GpSSTR3R141A had cAMP levels and GH mRNA expression similar to that of GpCon. GSK3-β phosphorylation at serine-9 was reduced in GpSSTR3WT, but not in GpSSTR3R141A, compared with that in control GpCon cells (Figure 3F). GpSSTR3WT probably exhibits GSK3-β activation, because the classic GSK3-β substrate β-catenin showed increased phosphorylation at GSK3-β target sites Ser33/37 and Thr42 (Figure 4A). Activation of GSK3-β by constitutive SSTR3 activity was rescued upon treatment with exogenous 8-Br-cAMP. GpSSTR3WT, but not GpCon, exhibited dose-dependent GSK3-βSer9 phosphorylation in response to stimulation with 8-Br-cAMP (Figure 4B).
Figure 3.

R141A mutation of the DRY motif rescues constitutive SSTR3 activity. A, hSSTR3 transgene expression in second-generation stable cells postsorting (n = 3). B and C, Dose dependence of intracellular cAMP levels with increasing doses of SRIF-14 in GpSSTR3WT, GpSSTR3R141A, and GpCon: SRIF-14 efficacy (B) and SRIF-14 potency (C), representative of 3 repeated experiments. D, Relative baseline cAMP levels in regenerated stable transfectants (n = 3). E, Relative basal rGH expression in regenerated stable transfectants (n = 3). F, Baseline GSK3-β Ser9 phosphorylation in stable transfectants. Relative band intensities of the phosphorylated form of GSK3-β were quantified by densitometry and the ratios of the phosphorylated to total signals are indicated below the blot (n = 3). A representative blot is depicted. *, P < .05; **, P < .01; N.S., not significant. N/A, not applicable.
Figure 4.
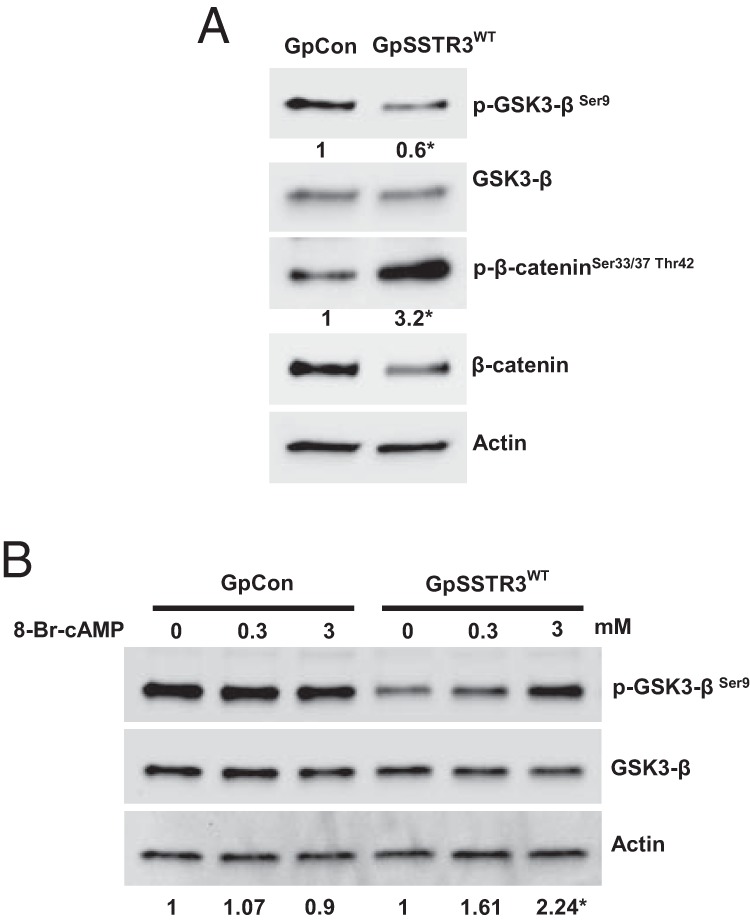
Constitutive SSTR3 mediates activation of GSK3-β. A, Western blot analysis of GSK3-β pathway activation. The phosphorylation status of GSK3-β Ser9 and the GSK3-β substrate β-catenin A, Ser33/37,Thr42, was assayed in untreated GpSSTR3WT and GpCon. Relative band intensities of the phosphorylated form of GSK3-β and β-catenin were quantified by densitometry, and the ratios of the phosphorylated to total signals are indicated below the individual phosphorylated blots (n = 3). A representative blot is depicted. B, Dose response of GSK3-β Ser9 phosphorylation state in response to 30-minute stimulation with 8-Br-cAMP in GpSSTR3WT and GpCon. Relative band intensities of the phosphorylated form of GSK3-β were quantified by densitometry, and the ratios of the phosphorylated to total signals are indicated below the blot (n = 3). Each condition is expressed as fold change from untreated, and a representative blot is depicted.
Constitutive hSSTR3 signaling suppresses GH transcription through GSK3-β activation
Despite suppressed baseline cAMP levels in GpSSTR3WT, CREB phosphorylation was not altered (Supplemental Figure 4). Furthermore, Pit-1 mRNA and protein expression were comparable in GpSSTR3WT and GpCon (Figure 5, A and B, respectively). In addition, expression of several GH transcriptional activators and coactivators including thyroid hormone receptor-α, thyroid hormone receptor-β, retinoic acid receptor-α, retinoid X receptor-α, estrogen receptor-α, estrogen receptor-β, glucocorticoid receptor, Zn-15, c-Fos, Egr1, and Zac1 were assayed by TaqMan real-time PCR, and all were comparable between GpSSTR3WT and GpCon (Supplemental Figure 5). To identify the potential targets of rGH transcriptional inhibition by SSTR3, rGH promoter deletion constructs were generated (Figure 5C). Both a 220-bp and a minimal 160-bp rGH promoter retained transcriptional inhibition in GpSSTR3WT (P < .5 and P < .01, respectively) (Figure 5D). Because the minimal 160-bp promoter contains two Pit-1 binding sites, quantitative ChIP for Pit-1 with the rGH promoter was performed using real-time PCR. GpSSTR3WT transfectants exhibited reduced Pit-1 binding to the rGH promoter compared with that of GpCon (Figure 6A).
Figure 5.
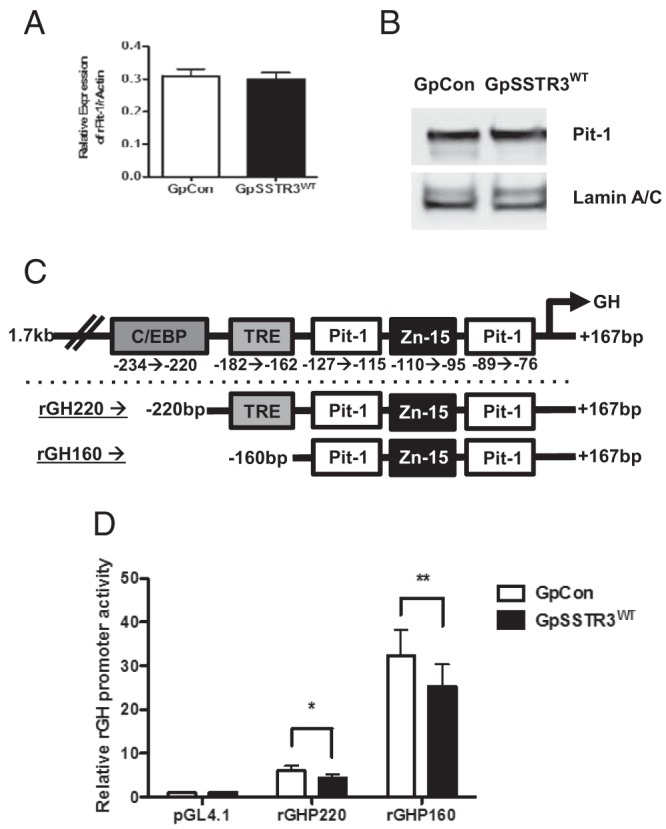
A minimal rGH promoter is required for SSTR3-dependent rGH transcriptional repression. A and B, Relative baseline Pit-1 mRNA expression (A, n = 3), and protein levels (B, representative blot) in untreated GpSSTR3WT and GpCon. C, Schematic diagram of the full-length (rGH1.7) rGH promoter as well as deletion constructs (rGHP220 and rGHP160); transcription factor binding sites are indicated. D, Relative promoter activity of rGH promoter deletion constructs in untreated GpSSTR3WT and GpCon (normalized to internal renilla luciferase control) (n = 5).
Figure 6.

Characterization of Pit-1 as a GSK3-β substrate. A, Quantitative ChIP analysis of Pit-1 occupancy on the rGH promoter (n = 4). B, Representative Western blot of basal serine and threonine phosphorylation state of Pit-1 in stable transfectants by immunoprecipitation including signal intensity quantification by densitometry analysis normalized to total Pit-1 levels. IP, immunoprecipitated; WB, Western blot (n = 3). C, Nuclear coimmunoprecipitation of cotransfected Myc-tagged Pit-1 and HA-tagged GSK3-β. GSK3-β fusion protein runs at 50-kDa and overlaps with IgG heavy chain. A representative blot is depicted. D, Effect of GSK3-β inhibition on rGH promoter activity in GpSSTR3WT and GpCon expressed as percent change from nontreated cells (n = 5). 99021, CHIR99021 (BioVision). **, P < .01; ***, P < .001.
Pit-1 was immunoprecipitated from GpCon and GpSSTR3WT and then was immunoblotted with total phospho-threonine or total phospho-serine antibodies. Densitometry analysis showed ∼1.7-fold increased basal phosphorylation of serine Pit-1 residues in GpSSTR3WT, whereas phosphorylation of threonine residues remained unchanged (Figure 6B). Pit-1 and GSK3-β interacted within the nucleus of 293T cells as evidenced by coimmunoprecipitation of cotransfected HA-tagged GSK3-β with Myc-tagged Pit-1 (Figure 6C). Finally, GpSSTR3WT transfectants treated with CHIR99021, a specific GSK3-β inhibitor, exhibited dose-dependent rebound in rGH promoter activity compared with that in GpCon (P < .001 and P < .01, respectively) (Figure 6D). Other less specific GSK3-β inhibitors including lithium chloride and SB 216763 were tested, and similar results were obtained (Supplemental Figure 6).
Discussion
We demonstrate here that SSTR3, in a ligand-independent manner, suppresses rGH synthesis and transcriptional activation through inhibition of cAMP/PKA signaling. Inhibition of cAMP and GH mRNA levels is specific to constitutive SSTR3 activity because they are abrogated upon selective mutation (R141A) within the SSTR3 DRY motif. Moreover, we identify GSK3-β activation as the mechanism, at least in part, by which constitutive SSTR3 activity mediates cAMP-dependent GH inhibition. GSK3-βSer9 dephosphorylation is reversed upon R141A mutation, and SSTR3WT transfectants exert rebound of rGH promoter activity after treatment with a selective GSK3-β inhibitor. Finally, we identify Pit-1 as a possible phosphorylation substrate of GSK3-β because the molecules were shown to interact by coimmunoprecipitation. Because SSTR3WT transfectants exhibit increased Pit-1 phosphorylation at serine residues as well as decreased occupancy of Pit-1 on the rGH promoter, the results suggest that GSK3-β activation by constitutive SSTR3 activity may mediate GH inhibition through Pit-1 phosphorylation and thus inhibition of GH transcription.
GC cells were chosen as a model to isolate and characterize exogenous SSTR3 function because they secrete GH and express low endogenous levels of rSstr2 yet do not express rSstr1, rSstr3, rSstr4, or rSstr5. Ligand-independent constitutive GPCR activity is an established phenomenon associated with the receptor expression level (12, 13, 36); therefore, it is critical to consider relative receptor transgene expression in the selection of stable transfectants for testing of physiologically relevant constitutive activity. Relative SSTR2 to SSTR3 abundance between ∼0.27- to 22-fold in human GH secreting pituitary tumors has been reported (37–39). Polyclonal GpSSTR3WT transfectants were antibiotic selected and sorted by flow cytometry for matching ZsGreen intensity to GpCon, and TaqMan gene expression analysis demonstrates that the relative expression of hSSTR3 transgene gene expression is ∼10-fold less than that of endogenous rSstr2 in GpCon, which falls within the reported physiological range.
GC cells do not express SSTR ligand (ie, SRIF or cortistatin) (18). Ligand-independent stable expression of WT SSTR3 in GC cells inhibited GH synthesis, as observed by suppressed GH mRNA expression and reduced exogenous rGH promoter activity. These results suggest that, in the absence of ligand, as previously demonstrated for SSTR2 (18), SSTR3 mediates basal regulation of GH synthesis; together these results suggest an overlapping role for ligand-independent SSTR action in regulation of GH production. We also observed reduced intracellular cAMP levels in GpSSTR3WT, consistent with our previous findings that knockdown of mouse (m) Sstr3 in AtT-20 corticotroph cells is associated with increased cAMP accumulation (14). Accordingly, basal activity of cAMP-dependent PKA was inhibited by SSTR3 in our ligand-free system.
mSstr2 expression inhibits cAMP accumulation in AtT-20 cells (14), and endogenous rSstr2 expression in GpSSTR3WT was found to be down-regulated and, therefore, was not likely to contribute to cAMP reduction. Overexpression of both rat and human SSTR2 and SSTR3 leads to formation of SSTR heterodimers in Hek293 cells (40, 41); SSTR2-SSTR3 heterodimerization inhibits the SSTR3 response to ligand, and ligand-mediated activation of cotransfected cells inhibits cell proliferation and induces apoptosis (40, 41). Although rSstr2-hSSTR3 heterodimerization cannot be excluded in our transfectants, low endogenous rSstr2 does not appear to hinder the ability of transfected SSTR3 to respond to ligand.
GHRH-induced GH expression is cAMP dependent (2, 16, 21–23) and, indeed, GpSSTR3WT exhibited rebound of rGH promoter activity when treated with forskolin, as well as a rebound in GH mRNA expression after both forskolin and 8-Br-cAMP treatments. The results suggest that inhibition of GH synthesis occurs downstream of SSTR3-dependent suppression of cAMP production. To elucidate whether suppression of cAMP and inhibition of GH synthesis were attributed to constitutive SSTR3 activity, we mutated the receptor at the DRY motif, a domain associated with GPCR stabilization, by replacing the arginine residue at position 141 with an alanine and recreated stable GC transfectants to compare GpSSTR3WT with GpSSTR3R141A. This specific mutation inhibits the constitutive activity of several class A GPCRs (34). After validation of equal transgene expression, GpSSTR3R141A did not inhibit either GH synthesis or intracellular cAMP production, further supporting an association between constitutive SSTR3 activity and GH inhibition. Interestingly, both GpSSTR3WT and GpSSTR3R141A cells exhibited down-regulation of endogenous rSstr2 expression, which was not observed when we had previously generated stable SSTR2 transfectants. Because GC cells only express a single endogenous SSTR subtype (rSstr2) and both SSTR3WT and SSTR3R141A respond to SRIF-14 ligand, as does SSTR2, introduction of an exogenous receptor with synonymous function may have resulted in down-regulation of endogenous rSstr2. Because both SSTR2 and SSTR3 regulate GH synthesis through their respective constitutive activity, as well as responding to SRIF ligand, the 2 receptors may exhibit compensatory pituitary function in vivo; compensatory SSTR3 action may explain why somatotroph tumors do not develop tachyphylaxis in response to somatostatin analog treatment in contrast with resistance developing in neuroendocrine tumors (6).
Multiple PKA phosphorylation substrates contribute to transcriptional regulation (42, 43); however, as of yet, the understanding of PKA-mediated gene transcription in pituitary cells is limited to regulation of CREB and GSK3-β. cAMP-dependent GHRH-induced GH synthesis is mediated via induction of CREB phosphorylation and Pit-1 expression and is inhibited by SRIF (2, 16, 21–23). In addition, cAMP/PKA signaling in AtT-20 corticotroph cells is mediated through GSK3-β; CRH-dependent inhibition of GSK3-β was effectively blocked upon activation of SSTR signaling (5). GSK3-β deacetylation is also associated with GH inhibition through suppression of CREB and Pit-1 expression (44). Although CREB phosphorylation and Pit-1 gene expression were unchanged, GpSSTR3WT, but not GpSSTR3R141A, exhibited suppressed GSK3-βSer9 phosphorylation. Because GSK3-β phosphorylation at serine-9 inhibits kinase activity (29), our results suggest that constitutive SSTR3 signaling increases GSK3-β activity; appropriately, the classic GSK3-β substrate β-catenin exhibits increased phosphorylation at the characterized GSK3-β target sites in ligand-free GpSSTR3WT. In addition, suppressed total β-catenin levels suggest increased proteolysis after β-catenin phosphorylation, consistent with previous reports of GSK3-β activation (33). Because GSK3-βSer9 dephosphorylation was also rescued by reintroduction of exogenous cAMP in GpSSTR3WT, activation of GSK3-β appears to require inhibition of cAMP levels.
Activation of GSK3-β, a serine/threonine protein kinase, is associated with regulation of gene expression by inhibition of transcription factor DNA binding through targeted phosphorylation (28, 45). A minimal 160-bp rGH promoter containing two Pit-1 binding sites retained transcriptional inhibition in GpSSTR3WT, and quantitative ChIP demonstrated decreased Pit-1 occupancy on the rGH promoter in GpSSTR3WT, suggesting a role for Pit-1 in GH inhibition by SSTR3. Pit-1 occupancy of the rGH promoter is sensitive to phosphorylation by PKA and PKC (26). Although Pit-1 has not heretofore been described to be a direct GSK3-β substrate, immunoprecipitation and densitometry analysis showed that GpSSTR3WT has enhanced phosphorylation of Pit-1 serine residues. Because PKA activity is also suppressed in GpSSTR3WT, increased basal Pit-1 serine phosphorylation could be explained by activation of another kinase. The results therefore suggest that Pit-1 is a likely GSK3-β phosphorylation substrate as both molecules coimmunoprecipitated. Furthermore, treatment of cells with a highly specific chemical GSK3-β inhibitor, CHIR99021 (46), caused dose-dependent rebound of rGH promoter activity in GpSSTR3WT, further indicating that GH inhibition by SSTR3 is mediated at least in part through GSK3-β activation and perhaps by enhanced Pit-1 phosphorylation by GSK3-β. Less selective inhibitors including lithium chloride and SB 216763 similarly exhibited a trend for rescued GH promoter activity in SSTR3 transfectants. Alternatively, SSTR3-dependent GH inhibition may be due to a combined effect of multiple GSK3-β phosphorylation substrates (29). Because SSTR3 is expressed in both prolactinomas and thyrotroph tumors (1, 47) and Pit-1 activation of prolactin and TSH is also phosphorylation dependent (26, 27, 48, 49), the role of constitutive SSTR3 signaling in mediating prolactin and TSH expression should be tested.
The use of recombinant GH therapy is standard practice for treatment of various GH deficiencies, however not without controversy or limitation. As we have described GH transcriptional suppression through both constitutive SSTR2 and SSTR3 signaling, specific inverse agonists, a class of drug that inhibits receptor constitutive activity, may prove to be an effective tool to increase endogenous levels of serum GH and should be further investigated.
In summary, we describe a novel mechanism defining a role for constitutive SSTR3 activity in regulating pituitary GH synthesis through cAMP inhibition and GSK3-β activation. We identify Pit-1 as a putative GSK3-β substrate, and the likely target of GSK3-β mediated GH inhibition by SSTR3. These findings support a role for SSTR3 in somatotroph physiology and suggest that SSTR3 may be a viable therapeutic target for regulation of GH production.
Additional material
Supplementary data supplied by authors.
Acknowledgments
We thank Dr Graeme Bell, Dr James Woodgett, and Dr Ronald Cohen for kindly providing plasmids, Dr Sandra Orsulic for providing 293T cells, and Patricia Lin of the Cedars-Sinai Medical Center flow cytometry core facility and Oxana Pichurin for their technical help.
This work was supported by the National Institutes of Health (Grant CA75979).
Disclosure Summary: A.B.-S. and S.M. are recipients of an investigator-initiated preclinical grant from Novartis. The other authors have nothing to disclose.
Footnotes
- ChIP
- chromatin immunoprecipitation
- CREB
- cAMP response element-binding protein
- GPCR
- G protein–coupled receptor
- GSK3-β
- glycogen synthase kinase
- h
- human
- m
- mouse
- PKA
- protein kinase A
- PKC
- protein kinase C
- r
- rat
- SRIF
- somatostatin
- SSTR
- somatostatin receptor subtype
- WT
- wild-type.
References
- 1. Ben-Shlomo A, Melmed S. Pituitary somatostatin receptor signaling. Trends Endocrinol Metab. 2010;21:123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bilezikjian LM, Vale WW. Stimulation of adenosine 3′,5′-monophosphate production by growth hormone-releasing factor and its inhibition by somatostatin in anterior pituitary cells in vitro. Endocrinology. 1983;113:1726–1731. [DOI] [PubMed] [Google Scholar]
- 3. Heisler S, Reisine TD, Hook VY, Axelrod J. Somatostatin inhibits multireceptor stimulation of cyclic AMP formation and corticotropin secretion in mouse pituitary tumor cells. Proc Natl Acad Sci USA. 1982;79:6502–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tentler JJ, Hadcock JR, Gutierrez-Hartmann A. Somatostatin acts by inhibiting the cyclic 3′,5′-adenosine monophosphate (cAMP)/protein kinase A pathway, cAMP response element-binding protein (CREB) phosphorylation, and CREB transcription potency. Mol Endocrinol. 1997;11:859–866. [DOI] [PubMed] [Google Scholar]
- 5. Khattak MN, Buchfelder M, Kleindienst A, Schöfl C, Kremenevskaja N. CRH and SRIF have opposite effects on the Wnt/β-catenin signalling pathway through PKA/GSK-3β in corticotroph pituitary cells. Cancer Invest. 2010;28:797–805. [DOI] [PubMed] [Google Scholar]
- 6. Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119:3189–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shimon I, Yan X, Taylor JE, Weiss MH, Culler MD, Melmed S. Somatostatin receptor (SSTR) subtype-selective analogues differentially suppress in vitro growth hormone and prolactin in human pituitary adenomas. Novel potential therapy for functional pituitary tumors. J Clin Invest. 1997;100:2386–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patel YC. Somatostatin and its receptor family. Front Neuroendocrinol. 1999;20:157–198. [DOI] [PubMed] [Google Scholar]
- 9. Zeyda T, Diehl N, Paylor R, Brennan MB, Hochgeschwender U. Impairment in motor learning of somatostatin null mutant mice. Brain Res. 2001;906:107–114. [DOI] [PubMed] [Google Scholar]
- 10. Zeyda T, Hochgeschwender U. Null mutant mouse models of somatostatin and cortistatin, and their receptors. Mol Cell Endocrinol. 2008;286:18–25. [DOI] [PubMed] [Google Scholar]
- 11. Low MJ, Otero-Corchon V, Parlow AF, et al. Somatostatin is required for masculinization of growth hormone-regulated hepatic gene expression but not of somatic growth. J Clin Invest. 2001;107:1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smit MJ, Vischer HF, Bakker RA, et al. Pharmacogenomic and structural analysis of constitutive G protein-coupled receptor activity. Annu Rev Pharmacol Toxicol. 2007;47:53–87. [DOI] [PubMed] [Google Scholar]
- 13. Conn PM, ed. Constitutive Activity in Receptors and Other Proteins, Part A. New York, NY: Academic Press; 2010. Methods in Enzymology, vol 484. [Google Scholar]
- 14. Ben-Shlomo A, Pichurin O, Barshop NJ, et al. Selective regulation of somatostatin receptor subtype signaling: evidence for constitutive receptor activation. Mol Endocrinol. 2007;21:2565–2578. [DOI] [PubMed] [Google Scholar]
- 15. Tuggle CK, Trenkle A. Control of growth hormone synthesis. Domest Anim Endocrinol. 1996;13:1–33. [DOI] [PubMed] [Google Scholar]
- 16. Tanner JW, Davis SK, McArthur NH, French JT, Welsh TH. Modulation of growth hormone (GH) secretion and GH mRNA levels by GH-releasing factor, somatostatin and secretagogues in cultured bovine adenohypophysial cells. J Endocrinol. 1990;125:109–115. [DOI] [PubMed] [Google Scholar]
- 17. Acunzo J, Thirion S, Roche C, et al. Somatostatin receptor sst2 decreases cell viability and hormonal hypersecretion and reverses octreotide resistance of human pituitary adenomas. Cancer Res. 2008;68:10163–10170. [DOI] [PubMed] [Google Scholar]
- 18. Ben-Shlomo A, Pichurin O, Khalafi R, et al. Constitutive somatostatin receptor subtype 2 activity attenuates GH synthesis. Endocrinology. 2013;154:2399–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clemmons DR. Modifying IGF1 activity: an approach to treat endocrine disorders, atherosclerosis and cancer. Nat Rev Drug Discov. 2007;6:821–833. [DOI] [PubMed] [Google Scholar]
- 20. Savage JJ, Yaden BC, Kiratipranon P, Rhodes SJ. Transcriptional control during mammalian anterior pituitary development. Gene. 2003;319:1–19. [DOI] [PubMed] [Google Scholar]
- 21. Dana S, Karin M. Induction of human growth hormone promoter activity by the adenosine 3′,5′-monophosphate pathway involves a novel responsive element. Mol Endocrinol. 1989;3:815–821. [DOI] [PubMed] [Google Scholar]
- 22. Brent GA, Harney JW, Moore DD, Larsen PR. Multihormonal regulation of the human, rat, and bovine growth hormone promoters: differential effects of 3′,5′-cyclic adenosine monophosphate, thyroid hormone, and glucocorticoids. Mol Endocrinol. 1988;2:792–798. [DOI] [PubMed] [Google Scholar]
- 23. Copp RP, Samuels HH. Identification of an adenosine 3′,5′-monophosphate (cAMP)-responsive region in the rat growth hormone gene: evidence for independent and synergistic effects of cAMP and thyroid hormone on gene expression. Mol Endocrinol. 1989;3:790–796. [DOI] [PubMed] [Google Scholar]
- 24. Soto JL, Castrillo JL, Dominguez F, Diéguez C. Regulation of the pituitary-specific transcription factor GHF-1/Pit-1 messenger ribonucleic acid levels by growth hormone-secretagogues in rat anterior pituitary cells in monolayer culture. Endocrinology. 1995;136:3863–3870. [DOI] [PubMed] [Google Scholar]
- 25. McCormick A, Brady H, Theill LE, Karin M. Regulation of the pituitary-specific homeobox gene GHF1 by cell-autonomous and environmental cues. Nature. 1990;345:829–832. [DOI] [PubMed] [Google Scholar]
- 26. Kapiloff MS, Farkash Y, Wegner M, Rosenfeld MG. Variable effects of phosphorylation of Pit-1 dictated by the DNA response elements. Science. 1991;253:786–789. [DOI] [PubMed] [Google Scholar]
- 27. Fischberg DJ, Chen XH, Bancroft C. A Pit-1 phosphorylation mutant can mediate both basal and induced prolactin and growth hormone promoter activity. Mol Endocrinol. 1994;8:1566–1573. [DOI] [PubMed] [Google Scholar]
- 28. Sutherland C. What are the bona fide GSK3 substrates? Int J Alzheimers Dis. 2011;2011:505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:40–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V. A chaperone-dependent GSK3β transitional intermediate mediates activation-loop autophosphorylation. Mol Cell. 2006;24:627–633. [DOI] [PubMed] [Google Scholar]
- 31. Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog Neurobiol. 2001;65:391–426. [DOI] [PubMed] [Google Scholar]
- 32. Sutherland C, Leighton IA, Cohen PA. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296(Pt 1):15–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci. 2010;35:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rovati GE, Capra V, Neubig RR. The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol Pharmacol. 2007;71:959–964. [DOI] [PubMed] [Google Scholar]
- 35. Ben-Shlomo A, Wawrowsky K, Melmed S. Constitutive activity of somatostatin receptor subtypes. Methods Enzymol. 2010;484:149–164. [DOI] [PubMed] [Google Scholar]
- 36. Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381–416. [DOI] [PubMed] [Google Scholar]
- 37. Taboada GF, Luque RM, Neto LV, et al. Quantitative analysis of somatostatin receptor subtypes (1–5) gene expression levels in somatotropinomas and correlation to in vivo hormonal and tumor volume responses to treatment with octreotide LAR. Eur J Endocrinol. 2008;158:295–303. [DOI] [PubMed] [Google Scholar]
- 38. Taboada GF, Luque RM, Bastos W, et al. Quantitative analysis of somatostatin receptor subtype (SSTR1–5) gene expression levels in somatotropinomas and non-functioning pituitary adenomas. Eur J Endocrinol. 2007;156:65–74. [DOI] [PubMed] [Google Scholar]
- 39. Casarini AP, Jallad RS, Pinto EM, et al. Acromegaly: correlation between expression of somatostatin receptor subtypes and response to octreotide-lar treatment. Pituitary. 2009;12:297–303. [DOI] [PubMed] [Google Scholar]
- 40. Pfeiffer M, Koch T, Schröder H, et al. Homo- and heterodimerization of somatostatin receptor subtypes. Inactivation of sst3 receptor function by heterodimerization with sst2A. J Biol Chem. 2001;276:14027–14036. [DOI] [PubMed] [Google Scholar]
- 41. War SA, Kumar U. Coexpression of human somatostatin receptor-2 (SSTR2) and SSTR3 modulates antiproliferative signaling and apoptosis. J Mol Signal. 2012;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shabb JB. Physiological substrates of cAMP-dependent protein kinase. Chem Rev. 2001;101:2381–2411. [DOI] [PubMed] [Google Scholar]
- 43. Fimia GM, Sassone-Corsi P. Cyclic AMP signalling. J Cell Sci. 2001;114:1971–1972. [DOI] [PubMed] [Google Scholar]
- 44. Monteserin-Garcia J, Al-Massadi O, Seoane LM, et al. Sirt1 inhibits the transcription factor CREB to regulate pituitary growth hormone synthesis. FASEB J. 2013;27:1561–1571. [DOI] [PubMed] [Google Scholar]
- 45. Glycogen synthase kinase-3 in neurological diseases. In: Hideyuki M, ed. Protein Kinase Technologies. New York: Humana Press; 2012:153–188. [Google Scholar]
- 46. An WF, Germain AR, Bishop JA, et al. Discovery of potent and highly selective inhibitors of GSK3b. In: Probe Reports from the NIH Molecular Libraries Program. Bethesda, MD: National Center for Biotechnology Information; 2010. [updated February 28, 2013]. http://www.ncbi.nlm.nih.gov/books/NBK133436/. [PubMed] [Google Scholar]
- 47. Yoshihara A, Isozaki O, Hizuka N, et al. Expression of type 5 somatostatin receptor in TSH-secreting pituitary adenomas: a possible marker for predicting long-term response to octreotide therapy. Endocr J. 2007;54:133–138. [DOI] [PubMed] [Google Scholar]
- 48. Jean A, Gutierrez-Hartmann A, Duval DL. A Pit-1 threonine 220 phosphomimic reduces binding to monomeric DNA sites to inhibit ras and estrogen stimulation of the prolactin gene promoter. Mol Endocrinol. 2010;24:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Steinfelder HJ, Radovick S, Wondisford FE. Hormonal regulation of the thyrotropin beta-subunit gene by phosphorylation of the pituitary-specific transcription factor Pit-1. Proc Natl Acad Sci USA. 1992;89:5942–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.