

ABSTRACT
Background: In beta thalassaemia major multiple blood transfusions, ineffective erythropoiesis and increased gastrointestinal iron absorption lead to iron overload in the body. Iron overload impairs the immune system, placing patients at greater risk of infection and illness. Iron overload can be determined by serum ferritin measurement.
Objective: The aim of the present study is to assess the serum ferritin levels in multi-transfused Thalassaemia major and Thalassaemia intermedia patients. The study was also done to estimate the present situation of awareness of iron overload in them.
Methods: Seventy two blood samples from clinically diagnosed thalassaemia major and intermedia patients were collected from different tertiary care hospitals in Bhopal, India for their serum ferritin estimation. Serum ferritin measurement was performed using indirect enzyme linked immune sorbent based serum ferritin assay kit. Data were analyzed to determine association between variables. The association between age, sex, and serum ferritin level were established.
Results: 87.4% of the beta thalassaemia major patients showed very high ferritin levels. The mean serum ferritin level was found to be 2767.52 ng/ml. 44.4% patients had serum ferritin between 1000 to 2500 ng/ml, while 43.05% patients had values above 2500 ng/ml. These levels reflect inadequate chelation and vulnerability to develop iron overload related complications.
Conclusion: There is an urgent need to rationalize the chelation therapy and to create awareness about the consequences of iron overload in the patients. The study showed high levels of serum ferritin beta thalassaemia major patients which give an overall bleak view.
INTRODUCTION
β-Thalassaemia major is a hereditary haemolytic anaemia that is treated with multiple blood transfusions (1). Studies have shown that the overall prevalence of β-thalassaemia in India is 3-4% with an estimate of around 8,000 to 10,000 new births with major disease each year (2). If a line is drawn between Mumbai and Kolkata on the Map of India, the region above the line has an incidence of 3 to 17%, whereas region below the line has incidence of less than 3% (3). Iron overload is a common complication of thalassaemia syndromes which could lead per se to the development of organ damage and increased mortality (4,5). In these patients, iron deposition in parenchymal tissues begins within 1 year of starting the regular transfusions (6).
Iron stores in the body exist primarily in the form of ferritin. In the body, small amounts of ferritin are secreted into the plasma. The concentration of this plasma (or serum) ferritin is positively correlated with the size of the total body iron stores in the absence of inflammation. Normal ferritin concentrations vary by age and sex. Concentrations are high at birth, rise during the first two months of life, and then fall throughout later infancy (7). At about one year of age, concentrations begin to rise again and continue to increase into adulthood (8). Beginning in adolescence, however, males have higher values than females; a trend that persists into late adulthood.
Although blood transfusions are important for patients with anemia, chronic transfusions inevitably lead to iron overload as humans cannot actively remove excess iron. The cumulative effects of iron overload lead to significant morbidity and mortality, if untreated (9). A unit of red blood cells transfused contains approximately 250 mg of iron (10), while the body cannot excrete more than 1 mg of iron per day. A patient who receives 25 units per year, accumulates 5 grams of iron per year in the absence of chelation (11). Add to this the increased intestinal iron absorption that is also seen in these patients. Research also indicates that iron overload can occur in patients with non-transfusion dependent thalassaemias (NTDT) (12). In Thalassaemia intermedia the increased gastro-intestinal absorption of iron, which is much higher than that in normal individuals is most likely due to a paradoxical suppression of hepcidin (13).
As iron loading progresses, the capacity of serum transferrin, the main transport protein of iron, to bind and detoxify iron may be exceeded. Thereafter, the non-transferrin-bound fraction of iron within plasma may promote generation of free hydroxyl radicals, propagators of oxygen-related damage (14).
Excess iron is extremely toxic to all cells of the body and can cause serious and irreversible organic damage, such as cirrhosis, diabetes, heart disease, and hypogonadism (15). Fibrosis of the liver correlates directly with the age, number of units transfused, and the liver iron concentration (16).
The iron burden on the body can be estimated by means of serum ferritin, iron and TIBC levels. Effective management of iron overload in thalassaemia requires monitoring both for iron toxicity and the effects of excessive chelation (17). The estimation of serum ferritin levels is the most commonly employed test to evaluate iron overload in Beta Thalassaemia Major. A target ferritin of approximately 1000 mg/l is generally recommended standard practice in thalassaemia major (TIF Guidelines, 2000) and other forms of iron overload resulting from blood transfusion. When the serum ferritin level reaches 1000 ng/l (usually after 10th to 12th transfusion), it is generally taken as the point to initiate iron chelation therapy.
Samples and Center of study
A total of 72 blood samples taken from β-thalassaemia major and thalassaemia intermedia were included in this prospective study. The study was conducted at School of Biotechnology, Rajiv Gandhi Proudyogiki Vishwavidyalaya, Bhopal, India.
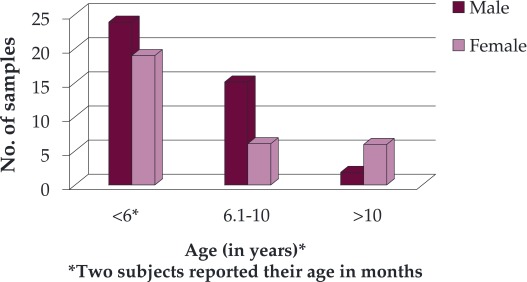
Sample collection: The known cases of β-thalassaemia major and thalassaemia intermedia that had been transfused irrespective of their age and gender were randomly selected. Subjects were classified into three age groups: <6 years, in between 6.1-10 years and >10 years (Figure 1).
Figure 1. Age and gender distribution of the study subjects.
Clinical Data: The clinical details of patients were recorded in a proforma, taking into account the age, gender, frequency of transfusions and estimation of serum ferritin levels.
Serum Ferritin Estimation
About 3 ml of patient's blood sample was collected by a clean venepuncture. The blood was allowed to clot. Serum was separated and stored at -20°C. Ferritin levels were performed by using indirect enzyme linked immunosorbent assay (ELISA) kit (Orgentec, Germany) along with normal and abnormal controls. Anti-human-ferritin antibodies were bound to microwells. ❑
RESULTS
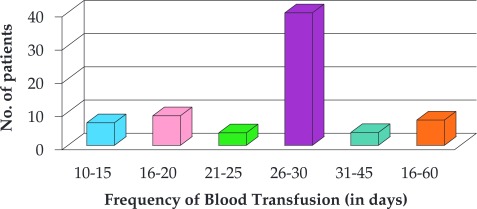
In this study, a total of 72 cases consisting of thalassaemia major and intermedia were examined, of which 56.9% were male and 43% female, with a male to female ratio of 1.32: 1. The mean age of males was 5.9 years whereas mean age of females was 6.2 years. The age of patients at the time of diagnosis of thalassaemia ranged from 10 months to 2 ½ years with a mean of 1 year and 4 months. The age at the time of serum ferritin estimation ranged between 10 months to 14 years. The interval between successive transfusions varied between 15 days and 8 weeks in different patients. In 55.6% patients (40), the frequency of transfusion was 30 days as suggested in Figure 2.
Figure 2. Clinical history of frequency of blood transfusion in the studied subjects.
Serum Ferritin Levels
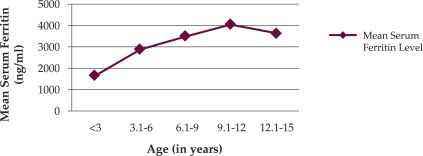
Figure 3 displays the range of serum ferritin levels observed in patients. The mean serum ferritin level was 2767.52 (SD 1849.1) ng/ml. Only nine patients (12.5%) had serum ferritin level less than 1000 ng/ml. Thirty two patients (44.4%) had serum ferritin level between 1000-2500 ng/ml, while 43.05% patients (31) had values more than 2500 ng/ml (Figure 3). The serum ferritin level increases as the frequency of blood transfusion and the age of the patient increases as depicted in Figure 4. The mean serum ferritin level in males was found to be 2900.8 ng/ml and it is comparable to the mean serum ferritin level in females i.e. 2591.2 ng/ml. Thalassaemia intermedia patients showed very low increase in their serum ferritin levels. ❑
Figure 3. Range of Serum ferritin level observed in subjects studied.
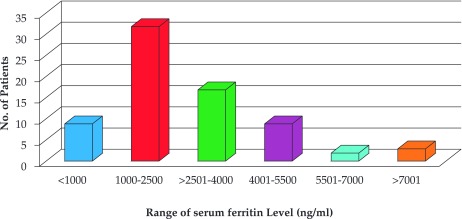
Figure 4. Relationship between age (in years) and serum ferritin Levels (expressed in ng/ml).
DISCUSSIONS
Thalassemias are common genetic disorders in the Indian subcontinent. Hyper-transfusion has improved the life expectancy of thalassaemic patient over the decades but iron overload is an unavoidable complication suffered by thalassaemia major patients as a consequence of an excessive number of blood transfusions. It is so common that it has been referred to as "second disease" during treatment of first (18) and it results in a number of other diseases and complications. Many studies concluded that cirrhosis of liver is associated with increase in serum ferritin levels (19).
Over the last 20 years, management of thalassemia major has improved to the point where patients' life expectancy will reach that of the normal population (20). These outcomes result from safer blood transfusions, the availability of three iron chelators, new imaging techniques that allow specific organ assessment of the degree of iron overload (20).
Transfused iron is deposited first within the reticuloendothelial cells prior to parenchymal iron loading within the heart and liver. However, as in primary iron overload, the majority of morbidity and mortality ultimately results from progressive heart and liver failure.
Effective management of iron overload requires frequent evaluation of the body iron stores (21). There is, therefore, a need for quantitative, non-invasive methods for measuring body iron that are safe, accurate and readily available. The iron status of the body in overload conditions can be assessed by different methods. Serum ferritin measurement, although easy to perform frequently, offers variable results, still at present, no other serum test is a better predictor (22).
The liver is the major site of iron overload, containing 70% or more of body iron content. Liver iron correlates closely with total body iron in transfusional iron overload and total body iron. Estimation of direct liver iron concentration is the most accurate method of estimation of iron overload. But in our set up this method was not available. Indirect method with serum ferritin level measurement is reliable, easy to perform, low cost, and had no side effects (23). In any event, when serum ferritin is greatly increased, whatever the reason, there is cause for concern and an increasingly aggressive iron chelation treatment should be given. In a study by Bandyopadhyay et al., patients even in the younger age group showed high serum ferritin levels. They found that in 1-5 years age group, average serum ferritin was 1750 ng/ml, and this increased to 3650 ng/ml in 11-15 years older patients (24). The serum ferritin level could not be controlled well as only few patients fully complied with recommended regimen at home (25). Similarly, in our study, the mean serum ferritin level was 2767.5 ng/ml, which is markedly higher than the normal recommended levels for normal individuals. Normal values of serum ferritin for men and women are 12-300 ng/mL and 12-150 ng/mL, respectively (20).
The values in our study are higher compared with similar regional and international studies. Cunningham et al in 2004 reported mean serum ferritin levels in beta thalassemia patients of North America to be 1696 ng/ml (26). However, Choudhry VP et al in India reported mean serum ferritin levels to be 6723 ng/ml (27) even higher than in our study.
The distribution of β-thalassaemia gene is not uniform in the Indian subcontinent. The highest frequency of β-thalassaemia trait is reported in Gujarat, followed by Sindh, Punjab, Tamil Nadu, South India and Maharashtra (28). In various parts of India, the prevalence of β-thalassemia is different: 6.5% in Punjab, 8.4% in Tamilnadu, 4.3% in South India, and 3.5% in Bengal. β-Thalassaemia has a high prevalence in some communities, such as Sindhi, Luvana, Tribes, and Rajputs. The incidence of β-thalassemia in Gujarat is 10% to 15% (28). Therefore, in these regions and communities proper management and care of thalassaemia patients should be done due to iron overload in their body. ❑
LIMITATIONS
The height and weight (and consequently the body mass index) of the patients was not obtained. The dose and frequency of Desferrioxamine infusions or any other iron chelator therapy is not investigated and is also a limitation. ❑
CONCLUSION
The estimation of serum iron can be done using ELISA based techniques. There is a strong need to create awareness among patients about the consequences of iron overload in their body. The high level of serum ferritin of beta thalassaemia major patients noted in this study supports the rationale for regular follow-up of transfusion dependent thalassaemic patients with respect to iron overload to ensure proper management of iron overload associated complications. Proper chelation of iron overload could improve the quality of life of these patients. The problems of poverty, low education level and inadequate provision of health care are the main stumbling blocks in effective treatment of iron overload in thalassaemic patients which is the main cause of morbidity and mortality in thalassaemia major. ❑
RECOMMENDATIONS
Routine measurements of serum ferritin levels, adherence to guidelines, awareness amongst the public for prevention of the disease and antenatal diagnosis are some of the methods which can decrease the suffering of not only the patients, but also of their families.
ACKNOWLEDGEMENTS
The corresponding author would like to gratefully acknowledge Indian Council of Medical Research, New Delhi, India for providing the Senior Research fellowship. The authors wish to thank all the thalassaemic patients for their cooperation in this study and the staff members of various associated hospital for their support, help and guidance.
CONFLICT OF INTEREST
none declared.
References
- 1.Argyropoulou MI, Astrakas L. MRI evaluation of tissue iron burden in patients with beta-thalassaemia major. Pediatr Radiol. 2007;37:1191–200. doi: 10.1007/s00247-007-0567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mohanty D, Colah RB, Gorakshakar AC, et al. Prevalence of beta-thalassemia and other haemoglobinopathies in six cities in India: a multicentre study. J Community Genet. 2013;4:33–42. doi: 10.1007/s12687-012-0114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinha A, Ray D, Kundu D, et al. Conservative management of Beta-thalassemia major cases in the sub-division level hospital of rural West Bengal, India. 2013;4:108–12. doi: 10.4103/0976-9668.107269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mariani R, Trombini P, Pozzi M, et al. Iron metabolism in thalassemia and sickle cell disease. Mediterr J Hematol Infect Dis. 2009;1:006–006. doi: 10.4084/MJHID.2009.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fung EB, Harmatz P, Milet M, et al. Morbidity and mortality in chronically transfused subjects with thalassemia and sickle cell disease: A report from the multi-center study of iron overload. Am J Hematol. 2007;82:255–65. doi: 10.1002/ajh.20809. [DOI] [PubMed] [Google Scholar]
- 6.Taksande A, Prabhu S, Venkatesh S. Cardiovascular Aspect of Beta-Thalassaemia. Cardiovasc Hematol Agents Med Chem. 2012;10:25–30. doi: 10.2174/187152512799201172. [DOI] [PubMed] [Google Scholar]
- 7.Domellof M, Dewey KG, Lonnerdal B, et al. The diagnostic criteria for iron deficiency in infants should be reevaluated. J Nutr. 2002;132:3680–6. doi: 10.1093/jn/132.12.3680. [DOI] [PubMed] [Google Scholar]
- 8.Gibson RS. Oxford University Press; 2005. Principles Of Nutritional Assessment. [Google Scholar]
- 9.Cappellini MD. Exjade(R) (deferasirox, ICL670) in the treatment of chronic iron overload associated with blood transfusion. Ther Clin Risk Manag. 2007;3:291–9. doi: 10.2147/tcrm.2007.3.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ozment CP, Turi JL. Iron overload following red blood cell transfusion and its impact on disease severity. Biochim Biophys Acta. 2009;1790:694–701. doi: 10.1016/j.bbagen.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Piomelli S. The management of patients with Cooley's anemia: transfusions and splenectomy. Semin Hematol. 1995;32:262–8. [PubMed] [Google Scholar]
- 12.Taher AT, Porter J, Viprakasit V, et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120:970–7. doi: 10.1182/blood-2012-02-412692. [DOI] [PubMed] [Google Scholar]
- 13.Gardenghi S, Marongiu MF, Ramos P, et al. Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood. 2007;109:5027–35. doi: 10.1182/blood-2006-09-048868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hershko C, Konijn AM, Link G. Iron chelators for thalassaemia. Br J Haematol. 1998;101:399–406. doi: 10.1046/j.1365-2141.1998.00726.x. [DOI] [PubMed] [Google Scholar]
- 15.Melchiori L, Gardenghi S, Rivella S. Beta-Thalassemia: HiJAKing Ineffective Erythropoiesis and Iron Overload. Adv Hematol. 2010;2010:938640–938640. doi: 10.1155/2010/938640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wintrobe MM, John P, Greer M. Wolters Kluwer Health. Lippincott Williams & Wilkins; 2009. Wintrobe's Clinical Hematology. [Google Scholar]
- 17.Porter JB, Davis BA. Monitoring chelation therapy to achieve optimal outcome in the treatment of thalassaemia. Best Pract Res Clin Haematol. 2002;15:329–68. [PubMed] [Google Scholar]
- 18.Riaz H, Riaz T, Khan MU, et al. Serum ferritin levels, socio-demographic factors and desferrioxamine therapy in multi-transfused thalassemia major patients at a government tertiary care hospital of Karachi, Pakistan. BMC research notes. 2011;4:287–287. doi: 10.1186/1756-0500-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knovich MA, Storey JA, Coffman LG, et al. Ferritin for the clinician. Blood Rev. 2009;23:95–104. doi: 10.1016/j.blre.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berdoukas V, Farmaki K, Carson S, et al. Treating thalassemia major-related iron overload: the role of deferiprone. J Blood Med. 2012;3:119–29. doi: 10.2147/JBM.S27400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jensen P-D. Evaluation of iron overload. Br J Haematol. 2004;124:697–711. doi: 10.1111/j.1365-2141.2004.04838.x. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal MB. Advances in management of thalassemia. Indian J Pediatr. 2009;76:177–84. doi: 10.1007/s12098-009-0048-7. [DOI] [PubMed] [Google Scholar]
- 23.Olivieri NF, Brittenham GM. Iron-Chelating Therapy and the Treatment of Thalassemia. Blood. 1997;89:739–61. [PubMed] [Google Scholar]
- 24.Bandyopadhyay U, Kundu D, Sinha A, et al. Conservative management of Beta-thalassemia major cases in the sub-division level hospital of rural West Bengal, India. J Nat Sci Biol Med. 2013;4:108–12. doi: 10.4103/0976-9668.107269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eleftheriou A. Thalassemia International Federation: Guidelines for the clinical management of thalassemia. Thalassemia International Federation Nicosia Cyprus. 2008 Mar; [PubMed] [Google Scholar]
- 26.Cunningham MJ, Macklin EA, Neufeld EJ, et al. Thalassemia Clinical Research N. Complications of beta-thalassemia major in North America. Blood. 2004;104:34–9. doi: 10.1182/blood-2003-09-3167. [DOI] [PubMed] [Google Scholar]
- 27.Choudhry VP, Pati HP, Saxena A, et al. Deferiprone, efficacy and safety. The. Indian J Pediatr. 2004;71:213–6. doi: 10.1007/BF02724272. [DOI] [PubMed] [Google Scholar]
- 28.Ambekar SS, Phadke MA, Mokashi GD, et al. Pattern of hemoglobinopathies in western Maharashtra. Indian Pediatr. 2001;38:530–4. [PubMed] [Google Scholar]