

Abstract
NADPH oxidase (Nox) enzymes are a significant source of reactive oxygen species, which contribute to glomerular podocyte dysfunction. Although studies have implicated Nox1, -2, and -4 in several glomerulopathies, including diabetic nephropathy, little is known regarding the role of Nox5 in this context. We examined Nox5 expression and regulation in kidney biopsies from diabetic patients, cultured human podocytes, and a novel mouse model. Nox5 expression increased in human diabetic glomeruli compared with nondiabetic glomeruli. Stimulation with angiotensin II upregulated Nox5 expression in human podocyte cultures and increased reactive oxygen species generation. siRNA-mediated Nox5 knockdown inhibited angiotensin II–stimulated production of reactive oxygen species and altered podocyte cytoskeletal dynamics, resulting in an Rac-mediated motile phenotype. Because the Nox5 gene is absent in rodents, we generated transgenic mice expressing human Nox5 in a podocyte-specific manner (Nox5pod+). Nox5pod+ mice exhibited early onset albuminuria, podocyte foot process effacement, and elevated systolic BP. Subjecting Nox5pod+ mice to streptozotocin-induced diabetes further exacerbated these changes. Our data show that renal Nox5 is upregulated in human diabetic nephropathy and may alter filtration barrier function and systolic BP through the production of reactive oxygen species. These findings provide the first evidence that podocyte Nox5 has an important role in impaired renal function and hypertension.
Albuminuria is a clinical marker of kidney dysfunction that arises in most glomerulopathies and is associated with poor prognoses for ESRD, hypertension, and cardiovascular mortality. Changes to the podocyte (e.g., foot process effacement, hypertrophy, detachment, and loss) underlie the development and progression of albuminuria and thereby highlight the critical role for these cells in upholding the glomerular filtration barrier.1,2 Therefore, identifying factors that induce podocyte injury and loss is essential to understanding the mechanisms of filtration barrier dysfunction.
Of the many factors implicated in podocyte dysfunction, excessive production of reactive oxygen species (ROS; oxidative stress) may be particularly important.3–6 Although sources of ROS are numerous, the NADPH oxidase (Nox) family of enzymes yields significant superoxide production in the kidney.7–10 Nox-induced ROS production has been closely linked to various glomerular pathologies. In animal models of minimal change disease, membranous nephropathy, and FSGS, inhibition of Nox activity is associated with decreased podocyte effacement and amelioration of albuminuria.11–14 In models of diabetic nephropathy, treatment with the Nox inhibitor apocynin, as well the antioxidant vitamin E, reduces oxidative stress, podocyte effacement and loss, and albuminuria.6,15,16 Noxs are regulated by many factors, including the renin angiotensin aldosterone system.5,9 Several studies have linked increased renin angiotensin aldosterone system activity to enhanced renal Nox activity and ROS generation.5,17 Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers slow progression of proteinuria in models of diabetes, and these effects may be, in part, independent of their effects on systemic BP,17–20 because direct activation of Nox enzymes through the angiotensin II (AngII)/AT1 receptor (AT1R) pathway leads to oxidative stress. In vitro studies in both human and rodent cell lines have also shown that Nox family member expression and activity are regulated by disease-associated factors, including AngII, ET-1, TGF-β, high glucose, mechanical stretch, and PDGF (factors that are upregulated in the diabetic milieu).3,21–23
The roles of Nox4, and to a lesser extent, Nox1 and -2, in the kidney have been examined, but nothing is known regarding the role of the most recently identified member of the Nox family, Nox5. The Nox5 gene is absent from the mouse and rat genomes, making the use of conventional animal models unfeasible. Unlike other Nox family members, Nox5 does not require membrane-bound or cytosolic components, such as p22phox or p47phox, for its activity, but is tightly regulated by changes in intracellular calcium levels.24,25 Nox5 has a large amino terminal EF hand-containing domain that plays a critical role in its calcium-dependent activation along with several phosphorylation sites that alter the sensitivity of Nox5 to intracellular calcium.26–29 Because AngII increases intracellular calcium concentrations, it seems to induce renal Nox5-dependent ROS generation, which was shown in human endothelial cells.23 Here, we show that (1) Nox5 is upregulated in human diabetic glomeruli; (2) AngII stimulates ROS generation in human podocytes in a Nox5-dependent manner, a process associated with actin cytoskeletal reorganization and activation of Rac GTPase, which promotes podocyte motility in vitro; (3) mice that express human Nox5 in a podocyte-specific manner (Nox5βpod+ mice) exhibit renal dysfunction, including albuminuria, podocyte effacement, glomerular basement membrane (GBM) thickening, interstitial fibrosis, and hypertension; and (4) Nox5pod+ mice subjected to streptozotocin (STZ)-induced diabetes develop a more severe kidney phenotype than nontransgenic littermates. These novel data indicate the potential importance of podocyte Nox5 in human renal pathologies, such as diabetic nephropathy.
Results
Nox5 Is Expressed in Human Podocytes
Consistent with a potential role in diabetic nephropathy, immunodetectable Nox5 was observed in glomeruli from diabetic renal biopsies but not nondiabetic samples (Figure 1B). Furthermore, Nox5 did not colocalize with TGF-β, which is produced mainly by mesangial cells in diabetic glomeruli, thereby suggesting that Nox5 induction was primarily in podocytes. Coimmunofluorescent localization with the podocyte marker nephrin was consistent with Nox5 expression in podocytes of diabetic glomeruli (Figure 1B). Nox4 expression was apparent in both nondiabetic and diabetic glomeruli, with greater expression observed in the latter (Supplemental Figure 1). In contrast, low levels of Nox1 and -2 were observed in glomeruli, with little difference in expression between diabetic and nondiabetic samples (Supplemental Figure 1). RT-PCR of RNA from conditionally-immortalized human podocytes (hPODs) revealed ample Nox5 transcript (Figure 1C). Nox1 and -4 were also detected in hPODs along with minimal Nox2 expression (Figure 1C). Examination of Nox5 splice variants by RT-PCR identified Nox5β as the predominant species in hPODs (Figure 1D). Likewise, Nox5β was the principal variant detected in human embryonic kidney cells and human proximal tubule epithelial cells. Low levels of Nox5α were detected in human embryonic kidney cells and proximal tubule epithelial cells but not hPODs, whereas Nox5δ and -5γ were absent from all cell lines examined (Figure 1D).
Figure 1.

Nox5 is expressed in human diabetic kidney biopsies and human podocytes. Characterization of Nox expression in human diabetic kidney and podocytes. (A) Immunofluorescence for Nox5 (green) and TGF-β (red) expression in nondiabetic and diabetic human kidney biopsies. Scale bar, 50 μm. (B) Costaining of human diabetic kidney for Nox5 (red) and nephrin (green) expression (4′,6-diamidino-2-phenylindole as a nuclear counterstain). (C) RT-PCR analyzing expression of Nox1, -2, -3, -4, and -5 in hPOD lysates. (D) Expression of Nox5 splice variants-α (327 bp), -β (270 bp), -δ (354 bp), and -γ (404 bp) in hPOD, human embryonic kidney (HEK) cells, and human proximal tubule epithelial cells (PTECs). Spleen and testis served as positive controls. (E) SYBR green-based quantitative PCR showing relative fold change of Nox5 expression in vehicle (white columns)- and AngII (500 nM; black columns)-treated human podocytes compared with untreated controls. Values are expressed as mean±SEM (n=3). *P=0.03; **P=0.008. (F) SYBR green-based quantitative PCR showing relative fold change of Nox5 expression in HG-treated (black columns) human podocytes compared with low-glucose (LG) controls (white columns) at 2 and 10 hours after stimulation. Values are expressed as mean ± SEM (n=3). (G) Western blot for Nox5 (86 kDa) on human podocyte lysates comparing untreated controls (cont) with vehicle (veh)- and AngII (500 nM)-treated cells at various time points after stimulation. β-Actin served as loading control. (H) Western blot for Nox5 (86 kDa) on human podocyte lysates comparing controls with HG-treated cells. β-Actin served as loading control.
Podocyte Nox5 Is Induced and Activated by Factors in the Diabetic Milieu
Previous reports showed increased ROS production in podocytes exposed to high glucose (HG).30,31 Furthermore, because podocytes express AT1Rs, AngII may induce Nox5 expression and activity in human podocytes.32 As determined by quantitative RT-PCR, Nox5 mRNA expression was significantly increased in response to AngII at both 2 and 8 hours (Figure 1E). Stimulation with HG media for 2 hours failed to increase Nox5 mRNA; however, 10 hours after stimulation, Nox5 mRNA levels trended upward (Figure 1F). Later time points were not examined. Only the β-variant of Nox5 was induced in response to AngII stimulation (Supplemental Figure 2). No significant changes in Nox1, -2, or -4 expression were observed after AngII stimulation over this time course (Supplemental Figure 2). AngII, but not HG, induced Nox5 protein expression in cultured hPODs. Similar to our observations in normal human kidney biopsies, Nox5 protein was mostly absent in unstimulated and vehicle-treated hPODs. By contrast, Nox5 protein expression was induced as early as 0.5 hours poststimulation in response to AngII and sustained for up to 24 hours (Figure 1, G and H).
To assess whether AngII-dependent increases in Nox5 protein expression resulted in a corresponding increase in ROS, superoxide production was assayed using lucigenin-based fluorescence. Unstimulated and vehicle-treated controls displayed minimal baseline ROS production (Figure 2A). AngII induced a significant increase in ROS production at all time points assayed (Figure 2A). Similarly, HG induced a significant increase in ROS at 10 hours (Figure 2B). Although not statistically significant compared with AngII or HG alone, costimulation with HG+AngII increased ROS production in an additive manner, with an increase of 167.7±9.0% of control (Figure 2B). To determine if AngII-mediated ROS production was Nox5-dependent, small interfering RNA (siRNA) oligonucleotides were used to knock down expression of either Nox5 or p22phox, a subunit required for activity of Nox1, -2, -3, and -4. Knockdown of Nox5 and p22phox was confirmed by Western blot (Figure 2D). Nox5 knockdown significantly blunted ROS production in response to AngII (91.2±4.2% of control), whereas scrambled control oligonucleotides (scrs) were without effect (126.4±20.2% for scr versus 121.1±9.6% for control) (Figure 2C); p22phox knockdown did not affect AngII-stimulated ROS production (125.3±10.0% for p22phox versus 126.4±20.2% for scr), suggesting that Nox5 activity predominates over other Nox isoforms in this context (Figure 2C).
Figure 2.

AngII induces Nox5-dependent ROS production in human podocytes. Nox5-dependent ROS production in human and mouse podocytes. (A) Lucigenin-based ROS assay measuring superoxide production in human podocytes stimulated with vehicle (Veh; white columns) or 500 nM AngII (black columns) for various times. Values are reported as percent of untreated controls (mean ± SEM; n=3). *P=0.04; P=0.03; P=0.05 at 2, 8, and 24 hours, respectively. (B) Lucigenin-based ROS assay measuring superoxide production in human podocytes stimulated with control (white columns) or HG (black columns). Values are reported as percent of controls (mean±SEM; n=3). *P<0.05; **P<0.01. (C) Lucigenin-based ROS assay on human podocytes treated for 8 hours with vehicle (white columns) or 500 nM AngII (black columns) after siRNA knockdown with nontargeting scr-, Nox5-, or p22phox-specific siRNA. Values are expressed as percent of untreated controls (Cont; mean±SEM; n=3; *P<0.05). (D) Western blots for Nox5 or p22phox in human podocytes transfected with nontargeting scr-, Nox5-, or p22phox-specific siRNA. Untransfected cells are controls. β-Actin is the loading control. (E) HA-7 Western blot on lysates from uninfected and adNox5βHA (86 kDa) -infected mouse podocytes. (F) Immunofluorescence using HA-7 on uninfected or adNox5βHA-infected mouse podocytes showing expression of Nox5 (green). Scale bar, 100 μm. (G) Lucigenin-based ROS assay comparing superoxide production in adGFP- and adNox5βHA-infected mouse podocytes treated for 8 hours with vehicle (white columns) or 500 nM AngII (black columns). Values are expressed as percent of untreated controls (mean ± SEM; n=3). *P=0.03; **P=0.009.
Heterologous Nox5 Expression Generates ROS and Alters Cytoskeletal Dynamics
To examine the role of Nox5 in podocyte function in an animal model, transgenic mice expressing Nox5 in a podocyte-specific manner were generated. As proof of principle for this approach, conditionally immortalized mouse podocytes (mPODs) were transduced with an adenovirus (ad) to express Nox5β. Nox5 expression was verified by Western blotting and immunofluorescence (Figure 2, E and F). Cells infected with adNox5β displayed greater basal ROS production than adGFP-infected cells (116.9±3.2% of control for adNox5β versus 87.5±5.4% of control for adGFP) (Figure 2G). Furthermore, ROS generation in mPODs expressing adNox5β was responsive to AngII (148.3±16.9% of control) in a manner similar to human podocytes (Figure 2G).
adNox5β-infected mPODs exhibited reduced surface area, more numerous cellular projections, and lamellipodia compared with uninfected and adGFP-infected cells (Figure 3A). Staining with fluorescently-conjugated phalloidin revealed a reduction in stress fibers and greater numbers of peripheral actin aggregates in Nox5-expressing cells (Figure 3, A–H). Incubation of Nox5-expressing mPODs with the Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor KN-93, which is known to reduce Nox5 activity, and the broad spectrum Nox inhibitor diphenyleneiodonium blunted these effects33 (Figure 3I). Furthermore, KN-93 significantly attenuated ROS production (199.6±19.7% of control for infected+vehicle versus 107.2±2.0% of control for infected+KN-93) (Figure 3J). Consistent with the enhanced lamellipodia presence, Rac1 activity in adNox5-infected cells was elevated compared with controls (Figure 3, K and L).
Figure 3.

Ectopic Nox5β induces ROS-dependent cytoskeletal rearrangement in mouse podocytes. Immunofluorescence for phalloidin (red) and Nox5βHA (green) in (A–D) uninfected and (E–H) adNox5βHA-infected mouse podocytes. (A, B, E, and F) Cells without inhibitors were compared with cells incubated for 30 minutes with either (C and G) 10 μM diphenyleneiodonium (DPI) or (D and H) 10 μM KN-93. Scale bar, 100 μm. (I) Graphical representation of motile phenotype of cells in A–H. ***P<0.001 for control versus adNox5βHA; *P<0.05 for adNox5βHA versus adNox5βHA+DPI and adNox5βHA versus adNox5βHA+KN-93. White columns, control; gray columns, adGFP; black columns, adNox5. (J) Lucigenin-based assay measuring superoxide production in adNox5βHA-infected mouse podocytes incubated for 30 minutes with or without KN-93. **P=0.01. Veh, vehicle. (K) Rac pull-down assay showing levels of active Rac1 in uninfected and adGFP- and adNox5βHA-infected mouse podocytes. CTRL, control; PBD-GST, p21-activated kinase coupled to glutathione-S-transferase. (L) Quantification of Rac1 activity by densitometry (n=3). *P=0.05. White columns, control; black columns, Nox5.
Albuminuria and Increased BP in Nox5pod+ Mice
Having verified that human Nox5β is active in mouse podocytes in vitro, we next examined its function in vivo. For this purpose, human Nox5β cDNA was cloned directly downstream of an 8.3-kb fragment of the mouse nephrin promoter (mNPHS1) used previously34,35 (Figure 4A). After pronuclear injection, 11 Nox5pod+ FVB/n founders were identified by PCR-based genotyping. Three separate founder colony lines were maintained that exhibited similar phenotypic characteristics. Nox5 was readily detectable in the renal cortexes of transgenic mice by both Western blotting and RT-PCR (Figure 4, B and C). Immunofluorescence of Nox5 revealed expression in glomerular structures, which is consistent with podocyte-specific localization (Figure 4D). Glomerular Nox5 activity was determined by measuring oxidative stress in vivo by dihydroethidium (DHE) staining. DHE-positive glomerular staining was greater for Nox5pod+ mice than nontransgenic (non-tg) littermates (Figure 4E). Weekly spot urine beginning at 6 weeks of age showed elevated albumin to creatinine ratios (ACRs) in Nox5pod+ mice compared with non-tg littermates. By 12 weeks of age, Nox5pod+ mice showed a mean ACR of 687±100 μg/mg compared with 259±70 μg/mg for non-tg littermates (Figure 5A). ACR values progressively rose in Nox5pod+ animals, reaching a mean of 1370±57 μg/mg by 40 weeks of age (Figure 5F). Systolic BP was also monitored by tail-cuff plethysmography. Nox5pod+ mice exhibited significant increases in systolic BP (120±3 mmHg) compared with non-tg littermates (108±2 mmHg) (Figure 5B) at 12 weeks of age. ACR levels directly correlated with changes in systolic BP in Nox5pod+ mice (Figure 5C).
Figure 4.
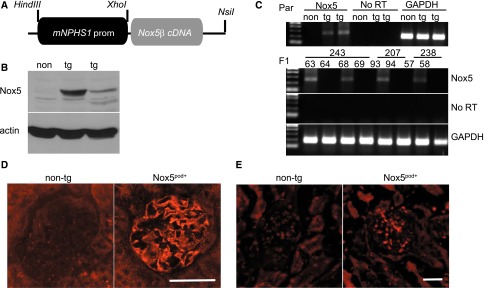
Characterization of Nox5pod+ tg mice expressing podocyte-specific Nox5β. (A) Schema showing targeting strategy used to generate Nox5pod+ tg mice specifically expressing Nox5β in podocytes. (B) Western blot for Nox5β (86 kDa) expression in non-tg and Nox5pod+ founder kidney cortex lysates. (C) RT-PCR for Nox5 expression in founders (Par) and subsequent F1 offspring of three independent founder lines. Non-tg and Nox5pod+ littermates are compared. No RT and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as negative and positive controls, respectively. (D) Immunofluorescence for Nox5 (red) on paraffin-embedded kidney sections of 20-week-old Nox5pod+ and non-tg littermates. Scale bar, 50 μm. (E) DHE staining on optimal cutting temperature (OCT) -embedded frozen kidney sections from 20-week-old Nox5pod+ and non-tg littermates. Scale bar, 50 μm.
Figure 5.

Nox5pod+ mice develop progressive albuminuria and increased systolic BP (SBP). (A) ELISA-based ACRs for non-tg (n=6) and Nox5pod+ (n=21) spot urine samples obtained at 12 weeks of age. *P=0.01. (B) SBP measured by tail-cuff plethysmography for non-tg (n=6) and Nox5pod+ (n=21) mice at 12 weeks of age. **P =0.005. (C) Linear correlation of (A) ACR to (B) SBP (r2=0.4159; P=0.002). (D) SYBR green-based quantitative PCR for Wilm’s tumor-1 mRNA levels on RNA lysates from non-tg and Nox5pod+ littermates at age 20 weeks (n=3). (E) SYBR green-based quantitative PCR for nephrin mRNA levels on RNA lysates from non-tg and Nox5pod+ littermates at age 20 weeks (n=3). (F) ELISA-based ACRs for non-tg (n=3) and Nox5pod+ (n=3) spot urine samples obtained at 40 weeks of age (*P<0.05).
Glomerular Filtration Barrier Damage in Nox5pod+ Mice
Podocyte number was unchanged by Nox5 expression, because Wilm’s tumor-1 cortex mRNA levels in Nox5βpod+ mice remained similar to those levels seen in non-tg mice (Figure 5D). However, podocyte injury was suggested, because nephrin levels were significantly reduced in Nox5pod+ mice (Figure 5E). To further assess progressive pathologic changes induced in glomeruli of Nox5pod+ animals, we examined periodic acid–Schiff (PAS)-stained sections of renal cortex. At 20 weeks of age, no gross morphologic changes were observed. However, at 40 weeks of age, a trend to larger total glomerular area and decreased glomerular tuft area was observed (Figure 6, A and B). Interestingly, these changes resulted in a 614-μm2 increase in overall Bowmann’s space area (1809±59 μm2 Nox5pod+ versus 1195±45 μm2 non-tg) (Figure 6, C and E–H). Evidence of tubular interstitial fibrosis was also observed in Nox5pod+ animals at 40 weeks of age. This result was indicated by α-smooth muscle actin staining in tubulointerstitial spaces in Nox5pod+ animals, which was limited to vascular structures in non-tg animals. Examination of Nox5pod+ glomerular and podocyte ultrastructure by electron microscopy revealed widespread foot process effacement and fusion as well as mesangial expansion, changes that were not observed in age-matched non-tg littermates (Figure 6, I–L). Furthermore, electron micrographs revealed a striking increase in GBM thickness for Nox5pod+ mice (Figure 6, I–L) (347.2±5.4 μm Nox5pod+ versus 232.2±2.1 μm non-tg) (Figure 6D).
Figure 6.

Nox5pod+ mice display GBM thickening, foot process effacement, and progressive interstitial fibrosis. (A) Total glomerular area for non-tg (white columns) and Nox5pod+ (black columns) littermates at ages 20 and 40 weeks (n=3). ***P<0.001. (B) Glomerular tuft area for non-tg (white columns) and Nox5pod+ (black columns) littermates at ages 20 and 40 weeks (n=3). ***P<0.001. (C) Bowmann’s space area for non-tg (white columns) and Nox5pod+ (black columns) calculated by subtracting (B) glomerular tuft area from (A) total glomerular area (n=3). ***P<0.001. (D) GBM thickness for non-tg (white columns) and Nox5pod+ (black columns). ***P<0.001. PAS staining on paraffin-embedded kidney sections from (E) non-tg mice and (F) Nox5pod+ littermates at 40 weeks of age. Scale bar, 100 μm. α-Smooth muscle actin staining (green) on paraffin-embedded kidney sections from (G) non-tg mice and (H) Nox5pod+ littermates at 40 weeks of age. Scale bar, 100 μm. Electron micrographs showing glomerular structure in (I and K) non-tg and (J and L) Nox5pod+ littermates at 40 weeks of age. Scale bar, 4 μm in I and J; 1 μm in K and L. Arrow in J indicates mesangial expansion. Arrowhead in L highlights extensive GBM thickening.
Nox5pod+ Mice Develop More Severe Renal Damage in Response to STZ-Induced Diabetes
We next examined the potential pathologic role of Nox5 in diabetic nephropathy. Nox5pod+ and non-tg littermates (8 weeks of age) were subjected to low-dose STZ injections and followed for 16 weeks. Albumin levels in 24-hour urine samples were increased in diabetic animals compared with controls, with Nox5pod+ animals having significantly higher albumin excretion rates than non-tg littermates (Figure 7A). Although systolic BP increased slightly in non-tg diabetic animals compared with controls, it did not reach statistical significance at any point (Figure 7B). In contrast, diabetic Nox5pod+ animals had significant elevations in systolic BP at 8 weeks post-STZ compared with nondiabetic non-tg controls (123±4 versus 100±5 mmHg) and 16 weeks post-STZ compared with nondiabetic non-tg and Nox5pod+ controls (137±7 versus 105±8 and 111±7 mmHg, respectively) (Figure 7B). Importantly, systolic BP increases in Nox5pod+ STZ animals did not occur until 4 weeks after the initial increases in albumin excretion in Nox5pod+ versus non-tg animals. Morphologic examination of PAS-stained sections revealed increased glomerulosclerosis (39.2±0.5% versus 34.9±0.7%) in diabetic Nox5pod+ animals compared with non-tg patients with diabetes (Figure 7, C–G). Although STZ-induced diabetes did result in a decrease in overall podocyte number, which was determined by Wilm’s tumor-1 staining, podocyte loss was not significantly increased in Nox5pod+ animals compared with non-tg controls (Figure 8, A and B). Examination of podocyte ultrastructure by electron microscopy (Figure 8C) revealed that diabetic Nox5pod+ animals displayed a significant increase in GBM width (Figure 8D) compared with non-tg patients with diabetes (272.3±13.1 versus 226.0±6.9 nm) as well as a trend to increased foot process width (534.6±54.1 versus 422.7±14.5 nm), which is indicative of increased foot process effacement (Figure 8, E and F).
Figure 7.

Nox5 exacerbates diabetic kidney damage in an STZ model. (A) ELISA-based albumin normalized to 24-hour urine volume for non-tg (white columns) control (CTRL; n=7), non-tg STZ (n=10), Nox5pod+ control (CTRL; n=7), and Nox5pod+ (black columns) STZ (n=12) obtained at indicated time points after STZ injection. Data presented as mean±SEM. *P<0.05. (B) Systolic BP measured by tail-cuff plethysmography for the groups in A. Solid lines are Nox5pod+, and hashed lines are non-tg controls. Squares are STZ-injected animals, and circles are control injections. Data are presented as mean±SEM. *P<0.05 versus non-tg controls; #P<0.05 Nox5pod+ STZ versus non-tg STZ. (C) Total glomerular area, (D) glomerular tuft area, and (E) Bowmann’s space area (n=3 mice per group; minimum of 30 glomeruli per mouse). White columns are non-tg, and black columns are Nox5pod+. Data are presented as mean±SEM. ***P<0.001. (F) PAS staining on paraffin-embedded kidney sections from non-tg and Nox5pod+ control and STZ littermates 16 weeks post-STZ. Original magnification, ×400. (G) Percent of total mesangial area exhibiting sclerosis as determined by PAS staining. White columns are non-tg STZ; black columns are Nox5pod+ STZ. Data are presented as mean±SEM. **P=0.01 (n=3).
Figure 8.

Podocyte loss is not significantly altered in diabetic Nox5pod+ mice. (A) Wilm’s tumor-1 (WT1; green) for glomerular podocytes in non-tg and Nox5pod+ animals subjected to STZ or control (CTRL) injections; 4′,6-diamidino-2-phenylindole was used to stain nuclei. (B) Graphical representation of total WT1-positive nuclei per glomerulus in A. White columns are non-tg, and black columns are Nox5pod+ (n=6 mice per group, with a minimum of 40 glomeruli per mouse). *P<0.05 non-tg STZ versus non-tg CTRL. (C) Electron micrographs of podocytes from non-tg and Nox5pod+ animals subjected to STZ or control injections. (D) GBM thickness for non-tg STZ (white columns) and Nox5pod+ STZ (black columns; n=3). *P=0.04. (E) Average foot process width for non-tg STZ (white columns) and Nox5pod+ STZ (black columns; n=3). (F) Percent foot process effacement for non-tg STZ (white columns) and Nox5pod+ STZ (black columns; n=3). *P=0.04.
Discussion
Renal oxidative stress contributes significantly to the progressive pathologic changes associated with nephropathy. Observations in animal models show that increased renal ROS generation promotes oxidative kidney damage and that treatment with antioxidants, such as N-acetyl cysteine, tempol, ebeselen, and vitamin E, decreases albuminuria and delays the progression of diabetic nephropathy.36–39
As a major cellular source of ROS, the role of Nox family members in mediating the renal complications of diabetes has become increasingly the focus of investigation. However, it remains unclear which of the Nox isoforms is the primary source of ROS in kidney disease. At the cellular level, Nox activity is linked to podocyte effacement through the AT1R, with AngII-induced actin cytoskeletal rearrangement in podocytes occurring in an ROS-dependent manner, which leads to increased cellular motility.5 Our in vitro studies show that de novo human Nox5 expression in mouse podocytes induces actin cytoskeleton rearrangement, Rac1 activation, and a lamellipodia-rich, ROS-dependent cellular phenotype reminiscent of the phenotype reported in the work by Hsu et al.5 It has been suggested that increased motility in vitro is analogous to podocyte effacement in vivo, an event closely associated with slit diaphragm deterioration and development of albuminuria.40 In agreement with this hypothesis and our in vitro results, Nox5pod+ tg mice develop albuminuria and podocyte effacement, highlighting a role for Nox5 both at the cellular level and in the broader context of filtration barrier dysfunction.
Several reports have shown that Nox inhibition in vivo through apocynin reduces albuminuria and podocytopenia in rodent models of diabetes.15,41 Furthermore, pharmacological inhibition of Nox1 and -4 reduces albuminuria and slows diabetic nephropathy progression in a db/db type 2 diabetic model.42 Although these studies support a role for Nox inhibition as a viable treatment for diabetic nephropathy, gene deletion studies of Nox isoforms in animal models have raised questions regarding the role of individual Nox members in diabetic nephropathy. Deletion of either Nox4 or -2 in STZ-induced diabetes is not beneficial and may even be deleterious in the case of Nox4, supporting studies showing that Nox4 may play a protective role in chronic kidney injury and endothelial dysfunction.43–46 However, the role of Nox4 remains controversial in the diabetic setting, because short-term pharmacological Nox4 inhibition reduces renal ROS generation, glomerular hypertrophy, and fibronectin expression.42 Unlike other Nox family members, nothing is known regarding the function of Nox5 in animal models of disease, owing mainly to its absence from the mouse and rat genome. In vitro studies show a role for Nox5 in human vascular endothelial cells, where it is responsive to several factors present in the diabetic milieu, including AngII, ET-1, and PDGF.22,23 Furthermore, other reports indicate that Nox5 is normally absent, but upregulated in the vascular smooth muscle cells of vessels in coronary artery disease and in intramyocardial blood vessels after myocardial infarction.47,48 In support of these studies, our findings with human renal biopsies indicate that Nox5 is readily detectable in diabetic but not nondiabetic glomeruli. In contrast, glomerular Nox1 and -2 expressions remained unchanged irrespective of diabetes, whereas Nox4 seemed to be induced in diabetes, particularly in fibrotic glomeruli. Furthermore, our study showed that select diabetic factors, including AngII and TGF-β (data not shown), induced expression and activity of Nox5 in cultured human podocytes. Although incubation with HG had little impact on Nox5 expression at the time points examined, we cannot rule out a role for hyperglycemic conditions in driving podocyte Nox5-generated ROS, because they provided an additive effect when combined with AngII. Alternatively, glucose-dependent ROS may involve other Nox isoforms, such as Nox4.
Based on these observations and our finding that Nox5-derived ROS is involved in podocyte function, we hypothesized that Nox5-induced ROS is critical to the progression of nephropathy. Mouse models have provided significant insight into the early pathology of kidney injury; however, most fail to recapitulate the late pathologic changes associated with CKD or diabetic nephropathy, including hypertension, GFR decline, and tubulointerstitial fibrosis.49 Surprisingly, even without induction of diabetes, Nox5pod+ mice develop many pathologic features at the functional and structural levels that are associated with the early phases of diabetic nephropathy, including albuminuria, mesangial expansion, podocyte effacement, and GBM thickening, as well as later changes not normally observed in most mouse diabetic nephropathy models, including tubulointerstitial fibrosis. Moreover, Nox5pod+ mice rendered diabetic with STZ exhibited more severe glomerular and tubular pathologies than their non-tg diabetic controls. These data strongly suggest that Nox5-induced ROS production in the podocyte participates in the progression of diabetic nephropathy.
In addition to renal dysfunction, Nox5pod+ mice exhibited increased BP from 12 weeks of age, which was likewise exacerbated by STZ-induced diabetes. This phenomenon may facilitate the progression of nephropathy in a manner similar to the manner seen in eNOS−/− mice: diabetic nephropathy is exacerbated in conjunction with a moderate increase in systolic BP, leading to a more progressive renal phenotype, including tubulointerstitial fibrosis.50–53 Podocyte-expressing Nox5 mice exhibit early increases in systolic BP accompanied by progressive tubulointerstitial fibrosis. Although the exact mechanism that elevates BP remains unclear, it is possible that increased superoxide production resulting from aberrant Nox5 activity may lead to decreased NO bioavailability, with consequent decreased vasodilation and increased vascular resistance and elevation of systolic BP. Alternatively, we cannot rule out ROS-induced activation of the renin-angiotensin system in these mice. Importantly, because albumin leakage developed well before BP increases in our diabetic Nox5pod+ animals, such hypertension is likely secondary to progressive renal dysfunction. Furthermore, these results do not preclude a role for Nox5 in other forms of nephropathy, such as FSGS, in which ROS production has been implicated.14 Given that enhanced Nox5 activity in podocytes is sufficient to induce filtration barrier dysfunction, it is possible that additional investigation may uncover a role for Nox5 in other forms of CKD.
In summary, we showed that podocyte Nox5 expression is enhanced in human diabetic nephropathy, that Nox5-derived ROS is critically involved in the regulation of podocyte function, and that de novo introduction of Nox5β in mouse podocytes in vivo recapitulates numerous features reminiscent of diabetic nephropathy and exacerbates the progression of renal dysfunction in a diabetic model. These novel data identify Nox5 as an important Nox isoform in the development of nephropathy. Nox5 may emerge as a novel therapeutic target for reducing progression of CKD.
Concise Methods
Cell Culture
Conditionally immortalized human and mouse podocytes, provided by Dr. Moin Saleem (University of Bristol) and Dr. Karlhans Endlich (University of Heidelberg, Germany), respectively, were grown at 33°C on type I collagen (BD Biosciences)-coated plastic culture dishes in RPMI-1640 medium (Invitrogen) supplemented with 10% FBS, 100 U/ml penicillin/streptomycin, and 10 U/ml recombinant γ-interferon. Differentiation was induced by maintaining the cells at 37°C in the above media without recombinant γ-interferon for 10–14 days. Cells were maintained in RPMI-1640 medium supplemented with 2% FBS and 100 U/ml penicillin/streptomycin for 2 days before stimulation with 500 nM AngII (Bachem). For glucose experiments, cells were growth-arrested for 36 hours in RPMI-1640 supplemented with 0.5% FBS, penicillin/streptomycin, and 5 mM d-glucose. Cells were then exposed to either 5 or 25 mM d-glucose for 10 hours using mannitol as an osmotic control before a 30-minute stimulation with 500 nM AngII, where appropriate. For inhibitor experiments, cells were maintained as above and treated with 10 μM KN-93 (Cayman Chemical) and 10 μM diphenyleneiodonium (Invitrogen) for 30 minutes before the addition of 500 nM AngII.
Human Biopsies
Archival biopsies were obtained from the Department of Pathology at the Ottawa Hospital. Nondiabetic samples were chosen from renal transplant recipients who had undergone a protocol biopsy and were determined to have no glomerular lesions, but in some cases, they showed evidence of focal acute tubular necrosis. Biopsies of diabetic patients all showed evidence of glomerular injury without evidence of nondiabetic renal disease. Sections were stained with Nox1, -2, -4, and -5 (Santa Cruz Biotechnology).
RNA Extraction and Quantitative PCR
Snap-frozen cortex was mechanically homogenized using the TP-103 Amalgamator COE Capmixer (GC America, Inc.). Cells were homogenized using Qiashredder columns (Qiagen).
RNA was extracted from snap-frozen kidney cortexes of human and mouse podocytes using the Qiagen RNEasy Minikit as per the manufacturer’s instructions. Extracted RNA was converted to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) with 500 ng starting material per reaction as per the manufacturer’s instructions. Quantitative PCR was performed on an ABI Prism 7000 Sequence Detection System using SYBR Advantage qPCR Premix (Clontech) according to the manufacturer’s instructions. Primers used are listed in Supplemental Table 1.
Western Blots
For protein extraction from tissue, snap-frozen cortex was mechanically homogenized using the TP-103 Amalgamator COE Capmixer (GC America). The powdered tissue was resuspended in 1× Laemmli lysis buffer of 62.5 mM Tris⋅HCl (pH 6.8), 2% wt/vol SDS, 10% glycerol, and 50 mM dithiothreitol with protease inhibitors (Sigma-Aldrich) and incubated on ice for 30 minutes. For cells, plates were rinsed in PBS before being scrapped in 1× Laemmli lysis buffer with protease inhibitors and placed on ice for 30 minutes. Extracted proteins were run on appropriate percentage SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were blocked in 5% nonfat dairy milk (NFDM) in 1× PBS containing 0.2% Triton X-100 (PBST) for 45 minutes before the addition of primary antibodies Nox4, Nox5 (1:750), HA-7 (1:2500), and β-actin (1:1000). Membranes were incubated in 5% NFDM in PBST with primary antibody overnight at 4°C. Membranes were washed three times in PBST before incubation with 2% horseradish peroxidase (1:5000) in 5% NFDM in PBST for 45 minutes. Membranes were washed in PBST, incubated with enhanced chemiluminescence (GE Healthcare), and exposed to film.
Lucigenin Assays
Treated cells were harvested in 70 μl ice cold phosphate buffer of 50 mM KH2PO4, 1 mM EGTA, and 150 mM sucrose (pH 7.4) with protease inhibitors; 50 μl cell lysate was added to 175 μl buffer and 1.25 μl 10−3 M lucigenin (ENZO Life Sciences). Baseline activity was measured. Cells were stimulated by the addition of 25 μl 1 mM NADPH, and active levels were measured. Baseline activity was subtracted, and adjusted activity was normalized to protein concentration.
siRNA Knockdown
Transient transfection of siRNA was performed using HiPerfect Transfection Reagent (Qiagen) as per the manufacturer’s instructions. Briefly, scrambled nontargeting control, Nox5 siRNA, or p22phox siRNA (Santa Cruz Biotechnology) was diluted in serum-free RPMI media and mixed with transfection reagent. The mixture was incubated for 10 minutes at room temperature to allow for lipid–RNA complex formation and then added dropwise to podocytes (2×105) seeded on type I collagen-coated 10-cm dishes. Cells were maintained for 48–72 hours before use.
DHE Staining
Frozen sections of tissue in optimal cutting temperature were cut to a thickness of 8 μm and placed on glass slides; 50 μl 2 μM dihydroethidium (Sigma-Aldrich) in Krebs solution (pH 7.4) of 11.8 mM NaCl, 0.465 mM KCl, 0.118 mM MgSO4, 0.118 mM KH2PO4, 14.702 mM CaCl2, 25 mM NaHCO3, 5.5 mM d-glucose, and 0.026 mM EDTA was placed on top of each section in a light-protected humidified chamber and incubated at 37°C for 30 minutes. Negative controls were incubated with Krebs only. Staining solution was removed, and slides were rinsed in Krebs solution. Slides were coverslipped using Fluormount G (Southern Biotech) and imaged.
Viral Preparation and Infection
Nox5β adenoviral vector obtained from Dr. David Fulton was transfected into 293T cells using Lipofectamine (Qiagen) per the manufacturer’s recommendations. Cells were allowed to round up and detach from the plate, and they were then gently washed from the plate with media. The media/cell suspension was collected in 50-ml Falcon tubes and centrifuged at 1500 rpm for 5 minutes. The supernatant was collected, and the pellet was resuspended in 2 ml serum-free DMEM (Invitrogen). The pellet was flash frozen in liquid nitrogen followed by thawing in a 37°C water bath. The freeze/thaw cycle was repeated a total of four times. The resulting suspension was centrifuged at 1500 rpm for 5 minutes, and the resulting supernatant was added to the already collected supernatant. The supernatant was then used to infect new plates of 293T cells. Viral propagation was continued until sufficient viral supernatant was obtained. Virus was purified using the Fast-Trap Adenovirus Purification and Concentration Kit (Millipore). GFP virus was obtained from Dr. David Park. Purified virus was used to infect podocytes at an moi of 5–20.
Phalloidin Staining
Phalloidin staining was performed as previously described.40 Briefly, cells were fixed with 4% paraformaldehyde and permeabilized in PBST. Cells were then incubated with Alexa Fluor 594 phalloidin (Molecular Probes) to visualize actin cytoskeleton.
Rac1–Glutathione-S-Transferase Pull-Down Assay
Pull-down assays were used to determine the state of Rac1 activation in mouse podocytes using the Rac1/CDC42 binding domain of p21-activated kinase coupled to glutathione-S-transferase. For in vitro pull-down assays in mouse podocytes, 500 μg cleared cell lysate was immediately added to 80 μg p21-activated kinase coupled to glutathione-S-transferase beads and incubated at 4°C for 45 minutes. Beads were sedimented, washed four times in PBS, and subjected to Western blotting. Samples were boiled and resolved by 15% SDS-PAGE, transferred to nitrocellulose membrane, and blotted for Rac1 using 1:1000 anti-Rac1 primary antibody (Cytoskeleton). Active (bead-bound) Rac1 was normalized to total Rac1 using 10% of input. Ponceau staining was used to verify loading.
Construct and Generation of Tgs
The human Nox5β cDNA (GeneCopoeia) was cloned into the pCDNA3.0 vector (Promega). An 8.3-kb HindIII/XhoI fragment of the NPHS1 promoter was inserted upstream of the Nox5β start site. An 11.3-kb NPHS1/Nox5β fragment was excised using a HindIII/NsiI digest, and the resulting band was gel-purified. The purified DNA was provided to the University of Ottawa Core Transgenic Facility for pronuclear injection. Subsequent Nox5pod+ founders were identified by PCR genotyping.
STZ Injection and 24-Hour Urine Collection
Mice ages 8–10 weeks were intraperitoneally injected with low-dose STZ (5 mg/kg) or sodium citrate control one time per day for 5 days as per the Animal Models of Diabetic Complications Consortium. For 24-hour urine collection, mice were individually housed in metabolic cages for 24 hours, and urine was collected. Urine volume was measured and recorded for normalization of 24-hour albumin excretion.
Immunohistochemistry and Renal Pathology
Nox5pod+ mice were anesthetized with isofluorane and perfused with 20 ml PBS through the left ventricle. The kidneys were decapsulated, and the poles were removed. Portions of each kidney were placed in either 4% paraformaldehyde/PBS for paraffin embedding or 2.7% gluteraldehyde for electron microscopy, or they were embedded in optimal cutting temperature compound (Sakura Finetek USA, Inc., Torrance, CA) for frozen sectioning; 4-µm paraffin sections were stained with PAS or Mason Trichrome and then analyzed by light microscopy. Analytical measurements were performed using Axiovision software.
ELISA
Urinary albumin and creatinine levels were analyzed from spot urine samples using the Mouse Albumin ELISA Kit (Bethyl Laboratories, Inc.) and The Creatinine Companion (Exocell), respectively. Urine samples were diluted to 1:2000 for albumin and 1:20 for creatinine, and they were processed as per the manufacturer’s instructions.
BP Measurements
Systolic BP was measured using tail-cuff plethysmography (BP-2000; Visitech Systems). Mice were placed in isolation chambers on a heated platform, and systolic BP measurements were obtained. Before recording measurements, mice were trained daily for a 5-day period. For measurements, mice were subjected to 5 preliminary measurements, which were not recorded, followed by 10 recorded measurements. Systolic BP is plotted as the average across 10 measurements at the given time point.
Transmission Electron Microscopy
Kidneys were immersion-fixed in cold 2.4% glutaraldehyde in PBS buffer, postfixed in 2% buffered osmium tetroxide, dehydrated in graded ethanols, and embedded in Spurr resin. Samples were sectioned at 70 nm, placed on copper for transmission electron microscopy, and stained with uranyl acetate and lead citrate. Percent foot process effacement was calculated as previously described.54 Samples were screened on a Hitachi H-7100 Transmission Electron Microscope. Analytical measurements were performed using Axiovision software.
Statistical Analyses
GraphPad PRISM software was used to analyze all experimental data. Values are reported as the mean±SEM. Statistical significance was determined using either t test or one-way ANOVA followed by Newman–Keuls multiple comparison test.
Disclosures
None.
Supplementary Material
Acknowledgments
The authors thank Anthony Carter and Julie Zhu for their excellent technical assistance. Pronuclear injections were performed by Dr. Adriana Gambarotta under the supervision of Dr. David Lohnes at the University of Ottawa Core Transgenic Facility. The authors also thank Dr. David Fulton, Dr. Moin Saleem, and Dr. Karlhans Endlich for providing reagents as well as Dr. Jeffrey Kopp of the National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health for the use of the podocin-rtTA mice.
C.E.H. is a recipient of the Kidney Foundation of Canada Post-Doctoral Fellowship Award–Agostino Monteduro Endowment Fund. M.E.C. is a recipient of an Australian Fellowship from the National Health and Medical Research Council of Australia and a Scholar’s Award from the Juvenile Diabetes Research Foundation. This work was supported by a grant from the Canadian Institutes of Health Research (to C.R.J.K.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2013040371/-/DCSupplemental.
References
- 1.Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, Camussi G: Nephrin expression is reduced in human diabetic nephropathy: Evidence for a distinct role for glycated albumin and angiotensin II. Diabetes 52: 1023–1030, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Remuzzi G, Benigni A, Remuzzi A: Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J Clin Invest 116: 288–296, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, Abboud HE: Mechanisms of podocyte injury in diabetes: Role of cytochrome P450 and NADPH oxidases. Diabetes 58: 1201–1211, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ha H, Hwang IA, Park JH, Lee HB: Role of reactive oxygen species in the pathogenesis of diabetic nephropathy. Diabetes Res Clin Pract 82[Suppl 1]: S42–S45, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Hsu HH, Hoffmann S, Endlich N, Velic A, Schwab A, Weide T, Schlatter E, Pavenstädt H: Mechanisms of angiotensin II signaling on cytoskeleton of podocytes. J Mol Med (Berl) 86: 1379–1394, 2008 [DOI] [PubMed] [Google Scholar]
- 6.Susztak K, Raff AC, Schiffer M, Böttinger EP: Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55: 225–233, 2006 [PubMed] [Google Scholar]
- 7.Gill PS, Wilcox CS: NADPH oxidases in the kidney. Antioxid Redox Signal 8: 1597–1607, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE: Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J Biol Chem 280: 39616–39626, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Gorin Y, Ricono JM, Kim NH, Bhandari B, Choudhury GG, Abboud HE: Nox4 mediates angiotensin II-induced activation of Akt/protein kinase B in mesangial cells. Am J Physiol Renal Physiol 285: F219–F229, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Kitada M, Koya D, Sugimoto T, Isono M, Araki S, Kashiwagi A, Haneda M: Translocation of glomerular p47phox and p67phox by protein kinase C-beta activation is required for oxidative stress in diabetic nephropathy. Diabetes 52: 2603–2614, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Kim EY, Anderson M, Dryer SE: Insulin increases surface expression of TRPC6 channels in podocytes: Role of NADPH oxidases and reactive oxygen species. Am J Physiol Renal Physiol 302: F298–F307, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kinugasa S, Tojo A, Sakai T, Tsumura H, Takahashi M, Hirata Y, Fujita T: Selective albuminuria via podocyte albumin transport in puromycin nephrotic rats is attenuated by an inhibitor of NADPH oxidase. Kidney Int 80: 1328–1338, 2011 [DOI] [PubMed] [Google Scholar]
- 13.Peng H, Takano T, Papillon J, Bijian K, Khadir A, Cybulsky AV: Complement activates the c-Jun N-terminal kinase/stress-activated protein kinase in glomerular epithelial cells. J Immunol 169: 2594–2601, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Wei X, Zhang Y, Ma X, Li B, Zhang S, Du P, Zhang X, Yi F: NADPH oxidase-derived ROS contributes to upregulation of TRPC6 expression in puromycin aminonucleoside-induced podocyte injury. Cell Physiol Biochem 24: 619–626, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Asaba K, Tojo A, Onozato ML, Goto A, Quinn MT, Fujita T, Wilcox CS: Effects of NADPH oxidase inhibitor in diabetic nephropathy. Kidney Int 67: 1890–1898, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Koya D, Hayashi K, Kitada M, Kashiwagi A, Kikkawa R, Haneda M: Effects of antioxidants in diabetes-induced oxidative stress in the glomeruli of diabetic rats. J Am Soc Nephrol 14[Suppl 3]: S250–S253, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS: Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: Effects of ACEI and ARB. Kidney Int 61: 186–194, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Anderson S, Rennke HG, Brenner BM: Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Clin Invest 77: 1993–2000, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Björck S, Nyberg G, Mulec H, Granerus G, Herlitz H, Aurell M: Beneficial effects of angiotensin converting enzyme inhibition on renal function in patients with diabetic nephropathy. Br Med J (Clin Res Ed) 293: 471–474, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lemley KV: When to initiate ACEI/ARB therapy in patients with type 1 and 2 diabetes. Pediatr Nephrol 25: 2021–2034, 2010 [DOI] [PubMed] [Google Scholar]
- 21.Hitomi H, Fukui T, Moriwaki K, Matsubara K, Sun GP, Rahman M, Nishiyama A, Kiyomoto H, Kimura S, Ohmori K, Abe Y, Kohno M: Synergistic effect of mechanical stretch and angiotensin II on superoxide production via NADPH oxidase in vascular smooth muscle cells. J Hypertens 24: 1089–1095, 2006 [DOI] [PubMed] [Google Scholar]
- 22.Jay DB, Papaharalambus CA, Seidel-Rogol B, Dikalova AE, Lassègue B, Griendling KK: Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic Biol Med 45: 329–335, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, Callera GE, He G, Krause KH, Lambeth D, Quinn MT, Touyz RM: Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res 106: 1363–1373, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bánfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnár GZ, Krause KH, Cox JA: Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5). J Biol Chem 279: 18583–18591, 2004 [DOI] [PubMed] [Google Scholar]
- 25.BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, Görlach A: NOX5 variants are functionally active in endothelial cells. Free Radic Biol Med 42: 446–459, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ: Novel mechanism of activation of NADPH oxidase 5. calcium sensitization via phosphorylation. J Biol Chem 282: 6494–6507, 2007 [DOI] [PubMed] [Google Scholar]
- 27.Pandey D, Patel A, Patel V, Chen F, Qian J, Wang Y, Barman SA, Venema RC, Stepp DW, Rudic RD, Fulton DJ: Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am J Physiol Heart Circ Physiol 302: H1919–H1928, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tirone F, Cox JA: NADPH oxidase 5 (NOX5) interacts with and is regulated by calmodulin. FEBS Lett 581: 1202–1208, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Tirone F, Radu L, Craescu CT, Cox JA: Identification of the binding site for the regulatory calcium-binding domain in the catalytic domain of NOX5. Biochemistry 49: 761–771, 2010 [DOI] [PubMed] [Google Scholar]
- 30.Khazim K, Gorin Y, Cavaglieri RC, Abboud HE, Fanti P: The antioxidant silybin prevents high glucose-induced oxidative stress and podocyte injury in vitro and in vivo. Am J Physiol Renal Physiol 305: F691–F700, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim NH, Rincon-Choles H, Bhandari B, Choudhury GG, Abboud HE, Gorin Y: Redox dependence of glomerular epithelial cell hypertrophy in response to glucose. Am J Physiol Renal Physiol 290: F741–F751, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, Pichler R, Griffin S, Couser WG, Shankland SJ: Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int 65: 30–39, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Pandey D, Gratton JP, Rafikov R, Black SM, Fulton DJ: Calcium/calmodulin-dependent kinase II mediates the phosphorylation and activation of NADPH oxidase 5. Mol Pharmacol 80: 407–415, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michaud JL, Stitt-Cavanaugh E, Endlich N, Endlich K, De Repentigny Y, Kothary R, Kennedy CR: Mice with podocyte-specific overexpression of wild type alpha-actinin-4 are healthy controls for K256E-alpha-actinin-4 mutant transgenic mice. Transgenic Res 19: 285–289, 2010 [DOI] [PubMed] [Google Scholar]
- 35.Stitt-Cavanagh EM, Faour WH, Takami K, Carter A, Vanderhyden B, Guan Y, Schneider A, Breyer MD, Kennedy CR: A maladaptive role for EP4 receptors in podocytes. J Am Soc Nephrol 21: 1678–1690, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee EA, Seo JY, Jiang Z, Yu MR, Kwon MK, Ha H, Lee HB: Reactive oxygen species mediate high glucose-induced plasminogen activator inhibitor-1 up-regulation in mesangial cells and in diabetic kidney. Kidney Int 67: 1762–1771, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Nakhoul FM, Miller-Lotan R, Awad H, Asleh R, Jad K, Nakhoul N, Asaf R, Abu-Saleh N, Levy AP: Pharmacogenomic effect of vitamin E on kidney structure and function in transgenic mice with the haptoglobin 2-2 genotype and diabetes mellitus. Am J Physiol Renal Physiol 296: F830–F838, 2009 [DOI] [PubMed] [Google Scholar]
- 38.Park NY, Park SK, Lim Y: Long-term dietary antioxidant cocktail supplementation effectively reduces renal inflammation in diabetic mice. Br J Nutr 106: 1514–1521, 2011 [DOI] [PubMed] [Google Scholar]
- 39.Peixoto EB, Pessoa BS, Biswas SK, Lopes de Faria JB: Antioxidant SOD mimetic prevents NADPH oxidase-induced oxidative stress and renal damage in the early stage of experimental diabetes and hypertension. Am J Nephrol 29: 309–318, 2009 [DOI] [PubMed] [Google Scholar]
- 40.Michaud JL, Chaisson KM, Parks RJ, Kennedy CR: FSGS-associated alpha-actinin-4 (K256E) impairs cytoskeletal dynamics in podocytes. Kidney Int 70: 1054–1061, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FY, Sourris KC, Penfold SA, Bach LA, Cooper ME, Forbes JM: Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes 57: 460–469, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Sedeek M, Gutsol A, Montezano AC, Burger D, Nguyen Dinh Cat A, Kennedy CR, Burns KD, Cooper ME, Jandeleit-Dahm K, Page P, Szyndralewiez C, Heitz F, Hebert RL, Touyz RM: Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of Type 2 diabetes. Clin Sci (Lond) 124: 191–202, 2013 [DOI] [PubMed] [Google Scholar]
- 43.Babelova A, Avaniadi D, Jung O, Fork C, Beckmann J, Kosowski J, Weissmann N, Anilkumar N, Shah AM, Schaefer L, Schröder K, Brandes RP: Role of Nox4 in murine models of kidney disease. Free Radic Biol Med 53: 842–853, 2012 [DOI] [PubMed] [Google Scholar]
- 44.Nlandu Khodo S, Dizin E, Sossauer G, Szanto I, Martin PY, Feraille E, Krause KH, de Seigneux S: NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J Am Soc Nephrol 23: 1967–1976, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM: Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol 31: 1368–1376, 2011 [DOI] [PubMed] [Google Scholar]
- 46.You YH, Okada S, Ly S, Jandeleit-Dahm K, Barit D, Namikoshi T, Sharma K: Role of Nox2 in diabetic kidney disease. Am J Physiol Renal Physiol 304: F840–F848, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, Harrison DG: Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol 52: 1803–1809, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hahn NE, Meischl C, Kawahara T, Musters RJ, Verhoef VM, van der Velden J, Vonk AB, Paulus WJ, van Rossum AC, Niessen HW, Krijnen PA: NOX5 expression is increased in intramyocardial blood vessels and cardiomyocytes after acute myocardial infarction in humans. Am J Pathol 180: 2222–2229, 2012 [DOI] [PubMed] [Google Scholar]
- 49.Brosius FC, 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T, Animal Models of Diabetic Complications Consortium : Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanetsuna Y, Takahashi K, Nagata M, Gannon MA, Breyer MD, Harris RC, Takahashi T: Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol 170: 1473–1484, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, Yuzawa Y, Atkinson MA, Johnson RJ, Croker B: Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol 18: 539–550, 2007 [DOI] [PubMed] [Google Scholar]
- 52.Zhang MZ, Wang S, Yang S, Yang H, Fan X, Takahashi T, Harris RC: Role of blood pressure and the renin-angiotensin system in development of diabetic nephropathy (DN) in eNOS-/- db/db mice. Am J Physiol Renal Physiol 302: F433–F438, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao HJ, Wang S, Cheng H, Zhang MZ, Takahashi T, Fogo AB, Breyer MD, Harris RC: Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol 17: 2664–2669, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiang ML, Hawkins EP, Berry PL, Barrish J, Hill LL: Diagnostic and prognostic significance of glomerular epithelial cell vacuolization and podocyte effacement in children with minimal lesion nephrotic syndrome and focal segmental glomerulosclerosis: An ultrastructural study. Clin Nephrol 30: 8–14, 1988 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.