

Abstract
RING3 is a novel, nuclear-localized, serine-threonine kinase that has elevated activity in human leukemias. RING3 transforms NIH/3T3 cells and is activated by mitogenic signals, all of which suggest that it may play a role in cell cycle-responsive transcription. We tested this hypothesis with transient transfection of RING3 into fibroblasts and assayed transactivation of the promoters of cyclin D1, cyclin A, cyclin E, and dihydrofolate reductase (dhfr) genes. RING3 transactivates these promoters in a manner dependent on ras signaling. A kinase-deficient point mutant of RING3 does not transactivate. Mutational analysis of the dhfr promoter reveals that transactivation also depends on the presence of a functional E2F binding site. Furthermore, ectopic expression of Rb protein, a negative regulator of E2F activity, suppresses the RING3-dependent transactivation of this promoter. Consistent with a potential role of E2F in RING3-dependent transcription, anti-RING3 immunoaffinity chromatography or recombinant RING3 protein affinity chromatography of nuclear extracts copurified a protein complex that contains E2F-1 and E2F-2. These data suggest that RING3 is a potentially important regulator of E2F-dependent cell cycle genes.
Introduction
Recent evidence has implicated the nuclear-localized kinase RING3 in transcriptional control. RING3 is ubiquitously expressed in mammalian cells and has high specific activity in acute leukemic blasts, although its activity is very low in normal peripheral blood lymphocytes (1). It was first identified as an Mr 85,000 autophosphorylating nuclear kinase activity in EL-4 6.1 C10 thymoma cells stimulated with IL3-1α and 70Z/3 cells stimulated with lipopolysaccharide (2). Autophosphorylation activity, which correlates with increased substrate-directed activity, was later shown to be increased in lymphocytes upon stimulation with mitogenic lectin in Chinese hamster ovary cells with IL-1α and in fibroblasts with platelet-derived growth factor-β (1), as well as in multiple organs of mice injected with epidermal growth factor, phorbol 12-myristate 13-acetate, or IL-1 α (3). The murine Mediator transcription complex, which contains homologues of the yeast transcriptional regulators Med6, Med7, Rgr1, and Srb7, also contains a protein that is homologous to human RING3 and its Drosophila homologue female sterile (1) homeotic (fsh) (4). This association led us to speculate that RING3, as a nuclear participant in signal transduction cascades, might have important transcriptional targets.
Apart from this circumstantial evidence implying a role in transcription, little is known about the mechanism of action of the RING3 protein (5). The gene is localized to the class II major histocompatibility locus on human chromosome 6p21.3 and encodes a member of a family of bromodomain-containing human proteins that include BRDT (6) and ORFX (7). There are reports of homologous genes in mice (8), frogs (9), and zebrafish (10). fsh is a temperature-sensitive, maternal effect gene required at two stages of development, identified by the trithorax phenotype. fsh activates the trithorax locus and interacts genetically with the Antennapedia and Ultrabithorax complexes (11). An fsh mutant called rancor (rnc) is embryonic lethal and affects head homeotic development (12), probably through the btd (Sp1)-dependent regulation of cnc, a bZIP transcription factor related to mammalian NF-E2.4 Intriguingly, a human homologue of Trithorax, ALL-1 or MLL, is a putative transcription factor that is damaged in leukemias associated with 11q23 chromosome breaks (13-15). By functional conservation with fsh, it is likely that RING3 contributes to the regulation of ALL-1 activity, i.e., improper signal transduction through RING3 and ALL-1 could lead to leukemia (1).
Two redundant genes in yeast, BDF1 and BDF2, are homologous to RING3 and fsh primarily in their NH2-terminal bromodomains (16, 17); at least one of the two genes is required for yeast viability. Bdf1 and RING3 (5) are related to the product of the mammalian cell cycle gene CCG1 (mammalian TAFII250); both gene products have COOH-terminal acidic domains and intrinsic kinase activity (18). Ccg1 and its yeast homologue Taf145 also possess histone acetyltransferase activity. Furthermore, the presence of two mutually related bromodomains in Ccg1, Bdf1 and Bdf2, RING3 and fsh suggests that this class of proteins may affect transcription through chromatin restructuring. Bromodomains (19) are commonly found in transcription factors (20) and the proteins that comprise chromatin-remodeling complexes (21-23).
In the present study, we explore possible mechanisms that might link increased RING3 activity with cancer, first with experiments focused on transformation activity and time course of activation and then with transcriptional analysis of potential target genes of RING3. We demonstrate that RING3 exhibits some characteristics of a mitogenic, signal-transducing kinase and that it transactivates the promoters of important E2F-responsive genes that regulate the cell cycle. Furthermore, E2F copurifies with RING3 in protein complexes isolated from nuclear extract. E2F proteins are pivotal regulators of mammalian cell cycle progression and differentiation. In relationship with members of the Rb family of proteins, they control the transcriptional activation or repression of numerous genes; destabilization of these control mechanisms can result in apoptosis, reversal of differentiation, or cancer (24, 25). These proposed links between RING3 and E2F-dependent transcription provoke further study of the relationship between chromatin-modifying complexes and cell cycle progression.
Results
Preliminary Observations
Transformation of NIH/3T3 Cells
The observation that serum stimulation of fibroblasts and mesenchymal cells or mitogenic lectin stimulation of lymphocytes increases RING3 kinase activity (1) suggested that RING3 might provide a link between mitogenic signals and proliferation. We hypothesized that improper expression of RING3 might destabilize mitogenic signal transduction and lead to transformation. We tested this idea with ectopic overexpression of RING3 protein in NIH/3T3 fibroblasts, which were either suspended in Matrigel or plated on tissue culture plastic to form confluent monolayers. In the case of semisolid media, foci were counted after 3 and 5 days, and in the case of monolayers, foci were counted after 3 weeks. Table 1 shows that foci did not form in either assay when cells were transiently transfected with empty expression vector alone or with constructs that encoded wild-type or the catalytically inactive (K574A) form of RING3 (1). Activated ras alone was sufficient to form foci, which is a well-known phenomenon (26). The amount of transfected expression construct for activated ras was titrated to give a minimal number of foci, and in this background, expression of wild-type RING3 protein significantly increased the number of foci formed. RING3 overexpressed with activated ras is therefore capable of transformation. Expression of the K574A mutant together with activated ras did not augment the number of foci. The transforming capability of RING3 was therefore dependent on its kinase activity in that the K574A mutant did not increase the number of foci above levels with ras alone.
Table 1.
Transformation of NIH/3T3 cells
| Transiently transfected cells were plated in Matrigel (semisolid medium) or on tissue culture plastic (adherent) and assayed for colony formation. Results are expressed as means of two or three experiments with SD in parentheses. |
| Cogene | No. of colonies |
|||
|---|---|---|---|---|
| Semisolid medium |
Adherent |
|||
| Vectora | H-rasa | Vectora | H-rasb | |
| Vector | 0 | 3 (0.7) | 0 | 4 (0) |
| RING3 (WT) | 0 | 15.3 (1.1) | 0 | 7 (1) |
| RING3 (K574A) | 0 | 4 (0.7) | 0 | 2 (1) |
n = 3.
n = 2.
Time Course of RING3 Activation
Growth factor treatment of fibroblasts or epithelial cells stimulates RING3 autophosphorylation activity within 15 min. Activity then rapidly declines to a basal level (1), which is characteristic of several “immediate early” type signal-transducing kinases. We wondered whether RING3 kinase activity was also elevated near the G1-S transition, similar to the response of p70S6K kinase (27) or ERK1 (28) to growth factors. Swiss/3T3 cells in a confluent monolayer were starved of serum to produce a quiescent population and then stimulated with 10% serum. Extracts were prepared at progressive times during the mitogenic program, and RING3 kinase activity was determined by autophosphorylation assay as described (1). As shown in Fig. 1 (above), RING3 autophosphorylation activity remained elevated through 16 h of mitogenic stimulation but then fell by 20 h and remained low throughout the remainder of the mitogenic program, even as αRING3-immunoreactive material accumulated (Fig. 1, below). This decrease in activity was well correlated in time with the onset of S phase, which suggests that changes in RING3 activity are coupled to the cell cycle.
Fig. 1.
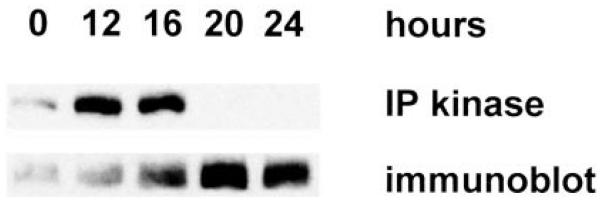
RING3 kinase activity and protein level after serum stimulation of fibroblasts. Extracts were prepared at progressive times from 107 serum-stimulated Swiss/3T3 cells and immunoprecipitated with αRING3 rabbit polyclonal antibody. Immune complexes were separated by SDS-PAGE, electroblotted to PVDF, and subjected to autophosphorylation assay (Ref. 1; IP kinase) or αRING3 immunoblot.
Transactivation of Cyclin Promoters
From these preliminary data, we reasoned that RING3 kinase might affect the activity of cellular genes that control growth. We therefore tested the RING3 dependence of transcription units known to regulate proliferation. Luciferase reporter vectors that contained synthetic, cis-acting E2F or CRE sites, or an enhancerless reporter as a negative control, were cotransfected into BALB/3T3 fibroblasts with an expression vector for RING3. Transfected cells were starved of serum overnight and then stimulated with either 10% serum (for E2F or enhancerless reporters) or 50 μM forskolin (for the CRE reporter) overnight. Coexpression of RING3 augmented the serum stimulation of the E2F reporter, whereas a RING3-dependent response was not observed for the CRE reporter after cAMP stimulation (Fig. 2). Although RING3 increased the basal transcription of the E2F reporter, the enhancerless reporter did not respond to RING3 or serum. These results confirmed that RING3 can affect serum-responsive, E2F-dependent transcriptional events but is probably not involved in cAMP-responsive transcriptional events.
Fig. 2.

Reporter screen. NIH/3T3 cells were transiently transfected in duplicate with luciferase reporters, either enhancerless (■) or containing CRE (□) or E2F sites (dark gray, hatched) and expression constructs for RING3 or empty vector control. Cells were starved of serum overnight (−) and then stimulated with 10% serum (for the E2F-reporter and enhancerless plasmid transfections) or 50 μM forskolin (for the CRE-reporter transfections) overnight (1) and then harvested for assay of luciferase. Bars, SE.
We expanded the analysis to the endogenous promoters of several well-characterized, cell cycle-regulated genes that contain E2F sites to determine whether RING3 could alter their transcriptional activity. NIH/3T3 cells were transiently transfected with luciferase reporter constructs that contained 5′ promoter elements from the murine genes cyclin D1, cyclin A, cyclin E, and dhfr. Overexpressed RING3 cDNA transactivated cyclin D1, A, E, and dhfr reporters but only in a context where activated ras was also present (Fig. 3A). Activated ras alone was capable of transactivating each promoter under these conditions, but the effect was dramatically enhanced (3–6-fold) when activated ras and RING3 were present together. Transfection into BALB/3T3 gave essentially the same results (not shown). We conclude that RING3 and ras activities combine to transactivate these promoters. MEKK, an effector of ras, was also tested for its ability to transactivate the dhfr promoter (Fig. 3B). As in the previous experiment, neither empty vector alone nor WT RING3 alone exhibited significant transactivation of the dhfr promoter, whereas constitutively active MEKK alone induced the dhfr reporter about 5-fold. Wild-type RING3 and MEKK together synergized to give a 60-fold induction. Activated Jun kinase and v-abl were incapable of transactivating the dhfr promoter in a RING3-dependent fashion, demonstrating specificity of the RING3 effect in the ras-MEKK pathway.
Fig. 3.
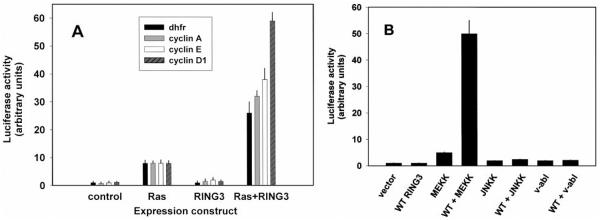
Transactivation of cyclin promoters. A, NIH/3T3 cells were transiently transfected with luciferase reporters for various cell cycle genes (cyclin D1, gray, hatched; cyclin A, gray; cyclin E, □; dhfr, ■) and expression constructs for H-ras, wild-type RING3, or empty vector control. Activity after 2 days was normalized for ras transactivation of each reporter, and a representative experiment is shown. Bars, SE. B, cells were cotransfected in duplicate with the dhfr reporter from A and various kinase constructs.
Having established that RING3 will transactivate a variety of cell cycle-responsive natural promoters, we turned to a more detailed analysis of the RING3-responsive element within the dhfr promoter. A construct in which the E2F binding sites had been mutated to render the dhfr promoter unresponsive to E2F-dependent transactivation (29) was tested (Fig. 4A). RING3 and MEKK together were no longer capable of transactivating this promoter, which strongly suggested that RING3-dependent transcriptional induction of the dhfr promoter required the binding of E2F proteins. To confirm this hypothesis, wild-type Rb protein was transiently overexpressed under conditions of maximal RING3-dependent transactivation. Excess Rb protein was predicted to reduce the “free” E2F available to transactivate this promoter (24, 25, 30), resulting in decreased luciferase activity. As expected, RING3-dependent transcription was attenuated as a dose-dependent function of overexpressed Rb (Fig. 4B). As a control for RING3 kinase activity, the (K574A) point mutant of RING3 was substituted for the wild-type enzyme (Fig. 4A). This mutant did not transactivate the wild-type or mutant dhfr reporters, either alone, or with activated MEKK.
Fig. 4.

E2F dependence of dhfr promoter transactivation. A, NIH/3T3 cells were transiently transfected in duplicate with a luciferase reporter that contained the wild-type E2F binding site of dhfr (from Fig. 3; ■) or a mutated E2F binding site (□) and various kinase constructs, including wild-type RING3 (WT), catalytically inactive RING3 (K574A), and constitutively active MEKK. B, the maximal RING3-dependent transcription in A was titrated with wild-type Rb protein and wild-type E2F binding site. Bars, SE.
A Macromolecular Complex That Contains RING3 and E2Fs
The cooperative actions of RING3 and E2F proteins on transcriptional processes raised the possibility that these two molecules might coexist in a transcriptional complex. Preliminary experiments with size-exclusion chromatography of HeLa nuclear extract revealed that RING3 kinase activity partitions into two fractions. The first fraction had an average apparent molecular weight of Mr ~90,000, which agrees with the average apparent molecular weight of the “free” kinase as determined by its apparent mobility during SDS-PAGE. The second fraction had an average apparent molecular weight of Mr ~330,000 (Fig. 5), suggesting that RING3 might participate in a complex with other nuclear proteins. The amount of RING3 detected in the complex could be reduced and the amount of free RING3 increased if chromatography was conducted in the absence of 5 mM magnesium ATP (results not shown), consistent with an ATP-dependent recruitment of RING3 into the complex.
Fig. 5.
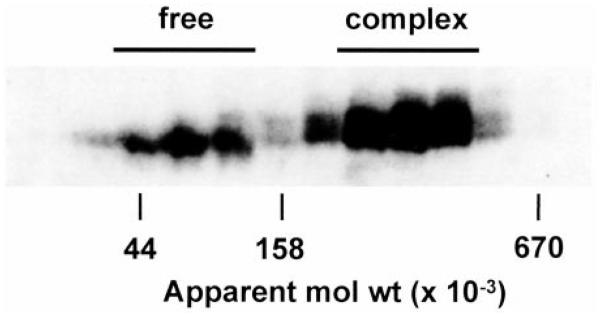
RING3 is part of a large nuclear complex. RING3 proteins in HeLa nuclear extract were resolved into free and complexed forms by size-exclusion chromatography on Superose-6. Proteins in column fractions were subjected to αRING3 immunoprecipitation, SDS-PAGE, and then autophosphorylation assay (1). The apparent molecular weights (mol wt) of protein standards are indicated (ovalbumin, Mr 44,000; aldolase, Mr 158,000; thyroglobulin, Mr 670,000).
The presence of copurified E2F proteins in a RING3 complex was first determined by anti-RING3 antibody affinity chromatography of nuclear extract. Columns were loaded with HeLa nuclear extract, washed extensively with loading buffer, and then eluted by reducing the pH to 2.5 with acetic acid, then raising it to 11.5 with sodium hydroxide. Proteins in the pooled column fractions were separated by SDS-PAGE and immunoblotted with anti-E2F-N antibody, which cross-reacts with all known E2Fs. Fig. 6B shows an immunoblot of the column input (Lane 1), the eluate of the non-immune IgG control column (Lane 2), and the eluate of a column composed of anti-RING3 antibody (Lane 3). An E2F protein was detected among the proteins that copurified from the anti-RING3 antibody column.
Fig. 6.

Nuclear complexes that contain E2Fs interact with RING3. A, diagram of COOH-terminal deletions of RING3 N-tagged with six histidines (ΔSnaBI, ΔBspMI, ΔBalI, ΔHindIII, ΔPstI, and ΔAccI), with the number of amino acids in RING3 shown on a scale (above). B, input HeLa nuclear extract (Lane 1) and eluted proteins (Lanes 2 and 3) from a nonimmune rabbit IgG affinity column (Lane 2) or αRING3 rabbit IgG affinity column (Lane 3) were separated by SDS-PAGE, electroblotted to PVDF, and probed with rabbit polyclonal antibody specific for all E2Fs (arrow). C, HeLa nuclear proteins were eluted from Ni-NTA columns charged with wild-type recombinant RING3 protein or with equal moles of the deletion mutants listed in A. Eluted fractions were immunoblotted with anti-E2F-1 or anti-E2F-2.
The association between RING3 and an E2F-containing protein complex implied that RING3 possesses domain(s) that permit interaction with the complex. Accordingly, serial deletions of recombinant histidine-tagged RING3 were constructed, beginning from the COOH terminus, based on a hypothesis that progressive deletion should eventually result in a protein fragment that is no longer able to interact with the complex. Separate Ni-NTA columns were charged with a series of truncated RING3 proteins, loaded with HeLa nuclear extract, washed extensively with loading buffer, and eluted with 1 M NaCl. Column fractions were immunoblotted with antibody to E2F-1 and E2F-2. We observed that the ability of RING3 to bind a nuclear complex that contained E2Fs was completely attenuated with the ΔSnaBI mutation (Fig. 6C), which removed the COOH-terminal 93 amino acids from this 754-amino acid protein as described previously (1). E2F-containing complexes were not detected in the eluate of any subsequent column composed of more extensively truncated RING3 derivatives.
Discussion
Early studies of RING3 linked its activity to proliferation or oncogenic transformation, particularly because mitogen or growth factor stimulation of mesenchymal cells, lymphocytes (1, 2), or whole animals (3) resulted in increased autophosphorylation of the kinase. Increased autophosphorylation is in turn correlated with increased substrate-directed trans phosphorylation (1, 31). Furthermore, very high levels of autophosphorylation activity are observed in human leukemic cell lines or leukemic blasts isolated from patients (1, 2). However, causal links were missing between RING3 activity and proliferation or oncogenesis. We therefore undertook the present studies to explore possible mechanisms of RING3-dependent transformation and to identify potential transcriptional targets of RING3.
In preliminary experiments, we observed that overexpressed RING3 cDNA transforms NIH/3T3 fibroblasts in a context where activated ras is also present (Table 1). This result is consistent with the earlier observation that bacterially expressed, recombinant RING3 is inactive and requires phosphorylation by a putative kinase kinase to become active (1). Activated ras may provide this signal. In later experiments, we isolated a clonal line of NIH/3T3 cells transformed with wild-type RING3 and injected them into athymic nude mice, which gave rise to tumors in 12 of 12 cases. However, a clonal line derived from K574A RING3 did not cause tumors in any of six cases.5 Taken together, these results suggest that increased RING3 kinase activity is likely to be a cause, not a consequence, of oncogenic transformation.
RING3 activation after serum stimulation of quiescent, synchronized fibroblasts (Fig. 1) suggests that it carries a mitogenic signal (1-3). By these criteria, RING3 might be classed with the mitogenic, signal-transducing kinases p70S6K, pp90RSK, MAPK, and ERK, which are also ras-responsive (27, 28). RING3 activity increases until the G1-S transition, which supports a hypothesis that RING3 is among the factors that link ras to E2F (32, 33), Rb (34), and G1-S progression. The cotransforming ability of RING3 with activated ras, its time course of activation by serum, and its nuclear localization in HeLa cells (1) suggested it might have transcriptional targets. We used transcriptional reporters to explore this idea and demonstrated that in fibroblasts RING3 transactivates promoters that contain E2F sites, from which we develop a hypothesis that RING3 promotes G1-S progression.
A synthetic, consensus E2F enhancer responded to overexpressed RING3 and serum, whereas a synthetic, consensus CRE enhancer did not respond to overexpressed RING3, although a control with forskolin alone responded properly. These results established promoter specificity; they implied that RING3 might participate in a serum-induced mitogenic program mediated through E2F activity, consistent with observations that cAMP-dependent signal transduction is generally not involved in fibroblast proliferation (35). Overexpressed RING3 transactivates the RING3-responsive reporter constructs under synchronized, serum-starved conditions and then serum-stimulated conditions, or in the continuous presence of serum. Under the latter condition, where the cells are not re-entering the cell cycle after a period of starvation, an additional upstream signal is required, such as activated ras or MEKK, an effector of ras. These observations support a hypothesis that RING3 participates in a mitogenic signal transduction pathway.
After demonstrating that promoters of the E2F-responsive cell cycle genes cyclin D1, cyclin A, cyclin E, and dhfr were also RING3 responsive, we focused our analysis on the dhfr promoter, which is an important and well-studied early player in E2F-mediated cell cycle progression. We successfully showed that RING3-dependent transactivation requires catalytically active RING3, E2F activity, and a functional E2F binding site. These observations raise the possibility that RING3 functions in a nuclear complex with E2Fs. We obtained evidence that RING3 is present in a large multiprotein complex by size-exclusion chromatography of HeLa nuclear extract (Fig. 5). We tested the idea that some of these proteins might be E2Fs by anti-RING3 immunoaffinity chromatography or RING3 protein affinity chromatography of nuclear extract, followed by anti-E2F immunoblot of the eluates. We detected copurified E2F-1 and E2F-2 protein in association with RING3 in these experiments. The timing of this association of E2Fs with RING3 during the cell cycle is a subject of our ongoing investigation.
There is evidence that signal transducing kinases such as Raf-1 can associate with Rb complexes after mitogenic stimulation and promote E2F-dependent G1-S progression (36) in a ras-dependent manner. A number of recent studies have explored E2F targets of the ras signal transduction pathway. In fibroblasts, the pathway typically originates with ligand activation of tyrosine kinase activity in a growth factor receptor or its deregulated activity in a tumor, e.g., the neu proto-oncogene (37), followed by the activation of a ras/Raf-1/MAPK signaling cascade (37). In the canonical pathway, this cascade leads to cyclin-dependent kinase 4/6 activation and the subsequent phosphorylation of Rb protein (38), which alleviates Rb/E2F repression of key cell cycle genes (39, 40). This derepression is a trigger event that initiates entry into S-phase (41). This pathway can be bypassed by viral oncoproteins such as human papilloma virus E7 (42) and SV40 large T-antigen (43) that bind Rb directly, thereby liberating E2F without regard to the stage of the cell cycle and provoking abnormal growth. We speculate that, like Raf-1, RING3 might partition into Rb/E2F-containing nuclear complexes at critical moments in the mitogenic program and facilitate proper transcription of cell cycle genes. RING3 is normally constitutively localized to the nucleus of exponentially growing cells such as HeLa, however in serum-starved fibroblasts it is delocalized throughout the cell and can be induced to translocate to the nucleus upon serum stimulation.6 Its regulated nuclear translocation is consistent with function at a step proximal to E2F activity. The recent identification of a RING3-like protein in the murine Mediator complex (4), where it associates with proteins that are homologous to the yeast transcriptional repressors Srb7 and Rgr1, and coactivator Med7, provides additional support for the hypothesis that RING3 functions at a transcriptional end point of mitogenic signal transduction.
Multiprotein complexes that contain RING3 and E2Fs are likely to be implicated in cell cycle progression, given the similar time course of RING3 and E2F mobilization. In subsequent experiments, we have been unable to detect a direct interaction between in vitro translated E2Fs and RING3 proteins, nor have we been able to demonstrate a RING3 effect on DNA binding by electrophoretic mobility shift assay with nuclear extract and a radiolabeled E2F probe. This result is perhaps not surprising because RING3 does not possess the protein motifs associated with chromatin-remodeling helicases or DNA binding activities or with Rb-like “pocket proteins” that bind directly to E2Fs. We speculate that RING3 probably exerts its effects on already assembled transcription complexes, consistent with the lack of effect of RING3 on enhancerless promoters (Fig. 2). We are currently investigating whether RING3 phosphorylates E2F, and if so, whether this phosphorylation affects its transcriptional activity. A recent two-hybrid screen with yeast Taf67, a homologue of mammalian TAFII55, identified Bdf1 and Bdf2 (the two yeast homologues of RING3; 18), which suggests that, in addition to E2F, RING3 complexes may also include TAFs and histone acetyltransferase functions.
Mammalian members of the RING3 family and its Drosophila homologue fsh contain two bromodomains of unknown function, although a link has been proposed (1) between RING3 and the bromodomain-containing protein ALL-1, a human leukemic homologue of the Drosophila developmental transcription factor encoded by the trithorax gene. Deregulation of a fsh/Trithorax pathway in flies, producing homeotic mutations, might be analogous to deregulation of a RING3/ALL-1 pathway in humans, producing leukemias. ALL-1 associates with chromatin remodeling complexes such as SWI/SNF (44), which in turn associate with Rb (23, 45, 46). SWI/SNF also associates with mammalian brahma (21, 47), which is homologous to a bromodomain protein of the Drosophila trithorax group. Mammalian brahma is necessary for Rb-dependent cell cycle arrest (23), mediated through histone deacetylase repression of cell cycle genes (39, 48, 49). The rnc mutant of fsh controls head development in Drosophila through a homologue of Sp1 (12),4 which in mammals also interacts with Rb/E2F (50, 51). A RING3/ALL-1 pathway might therefore influence E2F-dependent cell cycle events through SWI/SNF and its associated histone modification activities. In view of these functional links, we speculate that RING3 and fsh will also be identified in other bromodomain- or E2F-containing cocomplexes where it may be associated with transcriptional repression.
These findings of a novel kinase, which is activated upon mitogenic stimulation, transforms cells when overexpressed and transactivates cyclin gene expression through E2F sites, provide a new potential mechanism for modulation of mitogenic signals. We propose that RING3 is a member of the growing class of bromodomain-containing proteins that function in transcription complexes at the promoters of key E2F-dependent cell cycle genes. In concert with chromatin-modifying enzymes, these proteins enable the proper transcription of the genes for growth; deregulation of the RING3-dependent mitogenic signal transduction program may lead to destabilization of the cell cycle and neoplastic transformation.
Materials and Methods
Tissue Culture and Transfection
Low-passage NIH/3T3 cells were maintained in DMEM (Life Technologies, Inc.) supplemented with 10% donor calf serum, glutamine, penicillin, and streptomycin and transfected by the calcium phosphate method. The response of the luciferase reporter was optimized by titration of input plasmid DNAs. Transfections typically contained 30 μg of DNA per 100-mm tissue culture plate at 50% confluence, comprising 10 μg of reporter plasmid, 15 μg of expression vector, and 5 μg of balancing plasmid; transfection efficiencies were assessed by the activity of a cotransfected β-galactosidase expression vector. Unless otherwise noted, the enhancer-promoter of the immediate-early gene of human cytomegalovirus drove all expression constructs. For focus formation assays, cells were transfected with 2 μg of H-ras or 5 μg of MEKK expression vector, 10 μg of RING3 expression vector, balanced to 30 μg DNA with empty vector [pcDNA(I)Amp; Invitrogen], and plated in 80% (v/v) Matrigel basement membrane mixture (Becton Dickinson), and foci were counted by visual inspection or plated onto tissue culture plastic, where foci were visualized with Giemsa stain before counting. After transfection, cells were grown to confluence, which required 2 days, and then were harvested for luciferase assay. Monolayers were washed twice with PBS (pH 7.4), and then cells were scraped into 0.4 ml of luciferase lysis buffer (Promega) and passed through three freeze/thaw cycles. Extracts were cleared of debris by microcentrifugation and then 0.02 ml were mixed with luciferase assay reagent (Promega) to give a 0.1-ml reaction. Emitted light was quantified for 60 s in a TD-20/20 luminometer (Turner).
Plasmid Constructs
Expression vectors for RING3 were as described (1). Rb, constitutively active MEKK, and c-Jun NH2-terminal kinase kinase were from C. Lange-Carter (National Jewish Center for Immunology and Respiratory Medicine, Denver, CO), and v-abl was from R. Ren (Brandeis University, Waltham, MA). The CRE and E2F reporters (Clontech) comprised three copies or four copies, respectively, of a consensus CRE (5′-TGACGTCA-3′) or E2F-responsive element (5′-TTGGGCGCGTT-3′) upstream from the herpes simplex thymidine kinase promoter and the firefly luciferase gene. The luciferase reporter for cyclin D1 (−963) was from J. Nevins (Duke University Medical Center, Durham, NC), for cyclin A, from J. M. Blanchard (Institut de Genetique Moleculaire, CNRS, Montpellier, France), and for cyclin E, from P. Jansen-Durr (Forschungsschwerpunkt Angewandte Tumorvirologie, Heidelberg, Germany). The dhfr reporter with a wild-type E2F binding site (5′-CGATTTCGCGCCAAA-3′) and a mutated E2F binding site (5′-CGGCCCTATATCAAA-3′) were from J. Xiao (Boston University School of Medicine, Boston, MA) and were confirmed by sequencing. Serial truncations of recombinant histidine-tagged RING3 were constructed with convenient restriction sites in RSETA (Invitrogen). Excised fragments were bounded at the 3′ end by EcoRI and at the 5′ end by SnaBI, BspMI, BalI, HindIII, PstI, or AccI to give recombinant proteins of length 660, 578, 426, 298, 218, or 179 amino acids, respectively, not including the NH2-terminal histidine tag.
Chromatography
Size-exclusion chromatography was performed with Superose-6 media packed in a Pharmacia LKB XK 16/50 column and developed with an LCC-500 Plus liquid chromatography system (Amersham Pharmacia). A sample of 0.5 ml was applied to a column of dimensions 52 × 1.8 cm and chromatographed at 4°C and 0.1 ml/min in buffer A [50 mM sodium chloride, 20 mM Tris-HCl (pH 7.4), 50 mM disodium glycerol-2-phosphate, 10% glycerol, 5 mM magnesium chloride, 5 mM disodium ATP, 2 mM DTT, and the protease inhibitors phenylmethylsulfonyl fluoride, pepstatin, aprotinin, and leupeptin]. Proteins were immunoprecipitated with anti-RING3 polyclonal antibody (1) and solubilized in SDS sample buffer for 12% PAGE analysis. The column was calibrated with blue dextran (2,000,000), thyroglobulin (667,000), ferritin (440,000), catalase (232,000), aldolase (158,000), ovalbumin (44,000), and myoglobin (18,800; Amersham Pharmacia) under the same conditions. Autophosphorylation assay was performed as described previously (1).
Rabbit polyclonal antibodies were raised against recombinant RING3 (1) and purified from antisera by protein A affinity chromatography. A rabbit antibody affinity column (1.0 ml) was constructed by coupling 0.3 mg of periodate-oxidized antibody to a hydrazide-activated polymer (AvidChrom cartridge; Sigma). The control column of nonimmune rabbit immunoglobulin (IgG) agarose was from Sigma. HeLa nuclear extract (1.0 ml) was passed through a buffer exchange column charged with ice-cold buffer B [50 mM ammonium bicarbonate (pH 7.4), 5 mM disodium ATP, 5 mM magnesium chloride, 1 mM 2-mercaptoethanol, and protease inhibitors] and then applied to the antibody affinity columns at 0.1 ml/min. The columns were washed with 20 ml of ice-cold buffer B and eluted with 10 ml of acidic elution buffer (50 mM acetic acid, pH 2.5) and then 10 ml of basic elution buffer (120 mM sodium hydroxide pH 11.5). Eluted fractions were pooled, and proteins were precipitated with tRNA carrier and 10% trichloroacetic acid, washed with ice-cold acetone, and subjected to SDS-PAGE as above.
Recombinant RING3 protein affinity chromatography was performed by binding 0.5 ml of HeLa nuclear extract (5 mg protein) and 0.1 mg wild-type histidine-tagged RING3 protein (1) to 1.0 ml of Ni-NTA agarose (Qiagen) in ice-cold buffer C [50 mM disodium glycerol-2-phosphate, 30 mM imidazole (pH 7.4), 5 mM magnesium chloride, 5 mM disodium ATP, 1 mM 2-mercaptoethanol, 0.1% Tween 20, and protease inhibitors]. Column flow-through and washes (20 ml of buffer C) were collected, and then the columns were eluted with 5 ml of ice-cold 1 M NaCl in buffer C. Eluted proteins were precipitated as above. For experiments with truncated RING3 proteins, equal numbers of moles of each derivative were applied to the columns.
For immunoblotting experiments, proteins were separated by SDS-PAGE and electroblotted to a PVDF membrane (Bio-Rad), which was blocked with 5% nonfat dry milk in 120 mM sodium chloride, 10 mM Tris-HCl (pH 8), and 0.05% Tween 20 for 1 h, and then probed overnight at 4°C with an anti-E2F rabbit polyclonal antibody that detects all known E2Fs (Santa Cruz H-111), anti-E2F-1 mouse monoclonal antibody (Santa Cruz KH95), or an anti-E2F-2 rabbit polyclonal antibody (Santa Cruz C-20) diluted 1:2000 in the same solution. Primary antibody was detected with antimouse or antirabbit IgG secondary antibody conjugated to horseradish peroxidase (Boehringer Mannheim), diluted 1:10,000. Secondary antibody was visualized with Renaissance chemiluminescence reagent plus (New England Nuclear) and XB-1 blue film (Kodak). Prestained protein markers were from Bio-Rad and Amersham Pharmacia.
Acknowledgments
We thank J. M. Blanchard, C. Lange-Carter, P. Jansen-Durr, J. Nevins, R. Ren, and J. Xiao for expression and reporter plasmids and L. Recht (University of Massachusetts Medical Center, Worcester, MA) for assistance with athymic nude mice.
Footnotes
This work was supported by USPHS Grants CA75107 (to G. V. D.) and CA50459 (to D. V. F.) from the National Cancer Institute.
- IL
- interleukin
- RING3
- really interesting new gene 3
- dhfr
- dihydrofolate reductase
- MAPK
- mitogen-activated protein kinase
- MEKK
- MAPK kinase kinase
- CRE
- cAMP responsive element
- PVDF
- polyvinylidene difluoride.
B. Florence, personal communication.
G. V. Denis, unpublished data.
N. Guo, D. V. Faller, and G. V. Denis, J. Cell Sci., in press.
References
- 1.Denis GV, Green MR. A novel, mitogen-activated nuclear kinase is related to a Drosophila developmental regulator. Genes Dev. 1996;10:261–271. doi: 10.1101/gad.10.3.261. [DOI] [PubMed] [Google Scholar]
- 2.Rachie NA, Seger R, Valentine MA, Ostrowski J, Bomsztyk K. Identification of an inducible 85-kDa nuclear protein kinase. J. Biol. Chem. 1993;268:22143–22149. [PubMed] [Google Scholar]
- 3.Ostrowski J, Florio SK, Denis GV, Suzuki H, Bomsztyk K. Stimulation of p85/RING3 kinase in multiple organs after systemic administration of mitogens into mice. Oncogene. 1998;16:1223–1227. doi: 10.1038/sj.onc.1201624. [DOI] [PubMed] [Google Scholar]
- 4.Jiang YW, Veschambre P, Erdjument-Bromage H, Tempst P, Conaway JW, Conaway RC, Kornberg RD. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. USA. 1998;95:8538–8543. doi: 10.1073/pnas.95.15.8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beck S, Hanson I, Kelly A, Pappin DJC, Trowsdale J. A homologue of the Drosophila female sterile homeotic (fsh) gene in the class II region of the human MHC. DNA Seq. 1992;2:203–210. doi: 10.3109/10425179209020804. [DOI] [PubMed] [Google Scholar]
- 6.Jones MH, Numata M, Shimane M. Identification and characterization of BRDT: a testis-specific gene related to the bromodomain genes RING3 and Drosophila fsh. Genomics. 1997;45:529–534. doi: 10.1006/geno.1997.5000. [DOI] [PubMed] [Google Scholar]
- 7.Thorpe KL, Gorman P, Thomas C, Sheer D, Trowsdale J, Beck S. Chromosomal localization, gene structure and transcription pattern of the ORFX gene, a homologue of the MHC-linked RING3 gene. Gene (Amst.) 1997;200:177–183. doi: 10.1016/s0378-1119(97)00415-0. [DOI] [PubMed] [Google Scholar]
- 8.Taniguchi Y, Matsuzaka Y, Fujimoto H, Miyado K, Kohda A, Okumura K, Kimura M, Inoko H. Nucleotide sequence of the ring3 gene in the class II region of the mouse MHC and its abundant expression in testicular germ cells. Genomics. 1998;51:114–123. doi: 10.1006/geno.1998.5262. [DOI] [PubMed] [Google Scholar]
- 9.Salter-Cid L, Du Pasquier L, Flajnik M. RING3 is linked to the Xenopus histocompatibility locus. Immunogenetics. 1996;44:397–399. [PubMed] [Google Scholar]
- 10.Takami K, Zaleska-Rutczynska Z, Figueroa F, Klein J. Linkage of LMP, TAP, and RING3 with MHC class I rather than class II genes in the zebrafish. J. Immunol. 1997;159:6052–6060. [PubMed] [Google Scholar]
- 11.Mozer BA, Dawid IB. Cloning and molecular characterization of the trithorax locus of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA. 1989;86:3738–3742. doi: 10.1073/pnas.86.10.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Florence B, McGinnis W. A genetic screen of the Drosophila X chromosome for mutations that modify Deformed function. Genetics. 1998;150:1497–1511. doi: 10.1093/genetics/150.4.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71:691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- 14.Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, Croce CM, Canaani E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71:701–708. doi: 10.1016/0092-8674(92)90603-a. [DOI] [PubMed] [Google Scholar]
- 15.Rowley JD. Chromosomal translocations: dangerous liaisons. Leukemia. 1994;8:1–6. [PubMed] [Google Scholar]
- 16.Chua P, Roeder GS. Bdf1, a yeast chromosomal protein required for sporulation. Mol. Cell. Biol. 1995;15:3685–3696. doi: 10.1128/mcb.15.7.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lygerou Z, Conesa C, Lesage P, Swanson RN, Ruet A, Carlson M, Sentenac A, Seraphin B. The yeast BDF1 gene encodes a transcription factor involved in the expression of a broad class of genes including snRNAs. Nucl. Acids Res. 1994;22:5332–5340. doi: 10.1093/nar/22.24.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matangkasombut O, Buratowski RM, Swilling NW, Buratowski S. Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev. 2000;14:951–962. [PMC free article] [PubMed] [Google Scholar]
- 19.Jeanmougin F, Wurtz J-M, Le Douarin B, Chambon P, Losson R. The bromodomain revisited. Trends Biochem. Sci. 1997;22:151–153. doi: 10.1016/s0968-0004(97)01042-6. [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki H, Song J, Eckner R, Ugai H, Chiu R, Taira K, Shi Y, Jones N, Yamamoto KK. p300 and ATF-2 are components of the DRF complex, which regulates retinoic acid- and E1A-mediated transcription of the c-jun gene in F9 cells. Genes Dev. 1998;12:233–245. doi: 10.1101/gad.12.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elfring LK, Deuring R, McCallum CM, Peterson CL, Tamkun JW. Identification and characterization of Drosophila relatives of the yeast transcriptional activator SNF2/SWI2. Mol. Cell. Biol. 1994;14:2225–2234. doi: 10.1128/mcb.14.4.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peterson CL, Tamkun JW. The SWI-SNF complex: a chromatin remodeling machine? Trends Biochem. Sci. 1995;20:143–146. doi: 10.1016/s0968-0004(00)88990-2. [DOI] [PubMed] [Google Scholar]
- 23.Trouche D, Le Chalony C, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc. Natl. Acad. Sci. USA. 1997;94:11268–11273. doi: 10.1073/pnas.94.21.11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 25.Nevins JR. Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 1998;9:585–593. [PubMed] [Google Scholar]
- 26.Qiu RG, Chen J, Kirn D, McCormick F, Symons M. An essential role for Rac in ras transformation. Nature (Lond.) 1995;374:457–459. doi: 10.1038/374457a0. [DOI] [PubMed] [Google Scholar]
- 27.Simm A, Hoppe V, Karbach D, Leicht M, Fenn A, Hoppe J. Late signals from the PDGF receptors leading to the activation of the p70 S6-kinase are necessary for the transition from G1 to S phase in AKR-2B cells. Exp. Cell Res. 1998;244:379–393. doi: 10.1006/excr.1998.4200. [DOI] [PubMed] [Google Scholar]
- 28.Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem. J. 1997;326:61–68. doi: 10.1042/bj3260061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Means AL, Slansky JE, McMahon SL, Knuth MW, Farnham PJ. The HIP1 binding site is required for growth regulation of the dihydrofolate reductase gene promoter. Mol. Cell. Biol. 1992;12:1054–1063. doi: 10.1128/mcb.12.3.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature (Lond.) 1993;365:349–352. doi: 10.1038/365349a0. [DOI] [PubMed] [Google Scholar]
- 31.Hunter T. A thousand and one protein kinases. Cell. 1987;50:823–829. doi: 10.1016/0092-8674(87)90509-5. [DOI] [PubMed] [Google Scholar]
- 32.Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature (Lond.) 1997;387:422–426. doi: 10.1038/387422a0. [DOI] [PubMed] [Google Scholar]
- 33.Johnson DG, Cress WD, Jakoi L, Nevins JR. Oncogenic capacity of the E2F1 gene. Proc. Natl. Acad. Sci. USA. 1994;91:12823–12827. doi: 10.1073/pnas.91.26.12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peeper DS, Upton TM, Ladha MH, Neuman E, Zalvide J, Bernards R, DeCaprio JA, Ewen ME. Ras signaling linked to the cell-cycle machinery by the retinoblastoma protein. Nature (Lond.) 1997;386:177–181. doi: 10.1038/386177a0. [DOI] [PubMed] [Google Scholar]
- 35.McKenzie FR, Pouyssegur J. cAMP-mediated growth inhibition is fibroblasts is not mediated via mitogen-activated protein (MAP) kinase (ERK) inhibition. J. Biol. Chem. 1996;271:13476–13483. doi: 10.1074/jbc.271.23.13476. [DOI] [PubMed] [Google Scholar]
- 36.Wang S, Ghosh RN, Chellappan SP. Raf-1 physically interacts with Rb and regulates its function: a link between mitogenic signaling and cell cycle regulation. Mol. Cell. Biol. 1998;18:7487–7498. doi: 10.1128/mcb.18.12.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee RJ, Albanese C, Fu M, D’Amico M, Lin B, Watanabe G, Haines GK, III, Siegel PM, Hung MC, Yarden Y, Horowitz JM, Muller WJ, Pestell RG. Cyclin D1 is required for transformation activated by Neu and is induced through an E2F-dependent signaling pathway. Mol. Cell. Biol. 2000;20:672–683. doi: 10.1128/mcb.20.2.672-683.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 39.Iavarone A, Massagué J. E2F and histone deacetylase mediate transforming growth factor beta repression of cdc25A during keratinocyte cell cycle arrest. Mol. Cell. Biol. 1999;19:916–922. doi: 10.1128/mcb.19.1.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang HS, Postigo AA, Dean DC. Active transcriptional repression by the Rb-E2F complex mediates G1 arrest triggered by p16INK4a, TGFβ and contact inhibition. Cell. 1999;97:53–61. doi: 10.1016/s0092-8674(00)80714-x. [DOI] [PubMed] [Google Scholar]
- 41.He S, Cook BL, Deverman BE, Weihe U, Zhang F, Prachand V, Zheng J, Weintraub SJ. E2F is required to prevent inappropriate S-phase entry of mammalian cells. Mol. Cell. Biol. 2000;20:363–371. doi: 10.1128/mcb.20.1.363-371.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee J-O, Russo AA, Pavlevitch NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature (Lond.) 1998;391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- 43.Laiho M, DeCaprio JA, Ludlow JW, Livingston DM, Massagué J. Growth inhibition by TGF-β linked to suppression of retinoblastoma protein phosphorylation. Cell. 1990;62:175–185. doi: 10.1016/0092-8674(90)90251-9. [DOI] [PubMed] [Google Scholar]
- 44.Rozenblatt-Rosen O, Rozovskaia T, Burakov D, Sedkov Y, Tillib S, Blechman J, Nakamura T, Croce CM, Mazo A, Canaani E. The C-terminal SET domains of ALL-1 and Trithorax interact with the INI1 and SNR1 proteins, components of the SWI/SNF complex. Proc. Natl. Acad. Sci. USA. 1998;95:4152–4157. doi: 10.1073/pnas.95.8.4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bourachot B, Yaniv M, Murchardt C. The activity of mammalian brm/SNF2 a is dependent on high-mobility-group protein I/Y-like DNA binding domain. Mol. Cell. Biol. 1999;19:3931–3939. doi: 10.1128/mcb.19.6.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murchardt C, Yaniv M. The mammalian SWI/SNF complex and the control of cell growth. Semin. Cell Dev. Biol. 1999;10:189–195. doi: 10.1006/scdb.1999.0300. [DOI] [PubMed] [Google Scholar]
- 47.Papoulas O, Beek SJ, Moseley SL, McCallum CM, Sarte M, Shearn A, Tamkun JW. The Drosophila trithorax group proteins BRM, ASH1 and ASH2 are subunits of distinct protein complexes. Development (Camb.) 1998;125:3955–3966. doi: 10.1242/dev.125.20.3955. [DOI] [PubMed] [Google Scholar]
- 48.Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature (Lond.) 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 49.Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature (Lond.) 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 50.Karlseder J, Rotheneder H, Wintersberger E. Interaction of Sp1 with the growth- and cell cycle-regulated transcription factor E2F. Mol. Cell. Biol. 1996;16:1659–1667. doi: 10.1128/mcb.16.4.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin S-Y, Black AR, Kostic D, Pajovic S, Hoover CN, Azizkhan JC. Cell cycle-regulated association of E2F1 and Sp1 is related to their functional interaction. Mol. Cell. Biol. 1996;16:1668–1675. doi: 10.1128/mcb.16.4.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]