

Abstract
Background
Enhanced proliferation, resistance to apoptosis and metabolic shift to glycolysis of pulmonary arterial vascular smooth muscle cells (PAVSMC) are key pathophysiological components of pulmonary vascular remodeling in idiopathic pulmonary arterial hypertension (IPAH). The role of distinct mTOR complexes mTORC1 (mTOR-raptor) and mTORC2 (mTOR-rictor) in PAVSMC proliferation and survival in PAH and their therapeutic relevance is unknown.
Methods and Results
Immunohistochemical and immunoblot analyses revealed that mTORC1 and mTORC2 pathways are markedly up-regulated in small remodeled PAs and isolated distal PAVSMC from IPAH subjects that have increased ATP levels, proliferation and survival that depend on glycolytic metabolism. siRNA- and pharmacological-based analysis showed that while both mTORC1 and mTORC2 contributing to proliferation, only mTORC2 is required for ATP generation and survival of IPAH PAVSMC. mTORC2 down-regulated energy sensor AMPK allowing activation of mTORC1-S6 and increased proliferation, and deficiency of pro-apoptotic protein Bim and IPAH PAVSMC survival. Nox4 protein levels were increased in IPAH PAVSMC that was necessary for mTORC2 activation, proliferation and survival. Nox4 levels and mTORC2 signaling were significantly up-regulated in small PAs from hypoxia-exposed rats at days 2-28 of hypoxia. Treatment with the mTOR kinase inhibitor PP242 at days 15-28 suppressed mTORC2, but not Nox4, induced SM-specific apoptosis in small PAs and reversed hypoxia-induced pulmonary vascular remodeling in rats.
Conclusions
These data provide a novel mechanistic link of Nox4-dependent activation of mTORC2 via energy sensor AMPK to increased proliferation and survival of PAVSMC in PAH suggesting a new potential pathway for the therapeutic interventions.
Keywords: mTORC2, AMPK, IPAH, pulmonary vascular remodeling, energy metabolism, pulmonary vascular changes, proliferation, vascular smooth muscle, signal transduction
Introduction
Pulmonary arterial hypertension (PAH) is a multi-factorial disease with a poor prognosis that may be idiopathic, heritable, or associated with other diseases.1 All types of PAH share similar pathological manifestations such as remodeling of the small muscular PAs leading to increased right ventricular afterload and ultimately right heart failure and death.1 Increased cell proliferation and survival in the intima and media of small muscular PAs are key cellular events leading to pathological components of pulmonary vascular remodeling.2-4 We propose that the underlying mechanisms involve an interplay of metabolic adaptation with growth promoting cellular signals.
PA vascular smooth muscle cells (PAVSMC) in idiopathic PAH (IPAH) have increased expression of hypoxia-inducible factor 1-α (HIF1α) and metabolic glycolytic shift similar to the “Warburg effect” seen in human tumors supporting cancer cell growth and survival.2,5 Several of these characteristics are also present in hypoxic PH and cellular responses due to hypoxia. The majority of non-transformed cells, including human airway smooth muscle, respond to chronic hypoxia by up-regulating AMP-activated protein kinase (AMPK) that suppresses cell proliferation via inhibiting mammalian target of rapamycin complex 1 (mTORC1), a key positive regulator of protein, nucleotide and lipid synthesis.6 Cancer cells overcome this translational block by mutational up-regulation of phosphatidylinositol 3 kinase (PI3K)-Akt and Raf-extracellular signal-regulated kinases 1/2 (ERK1/2) pathways that activate mTORC1 via inhibiting or counter-balancing AMPK, increase HIF1α expression and/or transcriptional activity, stimulate glycolysis and protect cells from apoptosis.7 PAVSMC respond to chronic hypoxia by Akt and mTORC1 activation that is required for increased proliferation and VSM remodeling8-11 without changes in PI3K and ERK1/2 activities8 by the mechanism(s) that are currently unknown.
In addition to rapamycin-sensitive mTORC1, mTOR acts via functionally distinct mTORC2, which is rapamycin-resistant in most cell types including human VSMC8,12 and phosphorylates S-473 Akt.6-7 The only known activator of mTORC2 is growth factor- and insulin-induced PI3Ksignaling. Alternative mechanisms of mTORC2 activation and its function in pulmonary vasculature have not been studied. Several lines of evidence suggest that mTORC2 may act as a coordinator of the metabolic shift with proliferation and survival of PAVSMC in human PAH. First, chronic hypoxia induces PI3K-independent mTORC2 activation that is required for PAVSMC proliferation.8 Second, mTORC2 stimulates glycolysis and up-regulates expression of HIF1α in certain cell types.13-14 Third, NADPH oxidase Nox4, an important regulator of pulmonary vascular remodeling in PAH15, increases P-S473-Akt in human PAVSMC under chronic hypoxia16 and contributes to HIF1α expression in heart.17 Last, mTORC2 is required for cancer cell survival18-19 suggesting its possible role in regulating PAVSMC glycolytic metabolism, proliferation and survival in PAH.
In this study, we aimed to dissect the role of mTOR signaling in PAVSMC proliferation and survival in IPAH. Our data show a novel role for mTORC2 as a coordinator of PAVSMC energy metabolism, proliferation and survival in PAH and provide a novel mechanistic link of Nox4-dependent activation of mTORC2 via AMPK to increased proliferation and survival of PAVSMC in PAH suggesting a new potential pathway for the therapeutic interventions.
Methods
Methods are expanded in Online Data Supplement.
Human tissues and cell cultures
Lung tissues from four non-diseased (control) and four IPAH female lungs were provided by the Pulmonary Hypertension Breakthrough Initiative (PHBI) and National Disease Research Interchange (NDRI) under protocols approved by PHBI, NDRI, and the University of Pennsylvania and University of Colorado institutional review boards (Table S1). Distal (type III) PAVSMC (Table S1) were isolated and characterized as described (Online Data Supplement, Figures S1, S2). Each experiment was repeated using primary (3-8 passage) PAVSMC of the same passage from a minimum of three control and three IPAH subjects. LONZA media with 0.1% BSA used for serum-deprivation.
Immunohistochemical analysis was performed on lung tissue sections snap-frozen in OCT embedding compound (Tissue-Tek, Tokyo, Japan) as described.8
Transfection and immunoblot analysis were performed as described.8,19 siRNAs, pCMV6- Myc-DDK-Nox4 and pCMV6-Bim were purchased from Dharmacon (Lafayette, CO) and OriGene (Rockville, MD).
Apoptosis analysis was performed using In Situ Cell Death Detection Kit (Roche, Nutley, NJ) as described.19
DNA synthesis analysis was performed using BrdU incorporation assay as described.8,19
Cell growth and viability assay
Cells plated on 6-well cultured plates (180,000 cells/well) were placed in LONZA media supplemented with 0.1% BSA (day 0); cells were harvested at days 0, 5, and 10 and cell counts or viability measured using a Countess automated cell counter (Invitrogen, Grand Island, NY).
ATP analysis
Cell extracts were prepared as described in20. ATP colorimetric/fluorimetric assay (Abcam, Cambridge, UK) was performed according to manufacturer's protocol.
Animals
All animal procedures were in accordance with University of Pennsylvania Animal Care and Use Committee guidelines. 6-8-weeks-old male Sprague-Dawley rats randomly assigned to control and experimental groups (n=6/group). Experimental groups were exposed to hypoxia (10% O2) for 2, 14, or 28 days, or treated with PP242 (20 mg/kg, IP 5 days/week) or vehicle at days 15-28. Controls included normoxia-maintained animals.8 Animals were euthanized with pentobarbital overdose; the lungs were subjected to immunohistochemical or apoptosis analysis or stained with hematoxylin and eosin. Images were taken using Nikon TE2000 microscope; blinded morphometric analysis of PA medial wall thickness was performed as described.21 The lumen area at the level of the basement membrane and total vascular area at the adventitial border in muscular PA (25-150 μm outer diameter) per lung section were outlined, area sizes were measured using Image Pro-Plus 7. Medial wall thickness was calculated as [(total vascular area - lumen area)/total vascular area]x100. Analysis of fluorescent intensity in smooth muscle actin (SMA)-positive areas of small muscular PA was performed using Image-Pro 7. For visualization of pulmonary vascular tree, the lung vasculature of rats randomly selected from experimental groups was rinsed with PBS, inflated with AltaBlu reagent, and microCT analysis was performed by Numira Biosciences.
Data analysis
Data expressed as mean±SE using StatView software. Statistical comparisons between two groups were performed by the unpaired Student's t-test. Comparisons among ≥3 groups were performed with one-way, two-way, or three-way ANOVA without repeated measures as appropriate. Comparisons among ≥3 groups performed with one-way ANOVA followed by Dunnett's post-hoc test. Comparisons among ≥3 groups performed with two- or three-way ANOVA followed by stratified independent t-test with Bonferroni corrections for multiple comparisons. Statistical significance was defined as p≤0.05.
Results
mTORC1 and mTORC2 pathways are activated in PAVSMC in PAH
Immunohistochemical analysis of lung tissues from four IPAH and four non-diseased (control) subjects revealed a marked increase in P-S2481-mTOR, a marker for mTOR catalytic activity22, mTORC1-specific P-S235/236-S66,8 and mTORC2-specific P-S473-Akt7,19 in SMA-positive areas in small muscular remodeled PAs (50-250 μM outer diameter) in IPAH lungs (Figure 1A-D). Distal PAVSMC from IPAH patients demonstrated significant elevation of P-S2481-mTOR, P-S6 and P-S473-Akt in serum-replete conditions, which persisted after 48 h of serum-deprivation (Figure 2A-B) and associated with increased DNA synthesis, growth and viability (Figure 2C-E). These data show that PAVSMC from IPAH patients have activated mTORC1 and mTORC2 pathways and in vitro elevated proliferation and survival without mitogenic stimuli.
Figure 1.
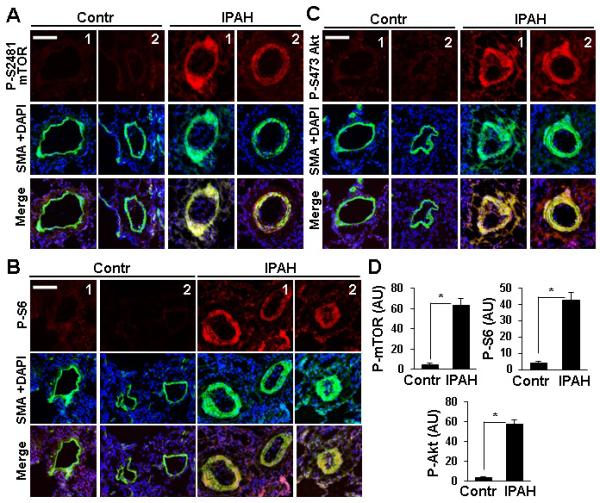
mTORC1 and mTORC2 pathways are activated in small PAs from IPAH lungs. Dual immunohistochemical analysis with anti-P-S2481-mTOR, anti-P-S6, anti-P-S473-Akt (red) and anti-SMA antibodies (green) and DAPI staining (blue) to detect nuclei of lung tissue specimens from four IPAH and four control subjects. A-C: Images were taken on a Nikon Eclipse 2000 microscope. Bar equals 100 μM. D: Statistical analysis of P-proteins in SMA-positive areas in small PAs from IPAH and control lungs. Data represent arbitrary units (AU). 4 human subjects/group; *p<0.01 by unpaired Student's t-test.
Figure 2.
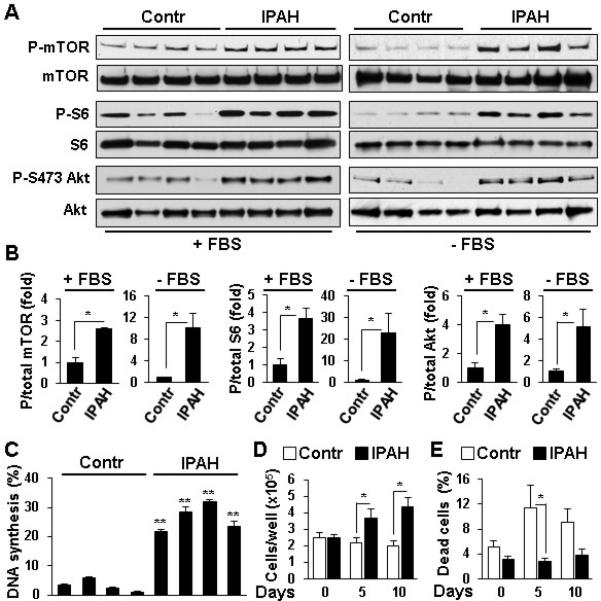
PAVSMC from IPAH lungs have activated mTORC1 and mTORC2 signaling, increased proliferation and survival. A,B: Distal PAVSMC from four non-diseased (control) and four IPAH subjects cultured in complete media (+FBS) or serum-deprived for 48 h (−FBS) were subjected to immunoblot analysis to detect indicated proteins. Data represent fold changes in P/total protein ratios; P/total ratio for controls taken as one fold. 4 subjects/group; *p<0.05 by unpaired Student t-test. C: DNA synthesis analysis of serum-deprived for 48 h PAVSMC (BrdU incorporation) from four control and four IAPH subjects, 3 measurements/subject, data from each subject presented as a separate bar. Data represent percentage of BrdU-positive cells per total number of cells. **p<0.001 vs. controls by unpaired Student t-test. D,E: Cell counts and viability of PAVSMC from four IPAH (squares) and four control subjects (circles) maintained in serum-free conditions. Data represent quantity of cells/well (D) and percentage of dead cells/total number of cells (E). 4 subjects/group; *p<0.05 by unpaired Student t-test.
Increased ATP generation, proliferation and survival of IPAH PAVSMC depend on glycolytic metabolism
Because glycolytic shift is proposed to play a role in pulmonary vascular cell proliferation in PAH5, we evaluated relative contributions of glycolytic versus mitochondrial metabolism to IPAH PAVSMC ATP generation, proliferation and survival. IPAH PAVSMC had ~2 fold higher cellular ATP content than controls in both serum-replete and serum-deplete conditions that was markedly reduced by the glycolytic inhibitor 2-deoxy-D-glucose (2-DG), while the mitochondrial respiratory chain inhibitor rotenone had modest effect (Figure 3A). 2-DG, but not rotenone, markedly decreased proliferation and promoted apoptosis in IPAH PAVSMC (Figure 3B,C). Contrarily, rotenone inhibited ATP levels, proliferation and survival of control PAVSMC while 2-DG had lesser effect (Figure 3A-C). These data show that, in contrast to non-diseased cells, increased ATP generation, proliferation and survival of IPAH PAVSMC depend predominantly on glycolytic metabolism.
Figure 3.
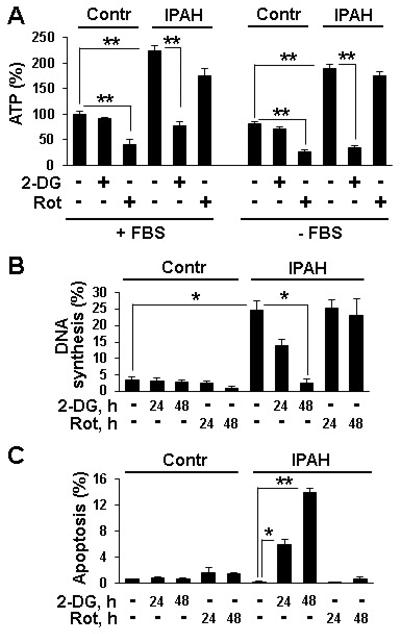
Increased ATP levels, proliferation and survival of IPAH PAVSMC depend on glycolytic metabolism. A: ATP assay performed on PAVSMC from three control and three IPAH subjects maintained in complete media (+FBS) or serum-deprived for 48 h (−FBS) and treated with 100 mM 2-DG, 10 μM rotenone or diluent for 24 h. ATP levels in control diluent-treated cells in complete media are taken as 100%. 3 subjects/group; **p<0.01 by 2-way ANOVA with a post hoc stratified independent t-test with corrections for multiple comparisons. B,C: DNA synthesis (B) and apoptosis analysis (C) of serum-deprived for 48 h cells treated with 100 mM 2-DG, 10 μM rotenone or diluent. Data represent percentage of BrdU-positive (B) (4 subjects/group) or TUNEL-positive cells (C) (3 subjects/group) per total number of cells. *p<0.05, **p<0.01 by 3-way ANOVA (IPAH/control, treatment, time) with a post hoc stratified independent t-test with corrections for multiple comparisons.
mTORC2 is required for elevated ATP generation, proliferation and survival of IPAH PAVSMC
To determine the specific roles of mTORC1 and mTORC2 in PAVSMC remodeling, we selectively disrupted the complexes using siRNA-induced knock-down of their specific regulatory proteins, raptor and rictor.6 siRNA raptor reduced mTORC1-specific P-S6 and IPAH PAVSMC proliferation without effects on mTORC2-specific P-S473-Akt, HIF1α protein levels, ATP content, and cell survival (Figures 4A-D, G, H). siRNA rictor suppressed both mTORC2-specific P-S473-Akt and mTORC1-specific P-S6, reduced cellular ATP and HIF1α levels, decreased proliferation and induced apoptosis in IPAH PAVSMC (Figures 4A-F, 5C, S3). These data demonstrate that mTORC2 is required for increased cellular ATP levels, mTORC1-S6 activity, proliferation and survival of IPAH PAVSMC, while mTORC1 contributes predominantly to increased proliferation. Comparison of the mTOR kinase inhibitor PP242 (inhibits both mTORC1 and mTORC2) and the allosteric mTORC1 inhibitor rapamycin8 revealed that both reduced proliferation, but only PP242 induced apoptosis in IPAH PAVSMC (Figure S4). Importantly, mTORC2 inhibition did not significantly affect ATP, proliferation and apoptosis rates of control PAVSMC (Figures 4B-D, S4) indicating specificity of mTORC2 up-regulation for diseased PAVSMC phenotype.
Figure 4.
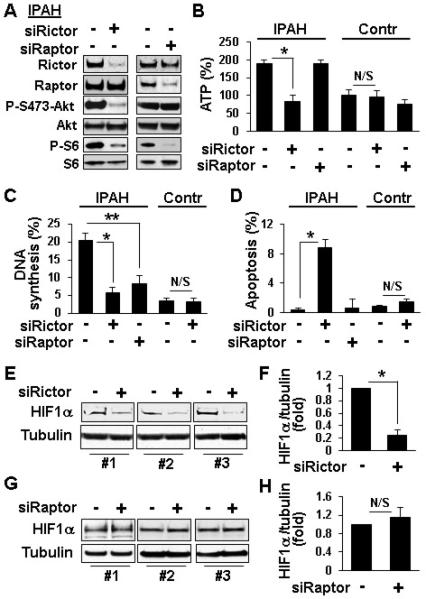
A-D: mTORC2 is required for maintenance of increased ATP levels, proliferation and survival of IPAH PAVSMC. A, B: Serum-deprived for 48 h PAVSMC were transfected with 100 nM control scrambled (−), rictor and raptor siRNAs followed by immunoblot analysis to detect indicated proteins (A) and ATP assay (B). ATP levels in control cells transfected with control siRNA are taken as 100%. 3 subjects/group; *p<0.01 by 2-way ANOVA with a post hoc stratified independent t-test with corrections for multiple comparisons. C,D: Serum-deprived for 48 h PAVSMC from IPAH and control subjects transfected with 100 nM control scrambled siRNA (−),rictor and raptor siRNAs were subjected to DNA synthesis (BrdU incorporation) (C) and apoptosis analysis (TUNEL) (D). Data are percentage of BrdU-positive (C) or TUNEL-positive cells (D) from total number of cells. 3 subjects/group; N/S - non-significant, *p<0.01 by 2-way ANOVA with a post hoc stratified independent t-test with corrections for multiple comparisons; **p<0.05 by unpaired Student t-test. E-H: siRNA rictor, but not siRNA raptor, decreases HIF1 α protein levels in IPAH PAVSMC. Serum-deprived for 48 h PAVSMC from three IPAH subjects were transfected with 100 nM control scrambled (−), rictor (E,F) or raptor (G,H) siRNAs followed by immunoblot analysis. Data represent fold changes in HIF1α/tubulin ratios; ratios for control siRNA-transfected cells taken as one fold. 3 subjects/group; N/S - non-significant, *p<0.001 by unpaired Student's t-test.
mTORC2 down-regulates AMPK enabling mTORC1 activation and proliferation of IPAH PAVSMC
Because mTORC2 positively regulates mTORC1-S6, we next determined whether mTORC2 controls mTORC1 activation and cell proliferation via energy sensor AMPK.20 IPAH PAVSMC showed a significant reduction of P-T172-AMPK and AMPK-specific P-acetyl CoA-carboxylase (ACC) (Figure 5A, B) demonstrating that AMPK signaling is down-regulated in IPAH PAVSMC. mTORC2 inhibition with siRNA rictor markedly increased P-ACC and P-AMPK while suppressing P-S6 (Figure 5C, D). siRNA-induced AMPK knock-down in IPAH PAVSMC rescued siRNA rictor-dependent inhibition of mTORC1-specific P-S6K1, P-S6 and proliferation (Figure 5E-G). In line with the role of AMPK, its activator AICAR decreased P-S6K1 and P-S6 without affecting mTORC2-specific P-S473-Akt (Figure S5). In aggregate, these data indicate that mTORC2 inhibition of AMPK leads to mTORC1 activation, which allows for proliferation of IPAH PAVSMC.
Figure 5.
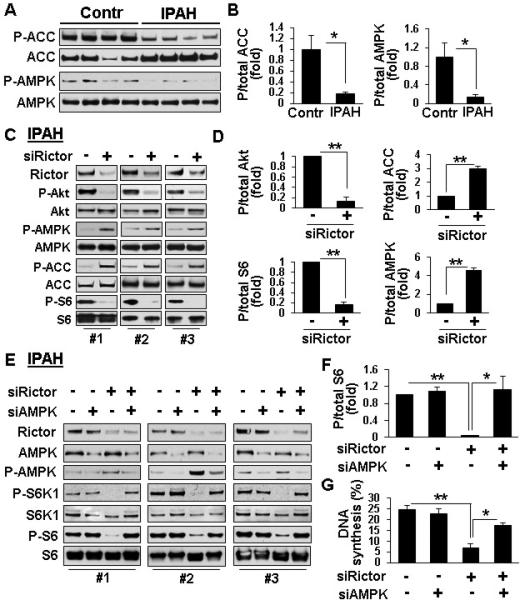
mTORC2 regulates mTORC1 signaling and IPAH PAVSMC proliferation via AMPK. A,B: Immunoblot analysis of serum-deprived for 48 h PAVSMC from four IPAH and four control subjects performed to detect indicated proteins. Data represent P/total protein ratios; mean ratio for control PAVSMC was taken as one fold. 4 subjects/group, *p<0.05 by unpaired Student's t-test. C-G: Serum-deprived PAVSMC from three IPAH subjects were transfected for 48 h with 100 nM rictor or control scrambled (−) siRNAs (C,D) or co-transfected with 50 nM siRNA rictor, siRNA AMPK, or control scrambled siRNA (−) (E-G) followed by immunoblot (C-F) and DNA synthesis (BrdU incorporation) (G) analyses. Data represent fold changes in P/total protein ratios; ratio for control siRNA-transfected cells taken as one fold (D,F) and percentage of BrdU-positive cells from total number of cells (G). D: 3 subjects/group; **p≤0.001 by unpaired Student t-test. F,G: 4 (F) and 3 (G) subjects/group; *p<0.05, **p≤0.001 by one-way ANOVA with a post hoc Dunnett's test.
mTORC2 regulates IPAH PAVSMC survival via AMPK and Bim
To determine the mechanisms by which mTORC2 stimulates IPAH PAVSMC survival, we tested whether mTORC2 regulates protein levels of Bim and Bcl2, its known downstream effectors in cancer cells.19,23 IPAH PAVSMC showed deficiency of pro-apoptotic Bim and elevated levels of anti-apoptotic Bcl2 (Figure 6A, B). mTORC2 inhibition with siRNA rictor, rather than mTORC1 suppression with siRNA raptor, markedly increased Bim levels without a significant effect on Bcl2 (Figure 6C-F). Since AMPK is shown to induce apoptosis via Bim24, we evaluated whether mTORC2 suppresses Bim levels and enhances IPAH PAVSMC survival via the inhibition of AMPK. siRNA AMPK prevented siRNA rictor-induced increase in Bim protein levels. Either siRNA AMPK or siRNA Bim rescued siRNA rictor-dependent apoptosis (Figures 6G-J, S6, S7). Bim overexpression induced apoptosis in IPAH PAVSMC (Figure 6K). These data demonstrate that mTORC2-dependent down-regulation of AMPK promotes Bim deficiency that is required for IPAH PAVSMC survival.
Figure 6.
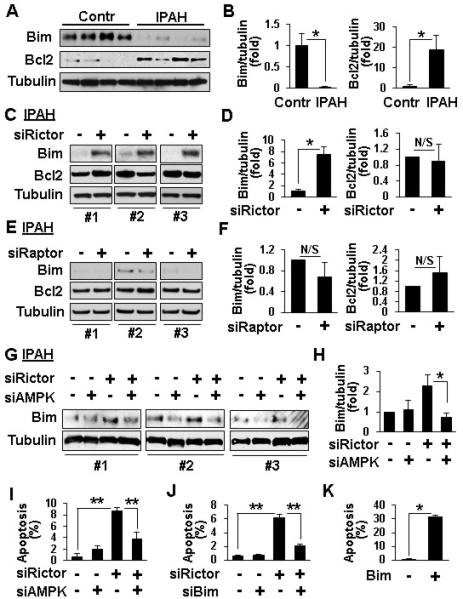
siRNA rictor regulates Bim protein levels and apoptosis via AMPK. A, B: Immunoblot analysis of serum-deprived for 48 h PAVSMC from four non-diseased (control) and four IPAH lungs. 4 subjects/group, *p<0.01 by unpaired Student t-test. C-F: Immunoblot analysis of serum-deprived for 48 h PAVSMC from three IPAH subjects transfected with 100 nM control scrambled siRNA (−), siRNA rictor, or siRNA raptor. 3 subjects/group, *p<0.05, N/S - non-significant by unpaired Student t-test. G-J: Serum-deprived for 48 h IPAH PAVSMC were transfected with 50 nM siRNA rictor, siRNA AMPK, siRNA Bim or control scrambled siRNA (−) separately or in combination followed by immunoblot (G,H) and apoptosis assay (I,J). 3 subjects/group, *p<0.05, **p<0.01 by one-way ANOVA with a post hoc Dunnett's test. K: Apoptosis analysis of IPAH PAVSMC from three subjects transfected with pCMV6-Bim (Bim) or mock-transfected (−). 3 subjects/group, *p<0.001 by unpaired Student t-test. Data represent Bim/tubulin or Bcl2/tubulin ratios with ratio for scrambled siRNA-transfected cells taken as one fold (B,D,F,H) and percentage of TUNEL-positive cells per total number of cells (I,J).
Activation of mTORC2 signaling, proliferation and survival of IPAH PAVSMC depend on Nox4
Nox4 contributes to PAH pathogenesis15 and to chronic hypoxia-induced S473-Akt phosphorylation in human PAVSMC.16 The link between Nox4 and mTORC2, however, have not been established. We found marked increase of Nox4 protein levels in human IPAH compared to control PAVSMC (Figure 7A). siRNA Nox4 decreased P-S2481-mTOR, mTORC2-specific P-S473-Akt, and mTORC1-specific P-S6, up-regulated P-AMPK, AMPK-specific P-ACC and Bim levels without affecting Bcl2 (Figure 7B-D), reduced proliferation and increased apoptosis of IPAH PAVSMC (Figures 7E, S8). Nox4 overexpression in non-diseased PAVSMC increased P-S473-Akt and P-S6, reduced P-ACC and Bim levels, and elevated cell proliferation (Figure 7F-H). These data show that Nox4 acts as an upstream positive regulator of mTORC2 signaling, proliferation and survival of IPAH PAVSMC.
Figure 7.
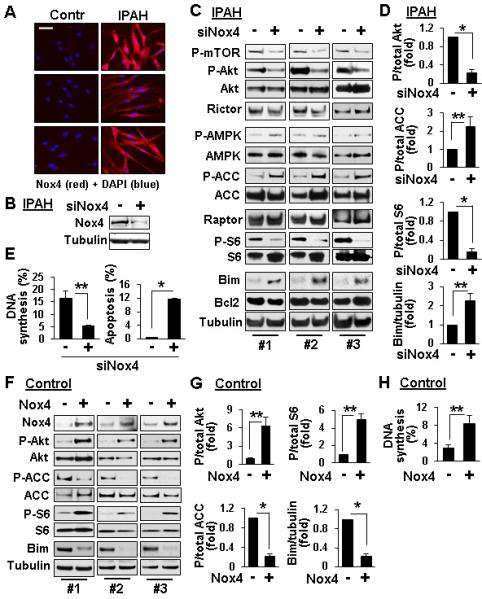
Nox4 contributes to up-regulation of mTORC2 signaling, PAVSMC proliferation and survival. A: Immunostaining with anti-Nox4 antibody (red) and DAPI nuclear staining (blue) performed on PAVSMC from three control and three IPAH subjects. Representative images were taken using a Nikon Eclipse 2000 microscope. Bar equals 50 μM. B-E: Immunoblot (B-D), DNA synthesis (BrdU incorporation) and apoptosis (TUNEL) analysis (E) of serum-deprived PAVSMC from IPAH subjects transfected for 48 h with siRNA Nox4 or scrambled siRNA (−). F-H: Immunoblot (F,G) and DNA synthesis (BrdU incorporation) analysis (H) of serum-deprived for 48 h PAVSMC from non-diseased subjects transfected with pCMV6-Myc-DDK Nox4 or mock-transfected (−). Data represent fold changes in phospho/total Akt, ACC, or S6, and Bim/tubulin ratios, ratios of control siRNA- (D) or mock-transfected cells (G) taken as one fold; and percentage of BrdU- (E,H) or TUNEL-positive cells (E) per total number of cells. Data are mean+SE from 3 subjects/group for E, D, G and 4 subjects/group for H; *p<0.001, **p<0.05 by unpaired Student t-test.
mTOR is required for PAVSMC survival and pulmonary vascular remodeling in vivo
To evaluate the role of mTORC2 and Nox4 in the development of pulmonary vascular remodeling, we performed immunohistochemical analysis of lung tissues from rats with chronic hypoxia-induced pulmonary vascular remodeling. We found significant up-regulation of P-S4281-mTOR, P-S473-Akt and Nox4 in SMA-positive areas of small muscular PAs (25-150 μM outer diameter) at day 2 of hypoxia exposure with further increase at day 14 (Figure 8A, B, D, E). Morphometric analysis under the same conditions showed significant SMC remodeling at day 14 of hypoxia with no changes in PA medial wall thickness at day 2 (Figure 8C). Thus, up-regulation of mTORC2 and Nox4 in distal PAVSMC occurs at the early stages of hypoxia preceding pulmonary vascular remodeling. Treatment with the mTOR kinase inhibitor PP242 at days 15-28 of hypoxia exposure markedly reduced P-S2481-mTOR and P-S473-Akt without significant effect on Nox4 (Figures 8F, G, S9) supporting our observations that Nox4 acts upstream of mTORC2. PP242 induced apoptosis in SMA-positive cells in small muscular PAs, which was associated with increase in Bim protein levels (Figures 8H, S10), and decreased PA medial wall thickness to levels comparable to normoxia-exposed controls (Figure 8I). MicroCT analysis showed improved pulmonary vascular density in PP242-treated rats compared to vehicle-treated animals (Figures 8J-K, Movies S1-S6). These data demonstrate that PP242 inhibits mTORC2 that induces SMA-specific apoptosis in small muscular PAs and reverses existing pulmonary vascular remodeling in vivo in a model relevant to pulmonary hypertension in humans.
Figure 8.
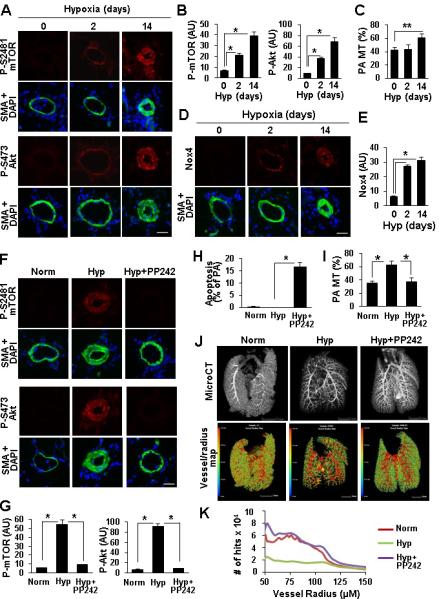
mTOR is required for PAVSMC survival and pulmonary vascular remodeling in rat chronic hypoxia PH model. A-E: Lung tissue sections from 6-8-week-old male Sprague-Dawley rats maintained under chronic hypoxia for 0, 2, 14 days were subjected to immunohistochemical (A,B,D,E) and PA medial wall thickness (MT) (C) analyses. B: 3 rats/group; C: 6 rats/group; *p<0.01, **p<0.05, by one-way ANOVA with a post hoc Dunnett's test. F-K: Immunohistochemical (F,G), apoptosis (H), PA medial wall thickness (MT) (I) and microCT (J,K) analyses performed on the lungs from rats maintained under normoxia or exposed to chronic hypoxia for 28 days and treated with PP242 or vehicle at days 15-28 of hypoxia. A, D, F: Bar equals 50 μM. B,E,G: Data represent arbitrary units (AU). H: Data represent % of small PAs with SM-specific apoptosis from total number of small PAs. G, H: 3 rats/group; I: 6 rats/group; *p<0.01 by one-way ANOVA with a post hoc Dunnett's test. J, K: Representative images of microCT analysis using μCT40 microCT scanner (J, top panel) and hit maps generated on the basis of microCT analysis using VHLab and SCIRun software (J, bottom panel). Bar equals 5 mm. K: Data represent number of hits/vessel radius.
Discussion
Increased proliferation and survival of PAVSMC in small PAs coupled with deregulated expression of HIF1α and 2α and glycolysis are critical components of the pathophysiology of pulmonary vascular remodeling in PAH. This study identifies mTORC2 as an important positive regulator of glycolysis-dependent proliferation and survival of PAVSMC in IPAH. We report the novel mechanistic link from mTORC2 via AMPK to the activation of mTORC1 signaling and increased proliferation, and Bim deficiency and survival of IPAH PAVSMC. We also show that Nox4 acts proximally to mTORC2-mediated effects to positively regulate IPAH PAVSMC proliferation and survival. Lastly, we demonstrate benefits of dual mTORC1/mTORC2 inhibition to reduce proliferation and promote apoptosis in IPAH PAVSMC, and reverse hypoxia-induced pulmonary vascular remodeling in rats (Figure S11) suggesting attractiveness of mTORC2 as a potential target to treat deregulated proliferation and survival in human PAH.
Metabolic shift to glycolysis, similar to the “Warburg effect” in cancer, contributes to increased PAVSMC proliferation and pulmonary vascular remodeling in PAH.2,5,8,25 Our data provide direct evidence that elevated ATP generation, proliferation and survival of PAVSMC from subjects with IPAH depend on glycolytic metabolism and can occur without the need of mitogenic stimuli indicating the critical role of glycolytic shift to PAVSMC proliferation and survival in IPAH.
Currently, the mechanisms coordinating the metabolic shift with increased vascular cell proliferation and survival in IPAH are not well understood. Recent studies in cancer indicate that the maintenance of a glycolytically active proliferative cell phenotype requires mutational activation of major proliferative pro-survival pathways, including PI3K-Akt and Raf-ERK1/2, that stimulate glycolysis, increased cell survival and enhanced mTORC1-dependent synthetic activity and cell growth.7,26 Our previous studies, however, have shown that hypoxia-induced PAVSMC proliferation was not associated with changes in PI3K activity and/or ERK1/2 signaling, but requires activation of mTORC1 and mTORC2 pathways.8 That led us to hypothesize about the critical role of mTORC2 in mediating glycolytic metabolism, increased proliferation and survival of PAVSMC in PAH. Here, we demonstrate that mTORC2 signaling is up-regulated in small remodeled PAs and in proliferative apoptosis-resistant distal IPAH PAVSMC without the need of exogenous mitogenic stimuli. Using both molecular and pharmacologic interventions, we subsequently showed that suppression of mTORC2 reduces cellular ATP levels, decreases P-S473-Akt and protein levels of HIF1α, two confirmed stimulators of glycolysis in other cell types5,7, inhibits proliferation and promotes apoptosis in IPAH PAVSMC, strongly suggesting the role for mTORC2 as an upstream positive regulator of glycolytic metabolism and PAVSMC growth in human IPAH.
The paucity of the understanding the role of mTORC2 in normal and diseased PAVSMC is likely attributable to the exclusive use of allosteric mTORC1 inhibitor rapamycin or its analogs (rapalogs) in prior studies. Rapalogs in the doses used for clinical applications have therapeutically proven cytostatic function with no appreciable pro-apoptotic effect in VSMC, including human PAVSMC.6,8,27 Indeed, we found that rapamycin inhibits proliferation, but does not induce IPAH PAVSMC apoptosis. Rapamycin in clinically-relevant doses attenuates development of pulmonary vascular remodeling in experimental PH, but has cytostatic effect on SM-like cells in clinical trials in pulmonary lymphangioleiomyomatosis and tuberous sclerosis, prevents apoptosis in rat carotid model of vascular injury, and does not reverse existing monocrotaline-induced PH in rats.9-10,19,28-29 High doses of rapamycin, however, attenuate pulmonary vascular remodeling and down-regulate both mTORC1-specific S6 and mTORC2-specific P-S473-Akt in same experimental model.30 Although not pharmacologically applicable to humans due to non-physiological doses of rapamycin, this study suggests potential link between mTORC2 and remodeling in experimental PH.
Our current findings show that mTORC1 pathway is activated in small PAs and in PAVSMC from IPAH lungs and is critical for IPAH PAVSMC proliferation. siRNA-based approach to selectively inhibit mTORC1 showed that mTORC1 inhibition does not affect cellular ATP levels and survival of IPAH PAVSMC. These data support the critical role of mTORC1 for cell proliferation, but strongly suggest that mTORC1 acts downstream of signaling pathways regulating IPAH PAVSMC energy metabolism.
mTORC1 is a homeostatic energy-triggered molecular relay and is activated by increased ATP levels.20 Hypoxic stress suppresses mTORC1 and cell proliferation via the energy sensor AMPK6 that has been recently identified as a key regulator of cardiovascular homeostasis, the dysfunction of which underlies several cardiovascular pathologies.31 In addition to growth-inhibitory effects, AMPK may act as a pro- or anti-apoptotic molecule in an isoform-specific manner32 by up-regulating p53 signaling or down-regulating the pro-apoptotic protein Bim.24,33 Moreover, we found evidence of mTORC2 activation of Akt, which may also contribute to overall activation of mTORC1. Akt inhibits AMPK via up-regulating cellular ATP levels,34-35 and both mTORC2 and Akt stimulate cell survival via suppressing Bim expression.19,36 These data raise the possibility that mTORC2 coordinates glycolytic ATP generation, proliferation and survival of IPAH PAVSMC via AMPK. Indeed, we found that, in IPAH PAVSMC, mTORC2 acts as an upstream negative regulator of AMPK signaling resulting in activation of mTORC1 and elevated proliferation, and in deficiency of Bim and increased cell survival.
The only confirmed regulator of mTORC2 is the PI3K/PTEN network.6 We did not detect significant differences in PI3K-specific mTORC2-independent T308-Akt phosphorylation as well as in PTEN protein levels between control and IPAH PAVSMC (data not shown) suggesting another mechanism(s) of mTORC2 activation. NADPH oxidase Nox4 is overexpressed in human IPAH lungs in the media of small PAs, and we recently reported an association of genetic variation in Nox4 with the risk of PAH in patients with portal hypertension.37-41 Hypoxia-induced Nox4 overexpression contributes to S473-Akt phosphorylation and increased proliferation of PAVSMC and pulmonary vascular remodeling in hypoxic PH.16,38-41 Our data indicate that Nox4 acts as an upstream positive regulator of mTORC2 signaling, proliferation and survival in IPAH PAVSMC providing a potential mechanism of mitogen-independent mTORC2 activation, increased cell proliferation and survival in PAH.
Recognizing that human PAH is a multifactorial disease, we anticipate that other factors such as dysregulation of BMPRII, PPARγ signaling and mitogen exposure might further impact on mTORC2-dependent regulation of proliferative apoptosis-resistant PAVSMC phenotype. Although there is no direct evidence exists linking BMPRII deficiency with mTORC2 activation, BMPRII downstream effector PPARγ inhibits mTORC1 signaling in cancer cells42-43 providing the link between BMPRII and PPARγ deficiency and mTOR activation. Of note, a recent report from Green et al shows that Nox4 modulates chronic hypoxia-induced expression of PPARγ and TGF-β144 suggesting potential cross-talk between Nox4, growth factors, BMPRII, PPARγ and mTORC2 signaling in human PAH that requires further investigation.
Collectively, our study demonstrates that mTORC2 coordinates preferential energy generation by glycolysis, increased proliferation and survival of PAVSMC in IPAH. The attractiveness of mTORC2 as a potential therapeutic target is further supported by our observations that inhibition of mTORC2 signaling by siRNA rictor and mTOR kinase inhibitor PP242 targets predominantly IPAH PAVSMC without significant effects on non-diseased cells. Similar selectivity of PP242 has already been demonstrated in a mouse leukemia model where PP242 showed improved therapeutic response compared to rapamycin, but only mildly affected normal lymphocytes.45 Although our study has focused on the IPAH PAVSMC and not other pulmonary vascular cell types, endothelial cells and adventitial fibroblasts in human IPAH also have a glycolytic phenotype,5,46 and hypoxia-induced endothelial cell proliferation requires constitutive mTORC2 activation.47 We recognize that our study has the limitations associated with small human sample size that arise from the nature of studied disease. The IPAH is rare disease that limits availability of human lung tissue specimens and primary human cell cultures of early passage for the mechanistic research of this type. However, given our current findings on pro-apoptotic effects of PP242 on pulmonary VSMC from both IPAH subjects and chronic hypoxia-exposed rats and recent advances in the pharmacological use of PP242 in animal models of cancer48-50, pre-clinical testing dual mTORC1/mTORC2 inhibitors on other human PAH cells and experimental PH models is worthy of further investigation.
Supplementary Material
Acknowledgments
The authors wish to thank the PHBI and NDRI for human lung specimens; Numira Biosciences for their microCT imaging and image analysis of the pulmonary vasculature; statisticians from the University of Pittsburgh Clinical and Translational Science Institute Design, Biostatistics and Epidemiology Core for their assistance with statistical analysis; Dr. John Blenis from the Harvard Medical School for productive discussion of data; and Mary McNichol from the University of Pennsylvana Pulmonary, Allergy and Critical Care Division for critical reading of the manuscript.
Funding Sources: This work is supported by NIH/National Heart, Lung and Blood Institute R01HL113178 (EAG), K24 HL103844 (SK), 1R01HL114085 (VPK) and UL1-TR-000005; and American Lung Association RG196551 (EAG). PHBI is supported by Cardiovascular Medical Research and Educational Fund (HD, SK, KIS).
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JXJ, Weir EK. Cellular and Molecular Basis of Pulmonary Arterial Hypertension. J Am Coll Cardiol. 2009;54:S20–S31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JGN, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–H578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 3.Masri FA, Xu W, Comhair SAA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand-Apte B, Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L548–L554. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 4.Das M, Dempsey EC, Bouchey D, Reyland ME, Stenmark KR. Chronic Hypoxia Induces Exaggerated Growth Responses in Pulmonary Artery Adventitial Fibroblasts. Potential Contribution of Specific Protein Kinase C Isozymes. Am J Respir Cell Mol Biol. 2000;22:15–25. doi: 10.1165/ajrcmb.22.1.3536. [DOI] [PubMed] [Google Scholar]
- 5.Tuder RM, Davis LA, Graham BB. Targeting Energetic Metabolism. Am J Respir Crit Care Med. 2012;185:260–266. doi: 10.1164/rccm.201108-1536PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu PP, Sabatini DM. Cancer Cell Metabolism: Warburg and Beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 8.Krymskaya VP, Snow J, Cesarone G, Khavin I, Goncharov DA, Lim PN, Veasey SC, Ihida-Stansbury K, Jones PL, Goncharova EA. mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J. 2011;25:1922–1933. doi: 10.1096/fj.10-175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, Tillmanns H, Kummer W, Braun-Dullaeus RC. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res. 2007;8:15. doi: 10.1186/1465-9921-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimura T, Faul JL, Berry GJ, Veve I, Pearl RG, Kao PN. 40-O-(2-Hydroxyethyl)-rapamycin Attenuates Pulmonary Arterial Hypertension and Neointimal Formation in Rats. Am J Respir Crit Care Med. 2001;163:498–502. doi: 10.1164/ajrccm.163.2.2006093. [DOI] [PubMed] [Google Scholar]
- 11.Luo C, Yi B, Bai L, Xia Y, Wang G, Qian G, Feng H. Suppression of Akt1 phosphorylation by adenoviral transfer of the PTEN gene inhibits hypoxia-induced proliferation of rat pulmonary arterial smooth muscle cells. Biochem Biophys Res Commun. 2010;397:486–492. doi: 10.1016/j.bbrc.2010.05.140. [DOI] [PubMed] [Google Scholar]
- 12.Goncharova EA. mTOR and vascular remodeling in lung diseases: current challenges and therapeutic prospects. FASEB J. 2013;27:1796–1807. doi: 10.1096/fj.12-222224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toschi A, Lee E, Gadir N, Ohh M, Foster DA. Differential Dependence of Hypoxia-inducible Factors 1alpha and 2alpha on mTORC1 and mTORC2. J Biol Chem. 2008;283:34495–34499. doi: 10.1074/jbc.C800170200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Rüegg MA, Hall MN. Hepatic mTORC2 Activates Glycolysis and Lipogenesis through Akt, Glucokinase, and SREBP1c. Cell Metab. 2012;15:725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 15.Griffith B, Pendyala S, Hecker L, Lee PJ, Natarajan V, Thannickal VJ. NOX enzymes and pulmonary disease. Antioxid Redox Signal. 2009;11:2505–2516. doi: 10.1089/ars.2009.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ismail S, Sturrock A, Wu P, Cahill B, Norman K, Huecksteadt T, Sanders K, Kennedy T, Hoidal J. NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-β1 and insulin-like growth factor binding protein-3. Am J Physiol Lung Cell Mol Physiol. 2009;296:L489–L499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsushima S, Kuroda J, Ago T, Zhai P, Ikeda Y, Oka S, Fong GH, Tian R, Sadoshima J. Broad Suppression of NADPH Oxidase Activity Exacerbates Ischemia/Reperfusion Injury Through Inadvertent Downregulation of Hypoxia-inducible Factor-1α and Upregulation of Peroxisome Proliferator–activated Receptor-α. Circ Res. 2013;112:1135–1149. doi: 10.1161/CIRCRESAHA.111.300171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evangelisti C, Ricci F, Tazzari P, Tabellini G, Battistelli M, Falcieri E, Chiarini F, Bortul R, Melchionda F, Pagliaro P, Pession A, McCubrey JA, Martelli AM. Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia. 2011;25:781–791. doi: 10.1038/leu.2011.20. [DOI] [PubMed] [Google Scholar]
- 19.Goncharova EA, Goncharov DA, Li H, Pimtong W, Lu S, Khavin I, Krymskaya VP. mTORC2 is Required for Proliferation and Survival of TSC2-Null Cells. Mol Cell Biol. 2011;31:2484–2498. doi: 10.1128/MCB.01061-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: A Homeostatic ATP Sensor. Science. 2001;294:1102–1105. doi: 10.1126/science.1063518. [DOI] [PubMed] [Google Scholar]
- 21.Ma W, Han W, Greer PA, Tuder RM, Toque HA, Wang KKW, Caldwell RW, Su Y. Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease. J Clin Invest. 2011;121:4548–4566. doi: 10.1172/JCI57734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soliman GA, Acosta-Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, Fingar DC. mTOR Ser-2481 Autophosphorylation Monitors mTORC-specific Catalytic Activity and Clarifies Rapamycin Mechanism of Action. J Biol Chem. 2010;285:7866–7879. doi: 10.1074/jbc.M109.096222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joha S, Nugues AL, Hetuin D, Berthon C, Dezitter X, Dauphin V, Mahon FX, Roche-Lestienne C, Preudhomme C, Quesnel B, Idziorek T. GILZ inhibits the mTORC2/AKT pathway in BCR-ABL+ cells. Oncogene. 2012;31:1419–1430. doi: 10.1038/onc.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Concannon CG, Tuffy LP, Weisová P, Bonner HP, Dávila D, Bonner C, Devocelle MC, Strasser A, Ward MW, Prehn JHM. AMP kinase–mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol. 2010;189:83–94. doi: 10.1083/jcb.200909166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, Dyck JRB, Michelakis ED. Fatty Acid Oxidation and Malonyl-CoA Decarboxylase in the Vascular Remodeling of Pulmonary Hypertension. Sci Transl Med. 2010:2, 44ra58. doi: 10.1126/scitranslmed.3001327. [DOI] [PubMed] [Google Scholar]
- 26.Kim D, Chung J. Akt: versatile mediator of cell survival and beyond. J Biochem Mol Biol. 2002;35:106–115. doi: 10.5483/bmbrep.2002.35.1.106. [DOI] [PubMed] [Google Scholar]
- 27.Marks AR. Sirolimus for the Prevention of In-Stent Restenosis in a Coronary Artery. New Engl J Med. 2003;349:1307–1309. doi: 10.1056/NEJMp038141. [DOI] [PubMed] [Google Scholar]
- 28.Reddy MK, Vasir JK, Sahoo SK, Jain TK, Yallapu MM, Labhasetwar V. Inhibition of Apoptosis Through Localized Delivery of Rapamycin-Loaded Nanoparticles Prevented Neointimal Hyperplasia and Reendothelialized Injured Artery. Circulation Cardiovasc Interv. 2008;1:209–216. doi: 10.1161/CIRCINTERVENTIONS.108.830018. [DOI] [PubMed] [Google Scholar]
- 29.McMurtry MS, Bonnet S, Michelakis ED, Bonnet S, Haromy A, Archer SL. Statin therapy, alone or with rapamycin, does not reverse monocrotaline pulmonary arterial hypertension: the rapamcyin-atorvastatin-simvastatin study. Am J Physiol Lung Cell Mol Physiol. 2007;293:L933–L940. doi: 10.1152/ajplung.00310.2006. [DOI] [PubMed] [Google Scholar]
- 30.Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, Saker M, Marcos E, Tissot CM, Dubois-Randé JL, Amsellem V, Adnot S. Rapamycin Reverses Pulmonary Artery Smooth Muscle Cell Proliferation in Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2013;48:568–577. doi: 10.1165/rcmb.2012-0429OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shirwany NA, Zou MH. AMPK in cardiovascular health and disease. Acta Pharmacol Sin. 2010;31:1075–1084. doi: 10.1038/aps.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ibe JCF, Zhou Q, Chen T, Tang H, Yuan JXJ, Raj JU, Zhou G. AMPK is Required for Pulmonary Artery Smooth Muscle Cell Survival and the Development of Hypoxic Pulmonary Hypertension. Am J Respir Cell Mol Biol. 2013;49:609–618. doi: 10.1165/rcmb.2012-0446OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-Activated Protein Kinase Induces a p53-Dependent Metabolic Checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 34.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 35.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt Activates the Mammalian Target of Rapamycin by Regulating Cellular ATP Level and AMPK Activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 36.Zhu S, Evans S, Yan B, Povsic TJ, Tapson V, Goldschmidt-Clermont PJ, Dong C. Transcriptional Regulation of Bim by FOXO3a and Akt Mediates Scleroderma Serum–Induced Apoptosis in Endothelial Progenitor Cells. Circulation. 2008;118:2156–2165. doi: 10.1161/CIRCULATIONAHA.108.787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberts KE, Fallon MB, Krowka MJ, Brown RS, Trotter JF, Peter I, Tighiouart H, Knowles JA, Rabinowitz D, Benza RL, Badesch DB, Taichman DB, Horn EM, Zacks S, Kaplowitz Nl, Kawut SM. Genetic Risk Factors for Portopulmonary Hypertension in Patients with Advanced Liver Disease. Am J Respir Crit Care Med. 2009;179:835–842. doi: 10.1164/rccm.200809-1472OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz A-C, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MAR, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HHHW, Weissmann N. Hypoxia-Dependent Regulation of Nonphagocytic NADPH Oxidase Subunit NOX4 in the Pulmonary Vasculature. Circ Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 39.Lu X, Murphy TC, Nanes MS, Hart CM. PPARγ regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-κB. Am J Physiol Lung Cell Mol Physiol. 2010;299:L559–L566. doi: 10.1152/ajplung.00090.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a Functionally Active gp91phox-Containing Neutrophil-Type NAD(P)H Oxidase in Smooth Muscle Cells From Human Resistance Arteries: Regulation by Angiotensin II. Circ Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 41.Wingler K, Wünsch S, Kreutz R, Rothermund L, Paul M, Schmidt HHHW. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Rad Biol Med. 2001;31:1456–1464. doi: 10.1016/s0891-5849(01)00727-4. [DOI] [PubMed] [Google Scholar]
- 42.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, Rabinovitch M. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han S, Zheng Y, Roman J. Rosiglitazone, an Agonist of PPARgamma, Inhibits Non-Small Cell Carcinoma Cell Proliferation In Part through Activation of Tumor Sclerosis Complex-2. PPAR Res. 2007:29632. doi: 10.1155/2007/29632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The Nox4 Inhibitor, GKT137831, Attenuates Hypoxia-Induced Pulmonary Vascular Cell Proliferation. Am J Respir Cell Mol Biol. 2012;47:718–726. doi: 10.1165/rcmb.2011-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C, Fruman DA. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, Tuder RM. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol. 2010;176:1130–1138. doi: 10.2353/ajpath.2010.090832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W, Petrimpol M, Molle KD, Hall MN, Battegay EJ, Humar R. Hypoxia-Induced Endothelial Proliferation Requires Both mTORC1 and mTORC2. Circ Res. 2007;100:79–87. doi: 10.1161/01.RES.0000253094.03023.3f. [DOI] [PubMed] [Google Scholar]
- 48.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-Site Inhibitors of mTOR Target Rapamycin-Resistant Outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e1000038. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, Cole DC, Ellingboe J, Ayral-Kaloustian S, Mansour TS, Gibbons JJ, Abraham RT, Nowak P, Zask A. Biochemical, Cellular, and In vivo Activity of Novel ATP-Competitive and Selective Inhibitors of the Mammalian Target of Rapamycin. Canc Res. 2009;69:6232–6240. doi: 10.1158/0008-5472.CAN-09-0299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.