

Abstract
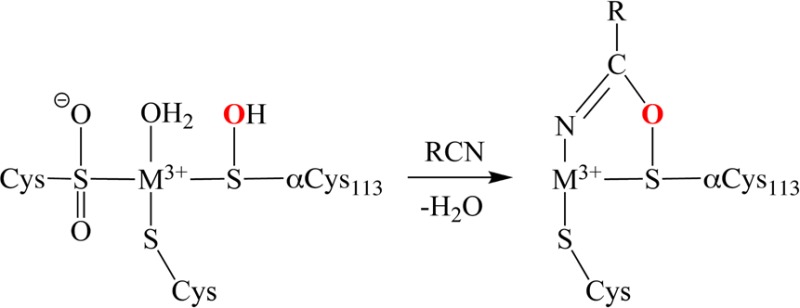
Nitrile hydratase (NHase) catalyzes the hydration of nitriles to their corresponding commercially valuable amides at ambient temperatures and physiological pH. Several reaction mechanisms have been proposed for NHase enzymes; however, the source of the nucleophile remains a mystery. Boronic acids have been shown to be potent inhibitors of numerous hydrolytic enzymes due to the open shell of boron, which allows it to expand from a trigonal planar (sp2) form to a tetrahedral form (sp3). Therefore, we examined the inhibition of the Co-type NHase from Pseudonocardia thermophila JCM 3095 (PtNHase) by boronic acids via kinetics and X-ray crystallography. Both 1-butaneboronic acid (BuBA) and phenylboronic acid (PBA) function as potent competitive inhibitors of PtNHase. X-ray crystal structures for BuBA and PBA complexed to PtNHase were solved and refined at 1.5, 1.6, and 1.2 Å resolution. The resulting PtNHase–boronic acid complexes represent a “snapshot” of reaction intermediates and implicate the cysteine-sulfenic acid ligand as the catalytic nucleophile, a heretofore unknown role for the αCys113–OH sulfenic acid ligand. Based on these data, a new mechanism of action for the hydration of nitriles by NHase is presented.
Nitrile hydratase (NHase, E.C. 4.2.1.84) is a metalloenzyme within the nitrile degradation pathway that catalyzes the hydration of nitriles to their corresponding amides under mild conditions (room temperature and physiological pH).1,2 NHases consist of two nonhomologous subunits, α and β, that form α2β2 heterotetramers.3−6 Several NHases have been structurally characterized and shown to contain either a low-spin nonheme Fe(III) ion (Fe-type) or a low-spin noncorrin Co(III) ion (Co-type) in their active site. The metal ion is coordinated by three cysteine sulfur atoms, two amide nitrogens, and a water molecule.7 Two of the active site cysteine residues are post-translationally modified to cysteine-sulfinic acid (Cys–SO2H) and cysteine-sulfenic acid (Cys–SOH) yielding an unusual metal coordination geometry, termed a “claw setting”.7 NHases have attracted substantial interest as biocatalysts in preparative organic chemistry and are used in several industrial applications such as the large scale production of acrylamide and nicotinamide.8,9 In addition, because of their exquisite reaction specificity, the nitrile-hydrolyzing potential of NHases is becoming increasingly recognized as a new type of “green” chemistry for the degradation of environmentally harmful nitriles such as bromoxynil, a nitrile-based pesticide.10
Several reaction mechanisms have been proposed for NHase enzymes; however, the precise catalytic pathway remains elusive. Based on single turnover stopped-flow data, the nitrile substrate nitrogen was shown to bind directly to the active site metal ion displacing the axial water molecule.11 Once bound, imidate is likely formed as the result of nucleophilic attack, which then isomerizes to the corresponding amide. However, the source of the nucleophile for this transformation is still under debate. To gain insight into the identity of the active site nucleophile, we examined the inhibition of the Co-type NHase from Pseudonocardia thermophila JCM 3095 (PtNHase) by boronic acids. The resulting X-ray crystal structures of PtNHase-boronic acid complexes are a “snapshot” of potential reaction intermediates and implicate the cysteine-sulfenic acid ligand as the catalytic nucleophile. The combination of these data with previously reported spectroscopic and X-ray crystallographic data have allowed a novel mechanism of action to be proposed for the metal-mediated nitrile hydration reaction catalyzed by NHase enzymes.
The ability of both 1-butaneboronic acid (BuBA) and phenylboronic acid (PBA) to inhibit PtNHase was examined by monitoring the hydration of acrylonitrile to acrylamide spectrophotometrically at 225 nm (ε = 2.9 mM–1 cm–1). In the absence of inhibitors, PtNHase catalyzed the hydration of acrylonitrile at pH 7.5 and 25 °C with a kcat value of 1310 ± 110 s1 and a Km value of 0.8 ± 0.1 mM. Inspection of the reaction time course for the hydration of acrylonitrile at pH 7.5 and 25 °C by PtNHase in the presence of BuBA or PBA indicates that both boronic acids function as potent competitive inhibitors of PtNHase. For BuBA, linear reaction progress curves were observed providing an inhibition constant (Ki) of 0.5 ± 0.1 μM. On the other hand, PBA displayed biphasic progress curves, which is characteristic of slow-binding inhibition (Figure S1).12
The slow-binding inhibition properties of PBA were analyzed based on the equilibrium shown in Scheme 1, where Ki (Ki = k1/k2) is the equilibrium inhibition constant for the formation of the initial complex, EI, and k3 and k4 are the forward and reverse rate constants for the equilibrium between the EI and the EI* complex.12 By measuring the reaction time course at different concentrations of PBA and substrate, the initial Ki was found to be 5.0 ± 0.9 μM. The overall Ki* of 0.04 ± 0.01 nM, where Ki* = Kik4/(k3 + k4), was determined by preincubating PtNHase with PBA for 1 h at pH 7.5 and 25 °C followed by reaction with various concentrations of acrylonitrile.
Scheme 1.
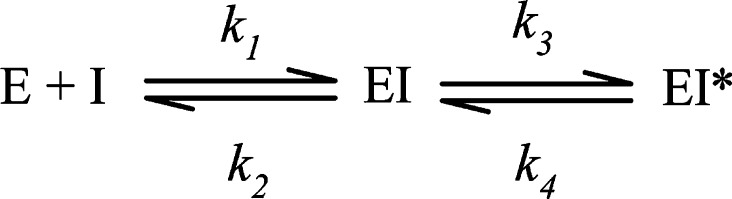
Since both BuBA and PBA function as competitive inhibitors of PtNHase, they must bind to the enzyme active site. In solution, BuBA was reported to be >99% in the boronic acid form (sp2 hybridized) at pH 8.0, with a reported pKa of 10.6.13 Similarly, the pKa of PBA is 8.9,14 so it too will be in the boronic acid form at pH 7.5. Therefore, both BuBA and PBA likely bind to PtNHase in their sp2 hybridized forms resulting in the displacement of the axial water molecule. This initial Co(III) binding step was hypothesized to be followed by attack of an active site nucleophile, such as the sulfenic acid ligand, on the electron deficient pz orbital of the boron atom, since it was recently suggested that boronic acids might inhibit NHases by interacting with the sulfenic acid ligand.15 Nucleophilic attack of the electron-deficient pz orbital of the boron atom is also consistent with previous studies of boronic acid inhibitors bound to hydrolytic enzymes, as hydrolytic reactions typically proceed through a transition state resulting from nucleophilic attack of the substrate.16,17
To confirm that BuBA binds directly to the low-spin Co(III) ion in the active site of PtNHase and to determine if BuBA has undergone nucleophilic attack, X-ray crystal structures of wild-type (WT) PtNHase and two PtNHase–BuBA complexes formed by either soaking or cocrystallization were solved and refined to 1.9, 1.5, and 1.6 Å resolution, respectively. Details of the data collection and refinement statistics are given in Table S1 of the Supporting Information (SI). The overall structure of WT PtNHase is identical to the previously reported structure (PDB code: 1IRE).18 The active site is the typical “claw setting” observed in all nitrile hydratases with an axial water molecule that forms hydrogen bonds with αSer112, the sulfinic acid ligand (αCys111–O2H), and the sulfenic acid ligand (αCys113–OH). The protonation states of the sulfenic and sulfinic acid ligands have previously been assigned based on sulfur K-edge XAS data, which indicated the presence of three types of Cys ligands in a WT Fe-type NHase.19 These XAS data combined with geometry-optimized DFT calculations suggested the presence of CysS–, CysSOH, and CysSO2– at pH 7.5.
Upon soaking a crystal of WT PtNHase in cryo-protectant containing 10 mM BuBA for 20 s followed by flash freezing in liquid nitrogen, electron density corresponding to the boronic acid inhibitor was present at the active site replacing the axial water molecule (Figure 1). This observation is consistent with the fact that BuBA is a competitive inhibitor of PtNHase. In the WT structure (not shown) the Co(III)–Owater bond distance is 2.3 Å compared to a Co(III)–O boronic acid oxygen bond distance of 2.2 Å. Interestingly, the O-atom of the sulfenic acid ligand (αCys113–OH) is covalently bound to the boron atom of BuBA (1.5 Å) (Figure 1). This covalent bond, which has not been observed previously, is the result of nucleophilic attack of the sulfenic acid O-atom on the empty pz orbital of the B-atom and the subsequent loss of a boronic acid O-atom. Even though the O-atom of αCys113-OH is covalently bound to boron, αCys113 remains ligated to the low-spin Co(III) ion with a bond distance of 2.2 Å identical to that observed in the WT enzyme. The resulting B-atom is essentially trigonal planar (sp2) with a dihedral angle of ∼170°.
Figure 1.
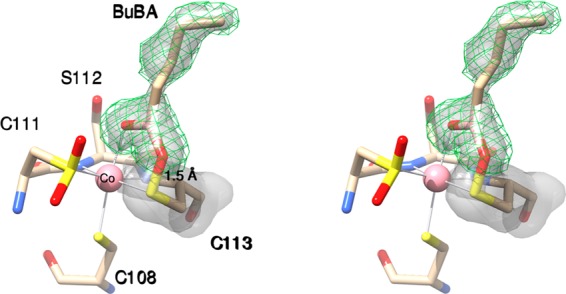
Stereoview of PtNHase bound by BuBA after soaking a crystal of WT PtNHase in cryo-protectant containing 10 mM BuBA for 20 s followed by flash freezing in liquid nitrogen. The 2fo – fc map is shown as a transparent gray surface at the 1.1 σ level around BuBA and αCys113. The simulated-annealing omit map (fo – fc) is shown around BuBA as a green mesh at 2.7 σ.
On the other hand, the PtNHase-BuBA structure obtained via cocrystallization of WT PtNHase and 10 mM BuBA reveals that the S–O boronic acid oxygen interaction is significantly diminished (Figure 2). BuBA binding displaces the axial water molecule resulting in a Co(III)–O bond distance of 2.2 Å; however, the second O-atom of BuBA is 2.9 Å away from the S-atom of Cys113. While this distance is still within the van der Waals radii of S and O, which is ∼3.3 Å, it is clear that the αCys113–OH interaction is considerably weakened compared to that observed in the PtNHase-BuBA structure obtained via soaking. This weak S–O interaction is likely due to the initial dissociation of boronic acid from the active site and not the initial binding step. If it were the initial binding step of a boronic acid, αCys113 would need to be in its fully reduced form which is not the case, as αCys113 is clearly oxidized to its sulfenic acid form in the WT PtNHase structure. Therefore, the observed S–O elongation is assigned to boronic acid dissociation. The αCys113sulfur remains bound to the Co(III) ion with a bond length of 2.3 Å. The B-atom of BuBA also remains nearly trigonal planner (sp2) with a dihedral angle of ∼160°.
Figure 2.
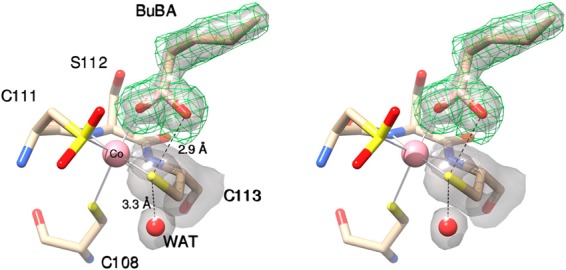
Stereoview of PtNHase bound by BuBA obtained via cocrystallization of WT PtNHase and 10 mM BuBA. The 2fo – fc map is shown as a transparent gray surface at the 1.1 σ level around BuBA and αCys113. The simulated-annealing omit map (fo – fc) is shown around BuBA as a green mesh at 2.8 σ.
These two structures represent a “snapshot” of two potential intermediate states in nitrile hydration by depicting nucleophilic attack by the sulfenic acid ligand and the initial stage of the product-release step. Product loss may occur as the result of a concomitant nucleophilic attack on the αCys113 ligand by a water molecule. This is consistent with the observation that a water molecule that is H-bound (2.9 Å) to the NH2 group of βArg157 is only 3.3 Å from the αCys113 ligand. This water molecule may represent the incoming O-atom required to reestablish the αCys113–OH ligand. Interestingly, no water molecule is observed within 4 Å of the B-atom in either BuBA structure (Figure 2), suggesting that a water molecule is not poised for nucleophilic attack on the B-atom facilitating boronic acid formation and product release.
Since PtNHase can hydrate both alkyl and aromatic nitriles,18 the X-ray crystal structure of the PtNHase–PBA complex also was obtained via cocrystallization of WT PtNHase and 10 mM PBA and refined to 1.2 Å resolution (Figures 3 and S2). Details of data collection and refinement statistics are given in Table S1 of the SI. Interestingly, electron density corresponding to the active site cobalt ion and the PBA suggests ∼80% occupancy. These data are consistent with inductively coupled atomic emission spectroscopy (ICP-AES), which typically indicates that only 0.8 to 0.9 cobalt ions are present per αβ dimer. Similar to the PtNHase-BuBA structure obtained via soaking, the structural model representing 80% occupancy contains a boronic acid O-atom that displaces the axial water molecule and binds directly to the active site Co(III) ion with a bond distance of 2.2 Å (Figures 3 and S2). Similar to the PtNHase-BuBA structure (Figure 1), the B-atom of PBA has undergone nucleophilic attack by the αCys113–OH O-atom forming a covalent bond with a B–O distance of 1.5 Å and a S–O bond distance of 1.8 Å. The S-atom of αCys113–OH remains a Co(III) ligand with a bond distance of 2.2 Å. The resulting B-atom is nearly trigonal planar with a dihedral angle of ∼160°, and the unbound O-atom of the original boronic acid moiety is lost. The structural model representing the remaining ∼20% of the observed density is similar to the previously reported apo-PtNHase (Figure S2).18 These data indicate that the PtNHase–PBA complex represents a covalently bound intermediate state for nitrile hydration, similar to the PtNHase–BuBA complexes, where the αCys113–OH ligand functions as the nucleophile.
Figure 3.
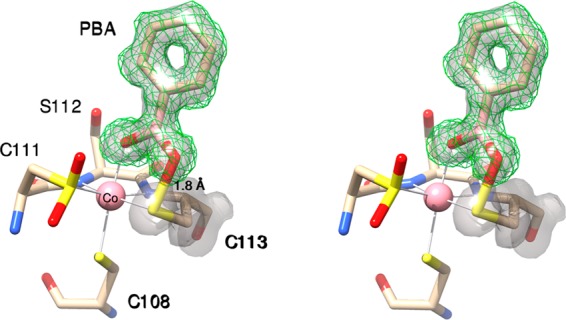
Stereoview of PtNHase bound by PBA at 1.2 Å resolution obtained via cocrystallization of WT PtNHase and 10 mM PBA. The 2fo – fc map of the structural model representing 80% occupancy is shown as a transparent gray surface at the 1.4 σ level around PBA and αCys113. The simulated-annealing omit map (fo – fc) is shown around PBA as a green mesh at 2.8 σ.
Based on the three structures reported herein, parallels can be drawn between boronic acid binding to PtNHase and a nitrile substrate that are consistent with the sulfenic acid ligand functioning as a nucleophile. Kinetic and X-ray crystallographic data indicate that proper oxidation of the αCys113 ligand to a cysteine-sulfenic acid is essential for NHase enzymes to be active catalysts suggesting that the S–OH bond is polarized and poised for nucleophilic attack.20−22 The catalytic function of the αCys113–OH is also consistent with the rate constants provided by single-turnover stopped-flow studies that indicate a fast pseudo-first-order step that involves substrate binding to the trivalent metal ion in the enzyme active site, while product release is the rate-limiting step consistent with a sulfenic acid covalent intermediate complex.11 Additional evidence for metal-ligated sulfenic acids that can function as nucleophiles can be gleaned from NHase model complex studies.23−25 A coordinatively saturated bis(sulfenato-S)Co(III) complex was shown to slowly hydrate acetonitrile to acetamide under acidic conditions while the bis(sulfinato)Co(III) complex was unable to hydrate nitriles. As neither of these compounds have an open coordination site to allow nitrile or water binding, these data implicated the Co(III) sulfenic acid ligand as the nucleophile.
In conclusion, it has long been assumed that a water molecule or a hydroxide ion in the NHase active site functions in the initial nucleophilic attack on an activated nitrile C-atom. However, in this work we present evidence that the αCys113–OH ligand can function as a nucleophile. The combination of these data with previously reported stopped-flow, kinetic, and X-ray crystallographic data allows a novel catalytic mechanism to be proposed for NHase enzymes with a heretofore unknown role for the αCys113–OH sulfenic acid ligand (Figure 4).11,26−28 Stopped-flow data indicate that the first step in catalysis involves the direct ligation of the nitrile to the active site low-spin, trivalent metal ion.11 Displacement of the metal-bound water molecule by a nitrile and coordination to the active site metal center activates the CN bond toward nucleophilic attack by the αCys113–OH sulfenic acid ligand. Once nucleophilic attack of the nitrile carbon occurs, two protons are transferred in the rate-limiting step for both Fe- and Co-type NHase enzymes.28,29 We propose that one proton transfer occurs between the αCys113–CH ligand and the nitrile N-atom, while the second transfer occurs between the water molecule that reforms αCys113–OH and the newly forming imidate N-atom, consistent with the observed normal isotope effect.28,29 That the αCys-OH ligand is protonated was suggested by S K-edge XAS and DFT studies.19 Once proton transfer occurs, the resulting covalently bound imidate can tautomerize to form an amide upon nucleophilic attack by a water molecule on αCys113 (pathway A). Alternatively, nucleophilic attack by water at the C-atom of the imidate intermediate could occur (pathway B), such that the O-atom of the amide product would be derived from H2O, not the sulfenic group. In this alternative, the Co(III) ion and sulfenic acid group work together to form and stabilize the imidate intermediate, which can then be hydrated with sulfenic acid functioning as the leaving group. Finally, the amide product can be displaced by a water molecule and thus provide the regenerated catalyst.
Figure 4.

Proposed catalytic mechanism for NHase.
Acknowledgments
The authors thank Dr. Miguel Ballicora for helpful discussions on enzyme kinetics. This work was supported by the National Science Foundation (CHE-1058357, RCH). GM/CA @ APS has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. DE-AC02-06CH11357.
Supporting Information Available
A detailed description of the materials, methods, kinetic analysis, and X-ray data collection and refinement, along with supplementary figures. This material is available free of charge via the Internet at http://pubs.acs.org. The reported coordinates and structure factors have been deposited in the PDB bank as Native PtNHase (PDB code: 4OB3), PtNHase-BuBA Co-crystal (PDB code: 4OB1), PtNHase-BuBA soaking (PDB code: 4OB2), and PtNHase-PBA Co-crystal (PDB code: 4OB0).
Author Contributions
⊥ S.M. and R.W. contributed equally.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Yamada H.; Kobayashi M. Biosci. Biotech. Biochem. 1996, 60, 1391. [DOI] [PubMed] [Google Scholar]
- Brady D.; Beeton A.; Zeevaart J.; Kgaje C.; van Rantwijk F.; Sheldon R. A. Appl. Microbiol. Biotechnol. 2004, 64, 76. [DOI] [PubMed] [Google Scholar]
- Huang W.; Jia J.; Cummings J.; Nelson M.; Schneider G.; Lindqvist Y. Structure 1997, 15, 691. [DOI] [PubMed] [Google Scholar]
- Nagashima S.; Nakasako M.; Dohmae N.; Tsujimura M.; Takio K.; Odaka M.; Yohda M.; Kamiya N.; Endo I. Nat. Struct. Biol. 1998, 5, 347. [DOI] [PubMed] [Google Scholar]
- Miyanaga A.; Fushinobu S.; Ito K.; Wakagi T. Biochem. Biophys. Res. Commun. 2001, 288, 1169. [DOI] [PubMed] [Google Scholar]
- Brodkin H. R.; Novak W. R. P.; Milne A. C.; D’Aquino J. A.; Karabacak N. M.; Goldberg I. G.; Agar J. N.; Payne M. S.; Petsko G. A.; Ondrechen M. J.; Ringe D. Biochemistry 2011, 50, 4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs J. A. Chem. Rev. 2004, 104, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrillo K. L.; Wu S.; Hann E. C.; Cooling F. B.; Ben-Bassat A.; Gavagan J. E.; Dicosimo R.; Payne M. S. Appl. Microbiol. Biotechnol. 2005, 67, 664. [DOI] [PubMed] [Google Scholar]
- Prasad S.; Bhalla C. Biotechnol. Adv. 2010, 28, 725. [DOI] [PubMed] [Google Scholar]
- Baxter J.; Cummings S. P. Antonie van Leeuwenhoek 2006, 90, 1. [DOI] [PubMed] [Google Scholar]
- Gumataotao N.; Kuhn M. L.; Hajnas N.; Holz R. C. J. Biol. Chem. 2013, 288, 15532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J. F.; Walsh C. T. In Advances in Enzymology and Related Areas of Molecular Biology; John Wiley & Sons, Inc.: 2006; p 201. [DOI] [PubMed] [Google Scholar]
- Baker J. O.; Wilkes S. H.; Bayliss M. E.; Prescott J. M. Biochemistry 1983, 22, 2098. [DOI] [PubMed] [Google Scholar]
- Springsteen G.; Wang B. Tetrahedron 2002, 58, 5291. [Google Scholar]
- Liu C. T.; Benkovic S. J. J. Am. Chem. Soc. 2013, 135, 14544. [DOI] [PubMed] [Google Scholar]
- DePaola C. C.; Bennett B.; Holz R. C.; Ringe D.; Petsko G. A. Biochemistry 1999, 38, 9048. [DOI] [PubMed] [Google Scholar]
- Smoum R.; Rubinstein A.; Dembitsky V. M.; Srebnik M. Chem. Rev. 2012, 112, 4156. [DOI] [PubMed] [Google Scholar]
- Miyanaga A.; Fushinobu S.; Ito K.; Shoun H.; Wakagi T. Eur. J. Biochem. 2004, 271, 429. [DOI] [PubMed] [Google Scholar]
- Dey A.; Chow M.; Taniguchi K.; Lugo-Mas P.; Davin S.; Maeda M.; Kovacs J. A.; Odaka M.; Hodgson K. O.; Hedman B.; Solomon E. I. J. Am. Chem. Soc. 2006, 128, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T.; Nojiri M.; Nakayama H.; Odaka M.; Yohda M.; Dohmae N.; Takio K.; Nagamune T.; Endo I. Protein Sci. 2000, 9, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura M.; Odaka M.; Nakayama H.; Dohmae N.; Koshino H.; Asami T.; Hoshino M.; Takio K.; Yoshida S.; Maeda M.; Endo I. J. Am. Chem. Soc. 2003, 125, 11532. [DOI] [PubMed] [Google Scholar]
- Arakawa T.; Kawano Y.; Katayama Y.; Nakayama H.; Dohmae N.; Yohda M.; Odaka M. J. Am. Chem. Soc. 2009, 131, 14838. [DOI] [PubMed] [Google Scholar]
- Heinrich L.; Mary-Verla A.; Li Y.; Vaissermann J.; Chottard J.-C. Eur. J. Inorg. Chem. 2001, 2203. [Google Scholar]
- Heinrich L.; Li Y.; Vaissermann J.; Chottard J.-C. Eur. J. Inorg. Chem. 2001, 1407. [Google Scholar]
- Yano T.; Wasada-Tsutsui Y.; Arii H.; Yamaguchi S.; Funahashi Y.; Ozawa T.; Masuda H. Inorg. Chem. 2007, 46, 10345. [DOI] [PubMed] [Google Scholar]
- Hashimoto K.; Suzuki H.; Taniguchi K.; Noguchi T.; Yohda M.; Odaka M. J. Biol. Chem. 2008, 105, 15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka Y.; Hashimoto K.; Ohtaki A.; Noguchi K.; Yohda M.; Odaka M. J. Biol. Inorg. Chem. 2010, 15, 655. [DOI] [PubMed] [Google Scholar]
- Mitra S.; Holz R. C. J. Biol. Chem. 2007, 282, 7397. [DOI] [PubMed] [Google Scholar]
- Rao S. N.; Holz R. C. Biochemistry 2008, 47, 12057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.