

Abstract
Diversely functionalized, fused aryl-alkyl ring systems hold a prominent position as well-established molecular frameworks for a variety of anti-cancer agents. The benzosuberene (6,7 fused, also referred to as dihydro-5H-benzo[7]annulene and benzocycloheptene) ring system has emerged as a valuable molecular core component for the development of inhibitors of tubulin assembly, which function as antiproliferative anti-cancer agents and, in certain cases, as vascular disrupting agents (VDAs). Both a phenolic-based analogue (known as KGP18, compound 39) and its corresponding amine-based congener (referred to as KGP156, compound 45), which demonstrate strong inhibition of tubulin assembly (low micromolar range) and potent cytotoxicity (picomolar range for KGP18 and nanomolar range for KGP156) are noteworthy examples of such benzosuberene-based compounds. In order to extend the structure-activity relationship (SAR) knowledge base related to benzosuberene anti-cancer agents, a series of eleven analogues (including KGP18) were prepared in which the methoxylation pattern on the pendant aryl ring as well as functional group incorporation on the fused aryl ring were varied. The synthetic approach to these compounds featured a sequential Wittig olefination, reduction, Eaton's reagent-mediated cyclization strategy to achieve the core benzosuberone intermediate, and represented a higher-yielding synthesis of KGP18 (which we prepared previously through a ring-expansion strategy). Incorporation of a fluorine or chlorine atom at the 1-position of the fused aryl ring or replacement of one of the methoxy groups with hydrogen (on the pendant aryl ring of KGP18) led to benzosuberene analogues that were both strongly inhibitory against tubulin assembly (IC50 approximately 1.0 M) and strongly cytotoxic against selected human cancer cell lines (for example, GI50 = 5.47 nM against NCI-H460 cells with fluorobenzosuberene analogue 37). A water-soluble phosphate prodrug salt of KGP18 (referred to as KGP265, compound 44) and a water-soluble serinamide salt (compound 48) of KGP156 were also synthesized and evaluated in this study.
Keywords: inhibitors of tubulin assembly, benzosuberene-based anti-cancer agents, vascular disrupting agents (VDAs), combretastatin analogues
1. Introduction
The discovery and development of small-molecule, anti-cancer agents that demonstrate pronounced cytotoxicity against human cancer cell lines remains an important goal in the search for new cancer treatment agents and related therapeutic strategies. An established approach involves the selective targeting of tumor vasculature and more specifically the tubulinmicrotubule protein system. Research efforts in this area have led to a class of therapeutics known as vascular targeting agents (VTAs)1,2 that is further sub-divided into angiogenic inhibiting agents (AIAs),3,4,5 which interfere with tumor neovascularization and vascular disrupting agents (VDAs),3,4,6-9 including both small-molecules and biologics, which selectively damage existing tumor vasculature. Tubulin-binding VDAs function through the inhibition of tubulin polymerization within endothelial cells lining tumor microvessels. A subsequent series of cell signaling events leads to morphological transformations (flat to round) of these endothelial cells and results in microvessel occlusion and vascular shutdown, which ultimately starves the tumor of necessary nutrients and oxygen.3,6,10,11 While still an active area of research inquiry, there is evidence indicating that activated endothelial cells (such as those lining the vasculature supplying tumors) are affected by VDAs to a greater extent than quiescent endothelial cells found in vasculature feeding normal tissue.12-14 A significant focal point centers on small-molecule VDAs that bind to the colchicine site15,16 on the -tubulin heterodimer. It is important to note that AIAs and VDAs function biologically through mechanistically distinct pathways.9,17
A progressive research agenda in our laboratory, focused on the design and synthesis of inhibitors of tubulin assembly, led to the establishment of functionalized benzosuberene analogues as promising compounds for further evaluation as anti-cancer agents.18-20 This exploration was guided, in part, by the molecular structures of colchicine and certain members of the combretastatin family of natural products along with our previously discovered dihydronaphthalene-based analogues18,21,22 (Fig. 1).
Figure 1.

Representative Small-Molecule Inhibitors of Tubulin Assembly That Bind to the Colchicine Site; Including: Benzosuberene Analogues (KGP18,18KGP15619), Dihydronaphthalene Analogues (KGP03,18,21-23KGP0522,23), and Combretastatin Analogues (CA4,24,25CA1,26 Ajinomoto Amine,27,28KGP06,10,29KGP0830).
Specifically, combretastatin A-4 (CA4)24,25 and combretastatin A-1 (CA1)26 are among the most potent colchicine site tubulin-binding agents and they were each further developed into their corresponding phosphate prodrug salts to improve aqueous solubility.31-33 Efforts to mimic the combretastatin molecular-framework and optimize a wide-variety of biological parameters, resulted in a cadre of structural modifications.34,35 A very limited sub-set of these molecules that were inspired by the combretastatins include benzophenone,36,37 dihydronaphthalene,18,21-23,38,39 indole,40-43 and benzosuberene18,19,20,44 analogues in which a single sp2 hybridized carbon atom bridges the two aromatic rings and maintains the pseudo cis-orientation45 that is important for enhanced biological activity. Two such benzosuberene-based compounds, referred to as KGP18 (phenol-based)18,20,46 and KGP156 (amine-based),19 have emerged as potential pre-clinical candidates (Fig. 1). In addition to their robust in vitro cytotoxicity against human cancer cell lines (picomolar for KGP18 and nanomolar for KGP156), preliminary studies have shown that these benzosuberene analogues function as vascular disrupting agents (VDAs).19,20,47 Our original synthetic route to these benzosuberene analogues included a ring expansion, reduction, selective oxidation sequence that while reliable, was somewhat limiting due to low reaction yields.18 A revised synthetic methodology that relies on an efficient ring-closing cyclization step was utilized in our synthesis19 of the amine analogue, KGP156, and a recent publication by Maderna and co-workers44 describes an efficient ring closing metathesis (RCM) step to assemble the benzosuberene molecular core followed by a Suzuki coupling reaction. In order to advance the known structure activity relationship (SAR) data, a collection of eleven benzosuberene analogues, selected primarily to explore functional group modification at the C-1 position, were prepared by chemical synthesis and evaluated for their cytotoxicity against selected human cancer cell lines, and for their ability to inhibit tubulin assembly.
2. Results and Discussions
2.1 Chemistry
A series of eleven functionalized benzosuberene-based analogues were prepared by chemical synthesis. The synthetic strategy relied on an intramolecular Friedel-Crafts annulation with Eaton's reagent48,49 to install the benzosuberone ring system (Scheme 1). The prerequisite carboxylic acid derivatives were prepared through a sequential Wittig olefination followed by catalytic hydrogenation sequence, which, overall, is nearly identical to the synthetic methodology described by Negoro et al.50 This synthetic strategy to prepare benzosuberone derivatives has proved to be highly proficient within our laboratory.19,46,51 Protecting group strategies were included when necessary. Additional modifications of benzosuberones 16 and 18 were achieved through selective demethylation with ionic liquid [TMAH][Al2Cl7]52 resulting in phenolic benzosuberones 21-22 which were subsequently converted to their corresponding silyl ethers 23-24 with TBSCl (Scheme 2). Confirmation of the regioselective demethylation to form benzosuberone intermediate 22 was provided by X-ray crystallographic analysis of the final compound KGP18 (compound 39) that resulted from further synthetic manipulation of intermediate 22 (see Supplementary Data). Similarly, the regioselective demethylation to form intermediate 21 was confirmed by HSQC and HMBC analysis, along with X-ray crystallography, of final compound 38 (see Supplementary Data). The functionalized pendant aryl ring was incorporated, in each case, through the addition of an appropriately functionalized aryllithium intermediate (prepared in situ from the corresponding aryl bromide) to the requisite benzosuberone derivative to generate the corresponding tertiary alcohol that underwent elimination to form the benzosuberene core structure (Scheme 3). This overall synthetic sequence proved to be quite robust and is complementary to other known synthetic routes toward benzosuberene ring systems18,44,53,54 (including Friedel-Crafts cyclization55). An alternative synthetic strategy for the preparation of KGP18 (compound 39) that utilized an intramolecular acid chloride mediated cyclization strategy was also successful (see Supplementary Data for details).
Scheme 1.

Synthetic route to benzosuberone intermediates 16-20.
Scheme 2.
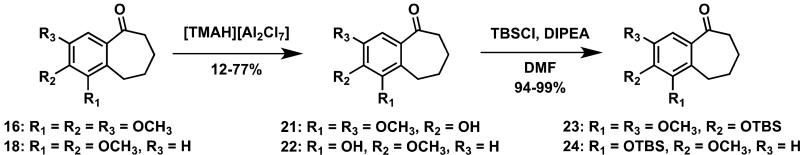
Synthetic modifications affording benzosuberone intermediates 23-24.
Scheme 3.

Synthetic route to benzosuberene analogues 34-41.
The preparation of benzosuberene analogues 42-44 required further synthetic manipulation. Analogue 42 was achieved through desilylation with TBAF and analogue 43 was obtained by removal of the tosyl protecting group upon treatment with NaOH (Scheme 4). In order to facilitate a variety of planned in vivo studies, the hydrophobic benzosuberene analogue 39 (KGP18) was converted to its corresponding water-soluble, disodium phosphate prodrug salt 44 (KGP265) through phosphorylation with POCl3 followed by treatment with NaOH. This phosphate prodrug strategy has proved to be highly effective for both combretastatin A-4P (CA4P)33 and combretastatin A-1P (CA1P).31
Scheme 4.

Synthetic route to benzosuberene analogues 42-44.
Previously described hydrophobic aniline analogue 45 (KGP156)19 was converted to its corresponding water-soluble, serinamide salt 48 through initial amide bond formation between benzosuberene 45 and acetyl-Fmoc protected serinamide to form serinamide 46 and subsequent treatment with NaOH to yield serinamide benzosuberene analogue 47 (Scheme 5). Treatment of serinamide 47 with HCl led to the corresponding hydrochloride serinamide salt 48. This chemistry is reminiscent of the synthetic strategy employed by Ohsumi and co-workers56 for the preparation of water-soluble amino acid (and related) prodrug salts of 3-amino-combretastatin (serinamide analogue referred to as AVE8062)57,58 along with our later studies of 2-amino-combretastatin10,29 and 2,3-diamino-combretastatin30 serinamide salts.
Scheme 5.

Synthetic route to benzosuberene analogues 47-48.
2.2 Biological Evaluation
The batch of KGP18 (prepared using the synthetic strategy described herein) along with ten new analogues (structures depicted in Fig. 2) incorporating functional group modifications were evaluated (Table 1) for their ability to inhibit tubulin assembly (cell free assay) and for their cytotoxicity against three human cancer cell lines (SK-OV-3, ovarian; NCI-H460, lung; and DU-145, prostate). As anticipated, the potent cytotoxicity (GI50 < 100 pM) observed for KGP18 (compound 39, bearing a 1-hydroxy group) in this study mirrors (within error-limits inherent to the assay) our previously reported data for KGP18 (prepared by a separate synthetic methodology).18 Replacement of the 1-hydroxy moiety with a fluorine atom resulted in a benzosuberene analogue 37 that was both strongly inhibitory against tubulin assembly (IC50 = 0.89 M) and potently cytotoxic (GI50 = 5.47 nM against NCI-H460 cells, for example). The chlorine atom congener 36 was equally active as an inhibitor of tubulin assembly and only slightly less cytotoxic. Fluorine atom substitution has been a productive strategy in certain structurally related combretastatin analogues.59-60 An analogue of KGP18 that replaced the pendant trimethoxyaryl ring with a dimethoxyaryl ring (compound 42) was also active against tubulin assembly and as a cytotoxic agent (GI50 = 33.4 nM against SK-OV-3 cells). Other structural modifications (compounds 35, 38, 41, and 43) around the pendant aryl ring resulted in benzosuberene analogues that were inactive (IC50 > 40 M) as inhibitors of tubulin assembly and decidedly less cytotoxic (against these three cell lines) thus underscoring the limited structural variation that is tolerated in these molecules. Data for both combretastatin A-4 (CA4)61 and our previously reported19 6-aminobenzosuberene analogue (KGP156, compound 45) are included in Table 1 for comparative reference. The water-soluble phosphate prodrug salt of KGP18, referred to as KGP265 (compound 44) was inactive (IC50 > 40 M) against tubulin as anticipated in this cell-free assay (which is devoid of enzymes necessary to cleave the inactive prodrug to its active parent compound (KGP18)), however it was active in terms of cytotoxicity (GI50 = 9.51 nM against DU-145 cells, for example). It is well-established that human cancer cell lines have one or more phosphatase enzyme at their cell surface,62 thus it was expected that the prodrug would be very effectively cleaved to yield the parent compound under these conditions. A water-soluble serinamide prodrug salt 48 of KGP156 (compound 45) proved to be inactive against tubulin assembly (as anticipated in this type of cell free assay) and also surprisingly inactive in terms of cytotoxicity. It is possible that either the level of requisite aminopeptidase enzymes secreted from these cells is not sufficient to cleave the prodrug construct, or that this particular benzosuberene prodrug (compound 48) itself is not a good substrate for the enzyme, since other (structurally non-related) serinamide prodrug salts do show cytotoxicity in this type of assay63 and some have been evaluated in the presence of exogenous peptidase enzymes to demonstrate enzyme-mediated hydrolysis with release of the parent amino-drug.56,64 Further study is necessary in this regard. It is well documented throughout the literature27,61 that the most active small-molecule inhibitors of tubulin assembly are typically in the low micromolar range (in terms of IC50) while the same compounds demonstrate in vitro cytotoxicity with GI50 values in the nanomolar to sub-nanomolar range. This activity difference (cell-free tubulin assay versus cell-based in vitro cytotoxicity assay) can be attributed to several possible factors including stoichiometry (inhibitor to tubulin heterodimer ratio) differences between the cell-free assay and what takes place in cells, the cell-based release (during microtubule disassembly) of molecular components (factors) that increase cytotoxicity through signal transduction pathways, and the practical lower limit inherent to this type of inhibition of tubulin assembly assay.65
Figure 2.

Molecular Structures of Target Benzosuberene Analogues
Table 1.
Inhibition of tubulin polymerization and cytotoxicity against human cancer cell lines SK-OV-3, NCI-H460, and DU-145.
| Compound | Inhibition of tubulin polymerization IC50 (M) | GI50 (M) SRB assaya | ||
|---|---|---|---|---|
| SK-OV-3 | NCI-H460 | DU-145 | ||
| CA4 | 1.0b | 0.00455 | 0.00223c | 0.00327c |
| CA4P | >40b | 0.00119 | 0.00194c | 0.00323c |
| 35 | >40 | >89.5 | >136.5 | 42.5 |
| 36 | 0.93 | 0.0444 | 0.0713 | 0.152 |
| 37 | 0.89 | 0.00751 | 0.00547 | 0.0189 |
| 38 | >40 | 7.13 | 18.0 | 15.7 |
| 39 | 1.4d | 0.0000543e | 0.0000418e | 0.0000249e |
| 41 | >40 | 19.1 | 32.5 | 22.0 |
| 42 | 0.74 | 0.0334 | 0.262 | 0.109 |
| 43 | >40 | 18.1 | 25.9 | 38.4 |
| 44 | >40 | 0.00772 | 0.00796 | 0.00951 |
| 45 | 1.2f | 0.000102g | 0.00280g | 0.00223g |
| 47 | ndh | 36.8 | 39.0 | 70.5 |
| 48 | ndh | 35.0 | 53.8 | 69.4 |
3. Conclusion
In summary, we have prepared eleven new benzosuberene-based analogues through an extension of our previously reported synthetic methodology19,20 directed towards these ring-fused systems. The most active compounds (in terms of inhibition of tubulin assembly and cytotoxicity against selected human cancer cell lines) feature fluorine atom (compound 37) or chlorine atom (compound 36) incorporation at position-1 of the fused aryl ring along with a dimethoxyaryl ring modification (compound 42) of the trimethoxyaryl ring bearing-phenolic analogue KGP18 (compound 39). Thus the known SAR for benzosuberene derivatives of this type has been extended. The two water-soluble prodrug salts (44 and 48) should prove useful for future in vivo studies.
4. Experimental Section
4.1 Chemistry
4.1.1 Materials and instrumentation
Methylene chloride (CH2Cl2), acetonitrile, methanol (MeOH), ethanol (EtOH), dimethylformamide (DMF), and tetrahydrofuran (THF) were used in their anhydrous form as obtained from the chemical suppliers. Reactions were performed under an inert atmosphere using nitrogen gas unless specified. Thin-layer chromatography (TLC) plates (pre-coated glass plates with silica gel 60 F254, 0.25 mm thickness) were used to monitor reactions. Reactions carried out under microwave irradiation were performed with a Biotage Initiator Microwave Synthesizer. Purification of intermediates and products was carried out with a Biotage Isolera 1 or 4 flash purification system using silica gel (200-400 mesh, 60 Å) or RP-18 prepacked columns. Intermediates and products synthesized were characterized on the basis of their 1H NMR (500 MHz), 13C NMR (125 MHz), 31P NMR (202 MHz), and 19F NMR (470 MHz) spectroscopic data. All the chemical shifts are expressed in ppm (δ), coupling constants (J) are presented in Hz, and peak patterns are reported as singlet (s), broad singlet (bs), doublet (d), triplet (t), quartet (q), pentet (p), septet (sept), and multiplet (m). Mass spectrometry was carried out under positive ESI (electrospray ionization) using a Thermo scientific LTQ Orbitrap Discovery instrument. Purity of the final compounds was further analyzed at 25 °C using a Agilent 1200 HPLC system with a diode-array detector ( = 190-400 nm), a Zorbax XDB-C18 HPLC column (4.6 mm × 150 mm, 5 μm), and a Zorbax reliance cartridge guard-column; eluents, solvent A: H2O, solvent B: acetonitrile; gradient, 90%A / 10%B to 0%A / 100%B over 0 to 40 min; flow rate 1.0 mL/min; injection volume 20 μL; monitored at wavelengths of 254, 280 and 300 nm. Column volume is represented by CV.
Experimental Procedures for Final Compound 38
4.1.2. (Z)/(E)- 5-(2′,3′,4′-Trimethoxyphenyl)pent-4-enoic acid (6).20
To a well-stirred solution of (3-carboxypropyl)triphenylphosphonium bromide (24.06 g, 56.05 mmol) in THF (400 mL) was added K-OtBu (12.30 g, 109.6 mmol). The reaction mixture was then cooled to 0 °C and stirred for 15 min. A solution of aldehyde 1 (9.84 g, 50.2 mmol) in THF (25 mL) was added dropwise and the reaction mixture was stirred and allowed to reach room temperature. The reaction mixture was diluted with H2O (50 mL) and extracted with Et2O (2 × 200 mL). The aqueous phase was acidified with 2 M HCl until the product precipitated making the solution cloudy and then becoming clear again. The acidified aqueous phase was extracted with EtOAc (3 × 100 mL). The combined organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure. Purification by flash chromatography using a prepacked 160 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 20%A / 80%B (1 CV), 20%A / 80%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] afforded the mixture of E/Z-isomers 6 (9.48 g, 35.6 mmol, 64%) as a pale yellow liquid. 1H NMR (Mixture of E and Z) (CDCl3, 500 MHz): δ 7.11 (1H, d, J = 8.7 Hz, H-6′), 6.94 (1H, d, J = 8.6 Hz, H-6′), 6.65 (1H, d, J = 8.7 Hz, H-5′), 6.65 (1H, d, J = 8.6 Hz, H-5′), 6.64 (1H, d, J = 16.1 Hz, H-5), 6.52 (1H, dt, J = 11.5, 2 Hz, H-5), 6.11 (1H, m, H-4), 5.63 (1H, dt, J = 11.5, 7 Hz, H-4), 3.88-3.83 (3 × 3H, s, OCH3-2′, -3′, -4′), 3.88-3.83 (3 × 3H, s, OCH3-2′, -3′, -4′), 2.59 (2H, m, CH2-2/3), 2.56-2.54 (4H, m, CH2-2, -3), 2.47 (2H, m, CH2-3/2). 13C NMR (CDCl33, 125 MHz) δ 178.9, 178.7, 152.9, 152.8, 151.7, 151.1, 142.3, 142.2, 129.5, 127.5, 125.4, 125.2, 124.4, 124.2, 123.9, 120.7, 107.7, 106.9, 61.1, 61.0, 60.95, 60.89, 56.0, 55.9, 34.0, 33.9, 28.3, 23.9.
4.1.3. 5-(2′,3′,4′-Trimethoxyphenyl)pentanoic acid (11).20,66
To a solution of 6 (9.15 g, 34.4 mmol) in MeOH (200 mL) was added 10% Pd-C. The flask was evacuated under vacuum and H2 gas was introduced via balloon. The reaction was stirred for 24 h and checked for completion by filtering a small amount of the reaction mixture through Celite®, evaporating the solvent, and recording the 1H NMR. On completion, the reaction mixture was filtered through Celite®, concentrated under reduced pressure, and the resulting pale yellow liquid was subjected to flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording 11 (8.76 g, 32.6 mmol, 95%) as a pale yellow liquid. 1H NMR (CDCl3, 500 MHz): δ 6.81 (1H, d, J = 8.5 Hz, H-6′), 6.60 (1H, d, J = 8.5 Hz, H-5'), 3.87 (3H, s, OCH3-2′), 3.86 (3H, s, OCH3-3′), 3.84 (6H, s, OCH3-4′), 2.57 (2H, t, J = 7.5 Hz, H-5), 2.39 (2H, t, J = 7.3 Hz, H-2), 1.68 (2H, m, H-3), 1.61 (2H, m, H-4). 13C NMR (CDCl3, 125 MHz): δ 179.9 (C=O, C-1), 152.0 (C, C-4′), 151.8 (C, C-2′), 142.3 (C, C-3′), 128.0 (C, C-1′), 123.7 (CH, C-6′), 107.2 (CH, C-5′), 60.9 (CH3, OCH3-2′), 60.7 (CH3, OCH3-3′), 56.0 (CH3, OCH3-4′), 33.9 (CH2, C-2), 30.2 (CH2, C-4), 29.3 (CH2, C-5), 24.5 (CH2, C-3).
4.1.4. 1,2,3-Trimethoxy-benzocycloheptan-5-one (16).18,20,66
Pentanoic acid 11 (2.68 g, 10 mmol) was dissolved in Eaton's reagent [40.2 mL, P2O5 (7.7 wt%) in methanesulfonic acid] and the reaction mixture was stirred for 12 h. The reaction mixture was poured over ice and the ice was allowed to melt. The aqueous phase was extracted with CH2Cl2 (2 × 100 mL) and the combined organic extract was washed with saturated NaHCO3 (2 × 200 mL). The organic extract was dried over Na2SO4, concentrated under reduced pressure, subjected to flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording 16 (2.34 g, 9.34 mmol, 93%) as a pale yellow liquid. 1H NMR (CDCl3, 500 MHz): δ 7.13 (1H, s, H-4), 3.93 (3H, s, OCH3-2), 3.88 (3H, s, OCH3-3), 3.84 (3H, s, OCH3-1), 2.94 (2H, dd, J = 6.9, 5.2 Hz, H-9), 2.72 (2H, m, H-6), 1.83 (2H, m, H-8), 1.81 (2H, m, H-7). 13C NMR (CDCl3, 125 MHz): δ 205.0 (C=O, C-5), 151.6 (C, C-3), 151.0 (C, C-1), 145.9 (C, C-2), 134.4 (C, C-10/11), 128.9 (C, C-10/11), 107.5 (CH, C-4), 61.4 (CH3, OCH3-1), 60.8 (CH3, OCH3-2), 56.0 (CH3, OCH3-3), 40.8 (CH2, C-6), 25.0 (CH2, C-8), 23.0 (CH2, C-9), 20.9 (CH2, C-7).
4.1.5. [TMAH][Al2Cl7].52
To a suspension of AlCl3 (26.71 g, 101.4 mmol) in 200 mL CH2Cl2 cooled to 0 °C, trimethylammonium chloride [TMAH] (9.55 g, 50.7 mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred for 2 h. The transparent yellow solution of ionic liquid was used as such for the deprotection of methyl ethers of benzosuberones.
4.1.6. 2-Hydroxy-1,3-dimethoxy-benzocycloheptan-5-one (21).20
To a solution of 16 (5.30 g, 21.2 mmol) in CH2Cl2 (50 mL) cooled to 0 °C, [TMAH][Al2Cl7] (36.00 mL, 23.32 mmol, 1.93 M in CH2Cl2) was added dropwise. The reaction was monitored by TLC and upon completion, ice cold water was added to the reaction. The aqueous layer was extracted with CH2Cl2 (2 × 100 mL). The organic extract was washed with brine, dried over MgSO4, filtered, concentrated, and subjected to flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm]; to afford phenol 21 (0.58 g, 2.45 mmol, 12%). NMR characterization took place after the TBS protection (see compound 23).
4.1.7. 2-[(tert-Butyldimethylsilyl)oxy]-1,3-dimethoxy-benzocycloheptan-5-one (23).20
To a solution of phenol 21 (0.58 g, 2.45 mmol) and DIPEA (2.00 mL, 11.5 mmol) in DMF (5 mL) at 0 °C was added TBSCl (0.82 g, 5.44 mmol) in portions. The reaction mixture was stirred for 6 h, diluted with H2O (5 mL), and extracted with Et2O (2 × 20 mL). The organic extract was washed with brine, dried over MgSO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 3%A / 97%B (1 CV), 3%A / 97%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] to afford ketone 23 (0.82 g, 2.34 mmol, 94%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 7.03 (1H, s, H-4), 3.72 (3H, s, OCH3-1/3), 3.65 (3H, s, OCH3-1/3), 2.85 (2H, m, H-9), 2.61 (2H, m, H-6), 1.71 (4H, m, H-7, -8), 0.93 (9H, (CH3)3), , 0.07 (6H, Si(CH3)2. 13C NMR (CDCl3, 125 MHz): δ 204.8 (C, C-9), 149.9 (C, C-3), 149.3 (C, C-1), 142.6 (C, C-2), 131.9 (C, C-10/11), 129.4 (C, C-10/11), 40.8 (CH, C-8), 107.2 (CH, C-4), 60.8 (CH3, OCH3-2/3), 55.3 (CH3, OCH3-2/3), 25.7 (CH3, (CH3)3), 25.1 (CH2, C-8), , 23.1 (CH2, C-9), 21.0 (CH2, C-7), 18.7 (C, (C(CH3)3), -4.6 (CH3, Si(CH3)2)
4.1.8. 2-[(tert-Butyldimethylsilyl)oxy]-1,3-dimethoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocycloheptan-5-ol (30).20
To a solution of 3,4,5-trimethoxyphenyl bromide (1.04 g, 4.21 mmol) in THF (50 mL) at −78 °C, n-BuLi (1.70 mL, 2.5 M) was added and the reaction mixture was stirred for 30 min. Ketone 23 (0.73 g, 2.08 mmol) in 5 mL THF was added using an addition funnel over a period of 15 min. The reaction mixture was stirred for 12 h and was allowed to warm to room temperature. The reaction mixture was diluted with H2O (25 mL) and extracted with EtOAc (2 × 25 mL). The organic extract was washed with brine, dried over MgSO4, filtered, concentrated under reduced pressure, subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 15%A / 85%B (1 CV), 15%A / 85%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] resulting in alcohol 30 (0.80 g, 1.54 mmol, 74%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 6.92 (1H, s), 6.28 (2H, s), 3.70-3.48 (15H, m), 2.97 (1H, m), 2.48 (1H, m), 2.41 (1H, m), 1.98 (2H, m), 1.77 (2H, m), 1.58 (1H, m), 0.87 (9H, s), 0.00 (6H, s).
4.1.9. 1,3-Dimethoxy-2-hydroxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocyclohept-5-ene (38).20
A solution of 30 (0.77 g, 10.6 mmol) in AcOH (20 mL) and H2O (20 mL) was heated to reflux at 110 °C for 24 h. The reaction mixture was cooled and the reaction mixture was concentrated under reduced pressure and subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] affording benzosuberene 38 (0.49 g, 5.48 mmol, 52%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 6.50 (2H, s, H-2′, H-6′), 6.38 (1H, s, H-4), 6.37 (1H, t, J = 12.0 Hz, H-6), 5.62 (1H, s, OH-2), 3.92 (3H, s, OCH3-1), 3.88 (3H, s, OCH3-4′), 3.81 (6H, s, OCH3-3′, -5′), 3.75 (3H, s, OCH3-1), 2.66 (2H, t, J = 7.0 Hz, H-9), 2.15 (2H, p, J = 7.1 Hz, H-8), 1.97 (2H, q, J = 7.2 Hz, H-7). 13C NMR (CDCl3, 125 MHz): δ 153.1 (C, C-3′, C-5′), 145.4 (C, C-3), 144.6 (C, C-1), 143.0 (C, C-5), 138.2 (C, C-1′), 137.7 (C, C-2), 137.5 (C, C-4′), 131.7 (C, C-10), 128.4 (C, C-11), 127.8 (CH, C-4), 108.3 (CH, C-6), 105.3 (CH, C-2′, C-6′), 61.5 (CH3, OCH3-1), 61.1 (CH3, OCH3-4′), 56.5 (CH3, OCH3-3′, -5′), 56.3 (CH3, OCH3-3), 35.3 (CH2, C-8), 25.8 (CH2, C-7), 23.9 (CH2, C-9). Analysis: Calculated for C23H26O6, C 68.38, H 6.78, O 24.84. Found: C 68.22, H 6.85. HRMS: m/z: observed 409.1629 [M + Na]+, calculated for C22H26O6Na+, 409.1622. HPLC: 14.68 min.
Experimental Procedures for Final Compounds 39 and 44
4.1.10. (Z)/(E)- 5-(2′,3′-Dimethoxyphenyl)pent-4-enoic acid (8).20
To a well-stirred solution of (3-carboxypropyl)triphenylphosphonium bromide (21.65 g, 50.43 mmol) in THF (500 mL) was added K-OtBu (11.3 g, 101 mmol). The reaction mixture was then cooled to 0 °C and stirred for 15 min. A solution of aldehyde 3 (8.42 g, 50.7 mmol) in THF (60 mL) was added dropwise and the reaction mixture was allowed to reach room temperature. The reaction mixture was diluted with H2O (50 mL) and extracted with Et2O (2 × 200 mL). The aqueous phase was acidified with 2 M HCl until the product precipitated making the solution cloudy and then becoming clear again. This acidified aqueous phase was extracted with EtOAc (3 × 100 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure and subjected to flash chromatography using a prepacked 160 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 20%A / 80%B (1 CV), 20%A / 80%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] affording a mixture of E/Z-isomers 8 (6.98 g, 29.5 mmol, 58%) as a pale yellow oil. 1H NMR (E/Z-isomers) (CDCl3, 500 MHz): δ 7.03-6.99 (3H, m), 6.85-6.74 (3H, m), 6.76 (1H, d, J = 15.9 Hz), 6.59 (1H, dt, J = 11.5, 1.5 Hz), 6.22 (1H, dt, J = 15.9, 6.0 Hz), 5.70 (1H, dt, J = 11.5, 7.5 Hz), 3.86 (3H, s), 3.85 (3H, s), 3.78 (3H, s), 3.76 (3H, s), 2.60-2.53 (6H, m), 2.46 (2H, m). 13C NMR (E/Z-isomers) (CDCl3, 125 MHz): δ 179.14, 179.12, 153.0, 152.8, 146.9, 146.3, 131.5, 131.4, 130.7, 129.5, 125.7, 125.4, 124.0, 123.6, 121.9, 118.0, 111.3, 111.0, 60.8, 60.6, 55.8, 55.8, 34.0, 33.8, 28.3, 24.0.
4.1.11. 5-(2′,3′-Dimethoxyphenyl)pentanoic acid (13).20,67
To a solution of 8 (6.78 g, 28.7 mmol) in MeOH (100 mL) was added 10% Pd-C (0.70 g). The flask was evacuated under vacuum and H2 gas was introduced via balloon and the reaction mixture was stirred for 24 h. The reaction was monitored by filtering a small amount of the reaction mixture through Celite®, evaporating the solvent, and recording the 1H NMR spectrum. Upon completion, the reaction mixture was filtered through Celite®, concentrated under reduced pressure and subjected to flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording compound 13 (6.71 g, 28.2 mmol, 98%) as a pale yellow oil. 1H NMR (CDCl3, 500 MHz): δ 6.97 (1H, t, J = 8.6 Hz, H-5′), 6.76 (2H, d, J = 8.6 Hz, H-4′, -6′), 3.85 (3H, s, OCH3-3′), 3.81 (3H, s, OCH3-2′), 2.65 (2H, t, J = 7.0 Hz, H-2), 2.38 (2H t, J = 7.0, H-5), 1.67 (4H, m, H-3, -4). 13C NMR (CDCl3, 125 MHz): δ 180.0 (C, C-1), 152.7 (C, C-3′), 147.1 (C, C-2′), 135.9 (C, C-1′), 123.8 (CH, C-5′), 121.9 (CH, C-6′), 110.2 (CH, C-4′), 60.6 (CH3, OCH3-2′), 55.7 (CH3, OCH3-3′), 34.0 (CH2, C-5), 30.1 (CH2, C-4), 29.3 (CH2, C-2), 24.5 (CH2, C-3). HRMS: m/z: observed 261.1100 [M+Na]+, calculated for C13H18O4Na+, 261.1097. HPLC: 10.67 min.
4.1.12. 1,2-Dimethoxy-benzocycloheptan-5-one (18).20,68
Pentanoic acid 13 (6.76 g, 28.4 mmol) was dissolved in Eaton's reagent [75 mL, P2O5 (7.7 wt%) in methanesulfonic acid] and stirred for 12 h. The reaction mixture was poured over ice and the ice was allowed to melt. The aqueous phase was extracted with CH2Cl2 (2 × 100 mL) and the organic extract was washed with saturated NaHCO3 (2 × 200 mL). The organic extract was dried over Na2SO4, concentrated under reduced pressure and subjected to flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording compound 18 (5.39 g, 24.5 mmol, 86%) as a pale yellow oil. 1H NMR (CDCl3, 500 MHz): δ 7.54 (1H, d, J = 8.5 Hz, H-4), 6.84 (1H, d, J = 8.5 Hz, H-3), 3.91 (3H, s, OCH3-1), 3.80 (3H, s, OCH3-2), 3.01 (2H, m, H-9), 2.70 (2H, m, H-6), 1.85 (2H, m, H-7), 1.81 (2H, m, H-8). 13C NMR (CDCl3, 125 MHz): δ 205.0 (C=O, C-5), 156.1 (C, C-1), 145.9 (C, C-2), 135.7 (CH, C-10/11), 132.8 (C, C-10/11), 125.5 (CH, C-4), 109.7 (CH, C-3), 61.1 (CH3, OCH3-1/2), 55.8 (CH3, OCH3-1/2), 40.6 (CH2, C-6), 24.9 (CH2, C-7), 23.3 (CH2, C-9), 20.9 (CH2, C-8). Analysis: Calculated for C13H16O3: C 70.89, H 7.32, O 21.79. Found: C 70.94, H 7.26. HPLC: 10.55 min.
4.1.13. 1-Hydroxy-2-methoxy-benzocycloheptan-5-one (22).20,37,69
To a solution of methyl aryl ether 18 (2.22 g, 10.1 mmol) in CH2Cl2 (5 mL) was added [TMAH][Al2Cl7] (13.00 mL, 1.93 M in CH2Cl2) and the reaction mixture was subjected to microwave irradiation at 80 °C for 1h. After the reaction was complete, water was added. The reaction mixture was stirred vigorously for 2 min and the organic layer was extracted with CH2Cl2 (2 × 25 mL). The organic extract was washed with brine, dried over MgSO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording phenol 22 (1.59 g, 7.70 mmol, 77%). 1H NMR (CDCl3, 500 MHz): δ 7.34 (1H, d, J = 8.6 Hz, H-4), 6.79 (1H, d, J = 8.6 Hz, H-3), 5.79 (1H, s, OH), 3.94 (3H, s, OCH3-2), 3.02 (2H, m, H-9), 2.71 (2H, t, J = 12.0 Hz, H-6), 1.85 (2H, m, H-8), 1.80 (2H, m, H-7). 13C NMR (CDCl3, 125 MHz): δ 205.0 (C, C-5), 149.2 (C, C-2), 142.4 (C, C-1), 133.3 (C, C-10/11), 127.7 (C, C-10/11), 120.8 (CH, C-4), 107.9 (CH, C-3), 56.1 (CH3, OCH3-2), 40.8 (CH2, C-6), 24.5 (CH2, C-8), 23.1 (CH2, C-9), 21.3 (CH2, C-7). Analysis: Calculated for C12H14O3: C 69.88, H 6.84. Found: C 69.93, H 6.86. HPLC: 7.08 min.
4.1.14. 1-[(tert-Butyldimethylsilyl)oxy]-2-methoxy-benzocycloheptan-5-one (24).20
To a solution of phenol 22 (6.36 g, 30.8 mmol) and DIPEA (5.75 g, 44.5 mmol) in DMF (25 mL) at 0 °C was added TBSCl (7.01 g, 46.5 mmol) in portions. The reaction mixture was stirred for 6 h and diluted with H2O (50 mL). The reaction mixture was extracted with Et2O (2 × 100 mL) and the organic extract was washed with brine, dried over MgSO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 0%A / 100%B (1 CV), 0%A / 100%B → 30%A / 70%B (10 CV), 30%A / 70%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording aldehyde 24 (9.80 g, 30.6 mmol, 99%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 7.38 (1H, d, J = 8.6 Hz, H-4), 6.78 (1H, d, J = 8.6 Hz, H-3), 3.84 (3H, s, OCH3-2), 3.01 (2H, dd, J = 7.5, 5 Hz, H-9), 2.70 (2H, t, J = 12.0 Hz, H-6), 1.82 (2H, m, H-7), 1.80 (2H, m, H-8), 1.02 (9H, (CH3)3), 0.19 (6H, Si(CH3)2). 13C NMR (CDCl3, 125 MHz): δ 205.3 (C, C-5), 153.2 (C, C-2), 141.8 (C, C-1), 133.10 (C, C-10/11), 133.08 (C, C-10/11), 122.3 (CH, C-4), 108.8 (CH, C-3), 54.8 (CH3, OCH3-2), 40.7 (CH2, C-6), 26.1 (CH3, (CH3)3), 24.7 (CH2,C-7), 24.0 (CH2,C-9), 21.2 (CH2,C-8), 18.9 (C, (C(CH3)3), -3.90 (CH3, Si(CH3)2). Analysis: Calculated for C18H28O3Si: C 67.46, H 8.81. Found: C 67.70, H 8.82. HPLC: 20.96 min.
4.1.15. 1-[(tert-Butyldimethylsilyl)oxy]-2-methoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocycloheptan-5-ol (31).20
To a solution 3,4,5-trimethoxyphenyl bromide (16.8 g, 68.0 mmol) in THF (400 mL) at -78 °C was added n-BuLi (27.2 mL, 2.5 M in hexanes) and the reaction mixture was stirred for 30 min. Benzosuberone 24 (9.80 g, 30.6 mmol) in 25 mL THF was added using an addition funnel over a period of 15 min. The reaction mixture was allowed to warm to room temperature over 12 h. Upon completion, the reaction mixture was diluted with H2O (50 mL) and extracted with EtOAc (2 × 100 mL). The organic extract was washed with brine, dried over MgSO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 15%A / 85%B (10 CV), 15%A / 85%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording alcohol 31 (11.6 g, 17.4 mmol, 57%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.16 (1H, d, J = 8.7 Hz), 6.70 (1H, d, J = 8.7 Hz), 6.50 (2H, s), 3.84 (3H, s), 3.80 (3H, s), 3.75 (6H, s), 3.37 – 3.23 (1H, m), 2.62 – 2.49 (1H, m), 2.30 – 2.22 (1H, m), 2.18 – 2.05 (1H, m), 1.99 – 1.85 (1H, m), 1.83 – 1.65 (2H, m), 1.47 – 1.34 (1H, m), 0.99 (9H, s), 0.17 (3H, s), 0.15 (3H, s). 13C NMR (CDCl3, 125 MHz): δ 152.4, 148.6, 145.2, 141.5, 136.5, 135.1, 130.0, 121.3, 109.4, 104.0, 75.5, 60.8, 56.1, 54.7, 41.0, 26.2, 24.8, 19.2, 19.0, -3.6, -3.8.
4.1.16. 1-Hydroxy-2-methoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocyclohept-5-ene (39).18,20,44
A solution of 31 (11.6 g, 10.6 mmol) in AcOH (150 mL) and H2O (100 mL) was heated to reflux at 110 °C for 12 h. The reaction mixture was cooled to room temperature, concentrated under reduced pressure and subjected to flash chromatography using a prepacked 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B (1 CV), 5%A / 95%B → 15%A / 85%B (10 CV), 15%A / 85%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording benzosuberene 39 (6.20 g, 17.4 mmol, 57%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 6.71 (1H, d, J = 8.6 Hz, H-3), 6.57 (1H, d, J = 8.6 Hz, H-4), 6.50 (2H, s, H-2′, H-6′), 6.33 (1H, t, J = 7.0 Hz, H-6), 5.74 (1H, s, OH), 3.91 (3H, s, OCH3-2), 3.86 (3H, s, OCH3-4), 3.80 (6H, s, OCH3-3′, -5′), 2.76 (2H, t, J = 7.0 Hz, H-9), 2.14 (2H, p, J = 7.0 Hz, H-8), 1.96 (2H, q, J = 7.0 Hz, H-7). 13C NMR (CDCl3, 125 MHz): δ 153.0 (C, C-3′, C-5′), 145.2 (C, C-2), 142.9 (C, C-1), 142.5 (C, C-5), 138.6 (C, C-1′), 137.4 (C, C-4′), 134.4 (C, C-10/11), 127.9 (C, C-10/11), 127.3 (CH, C-6), 121.0 (CH, C-4), 107.8 (CH, C-3), 105.4 (CH, C-2′, C-6′), 61.1 (CH3, OCH3-4′), 56.3 (CH3, OCH3-3′, -5′), 56.1 (CH3, OCH3-2), 33.7 (CH2, C-8), 25.8 (CH2, C-7), 23.7 (CH2, C-9). Analysis: Calculated for C21H24O5: C 70.77, H 6.79, O 22.45. Found: C 71.05, H 6.77. HRMS: m/z: observed 379.1565 [M+Na]+, calculated for C21H24O5Na+, 379.1516. HPLC: 15.59 min.
4.1.17. Disodium 2-Methoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocyclohept-5-ene-1-phosphate (44).20
To a solution of 39 (0.32 g, 0.83 mmol) in CH2Cl2 was added POCl3 (0.3 mL, 3.3 mmol) and pyridine (0.25 mL, 3.01 mmol) and the reaction mixture was stirred for 8 h. NaOH (5 mL, 2M) was added dropwise to the reaction mixture and the reaction was stirred for 5 min. The reaction mixture was extracted with CH2Cl2 (2 × 25 mL) and concentrated under reduced pressure. NaOH (5 mL, 2 M) was added to the viscous liquid obtained and the solution was stirred at 60 °C for 15 min. The aqueous phase was concentrated under reduced pressure and subjected to flash chromatography using a prepacked 25 g RP-18 silica column [solvent A: water; solvent B: CH3CN; gradient: 100%A / 0%B (1 CV), 100%A / 0%B → 60%A / 40%B (10 CV), 0%A / 100%B (3 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] affording 44 (0.245 g, 0.478 mmol, 58%) as white solid. 1H NMR (D2O, 500 MHz) δ 6.85 (1H, d, J = 8.5 Hz), 6.70 (3H, d, J = 8.5 Hz), 6.66 (2H, s), 6.46 (1H, t, J = 7.3 Hz), 3.84 (3H, s), 3.80 (6H, s), 3.77 (3H, s), 2.85 (2H, t, J = 10 Hz), 2.16 (2H, m), 1.92 (2H, q, J = 7.2 Hz). 13C NMR (D2O, 125 MHz): δ 152.1 (C, C-3′, C-5′), 151.0 (C, C-2), 141.8 (C, C-5), 140.5 (C, C-1), 139.4 (C, C-1′), 136.5 (C, C-10/11), 135.9 (C, C-4′), 133.4 (C, C-10/11), 128.7 (CH, C-6), 124.1 (CH, C-4), 109.4 (CH, C-3), 105.5 (CH, C-2′, C-6′), 60.9 (CH3, OCH3-4′), 55.9 (CH3, OCH3-3′, -5′), 55.5 (CH3, OCH3-2), 33.3 (CH2, C-8), 25.1 (CH2, C-7), 24.6 (CH2, C-9). 31P NMR (D2O, 202 MHz): δ 2.95. HRMS: m/z: observed 481.0999 [M+H]+, calculated for C21H24O8Na2P+, 481.0999. HPLC: 3.66 min.
Experimental Procedures for Final Compound 43
4.1.18. (Z)/(E)-5-(3′,4′-Dimethoxy-2′-(tosyloxy)phenyl)pent-4-enoic acid (7).20
To a well-stirred solution of (3-carboxypropyl)triphenylphosphonium bromide (3.82 g, 8.90 mmol) in THF (200 mL) at −50 °C was added n-BuLi (5.4 mL, 2.5 M in hexanes). The reaction mixture was allowed to warm to room temperature and stirred for 15 min and then cooled to −78 °C. Aldehyde 2 (2.01 g, 5.97 mmol) dissolved in THF (15 mL) was added dropwise and the reaction mixture was allowed to reach room temperature. H2O (50 mL) was added and the aqueous phase was extracted with EtOAc (3 × 200 mL). The organic extract was washed with brine, dried with MgSO4, concentrated under reduced pressure, and subjected to flash chromatography [silica gel, 40% EtOAc, 60% Hexanes] to obtain a mixture of E/Z-isomers 7 (1.03 g, 2.53 mmol, 42%) as an off-white solid. 1H NMR (Mixture of E and Z) (CDCl3, 500 MHz): δ 7.89 (2H, d, J = 8.2 Hz, H-2″, -6″), 7.87 (2H, d, J = 8.2 Hz, H-2″, -6″), 7.35 (2H, d, J = 8.2 Hz, H-3″, -5″), 7.32 (2H, d, J = 8.2 Hz, H-3″, -5″), 7.16 (1H, d, J = 8.8 Hz, H-5′/6′), 6.96 (1H, d, J = 8.6 Hz, H-5′/6′), 6.81 (1H, d, J = 8.6 Hz, H-5′/6′), 6.79 (1H, d, J = 8.8 Hz, H-5′/6′), 6.35 (1H, d, J = 16.0 Hz, H-5), 6.35 (1H, d, J = 11.0 Hz, H-5), 6.04 (1H, dt, J = 15.8, 6.3 Hz, H-4), 5.55 (1H, dt, J = 11.5, 6.9 Hz, H-4), 3.88-3.83 (2 × 3H, s, OCH3-3′, -4′), 3.88-3.83 (2 × 3H, s, OCH3-3′, -4′), 2.59 (2H, m, H-2/3), 2.56-2.54 (4H, m, H-2, -3), 2.47 (2H, m, H-2/3), 2.46 (6H, s, CH3-4′′). 13C NMR δ 178.1, 178.0, 152.74, 152.72, 144.9, 144.8, 142.5, 141.6, 141.0, 134.6, 134.58, 131.1, 129.5, 129.4, 128.9, 128.34, 128.30, 125.3, 125.0, 124.7, 124.4, 124.2, 120.3, 111.1, 110.46, 110.42, 60.71, 60.70, 56.2, 56.1, 33.6, 33.3, 27.9, 23.8, 21.7, 21.68. HRMS: m/z: observed 429.0977 [M+Na]+, calculated for C20H22O7NaS+, 429.0978. HPLC: 13.53 min.
4.1.19. 5-(3′,4′-Dimethoxy-2′-(tosyloxy)phenyl)pentanoic acid (12).20
To a solution of pentanoic acid 7 (1.25 g, 20.2 mmol) in MeOH (40 mL) and EtOH (15 mL) was added 10% Pd-C (400 mg). The flask was evacuated and H2 gas was introduced via balloons. The reaction mixture was stirred for 12 h and was checked for completion by filtering a small amount of the reaction mixture through Celite®, concentrating under reduced pressure, and recording the 1H NMR spectrum. Upon completion, the reaction mixture was filtered through Celite® and concentrated under reduced pressure to obtain 12 (0.94 g, 2.3 mmol, 75%) as an off-white solid. 1H NMR (CDCl3, 500 MHz): δ 7.93 (2H, d, J = 8.2 Hz, H-2″, -6″), 7.35 (2H, d, J = 8.2 Hz, H-3″, -5″), 6.89 (1H, d, J = 8.6 Hz, H-6′), 6.77 (1H, d, J = 8.6 Hz, H-5′), 3.82 (3H, s, OCH3-4′), 3.51 (3H, s, OCH3-3′), 2.58 (2H, m, H-5), 2.46 (3H, s, CH3-4″), 2.34 (2H, m, H-2), 1.61 (4H, m, H-3,-4). 13C NMR (CDCl3, 125 MHz): δ 179.0 (C, C-1), 151.8 (C, C-4′), 144.7 (C, C-4″), 142.3 (C, C-2′), 142.1 (C, C-3′), 134.9 (C, C-1″), 129.5 (CH, C-3″,-5″), 129.0 (C, C-1′), 128.1 (C, C-2″,-6″), 123.9 (CH, C-6′), 110.8 (CH, C-5′), 60.5 (CH3, OCH3-3′), 56.1 (CH3, OCH3-4′), 33.7 (CH2, C-2), 29.6 (CH2, C-5), 29.5 (CH2, C-4), 24.4 (CH2, C-3), 21.7 (CH3, CH3-4″).
4.1.20. 1-Tosyloxy-2,3-dimethoxy-benzocycloheptan-5-one (17).20
Pentanoic acid 12 (0.90 g, 2.2 mmol) was dissolved in Eaton's reagent [14 mL, P2O5 (7.7 wt%) in methanesulfonic acid] and the solution was stirred for 12 h. The reaction mixture was poured over ice and the ice was allowed to melt. The aqueous phase was extracted with CH2Cl2 (3 × 100 mL) and NaHCO3 powder was added in small amounts until neutralized. The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 25%A / 75%B (1 CV), 25%A / 75%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] affording 17, (0.70 g, 1.8 mmol, 81%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.93 (2H, d, J = 8.0 Hz, H-2′, -6′), 7.37 (2H, d, J = 8.0 Hz, H-3′, -5′), 7.29 (1H, s, H-4), 3.87 (3H, s, OCH3-2), 3.58 (3H, s, OCH3-3), 2.95 (2H, dd, J = 6.9, 4.9 Hz, H-9), 2.73 (3H, m, H-6), 2.48 (3H, s, CH3-4″), 1.84 (2H, m, H-8), 1.81 (2H, m, H-7). 13C NMR (CDCl3, 125 MHz) δ 204.0 (C, C-5), 151.3 (C, C-3), 145.4 (C, C-2), 145.0 (C, C-4′), 141.2 (C, C-1), 134.32 (C, C-10/11), 134.28 (CH, C-1′), 129.9 (C, C-10/11), 129.5 (CH, C-3′,-5′), 128.2 (CH, C-2′,-6′), 110.9 (CH, C-4), 60.5 (CH3, OCH3-3), 56.0 (CH3, OCH3-2), 40.7 (CH2, C-6), 24.7 (CH2, C-8), 24.5 (CH2, C-9), 21.7 (CH3, CH3-4′), 20.8 (CH2, C-7).
4.1.21. 1-Tosyloxy-2,3-dimethoxy-5-(3′,4′,5′-trimethoxyphenyl-benzocycloheptan-5-ol(26).20
To a solution of 3,4,5-triemethoxyphenyl bromide (0.85 g, 3.4 mmol) in THF (100 mL) at −78 °C was added n-BuLi (1.4 mL, 2.5 M in hexanes) and the reaction mixture was stirred for 30 min. Benzosuberone 17 (0.67 g, 1.7 mmol) in THF (15 mL) was added using an addition funnel over a period of 15 min. The reaction mixture was allowed to reach room temperature over 12 h. Upon completion, the reaction mixture was extracted with Et2O (150 mL) and EtOAc (15 mL). The organic extract was washed with brine, dried over MgSO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc, solvent B: hexanes; gradient: 25%A / 75%B (1 CV), 25%A / 75%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] afforded alcohol 26 (0.61 g, 1.1 mmol, 64%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 7.91 (2H, d, J = 8.3 Hz, H-2″, -6″), 7.37 (1H, s, H-4), 7.35 (2H, d, J = 8.3 Hz, H-3″, -5″), 6.46 (2H, s, H-2′, H-6′), 3.844 (3H, s, OCH3-4′), 3.840 (3H, s, OCH3-3), 3.78 (6H, s, OCH3-3′, -5′), 3.54 (3H, s, OCH3-2), 3.12 (1H, m, H-9), 2.62 (1H, m, H-6), 2.46 (3H, s, CH3-4″), 2.23 (1H, m, H-9), 2.13 (1H, m, H-6), 1.88 (1H, s, H-7/8), 1.72 (3H, m, H-7/8). 13C NMR (CDCl3, 125 MHz): δ 153.2 (C, C-3′, C-5′), 150.5 (C, C-3), 144.7 (C, C-4″), 141.7 (C, C-1), 141.4 (C, C-10/11), 140.7 (C, C-2), 139.9 (C, C-1′), 137.5 (CH, C-4′), 134.7 (C, C-1″), 129.4 (C, C-3″, C-5″), 128.4 (C, C-10/11), 128.1 (C, C-2″, C-6″), 110.2 (CH, C-4), 104.2 (CH, C-2′, C-6′), 80.2 (C, C-5), 60.8 (CH3, OCH3-4′), 60.5 (CH3, OCH3-2), 56.2 (CH3, OCH3-3′, -5′), 56.0 (CH3, OCH3-3), 41.1 (CH2, C-6), 26.7 (CH2, C-7/8), 26.6 (CH2, C-9), 26.3 (CH2, C-7/8), 21.7 (CH3, CH3-4″).
4.1.22. 1-Tosyloxy-2,3-dimethoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocyclohept-5-ene (34).20
Alcohol 26 (0.54 g, 0.97 mmol) was dissolved in AcOH (20 mL) and H2O (30 mL) and was heated to reflux at 180 °C for 12 h. The reaction mixture was cooled, concentrated under reduced pressure, subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 15%A / 85%B (1 CV), 15%A / 85%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] affording benzosuberene 34 (0.41 g, 0.75 mmol, 78%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 7.99 (2H, d, J = 8.3 Hz, H-2″, -6″), 7.37 (2H, d, J = 8.3 Hz, H-3″, -5″), 6.54 (1H, s, H-4), 6.48 (2H, s, H-2′, H-6′), 6.44 (1H, t, J = 7.4 Hz, H-6), 3.87 (3H, s, OCH3-4′), 3.82 (6H, s, OCH3-3′, -5′), 3.69 (3H, s, OCH3-3), 3.54 (3H, s, OCH3-2), 2.71 (2H, t, J = 6.5 Hz, H-9), 2.48 (3H, s, CH3-4″), 2.21 (2H, p, J = 7.0 Hz, H-8), 1.99 (2H, q, J = 7.1 Hz, H-7). 13C NMR (CDCl3, 125 MHz): δ 153.0 (C, C-3′, C-5′), 151.0 (C, C-3), 144.7 (C, C-4″), 141.9 (C, C-5), 141.6 (C, C-1), 140.8 (C, C-2), 137.6 (C, C-1′), 137.5 (C, C-4′), 136.1 (C, C-10/11), 134.8 (C, C-1″), 129.6 (C, C-10/11), 129.5 (CH, C-3″, C-5″), 129.4 (CH, C-6), 128.2 (CH, C-2″, C-6″), 112.0 (CH, C-4), 105.2 (CH, C-2′, C-6′), 60.94 (CH3, OCH3-4′), 60.93 (CH3, OCH3-2), 60.5 (CH3, OCH3-3′, -5′), 56.2 (CH3, OCH3-3) 34.6 (CH2, C-8), 25.5 (CH2, C-7), 25.1 (CH2, C-9), 21.7 (CH3, CH3-4″).
4.1.23. 1-Hydroxy-2,3-dimethoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocyclohept-5-ene (43).20
A solution of sulfonate ester 34 (0.250 g, 0.462 mmol) dissolved in NaOH (1 mL, 2 M) and methanol (4 mL) in a 5 mL microwave safe sealed vial was subjected to microwaved irradiation at 100 °C for 1h. Upon completion, the reaction mixture was neutralized (1 mL, 2 M HCl), concentrated under reduced pressure, and subjected to flash chromatography using a pre-packed 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 0%A / 100%B (1 CV), 0%A / 100% B → 40%A / 60%B (10 CV), 40%A / 60%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] affording phenol 43 (0.15 g, 0.388 mmol, 84%) as an off-white solid. 1H NMR (CDCl3, 500 MHz): δ 6.50 (2H, s, H-2′, H-6′), 6.39 (1H, t, J = 7.4 Hz, H-6), 6.18 (1H, s, H-4), 5.94 (1H, s, OH-1), 3.95 (3H, s, OCH3-2), 3.86 (3H, s, OCH3-4′), 3.81 (6H, s, OCH3-3′, -5′), 3.70 (3H, s, OCH3-3), 2.67 (2H, t, J = 6.9 Hz, H-9), 2.13 (2H, p, J = 6.9 Hz, H-8), 1.96 (2H, q, J = 7.1 Hz, H-7). 13C NMR (CDCl3, 125 MHz): δ 153.0 (C, C-3′, C-5′), 149.8 (C, C-3), 146.3 (C, C-1), 142.8 (C, C-5), 138.1(C, C-1′), 137.5 (C, C-4′), 136.3 (C, C-10/11), 134.3 (C, C-2), 128.5 (CH, C-6), 121.4 (C, C-10/11), 105.3 (CH, C-2′, C-6′), 105.1 (CH, C-4), 61.08 (CH3, OCH3-2), 61.06 (CH3, OCH3-4′), 56.3 (CH3, OCH3-3′, -5′), 56.1 (CH3, OCH3-3), 34.3 (CH2, C-8), 25.8 (CH2, C-7), 23.4 (CH2, C-9). HRMS: m/z: observed 387.1807 [M+H]+, calculated for C22H27O +6, 387.1802. HPLC: 15.16 min.
Experimental Procedures for Final Compound 37
4.1.24. (E)/(Z) 5-(2′-Fluoro-3′-methoxyphenyl)pent-4-enoic acid (10).20
To a well stirred solution of (3-carboxypropyl)triphenylphosphonium bromide (17.2 g, 40.1 mmol) in THF (250mL) was added K-OtBu (8.96 g, 79.9 mmol). The reaction mixture was then cooled to 0 °C and stirred for 15 min. A solution of 2-fluoro-3-methoxybenzaldehyde (3.08 g, 20.0 mmol) in THF (25mL) was added dropwise and the reaction mixture was allowed to warm to room temperature. The reaction mixture was extracted with Et2O (2 × 250 mL) and the aqueous phase was acidified with 2 M HCl until the product precipitated making the solution cloud and then becoming clear again. The acidified aqueous phase was extracted with EtOAc (3 × 100 mL). The combined organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, affording a mixture of the E/Z isomers 10 (4.14 g, 18.5 mmol, crude yield 92%) as a colorless liquid. The crude product was used in the next step without purification.
4.1.25. 5-(1′-Fluoro-2′-methoxyphenyl)pentanoic acid (15).20
To a solution of pentanoic acid 10 (1.25 g, 20.2 mmol) in EtOH (15 mL) was added 10% Pd-C (400 mg). The flask was evacuated and H2 gas was introduced via balloon. The reaction mixture was stirred for 12 h and checked for completion by filtering a small amount of the reaction mixture through Celite®, concentrating under reduced pressure and recording the 1H NMR spectrum. Upon completion, the reaction mixture was filtered through Celite® and concentrated under reduced pressure to obtain 15 (0.94 g, 2.3 mmol, 75%), as an off-white solid. 1H NMR (CDCl3, 500 MHz): δ 6.99 (1H, t, J = 8.2 Hz, H-4′), 6.82 (1H, t, J = 8.2 Hz, H-3′), 6.76 (1H, t, J = 8.6 Hz, H-5′), 3.88 (3H, s, OCH3-2′), 2.68 (2H, t, J = 6 Hz, H-5), 2.39 (2H, t, J = 7 Hz, H-2), 1.69 (4H, m, H-3,-4).13C NMR (CDCl3,125 MHz): δ 179.8 (C, C-1), 150.8 (CF, d, J = 243.7 Hz, C-1′), 147.7 (C, d, J = 11.2 Hz, C-2′), 129.7 (CH, d, J = 13.5 Hz, C-6′), 123.5 (CH, d, J = 4.6 Hz, C-5′/3′), 121.9 ( CH, d, J = 4.0 Hz, C-5′/3′), 110.9 (C, d, J = 1.7 Hz, C-4′), 56.2 (CH3, OCH3-2′), 33.8 (CH2, C-2), 29.4 (CH C-3/4), 28.5 (CH2, C-5), 24.2 (CH2, C-4/3). 19F NMR (CDCl3, 470 MHz): δ -141.9 (m). Analysis: Calculated for C12H15FO3, C 63.70, H 6.68. Found: C 63.77, H 6.70.
4.1.26. 1-Fluoro-2-methoxy-benzocycloheptan-5-one (20).20
Pentanoic acid 15 (0.90 g, 2.2 mmol) was dissolved in Eaton's reagent [14 mL, P2O5 (7.7 wt%) in methanesulfonic acid] and the solution was stirred for 12 h. The reaction mixture was poured over ice and the ice was allowed to melt. The aqueous phase was extracted with CH2Cl2 (3 × 100 mL) and NaHCO3 powder was added in small amounts until neutralized. The organic extract washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 25%A / 75%B (1 CV), 25%A / 75%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] affording 20 (0.70 g, 1.8 mmol, 81%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 7.57 (1H, dd, J = 8.7, 1.7 Hz, H-4), 6.88 (1H, t, J = 8.3, Hz, H-3), 3.93 (3H, s, OCH3-2), 3.00 (2H, m, H-9), 2.72 (2H, m, H-6), 1.87 (2H, m, H-8), 1.82 (2H, m, H-7). 13C NMR (125 MHz, CDCl3) δ 203.6 (C, d, J = 2.4 Hz, C-5), 150.9 (C, d, J = 12.4 Hz, C-2), 149.2 (CF, d, J = 243.3 Hz, C-1), 132.4 (CH, d, J = 1.3 Hz, C-4), 129.6 (C, d, J = 13.8 Hz, C-10/11), 125.0 (C, d, J = 4.2 Hz, C-10/11), 110.2 (CH, d, J = 2.1 Hz, C-3), 56.2 (CH3, OCH3-2) , 40.6 (CH2, C-6) , 24.4 (CH2, C-8) , 22.4 (CH2, d, J = 5.8 Hz, C-9), 20.9 (CH2, C-7) . 19F NMR (CDCl3, 470 MHz): δ -140.8 (m). Analysis: Calculated for C12H13FO2, C 69.22, H 6.29. Found: C 69.00, H 6.30. HRMS: m/z: observed 209.0974 [M+H]+, calculated for C12H14O2F+, 209.0972. HPLC: 12.57 min.
4.1.27. 1-Fluoro-2-methoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocycloheptan-5-ol (29).20
To a solution of 3,4,5-trimethoxyphenyl bromide (0.85 g, 3.4 mmol) in THF (100 mL) at -78 °C was added n-BuLi (1.4 mL, 2.5 M in hexanes) and the reaction mixture was stirred for 30 min. Benzosuberone 20 (0.67 g, 1.7 mmol) in THF (15 mL) was added using an addition funnel over a period of 15 min. The reaction mixture was allowed to reach room temperature over 12 h. Upon completion, the reaction mixture extracted with Et2O (150 mL) and EtOAc (15 mL). The organic extract was washed with brine, dried over MgSO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 25%A / 75%B (1 CV), 25%A / 75%B → 80%A / 20%B (10 CV), 80%A / 20%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] affording alcohol 29 (0.61 g, 1.1 mmol, 64%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 7.27 (1H, d, J = 8.8 Hz, H-4), 6.79 (1H, t, J = 8.7 Hz, H-3), 6.48 (2H, s, H-2′, H-6′), 3.90 (3H, s, OCH3-2), 3.85 (3H, s, OCH3-4′), 3.76 (6H, s, OCH3-3′, -5′), 3.16 (1H, m, H-9), 2.37 (1H, m, H-9), 2.57 (1H, m, H-8), 2.11 (1H, m, H-8), 1.94 (1H, s, H-6), 1.77 (2H, m, H-6, -7), 1.49 (1H, m, H-7). 13C NMR (CDCl3, 125 MHz): δ 153.1 (C, C-3′, C-5′), 149.6 (CF, d, J = 240.7 Hz, C-1), 146.7 (C, d, J = 13.4 Hz, C-2), 141.1 (C, C-1′), 138.8 (C, C-4′), 137.4 (CH, C-4), 129.1 (C, d, J = 12.7 Hz, C-10/11), 122.1 (C, d, J = 4.1 Hz, C-10/11), 109.4 (C, d, J = 2.36 Hz C-3), 104.2 (CH, C-2′, C-6′), 79.8 (C, d, J = 1.9 Hz, C-5), 60.8 (CH3, OCH3-4′), 56.1 (CH3, OCH3-3′, -5′), 56.0 (CH3, OCH3-2), 41.2 (CH, C-6), 26.6 (CH2, CH2-8/7), 26.2 (CH2, CH2-7/8), 24.2 (CH2, d, J = 7.8 Hz, CH2-9). 19F NMR (CDCl3, 470 MHz): δ -139.9 (m). Analysis: Calculated for C21H25FO5, C 67.01, H 6.69. Found: C 67.11, H 6.66.
4.1.28. 1-Fluoro-2-methoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocyclohept-5-ene (37).20
A solution of alcohol 29 (1.27 g, 0.97 mmol) in AcOH (20 mL) and H2O (30 mL) was heated to reflux at 150 °C for 12 h. The reaction mixture was cooled, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 50%A / 50%B (10 CV), 50%A / 50%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm] affording benzosuberene 37 (1.07 g, 0.75 mmol, 78%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 6.81-6.77 (2H, m, H-3, H-4), 6.47 (2H, s, H-2′, H-6′), 6.36 (1H, t, J = 7.4 Hz, H-6), 3.91 (3H, s, OCH3-2), 3.86 (3H, s, OCH3-4′), 3.80 (6H, s, OCH3-3′, -5′), 2.74 (2H, td, J = 2.2, 7.0 Hz, H-9), 2.16 (2H, p, J = 7.1 Hz, H-8), 1.97 (2H, q, J = 7.2 Hz, H-7). 13C NMR (CDCl3, 125 MHz): δ 153.0 (C, C-3′, C-5′), 149.6 (C, d, J = 242.1 Hz, C-1), 146.4 (C, d, J = 12.1 Hz, C-2), 142.2 (C, d, J = 2.0 Hz, C-5), 138.1 (C, C-1′), 137.6 (C, C-4′), 134.0 (C, d, J = 3.30 Hz, C-10/11), 129.6 (C, d, J = 13.83 Hz, C-10/11), 127.8 (CH, C-6), 124.9 (CH, d, J = 3.90 Hz, C-3), 110.1 (CH, d, J = 2.00 Hz, C-4), 105.3 (CH, C-2′, C-6′), 60.9 (CH3, OCH3-4′), 56.3 (CH3, OCH3-3′, -5′,-2), 33.8 (CH2, C-8), 25.7 (CH2, C-7), 23.3 (CH2, d, J = 4.37 Hz, C-9). 19F NMR (CDCl3, 470 MHz): δ -142.4 (m). HRMS: m/z: observed 381.1474 [M + Na]+, calculated for C21H23O4FNa+, 381.1473. HPLC: 18.20 min.
Experimental Procedures for Final Compound 36
4.1.29. (E)/(Z) 5-(2′-Chloro-3′-methoxyphenyl)pent-4-enoic acid (9).20
To a well stirred solution of (3-carboxypropyl)triphenylphosphonium bromide (13.1g, 30.5 mmol) in THF (250 mL) was added K-OtBu (6.99, 62.3 mmol). The reaction mixture was then cooled to 0° C and stirred for 15 min. A solution of 2-fluoro-3-methoxybenzaldehyde 5 (3.44 g, 20.2 mmol) in THF (25 mL) was added dropwise and the reaction mixture was allowed to warm to room temperature. H2O (50 mL) was added and the reaction mixture was extracted with Et2O (2 × 250 mL). The aqueous phase was acidified with 2 M HCl until the product precipitated making the solution cloudy, and then becoming clear again. The acidified aqueous phase was extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure affording a crude mixture of the E/Z isomers 9 (4.80 g, 19.9 mmol, crude yield 98%) as a colorless liquid. The crude product was used in the next step without further purification.
4.1.30. 5-(1′-Chloro-2′-methoxyphenyl)pentanoic acid (14).20
To pentanoic acid 9 (4.87 g, 20.2 mmol) in EtOH (50 mL) was added 10% Pd-C (729 mg). The flask was evacuated and H2 gas was introduced via balloons. The reaction was monitored for completion by filtering a small amount of the reaction mixture through Celite®, concentrating under reduced pressure, and recording the 1H NMR spectrum. Upon completion, the reaction mixture was filtered through Celite® and concentrated under reduced pressure to obtain 14 (2.55 g, 2.3 mmol, 75%) as an off-white solid. 1H NMR (CDCl3, 500 MHz): δ 7.14 (1H, t, J = 8.2, 7.7 Hz, H-4′), 6.84 (1H, d, J = 7.7 Hz, H-5′), 6.79 (1H, d, J = 8.2 Hz, H-3′), 3.89 (3H, s, OCH3-2′), 2.77 (2H, m, H-5), 2.40 (2H, t, J = 7.5 Hz, H-2), 1.70 (4H, m, H-3, -4). 13C NMR (CDCl3, 125 MHz): δ 179.3 (C, C-1), 155.2 (C, C-2′), 141.3 (C, C-6′), 126.8 (CH, C-4′), 122.2 (C, C-1′), 122.16 (CH, C-5′), 109.6 (C, C-3′), 56.2 (CH3, OCH3-2′), 33.8 (CH2, C-2), 33.3 (CH2, C-5), 29.0 (CH2, C-3/4), 24.4 (CH2, C-4/3). Analysis: Calculated for C12H15ClO3: C 59.39, H 6.23. Found: C 59.57, H 6.23. HRMS: m/z: observed 265.0603 [M+Na]+, calculated for C12H15O3ClNa+, 265.0602. HPLC: 11.99 min.
4.1.31. 1-Chloro-2-methoxy-benzocycloheptan-5-one (19).20
Pentanoic acid 14 (2.50 g, 10.3 mmol) was dissolved in Eaton's reagent (55 mL) and the solution was stirred for 12 h. The reaction mixture was poured over ice and the ice was allowed to melt. The aqueous phase was extracted with CH2Cl2 (3 × 150 mL) and NaHCO3 powder was added in small amounts until neutralized. The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording 19 (2.23 g, 9.93 mmol, 96%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 7.61 (1H, d, J = 8.7 Hz, H-4), 6.86 (1H, d, J = 8.7, Hz, H-3), 3.95 (3H, s, OCH3-2), 3.14 (2H, m, H-9), 2.69 (2H, m, H-6), 1.86 (2H, m, H-8), 1.78 (2H, m, H-7). 13C NMR (CDCl3, 125 MHz): δ 204.7 (C, C-5), 157.9 (C, C-2), 140.3 (C, C-1), 133.3 (C, C-10/11), 128.1 (C, C-10/11), 122.0 (C, C-4), 109.3 (C, C-3), 56.4 (CH3, OCH3-2), 40.5 (CH2, C-6), 27.8 (CH2, C-8), 23.8 (CH2, C-9), 20.7 (CH2, C-7). HRMS: m/z: observed 225.0680 [M+H]+, calculated for C12H14O2Cl+, 225.0677. HPLC: 13.86 min.
4.1.32. 1-Chloro-2-methoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocycloheptan-5-ol (28).20
To a solution of 3,4,5-trimethoxyphenyl bromide (3.60 g, 14.6 mmol) in THF (250 mL) at -78 °C was added n-BuLi (6.0 mL, 2.5 M in hexanes) and the reaction was stirred for 30 min. Benzosuberone 19 (1.87 g, 8.32 mmol) in THF (15 mL) was added using an addition funnel over a period of 15 min. The reaction mixture was allowed to warm to room temperature over 12 h. Upon completion, H2O (100 mL) was added and the reaction mixture was extracted with EtOAc (2 × 200 mL). The organic extract was washed with brine, dried over MgSO4, filtered, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm] affording alcohol 28 (2.41 g, 6.13 mmol, 74%) as an off-white solid. 1H NMR (CDCl3, 500 MHz): δ 7.50 (1H, d, J = 8.8 Hz, H-4), 6.80 (1H, d, J = 8.8 Hz, H-3), 6.49 (2H, s, H-2′, H-6′), 3.92 (3H, s, OCH3-2), 3.84 (3H, s, OCH3-4′), 3.76 (6H, s, OCH3-3′, -5′), 3.41 (1H, m, H-9), 2.58 (1H, m, H-6), 2.51 (1H, m, H-9), 2.12 (1H, m, H-6), 1.88 (2H, m, H-7), 1.76 (1H, m, H-8), 1.47 (1H, m, H-8). 13C NMR (CDCl3, 125 MHz): δ 154.1 (C, C-2), 153.1 (C, C-3′, C-5′), 141.3 (C, C-1′), 140.1 (C, C-10/11), 138.9 (CH, C-10/11), 137.5 (CH, C-4′), 125.9 (CH, C-4), 122.4 (C, C-1), 108.6 (CH, C-3), 104.1 (CH, C-2′, C-6′), 79.9 (C, C-5), 60.8 (CH3, OCH3-4′), 56.2 (CH3, OCH3-3′, -5′), 56.1 (CH3, OCH3-2), 41.1 (CH, C-6), 29.5 (CH2, C-9), 25.9 (CH2, C-7), 25.8 (CH2, C-8). Analysis: Calculated for C21H25ClO5: C 64.20, H 6.41. Found: C 64.41, H 6.45.
4.1.33. 1-Chloro-2-methoxy-5-(3′,4′,5′-trimethoxyphenyl)-benzocycloheptan-5-ene (36).20
A solution of alcohol 28 (2.37 g, 6.03 mmol) in AcOH (50 mL) and H2O (50 mL) was heated to reflux at 110 °C for 12 h. The reaction mixture was cooled, concentrated under reduced pressure, and subjected to flash chromatography using a prepacked 25 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 10%A / 90%B (1 CV), 10%A / 90%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 50 mL/min; monitored at 254 and 280 nm] affording benzosuberene 36 (2.18 g, 5.81 mmol, 97%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 6.92 (1H, d, H-4), 6.78 (1H, d, H-3), 6.48 (2H, s, H-2′, H-6′), 6.37 (1H, t, J = 7.4 Hz, H-6), 3.92 (3H, s, OCH3-2), 3.86 (3H, s, OCH3-4′), 3.80 (6H, s, OCH3-3′, -5′), 2.90 (2H, t, J = 7.0 Hz, H-9), 2.17 (2H, p, J = 7.0 Hz, H-8), 1.92 (2H, q, J = 7.2 Hz, H-7). 13C NMR (CDCl3, 125 MHz): δ 154.0 (C, C-2), 153.1 (C, C-3′, C-5′), 142.7 (C, C-5), 141.2 (C, C-10/11), 138.0 (C, C-1′), 137.4 (CH, C-4′), 134.2 (CH, C-10/11), 128.3 (C, C-4), 127.8 (CH, C-6), 121.5 (C, C-1), 109.1 (C, C-3), 105.2 (CH, C-2′, C-6′), 61.1 (CH3, OCH3-4′), 56.31 (CH3, OCH3-2), 56.28 (CH3, OCH3-3′, -5′), 33.8 (CH2, C-8), 28.7 (CH2, C-9), 25.6 (CH2, C-7). Analysis: Calculated for C21H23ClO4: C 67.29, H 6.18. Found: C, 67.40, H, 6.21. HRMS: m/z: observed 397.1180 [M + Na]+, calculated for C21H23O4ClNa+, 397.1177. HPLC: 19.26 min.
Experimental Procedures for Final Compound 35
4.1.34. 1,2-Dimethoxy-5-(3,4-dimethoxyphenyl)-benzocycloheptan-5-ol (27).
To a solution of 3,4-dimethoxyphenylbromide (0.455 g, 2.09 mmol) in THF (10 mL) at -78 °C was added n-BuLi (0.85 mL, 2.5 M in hexanes) and the reaction mixture was stirred for 1 h. Ketone 18 (0.453 g, 2.06 mmol) in THF (5 mL) was slowly added and the reaction mixture was allowed to warm to room temperature over 12 h. Upon completion, H2O (5 mL) was added and the reaction mixture was extracted with EtOAc (4 × 15 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure. The crude tertiary alcohol 27 (0.442 g, 1.23 mmol, crude yield 60%) was obtained as a clear oil. The crude product was used without further purification.
4.1.35. 1,2-Dimethoxy-5-(3,4-dimethoxyphenyl)-benzocyclohept-5-ene (35).
Tertiary alcohol 27 (0.442 g, 1.23 mmol) dissolved in EtOH (5 mL) and EtOAc (10 mL) was added 2 M HCl (5 mL) and the reaction mixture was stirred for 12 h. The reaction was extracted with EtOAc (4 × 20 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, evaporated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica gel column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B → 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (10 CV), 60%A / 40%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm]. Benzosuberene analogue 35 (0.302 g, 0.888 mmol, 72%) was obtained as a white solid. 1H NMR (CDCl3, 500 MHz): δ 6.81 (3H, m), 6.77 (1H, J = 8.6 Hz), 6.75 (1 H, d, J = 8.6 Hz), 6.60 (1H, t, J = 7.3 Hz), 3.88 (3H, s), 3.866 (3H, s), 3.865 (3H, s), 3.84 (3H, s), 2.75 (2H, t, J = 6.9 Hz), 2.16 (2H, p, J = 7.1 Hz), 1.96 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 125 MHz): δ 151.5, 148.7, 148.4, 146.2, 142.6, 136.0, 135.7, 134.2, 126.3, 125.3, 120.6, 111.3, 110.8, 109.3, 61.3, 56.04, 56.02, 55.7, 34.7, 25.7, 24.2. HRMS: m/z: observed 363.1568 [M+Na]+, calculated for C21H24O4Na+, 363.1567. HPLC: 17.63 min.
Experimental Procedures for Final Compound 42
4.1.36 1-((tert-Butyldimethylsilyl)oxy)-2-methoxy-5-(3,4-dimethoxyphenyl)-benzocycloheptan-5-ol (32).
To a solution of 3,4-dimethoxyphenylbromide (0.257 g, 1.51 mmol) in THF (11 mL) at -78 °C was added n-BuLi (0.52 mL, 2.5 M in hexanes) and the reaction mixture stirred for 1 h. Ketone 24 (0.312 g, 0.972 mmol) in THF (5 mL) was added and the reaction mixture was allowed to warm to room temperature over 12 h. Upon completion, H2O (3 mL) was added and the reaction mixture was extracted using EtOAc (4 × 15 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 50 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B → 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (12 CV), 60%A / 40%B (2 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm]. Tertiary alcohol 32 (0.344 g, 0.750 mmol, 77%) was obtained as a clear oil. 1H NMR (CDCl3, 500 MHz): δ 7.21 (1H, d, J = 8.8 Hz), 6.85 (1H, d, J = 2.0 Hz), 6.75 (1H, d, J = 8.3 Hz), 6.71 (2H, m), 3.85 (3H, s), 3.80 (3H, s), 3.79 (3H, s), 3.32 (1H, dd, J = 13.7, 6.8 Hz), 2.60 (1H, ddd, J = 14.2, 6.1, 2.9 Hz), 2.12 (3H, m), 1.89 (1H, m), 1.71 (2H, m), 1.35 (1H, m), 0.99 (9 H, s), 0.18 (3 H, s), 0.14 (3 H, s). 13C NMR (CDCl3, 125 MHz): δ 149.3, 148.9, 148.3, 142.0, 139.0, 138.4, 132.8, 119.7, 119.4, 110.8, 110.5, 108.0, 79.8, 56.0, 55.9, 54.8, 41.3, 27.2, 26.7, 26.2, 25.6, 19.0, -3.8, -4.0.
4.1.37. 1-((tert-Butyldimethylsilyl)oxy)-2-methoxy-5-(3,4-Dimethoxyphenyl)-benzocyclohept-5-ene (40).
Tertiary alcohol 32 (0.236 g, 0.515 mmol) was dissolved in AcOH (5 mL) and the reaction mixture was stirred for 12 h. The reaction mixture was concentrated under reduced pressure and subjected to flash chromatography using a pre-packed 25 g silica gel column [solvent A: EtOAc; solvent B: hexanes; gradient: 0%A / 100%B → 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (12 CV), 20%A / 80%B (2 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm]. Benzosuberene analogue 40 (0.156 g, 0.355 mmol, 69%) was obtained as a clear oil. 1H NMR (CDCl3, 500 MHz): δ 6.81 (3H, m), 6.69 (1H, d, J = 8.4 Hz), 6.61 (1H, d, J = 8.6 Hz), 6.30 (1H, t, J = 7.3 Hz), 3.88 (3H, s), 3.83 (3H, s), 3.81 (3H, s), 2.78 (2H, t, J = 7.0 Hz), 2.11 (2H, q, J = 7.0 Hz), 1.95 (2H, p, J = 7.2 Hz), 1.05 (9 H, s), 0.24 (6 H, s). 13C NMR (CDCl3, 125 MHz): δ 148.7, 148.6, 148.3, 142.8, 141.6, 136.0, 134.2, 133.4, 126.0, 122.3, 120.5, 111.4, 110.8, 108.4, 56.01, 55.96, 54.8, 34.1, 26.3, 25.7, 24.3, 19.1, -3.7.
4.1.38. 1-Hydroxy-2-methoxy-5-(3,4-dimethoxyphenyl)-benzocyclohept-5-ene (42).
To a solution of TBS-protected analogue 40 (0.156 g, 0.355 mmol) dissolved in THF (5 mL) was added TBAF (0.45 mL, 1 M in THF). The reaction mixture was stirred for 12 h at room temperature, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica gel column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B →7%A / 93%B (1 CV), 7%A / 93%B → 44%A / 56%B (10 CV); flow rate: 25 mL/min; monitored at 254 and 280 nm]. Phenol analogue 42 (0.100 g, 0.305 mmol, 86%) was obtained as a light brown solid. 1H NMR (CDCl3, 500 MHz): δ 6.81 (3H, m), 6.70 (1H, d, J = 8.3 Hz), 6.56 (1H, d, J = 8.3 Hz), 6.30 (1H, t, J = 7.5 Hz), 5.73 (1H, s), 3.91 (3H, s), 3.88 (3H, s), 3.83 (3H, s), 2.76 (2H, t, J = 7.0 Hz), 2.14 (2H, p, J = 7.0 Hz), 1.96 (2H, q, J = 7.2 Hz). 13C NMR (125 MHz, CDCl3) δ 148.6, 148.3, 145.1, 142.5, 142.4, 135.8, 134.7, 127.9, 126.4, 120.8, 120.6, 111.3, 110.8, 107.7, 56.1, 55.9, 55.0, 33.8, 25.8, 23.6. HRMS: m/z: observed 349.1411 [M+Na]+, calculated for C20H22O4Na+, 349.1410. HPLC: 15.58 min.
Experimental Procedures for Final Compound 41
4.1.39. 1-Hydroxy-5-(3,4,5-trimethoxyphenyl)-benzocyclohept-5-ene (33).20
To a solution of 3,4,5-trimethoxyphenylbromide (0.910 g, 3.68 mmol) in THF (40 mL) at -78 °C was added n-BuLi (1.5 mL, 2.5 M in hexanes) and the reaction mixture was stirred for 1 h. Commercially available benzocycloheptan-5-one 25 (0.536 g, 3.34 mmol) in THF (5 mL) was added and the reaction mixture was allowed to warm to room temperature over 12 h. Upon completion, H2O (5 mL) was added and the reaction mixture was extracted using EtOAc (4 × 15 mL). The organic extract was washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 100 g silica column [solvent A: EtOAc; solvent B: hexanes; gradient: 5%A / 95%B → 7%A / 93%B (1 CV), 7%A / 93%B → 60%A / 40%B (12.5 CV), 60%A / 40%B (1 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm]. Tertiary alcohol 33 (0.320 g, 0.794 mmol, 11%) was obtained as a clear oil. 1H NMR (CDCl3, 500 MHz): δ 7.57 (1H, dd, J = 9.0, 1.7 Hz), 7.22 (2H, pd, J = 7.3, 1.82 Hz), 7.12 (1H, dd, J = 6.6, 1.2 Hz), 6.48 (2H, s), 3.84 (3H, s), 3.74 (6H, s), 2.74 (1H, dd, J = 14.4, 6.8 Hz), 2.63 (1H, m), 2.57 (1H, s), 2.21 (1H, m), 2.14 (1H, m), 1.95 (1H, m) 1.78 (2H, m), 1.54 (1H, m). 13C NMR (CDCl3, 125 MHz): δ 153.0, 145.2, 141.3, 141.1, 137.3, 130.5, 127.6, 127.2, 126.2, 104.4, 80.1, 60.8, 56.1, 41.1, 36.3, 27.3, 26.1.
4.1.40. 5-(3,4,5-trimethoxyphenyl)-benzocyclohept-5-ene (41).20
Tertiary alcohol 33 (0.123 g, 0.375 mmol) was dissolved in AcOH (5 mL) and stirred for 12 h. H2O (40 mL) was added and the reaction mixture was extracted with EtOAc (3 × 15 mL). The organic extract was washed with H2O (3 × 20 mL), washed with brine, dried over Na2SO4, filtered, concentrated under reduced pressure, and purified by flash chromatography using a pre-packed 25 g silica gel column [solvent A: EtOAc; solvent B: hexanes; gradient: 0%A / 100%B → 2%A / 98%B (1 CV), 2%A / 98%B → 20%A / 80%B (11.5 CV), 20%A / 80%B (2.5 CV); flow rate: 40 mL/min; monitored at 254 and 280 nm]. Benzosuberene analogue 41 (0.082 g, 0.264 mmol, 70%) was obtained as a clear oil. 1H NMR (CDCl3, 500 MHz): δ 7.28 (1H, dd, J = 7.3, 1.7 Hz), 7.21 (2H, m), 7.05 (1H, dd, J = 7.1, 2.0 Hz), 6.49 (2H, s), 6.42 (1H, t, J = 7.3 Hz), 3.87 (3H, s), 3.80 (6H, s), 2.67 (2H, t, J = 7.1 Hz), 2.19 (2H, p, J = 7.1 Hz), 1.97 (2H, q, J = 7.2 Hz). 13C NMR (CDCl3, 125 MHz): δ 153.0, 143.1, 142.3, 140.1, 138.3, 137.5, 129.5, 128.7, 128.2, 127.3, 125.9, 105.4, 61.0, 56.3, 35.4, 32.6, 25.5. HRMS: m/z: observed 311.1646 [M+H]+, calculated for C20H23O +3, 311.1642. HPLC: 18.84 min.
Experimental Procedures for Final Compounds 47 and 48
4.1.41. 1-(O-acetyl-N-Fmoc-L-ser)amido-5-(3′,4′,5′-trimethoxyphenyl)-2-methoxy-benzocyclohept-5-ene(46)
To a well-stirred solution of amino-benzosuberene 45 (0.355 g, 1.00 mmol) in CH2Cl2 (25 mL) Fmoc-(Ac)-L-serine (0.553 g, 1.50 mmol), T3P (0.75 mL, 2.50 mmol), and Et3N (0.21 mL, 1.50 mmol) were added, and the reaction mixture was allowed to stir for 12 h at room temperature. H2O (100 mL) was added and the reaction mixture was extracted with EtOAc. The organic extract was washed with brine, dried with Na2SO4, concentrated under reduced pressure, and purified by flash column chromatography (35% EtOAc/hexanes) to give the desired Fmoc-L-serinamide acetate 46 (0.393 g, 0.56 mmol, 56%) as a white solid. 1H NMR (CDCl3, 500 MHz): δ 7.74 (2H, d, J = 7.0 Hz, ArH), 7.65 (1H, s, NH), 7.57 (2H, d, J = 7.5 Hz, ArH), 7.38 (2H, dd, J = 7.5, 7.0 Hz, ArH), 7.27 (2H, dd, J = 7.5, 7.0 Hz, ArH), 6.97 (1H, d, J = 8.5 Hz, ArH), 6.73 (1H, d, J = 8.5 Hz, ArH), 6.49 (2H, s, ArH), 6.36 (1H, t, J = 7.5 Hz, C=CH), 5.94 (1H, d, J = 6.5 Hz, NH), 4.82 (1H, bs, CH), 4.49 (3H, m, CH2), 4.23 (1H, t, J = 6.5 Hz, CH), 3.86 (3H, s, OCH3), 3.79 (6H, s, OCH3), 3.74 (3H, s, OCH3), 2.59 (2H, t, J = 7.5 Hz, CH2), 2.14 (2H, m, CH2), 2.10 (3H, s, CH3), 1.95 (2H, m, CH2). 13C NMR (CDCl3, 125 MHz): δ 170.8, 168.4, 156.2, 153.2, 152.9, 143.6, 143.5, 142.4, 141.2, 140.7, 138.3, 137.3, 133.4, 129.4, 127.8, 127.1, 125.0, 122.1, 120.0, 108.3, 105.3, 67.2, 64.5, 60.9, 56.6, 55.1, 54.2, 47.1, 33.8, 27.0, 25.6, 20.7. HRMS: m/z: observed 729.2794 [M+Na]+, calculated for C41H42N2O9Na+, 729.2783.
4.1.42. 1-(L-ser)amido-5-(3′,4′,5′-trimethoxyphenyl)-2-methoxy-benzocyclohept-5-ene (47)
To a solution of Fmoc-L-serinamide acetate in CH2Cl2:MeOH (3 mL/3 mL) was added 46 (0.393 g, 0.56 mmol) and 2 M NaOH (0.63 mL, 1.26 mmol). After stirring for 13 h at the reaction mixture concentrated under reduced pressure and water (10 mL) was added. The reaction mixture was extracted with EtOAc (15 mL × 3) and washed with brine, dried with Na2SO4, concentrated under reduced pressure, and purified by flash column chromatography (5% MeOH / 95% CH2Cl2) to give the desired serinamide 47 (0.19 g, 0.42 mmol, 75%) as a white solid. 1H NMR (CDCl3,500 MHz): δ 8.73 (1H, s, NH), 6.97 (1H, d, J = 8.5 Hz, ArH), 6.78 (1H, d, J = 8.5 Hz, ArH), 6.49 (2H, s, ArH), 6.36 (1H, t, J = 7.5 Hz, C=CH), 4.04 (1H, dd, J = 11, 4.5 Hz, CH2), 3.86 (3H, s, OCH3), 3.84 (6H, s, OCH3), 3.80 (3H, s, OCH3), 3.80 (1H, dd, J = 11, 4.5 Hz, CH2), 3.71 (1H, t, J = 4.5 Hz, CH), 2.62 (2H, t, J = 6.5 Hz, CH2), 2.16 (2H, m, CH2), 1.98 (2H, q, J = 7.0 Hz, CH2). 13C NMR (CDCl3, 125 MHz): δ 173.5, 153.1, 152.8, 142.5, 140.6, 138.3, 133.5, 129.1, 127.6, 122.5, 109.9, 108.4, 105.3, 65.9, 60.9, 56.3, 56.1, 55.8, 34.0, 27.0, 25.5. HRMS: m/z: observed 443.2190 [M+H]+, calculated for C24H31N2O6+, 443.2177. HPLC: 8.23 min.
4.1.43. 1-(L-ser)amido-5-(3′,4′,5′-trimethoxyphenyl)-2-methoxy-benzocyclohept-5-ene hydrochloride (48)
To a solution of Serinamide 47 (0.10 g, 0.23 mmol) in CH3OH/CH2Cl2 (3 mL/3 mL) was added 4 M HCl-dioxane (0.28 mL, 1.13 mmol). After stirring for 3 h, the reaction mixture was concentrated under reduced pressure and upon recrystallization in EtOAc/CH3OH gave the desired serinamide salt 48 (0.074 g, 0.15 mmol, 67%) as a white solid. 1H NMR (CD3OD, 500 MHz): δ 6.96 (1H, d, J = 8.5 Hz, ArH), 6.92 (1H, d, J = 8.5 Hz, ArH), 6.53 (2H, s, ArH), 6.39 (1H, t, J = 7.5 Hz, C=CH), 4.26 (1H, dd, J = 7.5, 4.5 Hz, CH), 4.19 (1H, dd, J = 11.5, 4.0 Hz, CH2), 3.99 (1H, dd, J = 11.5, 7.5 Hz, CH2), 3.82 (3H, s, OCH3), 3.76 (6H, s, OCH3), 3.75 (3H, s, OCH ), 2.62 (2H, m, CH2), 2.16 (2H, m, CH2), 1.92 (2H, m, CH2). 13C NMR (CD3OD, 125 MHz): δ 166.9, 153.9, 152.8, 142.5, 140.7, 138.4, 137.1, 133.3, 129.3, 126.9, 121.7, 108.5, 105.1, 60.7, 59.7, 55.18, 55.16, 54.8, 33.5, 26.2, 24.9. HRMS: m/z: observed 443.2214 [M-Cl]+, calculated for C24H31N2O6+, 443.2177. HPLC: 8.32 min.
4.2 Biological Evaluation
4.2.1. Inhibition of Tubulin Assembly
In brief, 160 uL of tubulin (1.25 mg/mL in 1M glutamate) and 8 uL of desired inhibitor concentration dissolved in either water or DMSO were mixed in a microfuge vial and incubated at 37 °C for 15 minutes. Microfuge vials were then placed on ice for 15 minutes. 32 uL of GTP (2.5 mM) was added to vials and contents of vials (200 uL) were placed in their appropriate cells. Cells were placed in the cell holder of the UV/Vis spectrophotometer and allowed to cool at 0 °C for 8 minutes. UV/Vis settings were as follows: absorption: 350 nm, length of experiment: 3800 s, measurement intervals: 30 s. Each experiment was initiated and UV/Vis spectrophotometer took readings for 100 seconds at 0 °C. At 100 s, the temperature was switched to 38 °C for 120 seconds, followed by a temperature change to 31 °C until 2600 total seconds had passed since initiation of the experiment. At 2600 seconds, the cells were cooled to 0 °C throughout the remainder of the experiment which ended once 3800 seconds had passed. Tubulin was purified from calf brain. For details regarding effects on tubulin assembly, see references 70 and 71.
4.2.2. SRB Assay72
We assessed inhibition of human cancer cell growth using the National Cancer Institute's standard sulforhodamine B assay, as previously described.72 Briefly, cancer cell lines in a 5% fetal bovine serum/RPMI1640 medium, 1% gentamicin solution were plated in 96-well plates and incubated for 24 h. Serial dilutions of the compounds were then added. After 48 h, the plates were fixed with trichloroacetic acid, stained with sulforhodamine B, and read with an automated Biotek plate reader. A growth inhibition of 50% (GI50 or the drug concentration causing a 50% reduction in the net protein increase) was calculated from optical density data.
Supplementary Material
Acknowledgements
The authors are grateful to the Cancer Prevention and Research Institute of Texas (CPRIT, grant no. RP100406 to K.G.P. and M.L.T.), the National Cancer Institute of the National Institutes of Health (grant no. 5R01CA140674 to K.G.P. and M.L.T.) and Oxigene Inc. (grant to K.G.P. and M.L.T.) for their financial support of this project, and to the NSF for funding the Varian 500 MHz NMR spectrometer (grant no. CHE- 0420802). The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health. The authors also thank Dr. James Karban and Dr. Michelle Nemec (Director) for the use of the shared Molecular Biosciences Center at Baylor University, Dr. Alejandro Ramirez (Mass Spectrometry Core Facility, Baylor University), and Dr. Kevin Klausmeyer (X-ray analysis). The authors are grateful to Mr. Akshar Chauhan for his valuable contributions to the synthesis of certain analogues.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Data.
Characterization data (1H NMR, 13C NMR, 19F NMR, HPLC, and HRMS) for final compounds, HMBC and HSQC data for compound 38, X-ray crystallography for compounds 38 and 39, mechanistic speculation regarding regioselective demethylation reactions for compounds 16 and 18, information regarding the synthesis of compound 2, an alternative synthetic route to compound 39, and cLogP data for compounds 39, 44, 45, and 48 can be found, in the online version, at .
References and notes
- 1.Siemann DW, Chaplin DJ, Horsman MR. Cancer. 2004;100:2491. doi: 10.1002/cncr.20299. [DOI] [PubMed] [Google Scholar]
- 2.Horsman MR, Bohn AB, Busk M. Exp. Oncol. 2010;32:143–148. [PubMed] [Google Scholar]
- 3.Tozer GM, Kanthou C, Baguley BC. Nat. Rev. Cancer. 2005;5:423–435. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]
- 4.Siemann DW, Bibby MC, Dark GG, Dicker AP, Eskens FA, Horsman MR, Marme D, Lorusso PM. Clin. Cancer Res. 2005;11:416–420. [PubMed] [Google Scholar]
- 5.Abdelrahim M, Konduri S, Basha R, Philip PA, Baker CH. Angiogenesis: An Update and Potential Drug Approaches (Review). Int. J. Oncol. 2010;36(1):5–18. [PubMed] [Google Scholar]
- 6.Kanthou C, Tozer GM. Expert Opin. Thera. Targets. 2007;11:1443–1457. doi: 10.1517/14728222.11.11.1443. [DOI] [PubMed] [Google Scholar]
- 7.Lippert JW., III Bioorg. Med. Chem. 2007;15:605–615. doi: 10.1016/j.bmc.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 8.Pinney KG. Molecular Recognition of the Colchicine Binding Site as a Design and Paradigm for the Discovery and Development of Vascular Disrupting Agents; In Vascular-Targeted Therapies in Oncology. John Wiley and Sons, Ltd; New York: 2006. pp. 95–121. [Google Scholar]
- 9.Mason RP, Zhao D, Liu L, Trawick ML, Pinney KG. Integr. Biol. 2011;3:375–387. doi: 10.1039/c0ib00135j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monk KA, Siles R, Hadimani MB, Mugabe BE, Ackely JF, Studerus SW, Edvardsen K, Trawick ML, Garner CM, Rhodes MR, Pettit GR, Pinney KG. Bioorg. Med. Chem. 2006;14:3231–3244. doi: 10.1016/j.bmc.2005.12.033. [DOI] [PubMed] [Google Scholar]
- 11.Tozer GM, Akerman S, Cross NA, Barber PR, Björndahl MA, Grecco O, Harris S, Hill SA, Honess DJ, Ireson CR, Pettyjohn KL, Prise VE, Reyes-Aldaroso CC, Ruhrberg C, Shima DT, Kanthou C. Cancer Res. 2008;68:2301–2311. doi: 10.1158/0008-5472.CAN-07-2011. [DOI] [PubMed] [Google Scholar]
- 12.Dark GG, Hill SA, Prise VE, Tozer GM, Pettit GR, Chaplin DJ. Cancer Research. 1997;57:1829–1834. [PubMed] [Google Scholar]
- 13.Tozer GM, Prise VE, Wilson J, Locke RJ, Vojnovic B, Stratford MRL, Dennis MF, Chaplin DJ. Cancer Research. 1999;59:1626–1634. [PubMed] [Google Scholar]
- 14.Kremmidiotis G, Leske AF, Lavranos TC, Beaumont D, Gasic J, Hall A, O'Callaghan M, Matthews CA, Flynn B. Mol Cancer Ther. 2010;9:1562. doi: 10.1158/1535-7163.MCT-09-0815. [DOI] [PubMed] [Google Scholar]
- 15.Lu Y, Chen J, Xiao M, Li W, Miller DD. Pharm. Res. 2012;29:2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 17.Vincent L, Kermani P, Young LM, Cheng J, Zhang F, Shido K, Lam G, Bompais-Vincent H, Zhu Z, Hicklin DJ, Bohlen P, Chaplin DJ, May C, Rafii SJ. J. Clin. Invest. 2005;115:2992–3006. doi: 10.1172/JCI24586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sriram M, Hall JJ, Grohmann NC, Strecker TE, Wootton T, Franken A, Trawick ML, Pinney KG. Bioorg. Med. Chem. 2008;16:8161–8171. doi: 10.1016/j.bmc.2008.07.050. [DOI] [PubMed] [Google Scholar]
- 19.Tanpure RP, George CS, Sriram M, Strecker TE, Tidmore JT, Hamel E, Charlton-Sevcik AK, Chaplin DJ, Trawick ML, Pinney KG. Med. Chem. Comm. 2012;3:720–724. doi: 10.1039/C2MD00318J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinney KG, Sriram M, George C, Tanpure RP. PCT Int. Appl. 2012 WO 2012068284 A2 20120524.
- 21.Ghatak A, Dorsey JM, Garner CM, Pinney KG. Tetrahedron Letters. 2003;44:4145–4148. [Google Scholar]
- 22.Subsequent to our original work with tubulin-binding dihydronaphthalene analogues (see ref. 18), a further report appeared that included our analogues and other dihydronaphthalene derivatives: Rasolofonjatovo E, Provot O, Hamze A, Rodrigo J, Bignon J, Wdzieczak-Bakala J, Desravines D, Dubois J, Brion J-D, Alami M. Eur. J. Med. Chem. 2012;52:22–32. doi: 10.1016/j.ejmech.2012.03.001.
- 23.Pinney KG, Mocharla VP, Chen Z, Garner CM, Ghatak A, Hadimani M, Kessler J, Dorsey JM, Edvardsen K, Chaplin DJUS. Pat. Appl. Publ. 2004 US 20040043969 A1 20040304.
- 24.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Experientia. 1989;45:680. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 25.Lin CM, Ho HH, Pettit GR, Hamel E. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 26.Pettit GR, Singh SB, Niven ML, Hamel E, Schmidt JM. J. Nat. Prod. 1987;50:119–131. doi: 10.1021/np50049a016. [DOI] [PubMed] [Google Scholar]
- 27.Ohsumi K, Nakagawa R, Fukada Y, Hatanaka T, Morinaga Y, Nihei Y, Ohishi K, Suga Y, Akiyama Y, Tsuji T. J. Med. Chem. 1998;41:3022–3032. doi: 10.1021/jm980101w. [DOI] [PubMed] [Google Scholar]
- 28.Pinney KG, Mejia MP, Villalobos VM, Rosenquist BE, Pettit GR, Verdler-Pinard P, Hamel P. Bioorg. Med. Chem. 2000;8:2417–2425. doi: 10.1016/s0968-0896(00)00176-0. [DOI] [PubMed] [Google Scholar]
- 29.Subsequent to our original work with 2-amino-combretastatin analogues (ref. 10), the following paper appeared: Chang J-Y, Yang M-F, Chang C-Y, Chen C-M, Kuo CC, Liou J-P. J. Med. Chem. 2006;49:6412. doi: 10.1021/jm060616k.
- 30.Siles R, Ackley JF, Hadimani MB, Hall JJ, Mugabe BE, Guddneppanavar R, Monk KA, Chapuis JC, Pettit GR, Chaplin DJ, Edvardsen K, Trawick ML, Garner CM, Pinney KG. J. of Nat. Prod. 2008;71:313–320. doi: 10.1021/np070377j. [DOI] [PubMed] [Google Scholar]
- 31.Pettit GR, Lippert JW., III Anti-Cancer Drug Des. 2000;15:203–216. [PubMed] [Google Scholar]
- 32.Pettit GR, Lippert JW., III Anti-Cancer Drug Des. 1998;18:183–191. [PubMed] [Google Scholar]
- 33.Pettit GR, Temple C, Jr., Narayanan VL, Varma R, Simpson MJ, Boyd MR, Rener GA, Bansal N. Anti-Cancer Drug Des. 1995;10:299–309. [PubMed] [Google Scholar]
- 34.Hsieh HP, Liou JP, Mahindroo N. Curr. Pharm. Des. 2005;11:1655–1677. doi: 10.2174/1381612053764751. [DOI] [PubMed] [Google Scholar]
- 35.Pinney KG, Pettit GR, Trawick ML, Jelinek C, Chaplin DJ. The Discovery and Development of the Combretastatins. In: Cragg GR, Kingston DGI, Newmann DJ, editors. In Anticancer Agents from Natural Products Second Edition. CRC Press/ Taylor & Francis; Boca Raton, FL: 2012. pp. 27–63. [Google Scholar]
- 36.Pettit GR, Toki B, Herald DL, Verdier-Pinard P, Boyd MR, Hamel E, Pettit RK. J. Med. Chem. 1998;41:1688–1695. doi: 10.1021/jm970644q. [DOI] [PubMed] [Google Scholar]
- 37.Liou J-P, Chang C-W, Song J-S, Yang Y-N, Yeh C-F, Tseng H-Y, Lo Y-K, Chang Y-L, Chang C-M, Hsieh H-P. J. Med. Chem. 2002;45:2556–2562. doi: 10.1021/jm010365+. [DOI] [PubMed] [Google Scholar]
- 38.Pinney KG. Molecular Recognition of the Colchicine Binding Site as a Design Paradigm for the Discovery and Development of Vascular Disrupting Agents. In: Siemann DW, editor. Vascular-Targeted Therapies in Oncology. John Wiley & Sons, Ltd; 2006. pp. 95–121. [Google Scholar]
- 39.Pinney KG, Mocharla VP, Chen Z, Garner CM, Ghatak A, Hadimani M, Kessler J, Dorsey J. U.S. Patent Application. 2003 Jul 15; filed on March 12, 2001. Patent Granted (Patent No. 6,593,374) [Google Scholar]
- 40.Hadimani M, Kessler RJ, Kautz KA, Ghatak A, Shirali AR, O'Dell H, Garner CM, Pinney KG. Acta Cryst., Sect. C. 2002;C58:330–332. doi: 10.1107/s0108270102003669. [DOI] [PubMed] [Google Scholar]
- 41.Pinney KG, Wang F, Hadimani M. US Patent 6849656. 2005 [Google Scholar]
- 42.Pinney K, Wang F, Del Pilar Mejia M. From PCT Int. Appl. 2001 WO 0119794 A2.
- 43.Patil SA, Patil R, Miller DD. Future Med. Chem. 2012;4(16):2085–2115. doi: 10.4155/fmc.12.141. [DOI] [PubMed] [Google Scholar]
- 44.Chen Z, O'Donnell CJ, Maderna A. Tetrahedron Lett. 2012;53:64–66. [Google Scholar]
- 45.Rajak H, Dewangan PK, Patel V, Jain DP, Singh A, Veerasamy R, Sharma PC, Dixit A. Curr. Pharm. Design. 2013;19:1923–1955. doi: 10.2174/1381612811319100013. [DOI] [PubMed] [Google Scholar]
- 46.Pinney KG, Trawick ML, Tanpure RP, George CS, Sriram M, Strecker TE, Charlton-Sevcik A, Liu L, Mason R, Siims BG, Chaplin DG. The Discovery and Development of Highly Potent Benzosuberene Anti-Cancer Agents; CPRIT, Innovations in Cancer Prevention and Research Conference; Austin, TX. November 17, 2010. [Google Scholar]
- 47.Liu L, Magnusson J, Mason R, Trawick ML, Pinney KG. Comparison of Antivascular Effects of Novel Vascular Disrupting Agents in Breast Cancer Using Dynamic Bioluminescence Imaging. CPRIT; Austin, Texas: Oct 24-26, p. 2012. [Google Scholar]
- 48.Eaton PE, Carlson GR, Lee JT. J. Org. Chem. 1973;38:4071. [Google Scholar]
- 49.Tanis VM, Moya C, Jacobs RS, Little RD. Tetrahedron. 2008;64:10649–10663. [Google Scholar]
- 50.Negoro N, Sasaki S, Mikami S, Ito M, Suzuki M, Tsujihata Y, Ito R, Harada A, Takeuchi K, Suzuki N, Miyazaki J, Santou T, Odani T, Kanzaki N, Funami M, Tanaka T, Kogame A, Matsunaga S, Yasuma T, Momose Y. ACS Med. Chem. Lett. 2010;1:290–294. doi: 10.1021/ml1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.George C. Ph.D. Dissertation. Baylor University; Waco, TX: 2012. [Google Scholar]
- 52.Kemperman G, Roeters T, Hilberink P. Eur. J. Org. Chem. 2003:1681–1686. [Google Scholar]
- 53.Fillion E, Fishlock D, Wilsily A, Goll JM. J. Org. Chem. 2005;70:1316–1327. doi: 10.1021/jo0483724. [DOI] [PubMed] [Google Scholar]
- 54.Patti RK, Waynant KV, Herndon JW. Org. Lett. 2011;13:2848–2851. doi: 10.1021/ol200825j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allinger NL, Jones ES. J. Org. Chem. 1962;27:70–76. [Google Scholar]
- 56.Oshumi K, Hatanaka T, Nakagawa R, Fukada Y, Morinaga Y, Suga Y, Nihei Y, Ohishi K, Akiyama Y, Tsuji T. Anti-Cancer Drug Design. 1999;14:539–548. [PubMed] [Google Scholar]
- 57.Delmonte A, Sessa C. Expert Opin, Investig. Drugs. 2009;18:1541. doi: 10.1517/13543780903213697. [DOI] [PubMed] [Google Scholar]
- 58.Hori K, Saito S, Kubota K. British J. of Cancer. 2002;86:1604–1614. doi: 10.1038/sj.bjc.6600296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lawrence NJ, Hepworth LA, Rennison D, McGown AT, Hadfield JA. J. Fluorine Chem. 2003;123:101–108. [Google Scholar]
- 60.Hall JJ, Sriram M, Strecker TE, Tidmore JK, Jelinek CJ, Kumar GDK, Hadimani MB, Pettit GR, Chaplin DJ, Trawick ML, Pinney KG. Bioorg. Med. Chem. Lett. 2008;18:5146–5149. doi: 10.1016/j.bmcl.2008.07.070. [DOI] [PubMed] [Google Scholar]
- 61.Pettit GR, Grealish MP, Herald DL, Boyd MR, Hamel E, Pettit RK. J. Med. Chem. 2000;43:2731–2737. doi: 10.1021/jm000045a. [DOI] [PubMed] [Google Scholar]
- 62.Benham FJ, Fogh J, Harris H. Int. J. Cancer. 1981;27:637–644. doi: 10.1002/ijc.2910270510. [DOI] [PubMed] [Google Scholar]
- 63.Pettit GR, Anderson CR, Herald DL, Jung MK, Lee DJ, Hamel E, Pettit RK. J. Med. Chem. 2003;46:525–531. doi: 10.1021/jm020204l. [DOI] [PubMed] [Google Scholar]
- 64.Pochopin NL, Charman WN, Stella VJ. Int. J. Pharm. 1995;121:157–167. [Google Scholar]
- 65.Hadimani MB, MacDonough MT, Ghatak A, Strecker TE, Lopez R, Sriram M, Nguyen BL, Hall JJ, Kessler RJ, Shirali AR, Liu L, Garner CM, Pettit GR, Hamel H, Chaplin DJ, Mason RP, Trawick ML, Pinney KG. [July 31, 2013];J. Nat. Prod. 2013 doi: 10.1021/np400374w. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haworth RD, Moore BP, Pauson PL. J. Chem. Soc. 1948:1045–1051. [PubMed] [Google Scholar]
- 67.Gardner P, Horton WJ, Thomson G, Theives RR. J. Chem. Soc. 1952;74:5527–5529. [Google Scholar]
- 68.Caunt D, Crow WD, Haworth RD, Vodoz CA. J. Chem. Soc. 1950:1631–1635. [Google Scholar]
- 69.Horton WJ, Pitchforth LL. J. Org. Chem. 1960;25:131–132. [Google Scholar]
- 70.Hamel E. Cell Biochem. Biophys. 2003;38:1–22. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 71.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Mol. Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 72.Monks A, Scudiero D, Skehan P, Shoemaker R, Paul K, Vestica D, Hose C, Langley J, Cronise P, Vaigro-Wolf A. J. Natl. Cancer Inst. 1991;83:767–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.