

Abstract
Heterozygous loss-of-function SCN1A mutations cause Dravet syndrome, an epileptic encephalopathy of infancy that exhibits variable clinical severity. We utilized a heterozygous Scn1a knockout (Scn1a+/−) mouse model of Dravet syndrome to investigate the basis for phenotype variability. These animals exhibit strain-dependent seizure severity and survival. Scn1a+/− mice on strain 129S6/SvEvTac (129.Scn1a+/−) have no overt phenotype and normal survival compared with Scn1a+/− mice bred to C57BL/6J (F1.Scn1a+/−) that have severe epilepsy and premature lethality. We tested the hypothesis that strain differences in sodium current (INa) density in hippocampal neurons contribute to these divergent phenotypes. Whole-cell voltage-clamp recording was performed on acutely-dissociated hippocampal neurons from postnatal day 21–24 (P21–24) 129.Scn1a+/− or F1.Scn1a+/− mice and wild-type littermates. INa density was lower in GABAergic interneurons from F1.Scn1a+/− mice compared to wild-type littermates, while on the 129 strain there was no difference in GABAergic interneuron INa between 129.Scn1a+/− mice and wild-type littermate controls. By contrast, INa density was elevated in pyramidal neurons from both 129.Scn1a+/− and F1.Scn1a+/− mice, and was correlated with more frequent spontaneous action potential firing in these neurons, as well as more sustained firing in F1.Scn1a+/− neurons. We also observed age-dependent differences in pyramidal neuron INa density between wild-type and Scn1a+/− animals. We conclude that preserved INa density in GABAergic interneurons contributes to the milder phenotype of 129.Scn1a+/− mice. Furthermore, elevated INa density in excitatory pyramidal neurons at P21–24 correlates with age-dependent onset of lethality in F1.Scn1a+/− mice. Our findings illustrate differences in hippocampal neurons that may underlie strain- and age-dependent phenotype severity in a Dravet syndrome mouse model, and emphasize a contribution of pyramidal neuron excitability.
Keywords: seizures, epilepsy, mouse model, voltage-gated sodium channel, electrophysiology, modifier genes
INTRODUCTION
Idiopathic epilepsies are a group of clinically diverse disorders with a strong genetic component to their pathogenesis. Although most epilepsies exhibit complex inheritance, some result from single gene mutations. Mutations in genes encoding neuronal voltage-gated sodium channels (NaV) result in genetic epilepsies with overlapping clinical characteristics but divergent clinical severity (Meisler & Kearney, 2005). Mutation of SCN1A, encoding the pore-forming subunit NaV1.1, is the most commonly discovered cause of monogenic epilepsies (Catterall et al., 2010). More than 800 heterozygous SCN1A epilepsy-associated mutations have been identified, with more than 70% occurring in patients with Dravet syndrome (DS), also known as severe myoclonic epilepsy of infancy (Claes et al., 2009; Lossin, 2009). While DS is typically characterized by seizure onset in the first year of life with an ensuing epileptic encephalopathy consisting of cognitive, behavioral, and motor impairments, the severity of its presentation and progression can be variable (Brunklaus et al., 2012; Zuberi et al., 2011). However, it remains unclear why individuals bearing the same heterozygous SCN1A mutation exhibit divergent seizure phenotypes, even within the same family (Kimura et al., 2005; Pineda-Trujillo et al., 2005; Goldberg-Stern, 2013).
Genetic modifiers may contribute to the variable expressivity of SCN1A mutations in DS patients and this notion is further suggested by investigations of Scn1a knockout (Scn1a+/−) and Scn1a-R1407X knock-in mouse models of DS (Ogiwara et al., 2007; Yu et al., 2006; Miller et al, 2013). Heterozygous DS mice display spontaneous seizures and premature death as well as cognitive and motor impairments (Han et al., 2012; Ito et al., 2012; Kimura et al., 2005). Reduced sodium current (INa) density in morphologically identified hippocampal interneurons correlated with impaired excitability, whereas INa density was not different in excitatory pyramidal neurons (Bechi et al., 2012; Yu et al., 2006). These studies suggested that dysfunctional inhibitory circuits may underlie the pathophysiology of DS. Importantly, the phenotype severity observed in Scn1a+/− mice is influenced by genetic background. Scn1a+/− mice maintained on the 129 strain background have a normal lifespan with no seizures. By contrast, crossing 129.Scn1a+/− mice to C57BL/6 animals generates offspring with overt seizures and decreased lifespan (Yu et al., 2006; Miller et al, 2013). The neurophysiological basis of strain-dependent seizure severity observed in Scn1a+/− mice has not been investigated, but represents an opportunity to elucidate neuronal mechanisms responsible for variable disease expression.
In this study, we compared the properties of INa in hippocampal neurons from Scn1a+/− mice on different genetic backgrounds in order to test the hypothesis that variable sodium current compensation in hippocampal neurons accounts for strain-dependent phenotype differences. We observed significant differences in both interneuron and pyramidal neuron INa densities that correlated with strain- and age-dependent phenotypes. Our findings contribute a plausible explanation for the divergent seizure phenotypes observed in DS and offer new opportunities to connect genetic modifiers with neurophysiological mechanisms with relevance to epileptogenesis.
MATERIAL AND METHODS
Generation of Scn1a+/− mice
The Scn1aTmKea targeted null allele was generated by homologous recombination in TL1 ES cells (129S6/SvEvTac). Exon 1 of the mouse Scn1a gene was replaced by a selection cassette as described (Miller et al., 2013). The resultant Scn1a+/− mouse line (129.Scn1a+/−) was then maintained on the 129S6/SvEvTac (129) inbred strain by continuous backcrossing to 129. Strain C57BL/6J (B6) was crossed to 129.Scn1a+/− to generate (129.Scn1a+/− x B6) F1 offspring (designated as F1.Scn1a+/−) for experiments. For genotyping, mice were tail biopsied on postnatal day 12 (P12) and DNA was isolated using the Gentra Puregene Mouse Tail kit according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA). The Scn1a genotype was determined by multiplex PCR as described (Miller et al., 2013). All studies were approved by the Vanderbilt University Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Electroencephalography (EEG)
Male and female 129.Scn1a+/− and F1.Scn1a+/− mice and wild-type littermates were anesthetized with isoflurane and implanted with prefabricated headmounts (Pinnacle Technology, Inc., Lawrence, KS, USA) for video-EEG monitoring as previously described (Hawkins et al., 2011). Following 5–7 days of recovery, video-EEG data was collected from freely-moving mice in 13-hour overnight sessions once per week for up to 2 months. Digitized data were acquired and analyzed with Sirenia software (Pinnacle Technology, Inc.) along with contemporaneous video recordings. Seizure activity was scored manually.
Seizure threshold testing
Thresholds to induced seizures in P28–35 male and female 129.Scn1a+/− mice and wild-type littermates were determined as previously described using flurothyl (2,2,2-trifluroethylether; 10 ml/min) (Hawkins et al., 2011). Data were assessed for statistical significance using an unpaired, two-tailed Student’s t-test.
Acute dissociation of hippocampal neurons
Male and female P14–P24 day old mice were deeply anesthetized with isoflurane then rapidly decapitated. The brain was promptly removed under aseptic conditions and placed in ice-cold dissecting solution containing (in mM) 2.5 KCl, 110 NaCl, 7.5 MgCl2·6H2O, 10 HEPES, 25 dextrose, 75 sucrose, 1 pyruvic acid, and 0.6 ascorbic acid with pH adjusted to 7.35 with NaOH. Coronal slices (400 μm) were made through the hippocampi using a Leica VT 1200 vibratome (Leica Microsystems Inc., Buffalo Grove, IL, USA) in ice-cold dissecting solution bubbled with 95% O2/5% CO2. Slices were incubated for one hour at 30°C in artificial CSF (ACSF, in mM: 12 4 NaCl, 4.4 KCl, 2.4 CaCl2·2H2O, 1.3 MgSO4, 1 NaH2PO4, 26 NaHCO3, 10 glucose, and 10 HEPES with pH adjusted to 7.35 with NaOH) bubbled with 95% O2/5% CO2.
Hippocampi were dissected from the slices with micro-forceps and digested for 15 min at room temperature with proteinase (2 mg/ml) from Aspergillus melleus Type XXIII in dissociation solution (in mM: 82 Na2SO4, 30 K2SO4, 5 MgCl2·6H2O, 10 HEPES, and 10 dextrose with pH adjusted to 7.35 with NaOH), then washed multiple times with dissociation solution containing 1 mg/ml bovine serum albumin and allowed to recover for ≥10 min in the same solution before mechanical dissociation by gentle trituration with sequentially fire-polished Pasteur pipettes of decreasing diameter. The cell suspension was allowed to settle on coverslips for 15 min before experiments. Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Voltage-clamp recording
All electrophysiological experiments and data analysis were done blinded to strain and genotype. Glass coverslips with plated hippocampal neurons were placed into the recording chamber of an inverted microscope equipped with a Moticam 3000 digital camera. Individual neurons were photographed, then whole-cell voltage-clamp recordings were made at room temperature using an Axopatch 200B amplifier (Molecular Devices, LLC, Sunnyvale, CA, USA) in the absence and presence of 500 nM tetrodotoxin (TTX). Patch pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus Ltd., Edenbridge, Kent, UK) with a multistage P-97 Flaming-Brown micropipette puller (Sutter Instruments Co., San Rafael, CA, USA) and fire-polished using a microforge (Narashige MF-830; Tokyo, Japan) to a pipette resistance of 1.5–2.5 MΩ. The pipette solution consisted of (in mM) 5 NaF, 105 CsF, 20 CsCl, 2 EGTA, and 10 HEPES with pH adjusted to 7.35 with CsOH and osmolarity adjusted to 290 mOsmol/kg with sucrose. The recording chamber was continuously perfused with a bath solution containing in (mM) 20 NaCl, 100 N-methyl-D-glucamine, 10 HEPES, 1.8 CaCl2·2H2O, 2 MgCl2·6H2O, and 20 tetraethylammonium chloride with pH adjusted to 7.35 with HCl and osmolarity adjusted to 310 mOsmol/kg with sucrose. The reference electrode consisted of a 2% agar bridge with composition similar to the bath solution. Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Voltage-clamp pulse generation and data collection were done using Clampex 10.2 (Molecular Devices, LLC). Cells were allowed to stabilize over a period of three minutes after achieving whole-cell configuration in order to allow whole-cell current amplitude to reach steady-state. Whole-cell capacitance was determined by integrating capacitive transients generated by a voltage step from −120 mV to −110 mV filtered at 100 kHz low pass Bessel filtering. Series resistance was compensated with prediction >70% and correction >85% to assure that the command potential was reached within microseconds with a voltage error <3 mV. Leak currents were subtracted by using an online P/4 procedure. All whole-cell currents were filtered at 5 kHz low pass Bessel filtering and digitized at 50 kHz. All voltage-clamp experiments were conducted from a holding potential of −120 mV at room temperature (20–23°C).
Current clamp recording
Whole-cell recordings of neuronal cell bodies were made in current clamp mode using an Axon MultiClamp 700B amplifier. Patch pipettes were fabricated from borosilicate glass to have pipette resistance 2.0 – 2.5 MΩ when filled with a solution containing (in mM: 110 K-gluconate, 10 KCl, 10 HEPES, 10 dextrose, 10 sucrose, 10 phosphocreatine-Na2, 5 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 0.1 CaCl2, pH 7.35, 300 mOsmol/kg). Bath solution consisted of (in mM) 155 NaCl, 3.5 KCl, 1 MgCl2, 1.5 CaCl2, 10 HEPES, pH 7.35. The reference electrode consisted of a 2% agar bridge with composition of the bath solution. Recordings were low-pass Bessel filtered at 5 kHz and digitized at 50 kHz. Membrane potential was clamped to −80 mV for both spontaneous and evoked action potential firing measurements. Action potentials were evoked by a 500 ms depolarizing current injection in 10 pA increments. All current clamp experiments were conducted at room temperature (20–23°C).
Electrophysiological data analyses
Data were analyzed using a combination of Clampfit 10.2 (Molecular Devices), Excel 2007 (Microsoft), and SigmaPlot 10.0 (Systat Software, Inc., San Jose, CA, USA). The peak current was normalized for cell capacitance and plotted against step voltage to generate a peak current density-voltage relationship. Whole-cell conductance (GNa) was calculated as GNa = I/(V − Erev), where I is the measured peak current, V is the step voltage, and Erev is the calculated sodium reversal potential. GNa at each voltage step was normalized to the maximum conductance between −80 mV and 20 mV. To calculate voltage dependence of activation (protocol described in figure insets), normalized GNa was plotted against voltage and fitted with the Boltzmann function G/Gmax= (1 + exp[(V − V1/2)/k])−1, where V1/2 indicates the voltage at half-maximal activation and k is a slope factor describing voltage sensitivity of the channel. Voltage dependence of steady-state inactivation was assessed by plotting currents generated by the −10 mV post-pulse voltage step normalized to the maximum current against pre-pulse voltage step from −140 to −10 mV in 10 mV increments. The plot was fitted with the Boltzmann function.
Spontaneous action potential firing was measured as the number of action potentials fired over a period of 3 to 8 minutes. Action potentials were counted in pClamp 10 using threshold search, with an overshoot crossing 0 mV being counted as an action potential. Spontaneous firing frequency was determined by dividing the number of action potentials fired by the measured time period (3 to 8 minutes). Evoked action potential firing was measured in pClamp10 as the number of action potentials evoked over 500 ms as a function of current injection.
Results are presented as mean ± SEM. Statistical comparisons were performed using unpaired two-tailed Student’s t-test or one-way ANOVA. P < 0.05 was considered statistically significant.
Single-cell RT-PCR
Glass capillaries were baked at 200°C for an hour before pulling them into patch pipettes. Bath and pipette solutions were filtered at 0.2 μm. Neurons plated on coverslips were copiously washed with bath solution before placing them into the recording chamber. After voltage-clamp experiments, cell contents were aspirated into the patch pipette containing 15 μl of pipette solution. The pipette contents were then promptly emptied into an ice-cold 0.5 ml microtube containing 2 μl rRNasin (40 U/μl; Promega Corp., Madison, WI, USA), 3 μl random hexamers (50 μM; Applied Biosystems, Carlsbad, CA, USA), and 4 μl RNase-free water for reverse transcription (RT). The PCR tube was immediately incubated at 65°C for 5 min, then cooled to 5°C and placed on ice. Half (12 μl) of the volume was transferred into another tube to serve as a negative RT control. To each PCR tube, 4 μl Moloney Murine Leukemia Virus (M-MLV) reverse transcriptase buffer (5X; Promega Corp.), 4 μl dithiothreitol (100 mM), and 1 μl PCR grade dNTP mix (10 mM per dNTP; Roche, Basel, Switzerland) were added with 1 μl M-MLV reverse transcriptase (200 U/μl; Promega Corp.) added to one PCR tube and 1 μl RNase-free water to the negative RT control. The final volume of each PCR tube was 20 μl. Reverse transcription reactions were performed at 42°C for 50 min, then at 70°C for 15 min.
A first round multiplex PCR was performed using 5 μl of cDNA template and primers for Gad67 and Gfap (Table 1). First round amplification was initiated by denaturation for 3 min at 94°C followed by 21 cycles of 30 sec at 94°C, 30 sec at 60°C, and 1 min at 72°C. Second round PCR (35 cycles) was performed separately using nested primers for each gene separately (Table 1) with 5 μl of first round PCR product as template. The product of the second round PCR was electrophoresed on 2% agarose gel and visualized by ethidium bromide staining.
Table 1.
Primer sequences of genes used to perform single-cell nested RT-PCR and the Scn1a real-time quantitative RT-PCR with resulting PCR product lengths.
| Gene (NCBI Accession) | Primer Sequence (5′ → 3′)1 | Product Length (bp) | |
|---|---|---|---|
|
Gad67 NM_008077.4 |
Outer | TACGGGGTTCGCACAGGTC CCCCAGGCAGCATCCACAT |
599 |
| Nested | TACGGGGTTCGCACAGGTC TCCCCCAGGAGAAAATATCC |
225 | |
|
Gfap NM_010277.3 |
Outer | AGAACAACCTGGCTGCGTAT CTCACATCACCACGTCCTTG |
807 |
| Nested | AGAAAGGTTGAATCGCTGGA CCAGGGCTAGCTTAACGTTG |
517 | |
|
Scn1a NM_018733.2 |
Primer set | TGATGAAAATGGCCCAAAGC ACTGAACCGGAAGATGGCC |
178 |
| Probe | 6FAM-CCTGGACCCCTACTATA-MGBNFQ |
Abbreviations: 6FAM, 6-carboxyfluorescein; MGBNFQ, molecular-groove binding non-fluorescence quencher
Outer and nested primers (Table 1) for Gad67, a marker for GABAergic neurons, and Gfap, an astrocytic glial marker, were designed to span introns. Final second round PCR products were sequence verified as the gene of interest. All PCR reactions were tested to ensure absence of genomic DNA amplification with negative RT controls. The specificity of these markers was confirmed by performing single-cell nested RT-PCR on morphologically identified hippocampal pyramidal cells (n = 10; Figure 1), which did not express Gad67 or Gfap at detectable levels. Furthermore, routine sham experiments (n = 10; Figure 1) were performed where bath solution surrounding plated hippocampal neurons on a coverslip in the recording chamber was sampled with a patch pipette and tested with the single-cell nested RT-PCR protocol to ensure absence of RNA contamination.
Figure 1. Morphologic and molecular identification of hippocampal neurons.

Top row illustrates representative bright-field images of acutely dissociated hippocampal pyramidal and bipolar neurons. Lower rows are representative agarose gel electrophoresis of single-cell RT-PCR reactions performed to detect Gad67 and Gfap expression. Bath fluid in the recording chamber was sampled as a negative control (column 1; n = 10). Pyramidal neurons (column 2) were identified by a long apical process and pyramidal shaped soma, and did not exhibit Gad67 or Gfap expression. Fusiform shaped cell bodies with bipolar processes (columns 3, 4) were classified as GABAergic interneurons only when Gad67 but not Gfap expression was detected (column 3).
Real-time quantitative RT-PCR
Intact hippocampi were dissected from male and female P14 and P21 mice. RNA was extracted from hippocampi using the TRIzol reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized from 2 μg of total RNA using the RT protocol described above. Real-time quantitative PCR was performed using TaqMan Gene Expression Assays for Scn2a (Applied Biosystems Assay ID: Mm01270359_m1), Scn3a (Mm00658167_m1), Scn8a (Mm00488110_m1) and TATA box binding protein (Tbp) (Mm00446971_m1), and a custom primer and probe set for Scn1a (Table 1). Real-time PCR was performed in triplicate (40 cycles) using TaqMan Fast Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA) on a 7900HT Fast Real-Time PCR System with Sequence Detection System 2.2 software (Applied Biosystems). All assays lacked detectable signal in no RT-controls and no template controls. Relative transcript levels were assessed using the 2−ΔΔCT method with Tbp as a reference gene (Livak & Schmittgen, 2001). Experiments for P14 and P21 were performed separately. For each age, statistical comparison between groups was made by a one-way ANOVA with a four level independent variable. Data are presented as mean fold change ± SE of four biological replicates.
RESULTS
Strain-dependent seizure severity and survival of Scn1a+/− mice
Heterozygous F1.Scn1a+/− mice exhibited spontaneous seizures beginning at P18 and premature lethality, similar to the phenotypes reported for the Scn1a exon 26 knockout and Scn1a-R1407X knock-in heterozygotes (Ogiwara et al., 2007; Yu et al., 2006; Miller et al, 2013). However, heterozygous 129.Scn1a+/− mice did not exhibit any overt phenotype (Miller et al, 2013).
Lifespan was significantly influenced by strain background with F1.Scn1a+/− mice exhibiting substantially reduced survival compared to 129.Scn1a+/− animals. On the F1 strain background, more than 50% of F1.Scn1a+/− mice died by 5 weeks of age, while on the 129 strain background more than 85% of mice survived beyond 14 weeks of age (Miller et al., 2013). Deaths in F1.Scn1a+/− heterozygotes that were occasionally witnessed followed a short-duration, generalized seizure with tonic hind limb extension.
By video-EEG monitoring we observed spontaneous generalized tonic-clonic seizures in 50% of F1.Scn1a+/− mice (Table 2), with an average seizure frequency of 1 per 31 ± 6 hours. On the 129 strain, we did not observe any spontaneous seizures in 129.Scn1a+/− mice in 403 hours of video-EEG monitoring (Table 2). Spontaneous seizures were never observed in wild-type 129 or F1 control mice (Table 2). Although they did not exhibit spontaneous seizures, 129.Scn1a+/− mice did exhibit a shorter latency to first generalized tonic-clonic seizure compared with age-matched wild-type littermates following exposure to the volatile convulsant drug flurothyl (129.WT: 293.5 ± 5.1 sec (n=18), 129.Scn1a+/−: 223.5 ± 5.7 sec (n=16); p < 0.0001). These data effectively demonstrate that 129.Scn1a+/− mice have a latent phenotype.
Table 2.
Spontaneous generalized tonic-clonic seizures (GTCS) recorded from Scn1a+/− and wild-type control mice on the 129 and F1 strain backgrounds.
| Genotype | GTCS | Total Hours of Monitoring | Age Range (days) | Number of Mice with Seizures (total n) |
|---|---|---|---|---|
| F1.Scn1a+/− | 8 | 401.5 | 20–101 | 2 (4) |
| 129.Scn1a+/− | 0 | 403 | 24–184 | 0 (8) |
| F1.WT | 0 | 221 | 22–102 | 0 (5) |
| 129.WT | 0 | 221 | 25–88 | 0 (3) |
Strain-dependence of interneuron sodium currents
Previous studies demonstrated that impaired excitability of GABAergic hippocampal interneurons contributes significantly to the hyperexcitability that leads to epilepsy in the heterozygous Scn1a+/− mouse model of Dravet syndrome (Bechi et al., 2012; Yu et al., 2006). We, therefore, examined INa density in GABAergic hippocampal interneurons from 129.Scn1a+/− and F1.Scn1a+/− mice to investigate whether differences in interneuron INa density correlate with strain-dependent seizure severity.
Electrophysiological recordings were made on acutely dissociated, fusiform-shaped hippocampal neurons with bipolar processes from postnatal day 21–24 old (P21–24) mice. Post-hoc single-cell RT-PCR was performed on all recorded neurons to detect expression of Gad67, a marker of GABAergic neurons not expressed in pyramidal neurons, and Gfap, an astrocytic glial marker. Cells were identified as GABAergic inhibitory interneurons only when Gad67 but not Gfap expression was detected (Figure 1). By this criterion, 48% of morphologically identified bipolar hippocampal neurons were Gad67 positive, and only these cells were used in data analysis.
Sodium current densities in GABAergic inhibitory interneurons from wild-type mice on F1 and 129 strain backgrounds were not statistically different. No significant sex-dependent differences in INa density were noted in wild-type or Scn1a+/− mice on F1 and 129 strain backgrounds (n = 5; all subgroups). GABAergic inhibitory interneurons from 129.Scn1a+/− mice, which have a phenotype virtually indistinguishable from wild-type animals, exhibited INa densities similar to their wild-type littermates (−415 ± 57 pA/pF vs. −418 ± 53 pA/pF, respectively; n = 10 each; Figure 2A, B, E). However, interneurons from phenotypically more severe F1.Scn1a+/− mice had significantly lower INa density compared to their wild-type littermates (−285 ± 45 pA/pF vs. −504 ± 53 pA/pF, respectively; p < 0.01; n = 10 each; Figure 2C, D, F), consistent with previous electrophysiological studies performed with P14–16 mice (Yu et al., 2006). These data suggest that differences in interneuron INa are a contributing factor to the strain-dependent seizure phenotype observed between Scn1a+/− mice on F1 129 strain backgrounds.
Figure 2. Strain dependence of hippocampal interneuron sodium current.

Tetrodotoxin (500 nM) subtracted, whole-cell voltage-clamp recordings from hippocampal GABAergic interneurons from postnatal day 21–24 old 129.Scn1a+/− (n = 10; 5 male), F1.Scn1a+/− (n = 10; 5 male), and their respective wild-type littermate mice (n = 10 each; 5 male). Representative whole-cell INa from (A) 129.WT, (B) 129.Scn1a+/−,(C) F1.WT, and (D) F1.Scn1a+/− were elicited by voltage steps from −80 to 60 mV in 10 mV increments from a holding potential of −120mV (inset in C). Peak current normalized to cell capacitance (current density) at tested potentials from (E) 129 and (F) F1 mice. Error bars represent SEM. Closed and open symbols represent wild-type and Scn1a+/− genotypes, respectively.
Thus, significant loss of interneuron INa density correlated with the more severe phenotype observed for F1.Scn1a+/− mice compared to 129.Scn1a+/− animals. We also investigated biophysical parameters describing voltage dependence of activation and steady-state inactivation and found that these measures of sodium channel activity were not different among the four subgroups (Table 3). Further, qualitative inspection of the time course of inactivation showed no overt differences between genotypes or strains in Gad67 positive neurons. These results suggest that gating kinetics and voltage dependence of sodium currents do not contribute to strain-dependent phenotypes. Differences in INa density may contribute to strain-dependent seizure severity, however, this divergent property alone does not explain the lower flurothyl induced seizure threshold measured for 129.Scn1a+/− mice
Table 3.
Biophysical parameters for activation and steady-state inactivation of whole-cell currents in hippocampal GABAergic interneurons.
| Voltage-dependence of activation
|
Steady-state inactivation
|
|||||
|---|---|---|---|---|---|---|
| V1/2 (mV) | k (mV) | n | V1/2 (mV) | k (mV) | n | |
| F1.WT | −26.1 ± 1.1 | 8.9 ± 0.4 | 10 | −64.6 ± 1.5 | 7.3 ± 0.3 | 10 |
| F1.Scn1a+/− | −22.5 ± 1.3 | 7.8 ± 0.3 | 10 | −61.1 ± 2.6 | 7.7 ± 0.3 | 10 |
|
| ||||||
| 129.WT | −25.3 ± 1.9 | 9.0 ± 0.5 | 10 | −65.6 ± 0.9 | 7.2 ± 0.1 | 10 |
| 129.Scn1a+/− | −26.2 ± 2.1 | 8.2 ± 0.3 | 10 | −62.8 ± 2.2 | 7.2 ± 0.1 | 10 |
Strain-dependence of pyramidal neuron sodium currents
In order to determine whether divergent properties of excitatory pyramidal cells may also contribute to strain-dependent epilepsy observed for Scn1a+/− animals, we performed electrophysiological recordings on acutely dissociated pyramidal hippocampal neurons from P21–24 wild-type and Scn1a+/− mice on F1 and 129 strain backgrounds. Sodium current densities in excitatory pyramidal neurons from wild-type mice were not significantly different between strains, and there were no significant sex-dependent differences in INa density noted among wild-type or Scn1a+/− mice on F1 and 129 strain backgrounds (n = 5; all subgroups). Excitatory pyramidal neurons from the phenotypically less severe 129.Scn1a+/− mice had significantly greater INa density compared to their wild-type littermates (−386 ± 46 pA/pF vs. −270 ± 23 pA/pF, respectively; p = 0.03; n = 10 each; Figure 3A, B, E). Similarly, pyramidal neurons from the phenotypically more severe F1.Scn1a+/− mice had significantly greater INa density compared to their wild-type littermates (−490 ± 31 pA/pF vs. −310 ± 26 pA/pF, respectively; p < 0.001; n = 10 each; Figure 3C, D, F). Interestingly, pyramidal neurons from F1.Scn1a+/− display a significantly larger increase in current density compared to the less severe 129.Scn1a+/− strain, suggesting both excitatory and inhibitory networks may contribute to strain-dependent seizure severity. Biophysical parameters describing voltage dependence of activation and steady-state inactivation were not different among the four subgroups (Table 4), and similarly, qualitative inspection of the time course of inactivation showed no overt differences between genotypes or strains.
Figure 3. Strain dependence of hippocampal pyramidal neuron sodium current.

Tetrodotoxin (500 nM) subtracted, whole-cell voltage-clamp recordings from hippocampal pyramidal neurons from postnatal day 21–24 old 129.Scn1a+/− (n = 10; 5 male), F1.Scn1a+/− (n = 10; 5 male), and their respective wild-type littermate mice (n = 10 each; 5 male). Representative whole-cell INa from (A) 129.WT, (B) 129.Scn1a+/−, (C) F1.WT, and (D) F1.Scn1a+/− were elicited by voltage steps from −80 to 60 mV in 10 mV increments from a holding potential of −120mV (inset in C). Peak current normalized to cell capacitance (current density) at tested potentials from (E) 129 and (F) F1 mice. Error bars represent SEM. Closed and open symbols represent wild-type and Scn1a+/− genotypes, respectively.
Table 4.
Biophysical parameters for activation and steady-state inactivation of whole-cell currents in hippocampal pyramidal neurons from postnatal day 14–15 and 21–24 mice.
| Voltage-dependence of activation
|
Steady-state inactivation
|
|||||
|---|---|---|---|---|---|---|
| V1/2 (mV) | k (mV) | n | V1/2 (mV) | k (mV) | n | |
| Postnatal Days 14–15 | ||||||
| F1.WT | −26.6 ± 1.2 | 9.3 ± 0.4 | 10 | −58.2 ± 2.0 | 6.4 ± 0.2 | 10 |
| F1.Scn1a+/− | −24.6 ± 1.1 | 8.8 ± 0.4 | 10 | −64.3 ± 2.8 | 7.1 ± 0.2 | 10 |
|
|
||||||
| Postnatal Days 21–24 | ||||||
| F1.WT | −26.9 ± 1.1 | 8.7 ± 0.5 | 10 | −65.2 ± 1.3 | 7.3 ± 0.3** | 10 |
| F1.Scn1a+/− | −30.1 ± 1.3** | 9.0 ± 0.5 | 10 | −66.5 ± 1.2 | 7.1 ± 0.2 | 10 |
|
| ||||||
| 129.WT | −26.3 ± 1.4 | 8.0 ± 0.4 | 10 | −67.2 ± 1.4 | 7.8 ± 0.1 | 10 |
| 129.Scn1a+/− | −27.5 ± 2.0 | 7.7 ± 0.5 | 10 | −65.8 ± 2.0 | 7.9 ± 0.4 | 10 |
indicates p < 0.05 compared to respective genotype at P14–15 day old; statistical comparison made using one-way ANOVA followed by Tukey’s post test.
As shown above, excitatory pyramidal cells isolated from both Scn1a+/− strains at P21 – P24 exhibited increased peak sodium current density compared to wild-type controls. We hypothesized that persistent sodium current, a common biophysical defect identified in sodium channel mutations associated with epilepsy, might also be augmented in these cells. However, we found no difference in persistent sodium current among the four subgroups (129.WT: 0.32 ± 0.16%; 129.Scn1a+/−: 0.30 ± 0.16%, F1.WT: 0.29 ± 0.08%, 129.Scn1a+/−: 0.32 ± 0.09%; n = 9 – 11 for all strains) We concluded that an exaggerated increase in pyramidal neuron INa density correlated with a more severe phenotype in F1.Scn1a+/− mice.
Age-dependence of pyramidal neuron sodium currents
Our results demonstrate that pyramidal neurons from P21–24 Scn1a+/− mice have increased INa density. Previous studies performed with younger Scn1a+/− mice did not find a difference in pyramidal neuron INa density between heterozygous and wild-type mice (Bechi et al., 2012; Yu et al., 2006). In order to investigate age-dependence of pyramidal neuron INa density, we examined INa in acutely dissociated, pyramidal neurons from the phenotypically more severe F1.Scn1a+/− mice at P14–15.
Sodium current densities recorded in pyramidal neurons from P14–15 F1.Scn1a+/− mice were not significantly different from neurons of age-matched wild-type littermates (−304 ± 41 pA/pF vs. −327 ± 28 pA/pF, respectively; n = 10 each; Figure 4A). Further, pyramidal neuron INa density was not different in F1 wild-type mice as they aged from P14–15 to P21–24 (−327 ± 28 pA/pF to −310 ± 26 pA/pF; n = 10 each; Figure 4B). However, pyramidal neurons from P21–24 F1.Scn1a+/− mice exhibited a significantly greater INa density than neurons from P14–15 F1.Scn1a+/− mice (−490 ± 31 pA/pF vs. −304 ± 41 pA/pF, respectively; p < 0.002; n = 10 each; Figure 4B). The voltage dependence of activation in F1.Scn1a+/− mice demonstrated a significant hyperpolarizing shift from −24.6 mV at P14–15 to −30.1 mV at P21–24 (p = 0.02; Table 4). Steady-state inactivation was not significantly different between P21–24 and P14–15 pyramidal neurons from the same genotypes in the F1 strain (Table 4). Hence, we observed significant age-dependent changes in pyramidal INa density as well as the voltage dependence of activation in F1.Scn1a+/− mice.
Figure 4. Age dependence of hippocampal pyramidal neuron sodium current.
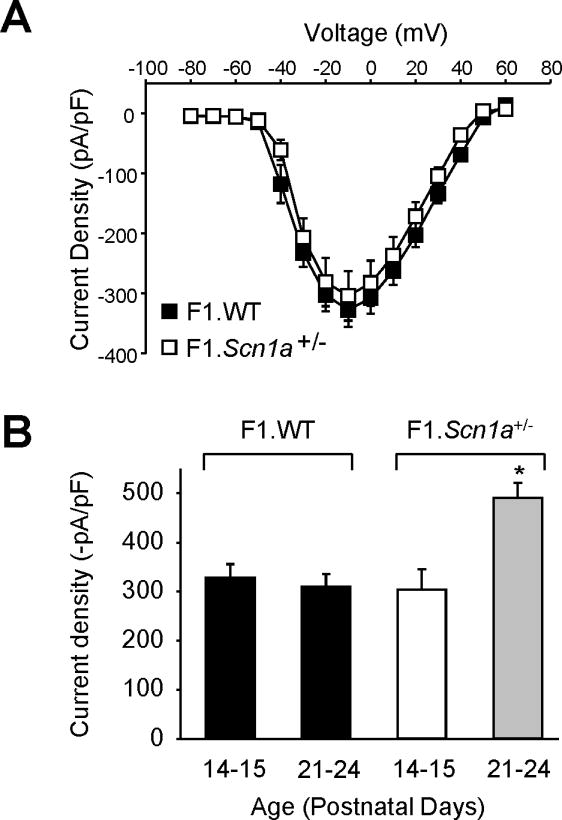
Tetrodotoxin (500 nM) subtracted, whole-cell voltage-clamp data from hippocampal pyramidal neurons from postnatal day 14–15 old F1.Scn1a+/− (n = 10; 5 male) and their wild-type littermate mice (n = 10 each; 5 male). (A) Peak current normalized to cell capacitance (current density) at tested potentials. Closed and open symbols represent wild-type and Scn1a+/− genotypes, respectively. (B) Average peak pyramidal neuron INa density from F1 wild-type and Scn1a+/− mice at postnatal day 14–15 (from Figure 4A) and 21–24 old (from Figure 3E) (*, p < 0.002)
Pyramidal neuron excitability in Scn1a+/− mice
The striking strain-dependent differences in pyramidal neuron INa density in Scn1a+/− mice prompted us to investigate the excitability of these neuronal populations. Pyramidal neurons from P21–24 F1.Scn1a+/− mice exhibited high frequency spontaneous action potential firing in contrast with neurons from wild-type animals that had very infrequent firing (2.99 ± 0.65 Hz, n = 8, vs 0.09 ± 0.5 Hz, n = 4, p < 0.001; Figure 5). Pyramidal neurons from the phenotypically less severe P21–24 129.Scn1a+/− mice also exhibited greater spontaneous firing frequencies than age-matched wild-type littermates (0.78 ± 0.17 Hz, n = 6, vs 0.14 ± 0.04 Hz, n=8; Figure 5), but the rate of firing was much less than F1.Scn1a+/− animals (p = 0.007 compared to F1.Scn1a+/− mice).
Figure 5. Spontaneous firing of pyramidal neurons.

Representative current clamp recordings of spontaneous action potentials from P21–24 hippocampal pyramidal neurons from (A) 129.WT, (B) 129.Scn1a+/−, (C) F1.WT, and (D) F1.Scn1a+/− mice. (E). Summary data plotting spontaneous action potential firing frequency calculated over 3 minutes of recording for 129 and F1 strains. Data are plotted as mean ± SEM for n = 4 – 8 cells.
In addition to spontaneous action potential firing, we also examined evoked action potential firing from each of the four subgroups by stepping across a range of 500 ms depolarizing current injections in 10 pA increments. Pyramidal neurons from both 129 and F1 wild-type show increased firing across the current injection range, reaching a peak before action potential number declined with further increases in current injection (Figure 6E, F). In contrast, while pyramidal neurons from the phenotypically less severe 129.Scn1a+/− strain showed a similar firing pattern compared to wild-type, F1.Scn1a+/− pyramidal neurons showed greater excitability across the range of current injection, with high frequency firing even at highly depolarizing current injection steps (Figure 6F).
Figure 6. Evoked firing of pyramidal neurons.

Representative current clamp recordings of evoked action potentials at increasing current injections from P21–24 hippocampal pyramidal neurons from (A) 129.WT, (B) 129.Scn1a+/−, (C) F1.WT, and (D) F1.Scn1a+/− mice. Summary data plotting action potential number vs. current injection for (E) 129 and (F) F1 mice. Data are plotted as mean ± SEM for n = 6 – 11 cells.
Hippocampal sodium channel expression in Scn1a+/− mice
To probe the molecular basis for strain-dependent differences in pyramidal neuron INa density in Scn1a+/− mice, we performed real-time quantitative RT-PCR on RNA isolated from P14 and P21 dissected hippocampi to assess relative expression of Scn1a, Scn2a, Scn3a, and Scn8a. For each age, we compared sodium channel transcript expression between groups using one-way ANOVA. As expected, there were significantly lower hippocampal Scn1a transcript levels in 129.Scn1a+/− and F1.Scn1a+/− mice compared to wild-type in both strains at P14 [F(3,12)=125.75, p<0.0001] and P21 [F(3,12)= 67.64 p<0.0001]. At P14, the reduction was smaller in F1.Scn1a+/− compared to 129.Scn1a+/− mice, with a 2.7-fold expression difference between strains (Table 5). However, by P21 levels of Scn1a transcript are equivalent in 129.Scn1a+/− and F1.Scn1a+/− mice, with both expressing approximately half of wild-type levels (Table 5). At P14, 129.Scn1a+/− mice exhibit significantly lower hippocampal Scn2a expression [F(3,12)=10.47, p<0.002] and Scn3a [F(3,12)=3.55, p<0.05] compared to 129 wildtype mice, while F1.Scn1a+/− mice express 1.8 fold more Scn8a transcript than 129.Scn1a+/− mice [F(3,12)=3.92, p<0.04] (Table 5). At P21, there was a trend toward higher Scn2a (p=0.0506) and Scn8a (p=0.0875) transcript expression in F1.Scn1a+/− mice compared to 129.Scn1a+/− mice, although these did not reach statistical significance. At both ages, there were no significant differences in sodium channel transcript levels between strains in wild-type mice (Table 5).
Table 5.
Relative hippocampal expression of voltage-gated sodium channel transcripts. Data are mean fold change ± SEM (n=4 per group). Statistical comparisons were made using one-way ANOVA with Fisher’s PLSD post-hoc tests.
| Postnatal Day 14 | Postnatal Day 21 | |||||||
|---|---|---|---|---|---|---|---|---|
| Scn1a | Scn2a | Scn3a | Scn8a | Scn1a | Scn2a | Scn3a | Scn8a | |
| F1.WT relative to 129.WT | 1.06 (0.97–1.17) | 0.90 (0.82–0.99) | 1.11 (0.97–1.28) | 1.40 (1.14–1.72) | 0.89 (0.84–0.96) | 0.944 (0.91–0.98) | 0.87 (0.83–0.92) | 0.89 (0.84–0.94) |
| F1.Scn1a+/− relative to 129.Scn1a+/− | 2.74† (2.44–3.06) | 1.24* (1.14–1.32) | 1.20 (1.02–1.42) | 1.82* (1.48–2.25) | 1.03 (0.95–1.12) | 1.17 (1.06–1.28) | 1.01 (0.93–1.11) | 1.17 (1.05–1.30) |
| 129.Scn1a+/− relative to 129.WT | 0.18† (0.16–0.20) | 0.66** (0.61–0.72) | 0.70* (0.61–0.81) | 0.77 (0.59–0.99) | 0.44† (0.42–0.47) | 0.95 (0.88–1.02) | 0.91 (0.85–0.99) | 0.88 (0.80–0.96) |
| F1.Scn1a+/− relative to F1.WT | 0.47† (0.42–0.52) | 0.90 (0.84–0.97) | 0.76 (0.65–0.89) | 1.00 (0.86–1.16) | 0.51† (0.47–0.56) | 1.17 (1.10–1.25) | 1.06 (1.00–1.13) | 1.15 (1.07–1.24) |
Significance is indicated with the following symbols:
p<0.05;
p< 0.001
p<0.0001.
DISCUSSION
Genetic epilepsies caused by mutations in genes encoding voltage-gated sodium channels exhibit variable expressivity, a common phenomenon among monogenic disorders and often ascribed to the action of genetic modifiers. Strain-dependence of phenotype severity in genetically engineered mice can provide a tractable model for investigating the genetic basis of variable disease expression (Kearney, 2011). In this study, we exploited a DS mouse model to investigate the neurophysiological basis for stain-dependent epilepsy severity.
DS has been successfully modeled in heterozygous Scn1a+/− knockout mice and in mice with a targeted nonsense Scn1a mutation (R1407X) (Ogiwara et al., 2007; Yu et al., 2006). We recently targeted exon 1 of Scn1a to generate a null allele on a pure 129S6/SvEvTac background. Our model recapitulated these earlier findings when observed on a mixed 129:B6 background (Miller et al., 2013). In all three mouse models of DS, a substantial strain-dependence of the phenotype is evident. The primary defect in DS associated with SCN1A mutations is loss of NaV1.1 function owing to truncating mutations, non-truncating alleles that confer impaired trafficking, or other potential mechanisms. As a consequence, diminished INa density specific to GABAergic interneurons contributes to reduced activity of inhibitory networks and gains in neuronal excitability presumably because of an excitatory-inhibitory imbalance. However, the neurophysiological basis for less severe phenotype in DS mice on specific genetic backgrounds was unknown.
Hippocampal neuron sodium current density and variable phenotypes
Our electrophysiological study of GABAergic and pyramidal hippocampal neurons from two different strains of heterozygous Scn1a+/− mice provides a neurophysiological explanation for their divergent seizure phenotype and advances our understanding of the contribution of pyramidal neurons to phenotype expression in these mice. We observed that INa density in GABAergic interneurons was lower in F1.Scn1a+/− than 129.Scn1a+/− mice, which correlates inversely with phenotype severity. Further, F1.Scn1a+/− mice exhibit a greater increase in pyramidal neuron INa density than 129.Scn1a+/−. These observations suggest that INa density in excitatory neuronal populations may contribute to the phenotype observed in DS mice. This is consistent with recent work demonstrating that selective deletion of Scn1a in forebrain interneurons resulted in a more severe phenotype than global deletion, while coupled deletion of Scn1a in forebrain excitatory and inhibitory neurons more closely approximated the global deletion phenotype (Ogiwara et al., 2013). We speculate that preserved interneuron INa in 129.Scn1a+/− mice is sufficient to protect animals from spontaneous seizures despite a moderate elevation in pyramidal neuron INa density..
We also observed an age-dependent elevation of pyramidal neuron INa density in F1.Scn1a+/− mice coupled with a hyperpolarizing shift in activation voltage dependence that suggest a plausible explanation for the age-dependence of mortality in these animals. Premature death in F1.Scn1a+/− mice begins at P19 and is attributed to severe seizures. The convergence of reduced INa in GABAergic interneurons with an elevated and more negatively activating pyramidal neuron INa at age P21–24 likely creates a substantial inhibitory-excitatory imbalance, promoting more frequent life-threatening seizures around this developmental time point.
Consistent with our findings, recent electrophysiological studies performed in brain slices of Scn1a+/− mice reveal hyperexcitability. Hippocampal CA1 and prefrontal cortex slices from P21–25 Scn1a+/− mice have increased frequency of spontaneous excitatory postsynaptic currents (EPSC), where CA1 pyramidal neurons exhibited a specific increase in AP-dependent EPSCs (Han et al., 2012). Hyperexcitability in hippocampal slices has also been noted to be age-dependent as Scn1a+/− mice age from P14–18 to P30–36 (Liautard et al., 2013). Further evidence for hyperexcitability comes from studies of pyramidal neurons differentiated from Dravet syndrome patient-derived IPSCs which also show increased INa and spontaneous firing compared to control-derived neurons (Liu et al., 2013). Our results corroborate data observed in slices and patient-derived neurons, and provide insight into how anti-homeostatic alterations to voltage dependent sodium currents at the single cell level may induce large changes in network excitability.
Transcriptional mechanisms underlying strain-dependent epilepsy severity
We observed preservation of INa density in 129.Scn1a+/− interneurons and elevated pyramidal INa density in both knockout strains, suggesting possible changes in sodium channel expression. We investigated global patterns of sodium channel gene expression in isolated hippocampal tissue (Table 5) and discovered some notable differences between strains. Specifically, at P14 we detected a significant difference in response to Scn1a deletion in 129.Scn1a+/− mice compared to F1.Scn1a+/− mice, with 129.Scn1a+/− mice exhibiting a larger than expected reduction. Although this difference resolved by P21, it suggests that there may be strain differences in compensatory response to Scn1a deletion. In addition, we detected a trend toward upregulation of Scn2a and Scn8a transcripts in F1.Scn1a+/− mice compared to wild-type. The phenotypically less severe 129.Scn1a+/− animals showed significant downregulation of Scn2a or Scn3a expression relative to wild-type at P14, while there were no differences in Scn2a, Scn3a or Scn8a expression relative to wild-type at P21. Previous studies have demonstrated that NaV1.2, encoded by Scn2a, and NaV1.6, encoded by Scn8a are highly expressed in pyramidal neurons by P21 (Krzemien et al., 2000; Schaller and Caldwell, 2000; Westenbroek et al., 1989). Although our experiment does not directly address cell specific expression changes, pyramidal neurons are far more numerous than interneurons in mouse hippocampus (Jinno & Kosaka, 2010) and constitute the majority (over 90%) of the neurons in the human hippocampus (Heckers & Konradi, 2010; Konradi et al., 2011). Elevated expression of Scn2a and/or Scn8a sodium channel subunits could account for increased INa in pyramidal neurons from phenotypically severe F1.Scn1a+/− mice observed at P21. The difference in Scn8a expression in F1.Scn1a+/− mice is intriguing given that gain-of-function mutation in this sodium channel is associated with early-onset epileptic encephalopathy, while loss-of-function mutations restore normal seizure thresholds and rescue survival of Scn1a+/− mice (Martin et al., 2007; Veeramah et al, 2012; O’Brien and Meisler, 2013). Similarly, transgenic expression of a gain-of-function Scn2a mutation exacerbates the phenotype of GEFS+ mice, another model for Scn1a-based epilepsy (Hawkins et al., 2011). Future studies examining expression of sodium channel subunits at the cellular level will be necessary to address cell-specific expression changes.
A thorough exploration of the developmental origins of altered neuronal INa density in Scn1a+/− mice is beyond the scope of this study. Future experiments will need to focus on the developmental time course of sodium channel isoform expression in GABAergic interneurons and pyramidal neurons, and on determining the contribution of synaptic activity to shaping final INa levels in fully differentiated neurons in the wild-type and disease states. Evidence from rodent studies indicates that GABAergic synaptic development precedes that of glutamatergic synapses (Kuzirian & Paradis, 2011). Conceivably, impaired GABAergic signaling during early development may alter the trajectory of sodium channel expression in both inhibitory and/or excitatory neurons. Our findings of differences in INa density and firing frequency may represent the persistent effects of electrical remodeling events during neuron development occurring in the absence of normal GABAergic signaling.
Conclusions
We have shown that strain-dependent epilepsy observed in Scn1a+/− mice on F1 and 129 strain backgrounds results from a combination of a loss-of-function phenotype in inhibitory interneurons, and a gain-of-function phenotype in excitatory pyramidal cells. We conclude that age-dependent changes in sodium channel currents contribute to variable excitability profiles of these cells, and strain-dependent seizure phenotypes that may correlate with strain-dependent changes in sodium channel gene expression.
HIGHLIGHTS.
Scn1a+/− mice have a more severe phenotype on the (129xB6)F1 strain compared to 129.
Interneuron sodium current density is reduced in affected F1.Scn1a+/− mice.
Interneuron sodium current density is unchanged in unaffected 129.Scn1a+/− mice.
Scn1a+/− pyramidal neurons exhibit age-dependent elevation of sodium current density.
Elevation of pyramidal neuron sodium current density correlates with premature lethality peak.
Acknowledgments
We thank Andrew Tapper, Ph.D., for providing PCR primer sequences of Gad67 gene, Clint McCollom for mouse husbandry, Jennifer Kunic for TaqMan probe and primer design, and Danny Winder for assistance with current clamp experimental design and analysis. This work was supported by National Institutes of Health grants [NS032387 to A.L.G., NS053792, NS063097 to J.A.K.]; and Howard Hughes Medical Institute Medical Research Fellowship to A.M.M.
ABBREVIATIONS
- 129
129S6/SvEvTac
- B6
C57BL/6J
- DS
Dravet syndrome
- EEG
electroencephalography
- EPSC
excitatory post-synaptic current
- Flurothyl
2,2,2-trifluroethylether
- GNa
sodium whole-cell conductance
- INa
sodium current
- M-MLV
Moloney Murine Leukemia Virus
- NaV
voltage-gated sodium channel
- TTX
tetrodotoxin
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bechi G, Scalmani P, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Pure haploinsufficiency for Dravet syndrome Na(V)1. 1 (SCN1A) sodium channel truncating mutations. Epilepsia. 2012;53:87–100. doi: 10.1111/j.1528-1167.2011.03346.x. [DOI] [PubMed] [Google Scholar]
- Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain. 2012;135:2329–2336. doi: 10.1093/brain/aws151. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Kalume F, Oakley JC. NaV1. 1 channels and epilepsy. J Physiol (London) 2010;588:1849–1859. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes LR, Deprez L, Suls A, Baets J, Smets K, Van DT, et al. The SCN1A variant database: a novel research and diagnostic tool. Hum Mutat. 2009 doi: 10.1002/humu.21083. [DOI] [PubMed] [Google Scholar]
- Goldberg-Stern H, Aharoni S, Afawi Z, Bennett O, Appenzeller S, Pendziwiat M, Biochem D, Kuhlenbäumer G, Basel-Vanagaite L, Shuper A, Korczyn AD, Helbig I. Broad Phenotypic Heterogeneity due to a Novel SCN1A Mutation in a Family With Genetic Epilepsy With Febrile Seizures Plus. J Child Neurol. 2013 Nov 20; doi: 10.1177/0883073813509016. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, et al. Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature. 2012;489:385–390. doi: 10.1038/nature11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, Martin MS, Frankel WN, Kearney JA, Escayg A. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiol Dis. 2011;41:655–660. doi: 10.1016/j.nbd.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Konradi C. Hippocampal pathology in schizophrenia. Curr Top Behav Neurosci. 2010;4:529–553. doi: 10.1007/7854_2010_43. [DOI] [PubMed] [Google Scholar]
- Ito S, Ogiwara I, Yamada K, Miyamoto H, Hensch TK, Osawa M, et al. Mouse with Na(v)1. 1 haploinsufficiency, a model for Dravet syndrome, exhibits lowered sociability and learning impairment. Neurobiol Dis. 2012;49C:29–40. doi: 10.1016/j.nbd.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Jinno S, Kosaka T. Stereological estimation of numerical densities of glutamatergic principal neurons in the mouse hippocampus. Hippocampus. 2010;20:829–840. doi: 10.1002/hipo.20685. [DOI] [PubMed] [Google Scholar]
- Kearney JA. Genetic modifiers of neurological disease. Curr Opin Genet Dev. 2011;21:349–353. doi: 10.1016/j.gde.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Sugawara T, Mazaki-Miyazaki E, Hoshino K, Nomura Y, Tateno A, et al. A missense mutation in SCN1A in brothers with severe myoclonic epilepsy in infancy (SMEI) inherited from a father with febrile seizures. Brain Dev. 2005;27:424–430. doi: 10.1016/j.braindev.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Konradi C, Zimmerman EI, Yang CK, Lohmann KM, Gresch P, Pantazopoulos H, et al. Hippocampal interneurons in bipolar disorder. Arch Gen Psychiatry. 2011;68:340–350. doi: 10.1001/archgenpsychiatry.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzemien DM, Schaller KL, Levinson SR, Caldwell JH. Immunolocalization of sodium channel isoform NaCh6 in the nervous system. J Comp Neurol. 2000;420:70–83. [PubMed] [Google Scholar]
- Kuzirian MS, Paradis S. Emerging themes in GABAergic synapse development. Prog Neurobiol. 2011;95:68–87. doi: 10.1016/j.pneurobio.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liautard C, Scalmani P, Carriero G, de CM, Franceschetti S, Mantegazza M. Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome. Epilepsia. 2013;54:1251–1261. doi: 10.1111/epi.12213. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, O’Malley HA, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol. 2013 doi: 10.1002/ana.23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lossin C. A catalog of SCN1A variants. Brain Dev. 2009;31:114–130. doi: 10.1016/j.braindev.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of Severe Myoclonic Epilepsy of Infancy. Hum Mol Genet. 2007;16:2892–2899. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010–2017. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AR, Hawkins NA, McCollom CE, Kearney JA. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 2013 doi: 10.1111/gbb.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JE, Meisler MH. Sodium channel SCN8A (Nav1. 6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet. 2013;4:213. doi: 10.3389/fgene.2013.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet. 2013 Aug 6; doi: 10.1093/hmg/ddt331. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, et al. NaV1. 1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda-Trujillo N, Carrizosa J, Cornejo W, Arias W, Franco C, Cabrera D, et al. A novel SCN1A mutation associated with severe GEFS+ in a large South American pedigree. Seizure. 2005;14:123–128. doi: 10.1016/j.seizure.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Schaller KL, Caldwell JH. Developmental and regional expression of sodium channel isoform NaCh6 in the rat central nervous system. Journal of Comparative Neurology. 2000;420:84–97. [PubMed] [Google Scholar]
- Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90:502–510. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Merrick DK, Catterall WA. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 1989;3:695–704. doi: 10.1016/0896-6273(89)90238-9. [DOI] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nature Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology. 2011;76:594–600. doi: 10.1212/WNL.0b013e31820c309b. [DOI] [PubMed] [Google Scholar]