

Abstract
The use of decellularized tendon tissue as a scaffold for tendon tissue engineering provides great opportunities for future clinical and current research applications. The aim of this study was to assess the effect of repetitive freeze-thaw cycles and two different detergents, t-octyl-phenoxypolyethoxyethanol (Triton X-100) and sodium dodecyl sulfate (SDS), on decellularization effectiveness and cytocompatibility in large tendons. Freshly collected equine superficial and deep digital flexor tendons were subjected to decellularization according to four different protocols (1 and 2: freeze-thaw cycles combined with either Triton X-100 or SDS; 3 and 4: Triton X-100 or SDS). Decellularization effectiveness was assessed based on the reduction of vital cell counts, histologically visible nuclei, and DNA content. Transmission electron microscopy was performed to evaluate cellular and extracellular matrix integrity. Further, cytocompatibility of scaffolds that had been decellularized according to the protocols including freeze-thaw cycles (protocols 1 and 2) was assessed by seeding the scaffolds with superparamagnetic iron oxide labeled mesenchymal stromal cells and monitoring the cells histologically and by magnetic resonance imaging for two weeks. Decellularization was significantly more effective when using the protocols including freeze-thaw cycles, leaving only roughly 1% residual nuclei and 20% residual DNA, whereas samples that had not undergone additional freeze-thaw cycles contained roughly 20% residual nuclei and 40% residual DNA. No morphological extracellular matrix alterations due to decellularization could be observed. Scaffolds prepared by both protocols including freeze-thaw cycles were cytocompatible, but the cell distribution into the scaffold tended to be better in scaffolds that had been decellularized using freeze-thaw cycles combined with Triton X-100 instead of SDS.
Introduction
Tendon and ligament pathologies are common in athletes and elderly people, Achilles tendinopathy and traumatic rupture of the anterior cruciate ligament being examples of the most frequently occurring conditions.1,2 Tissue engineering has become an important discipline in this field, aiming to improve the currently available therapeutic approaches that, until today, are often not evidence-based and fail to prevent re-injuries effectively.2–6 For treatment of tendinopathy, approaches in the sense of in vivo tissue engineering are currently under investigation, among others the local application of stem cells or growth factors,7–9 although the underlying mechanisms are not yet completely understood. Tendon or ligament ruptures, however, often require surgical replacement and thus a graft based on autologous, allo- or xenogeneic biological, or synthetic materials.6,10–12 Tendons and ligaments are highly specialized tissues, characterized by high extracellular matrix density, relatively low cellularity and vascularity, and outstanding mechanical properties combining stiffness and elasticity, which makes the design of appropriate bioartificial scaffolds demanding.10 The ideal scaffold should not only be biocompatible and match the natural biomechanical properties, but should also comprise naturally structured, extracellular matrix proteins that can interact with the repopulating cells and direct them toward tenogenic differentiation and matrix remodeling.10,11 Decellularization of tendon tissue offers the unique opportunity of obtaining a scaffold with a natural extracellular matrix structure that is hypoimmunogenic13,14 and displays biomechanical properties very similar to the original tissue,15–18 providing great advantages for potential clinical application and research use.
Different protocols for decellularization of tendon or ligament tissue have been investigated, using physical or chemical and/or enzymatical methods.15–21 Results obtained after applying different detergent-based protocols remain conflicting. While t-octyl-phenoxypolyethoxyethanol (Triton X-100) did not lead to satisfactory results in an early study,15 it was found to be most effective by others.19 Sodium dodecyl sulfate (SDS) was reported to be effective but to reduce cytocompatibility due to matrix alterations; however, the protocol used in these studies did not only include incubation in SDS, but also in Triton X-100.11,17,22,23 Further, several groups performed detergent-based decellularization with samples that had previously been frozen or freeze-dried15,16,24; it should be acknowledged that this may have had an additional effect on decellularization effectiveness.
Freeze-thaw cycles alone were used to prepare cell-free tendon matrices.25 However, our own unpublished data showed that this procedure, although reducing the content of vital cells, did not remove DNA, which was recently supported by others.18 Freeze-thaw cycles with subsequent nuclease treatment led to nearly complete removal of cells and DNA,18 but nuclease treatment was performed on sliced tendon only,18,21 thus, this protocol does not appear applicable for decellularization of whole, large tendon constructs.
The combination of freeze-thaw cycles and detergents for decellularization of tendon tissue was not investigated so far. Further, the effectiveness of current published decellularization protocols was commonly assessed using tendons of small diameter, including rat tail tendons,15 human forearm tendons,24,26 or tendon slices.18,21 Decellularization of large tendons appears challenging due to the high density of the tissue, which might prevent penetration of chemicals into the core. We hypothesized that the combination of freeze-thaw cycles and detergents would be effective for the preparation of large, cell-free tendon constructs. To investigate this hypothesis, in the present study, we assessed the effect of freeze-thaw cycles combined with subsequent incubation in either Triton X-100 or SDS on decellularization of large tendon structures.
Materials and Methods
Study design
Samples from six horses were recovered to evaluate the effectiveness of four different decellularization protocols using two large tendon structures of the distal limb that differ in diameter (superficial and deep digital flexor tendons; SDFT and DDFT [Fig. 1]). All protocols were carried out with both SDFT and DDFT from each animal to allow paired comparisons between the two tendon structures on the one hand and the four protocols on the other hand. The evaluated parameters included vital cell counts, histologically visible nuclei, and DNA content. Further tendons from three animals were used to assess cellular and extracellular matrix integrity by transmission electron microscopy (TEM) subsequent to decellularization according to the four protocols. Thereafter, samples from three animals were used to investigate the cytocompatibility of tendons that had undergone decellularization protocols including freeze-thaw cycles.
FIG. 1.

Large tendon structures used in the study: the equine superficial digital flexor tendon (SDFT) has a crescent-shaped cross-section with a width of roughly 0.5 cm, while the equine deep digital flexor tendon (DDFT) displays a circular shape of cross-section of roughly 1 cm diameter. Color images available online at www.liebertpub.com/tec
Sample collection
Fresh cadaver limbs of thoroughbred and warm-blood horses were obtained from an abatoir. Within 6 h after slaughter, 10 cm long pieces of SDFT and DDFT were aseptically recovered from the metacarpal region of all four matched limbs and stored in phosphate buffered saline (PBS; PAA Laboratories) overnight.
Decellularization
Two cm of each tendon were set aside to be used as internal controls. The remaining 8 cm long tendons were subjected to decellularization according to four protocols, which differed either in the application of freeze-thaw cycles or in the detergent used (Triton X-100 or SDS). Group 1 and 2, herein referred to as Cryo-Triton and Cryo-SDS, underwent five cycles of 2 min freezing in liquid nitrogen and 10 min thawing in PBS at 37°C21 before further decellularization. Group 3 and 4, herein referred to as Triton and SDS, did not undergo freeze-thaw cycles. All further decellularization and washing steps were performed at room temperature and under continuous agitation. Tendons of all groups were placed in hypotonic solution (aqua dest.) for 48 h. Thereafter, tendons were incubated for 48 h in Tris buffer (Carl Roth) (pH 7.6) containing either 1% Triton X-100 (Carl Roth) (Cryo-Triton and Triton groups) or 1% SDS (Carl Roth) (Cryo-SDS and SDS groups). All protocols were concluded by the following washing steps: 2×15 min in aqua dest., overnight in cell culture medium (DMEM 1 g glucose/l (Invitrogen), supplemented with 10% fetal bovine serum (FBS; Sigma Aldrich), 1% penicillin-streptomycin (PAA Laboratories), and 0.1% gentamycin (Invitrogen), and again overnight in PBS.
Assessment of decellularization effectiveness
For each of the analyses, two central pieces of each decellularized sample were used, of which one was obtained from the middle of the tendon length and the other at 1 cm distance from the peripheral ends, and a nondecellularized piece of the same tendon, serving as internal control. As no differences were evident between the results obtained from the two different decellularized sample pieces, their mean values were used for statistical analyses.
Collagenase digestion and vital cell count
One gram of tissue was minced into 1 mm3 pieces. Minced pieces were washed in Hank's balanced salt solution (HBSS; PAA Laboratories) and then incubated in collagenase I (Invitrogen) solution (5.6 mg/mL HBSS) for 8 h under permanent shaking at 37°C. Released cells were pelleted and then resuspended for counting by use of a hemocytometer and trypan blue for exclusion of nonvital cells.
Histology and nuclei count
Samples were fixed in 4% paraformaldehyde and embedded in paraffin. Longitudinal 6 μm sections were prepared and stained with hematoxylin and eosin. Randomly chosen regions of the slides were photographed at 20×magnification (IX51 research microscope; CC-12 digital color camera; Cell^A software; Olympus Soft Imaging System GmbH). Visible cell nuclei were counted in three images obtained from each sample piece, thus in a total of six images per decellularized sample.
DNA content
Two hundred milligram of tissue were minced into 1 mm3 pieces, washed in PBS, and subjected to papain (Sigma Aldrich) digestion at a concentration of 0.125 mg/mL at 60°C for 24 h. Digested tissue samples were stored at −20°C until further analysis. DNA content was then determined using the Quant-iT™ PicoGreen® dsDNA assay kit (Invitrogen). Samples were pipetted into 96-well plates and an equal volume of PicoGreen reagent working solution was added, followed by incubation for 5 min at room temperature, protected from light. Fluorescence at an excitation wavelength of 480 nm was measured by a microplate reader (Tecan Safire™, Magellan™ Software; Tecan Group Ltd.). DNA content was calculated using the standard curves obtained from DNA standards measured on the same plates.
Vital cell counts, visible nuclei, and DNA content were normalized to the respective controls and are given as percentages of residual cells, nuclei, or DNA.
Transmission electron microscopy
For TEM, tendon (SDFT) samples recovered from horses euthanized for unrelated reasons were used, as this allowed to prepare additional, nonstored control samples, which were taken immediately after euthanasia and subjected to processing within 15 min. This was deemed important due to the sensitivity of TEM imaging to tissue degradation during storage. Decellularized samples and controls stored in PBS were prepared as described above.
Pieces from the central regions of the tendons were excised to a dimension of 10×2×2 mm (two from each decellularized and control sample) and fixed for 24 h with 4% glutardialdehyde and 4% paraformaldehyde in 0.1 M sodium cacodylate trihydrate buffer (pH 7.2), then washed and postfixed in cacodylate buffered 1% osmium tetroxide for 120 min. After washing and dehydration in graded methanol, using block staining with 1% uranyl acetate in 50% methanol, samples were embedded in Glycidether100™ (formerly Epon 812; Carl Roth). Ultrathin cross- and longitudinal-sections were cut with a diamond knife on an Ultracut UCT (Leica Microsystems), collected on 300 mesh nickel grids and inspected using a Zeiss EFTEM Libra 120 electron microscope operated at 120 kV. Digital images were captured with a Sharpeye 2k CCD camera (TRS; Troendle).
Two longitudinal and two cross-sectional images of each sample were evaluated by two blinded observers using score systems for cell degradation and extracellular matrix integrity (Table 1).
Table 1.
Score System for Evaluation of Cellular and Extracellular Matrix Integrity on Transmission Electron Microscopy Images
| Cell score | |||||
|---|---|---|---|---|---|
| Membranes | Organelles | Electron density and cell structure | |||
| Not preserved | 0 | Not preserved; empty space between fibrils | 0 | Not preserved; no cellular shape definable | 0 |
| Fragmented | 1 | Not preserved; cellular debris between fibrils | 1 | Hardly preserved; cellular shape still definable | 1 |
| Recognizable | 2 | Loss of structure but mostly recognizable | 2 | Low electron density, cell hardly structured | 2 |
| Definable | 3 | Definable | 3 | Average electron density, definable cell structure | 3 |
| Clearly definable | 4 | Clearly definable | 4 | High electron density, clearly definable cell structure | 4 |
| Extracellular matrix score | |||
|---|---|---|---|
| Collagen cross striation (longitudinal sections) | Collagen fibrils (cross sections) | ||
| Not definable | 0 | Not definable | 0 |
| Definable in most areas | 1 | Definable in most areas | 1 |
| Overall clearly definable | 2 | Overall clearly definable | 2 |
Assessment of cytocompatibility
Scaffold preparation
Tendons (SDFT) were collected and decellularized as described above, using either the Cryo-Triton or the Cryo-SDS protocol. Subsequently, tendons were cut into 2 mm thick slices using razor blades to expose the collagen fibers in the core of the decellularized tendon. The slices were then trimmed into segments with a surface area of 3 cm2.
Stromal cell isolation and characterization
Equine adipose-derived multipotent mesenchymal stromal cells (MSC) were isolated by explant technique and expanded until passage 3. Plastic-adherence, trilineage differentiation potential, and expression of MSC-related surface markers were confirmed before the cells were used for the seeding experiments (data not shown).
Cell labeling and scaffold seeding
MSC were labeled with superparamagnetic iron oxide (SPIO) particles (Molday ION Rhodamine B™; Biopal), which allows microscopical identification and magnetic resonance (MR) imaging of the cells. Labeling was performed at an iron concentration of 25 μg/mL culture medium27 for 12 h at 37°C and 5% CO2. Subsequently, cells were thoroughly rinsed with PBS with calcium and magnesium and then passaged by trypsinization. Part of the cells were seeded into well plates and underwent DAPI and Prussian blue staining for microscopical confirmation of successful labeling. The remaining cells were suspended in cell culture medium and seeded on the surface of the decellularized tendon scaffolds (130,000 cells in 30 μL/cm2 scaffold surface). After 4 h of incubation at 37°C and 5% CO2, scaffolds were covered with cell culture medium and further incubated at 37°C and 5% CO2 for 7 or 14 days, with a medium change twice weekly.
Histology and LIVE/DEAD® staining
Paraffin sections were prepared from day 7 and day 14 samples as described above and stained with hematoxylin and eosin, DAPI, and Prussian blue. LIVE/DEAD staining, using the staining kit (Invitrogen) according to the manufacturer's instructions, was performed on the surface of unsectioned sample pieces at day 7 and 14 as well.
MR imaging
At day 1, 7, and 14, seeded and nonseeded control samples were placed in 15 mL tubes filled with PBS and subjected to MR imaging (0.27 T; Hallmarq) with a resolution of 0.82 mm. Samples were positioned in an angle of 55° to enable hyperintense visualization of the tendon tissue when using the T1 weighted (T1w) sequence,28 which was important for identifying the exact position of the constructs in the tubes. T2* weighted (T2*w) sequences were then used to enable visualization of the susceptibility artefacts generated by the SPIO-labeled MSC.27 The technique of MR imaging used in the current study had previously been evaluated by the authors. The evaluation included imaging of different concentrations of labeled cells suspended in agarose gels by low- and high-field MR over 28 days (unpublished data).
T1w images were rigidly registered onto T2*w images. Constructs in the registered T1w images were segmented using the Bernsen local adaptive binarization algorithm29 with an empirically determined filter window width of 65 pixels. Segmented construct volumes were used as a mask to extract signal intensities of the corresponding construct volumes from the T2*w images. MR seeding indices were calculated by subtracting the median signal intensities of the whole seeded constructs from the median signal intensities of the whole nonseeded control constructs. Mathematica 8 (Wolfram Research) was used for all image processing and calculations.
Statistical analysis
Using PASW Statistics 18 software (IBM), Wilcoxon signed-rank tests were performed for comparing the decellularization effectiveness in the two different tendon structures, for each protocol separately. For comparison of effectiveness of the four different decellularization protocols, Friedman tests and, in case of significance, Wilcoxon signed-rank tests were performed, for each tendon structure separately. The level of significance was set at p=0.05.
Results
Effectiveness of decellularization
All protocols led to a significant reduction of cell and DNA content in both SDFT and DDFT (p<0.01), with no significant differences between the different tendon structures. Nevertheless, there was a tendency that when using protocols without freeze-thaw cycles, decellularization was less effective in the DDFT, which was most evident regarding the residual nuclei (p=0.094), and differences between decellularization protocols became more distinct in the DDFT than in the SDFT (Fig. 2).
FIG. 2.

Box plots representing (A) vital cells, (B) histologically visible nuclei, and (C) DNA in % relative to the controls (n=6). Circles represent outliers and stars indicate significant differences (p<0.05).
In the SDFT, vital cell counts relative to the respective controls, determined after collagenase digestion, were low, which was similar among all decellularization groups. Histology showed very few visible nuclei in all groups except the Triton group, in which significantly higher counts were obtained compared to the Cryo-SDS and the SDS group (p<0.05). Median DNA content relative to the controls was lower in both Cryo groups compared with the Triton and the SDS group, but this difference was not significant (p>0.05) (Figs. 2 and 3).
FIG. 3.
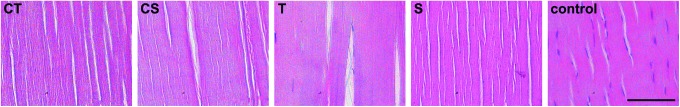
Representative images of hematoxylin and eosin stained decellularized and control superficial digital flexor tendons. CT: Cryo-Triton; CS: Cryo-SDS; T: Triton; S: SDS. Scale bar=100 μm. Color images available online at www.liebertpub.com/tec
In the DDFT, vital cell counts displayed similar median values; however, significant differences were found when comparing the SDS group to the Cryo-SDS and the Triton group (p<0.05). Histology revealed that again, very few nuclei were visible in both Cryo groups, while higher counts were obtained in the SDS and the Triton group (p<0.05 for Cryo-Triton versus Triton). Accordingly, DNA content was lower in both Cryo groups than in the Triton and the SDS group (p<0.05 for Triton versus Cryo-Triton and Cryo-SDS) (Fig. 2).
Transmission electron microscopy
TEM imaging supported the results obtained for decellularization effectiveness. Immense loss of cellular integrity was evident in all decellularized samples, although the extent differed between the decellularization protocols. In both Cryo groups and also in the SDS group, the cellular structures had disappeared, leaving empty spaces between the collagen fibers with very little remaining cell debris. In the Triton group, although membranes and organelles were hardly recognizable, the cellular structures were more maintained. This difference is illustrated by the results of the cell scoring, at which Triton group samples had scored higher than all other decellularized samples, indicating an incomplete decellularization. In contrast, the Cryo-Triton group samples had scored overall lowest, indicating complete loss of cellular integrity and removal of cellular debris. Cells in stored control samples partly showed signs of apoptosis, however, their overall morphology resembled that of the fresh controls more than that of the decellularized samples (Fig. 4).
FIG. 4.

Representative (A) cross-sectional and (B) longitudinal transmission electron microscopy images of decellularized and control superficial digital flexor tendons. The additional image of the stored control (inset) represents an example of beginning apoptosis, with reduced membrane integrity, blebbing, and chromatin condensation. Scale bars in A=1 μm; scale bar in B=2 μm. (C) Dot plots representing the score points for cell and extracellular matrix integrity achieved by the respective samples (n=3); high scores indicate a high preservation of integrity.
Further, TEM images revealed no effect of decellularization on extracellular matrix morphology. Collagen fibrils were definable and showed striated patterns in all groups. However, areas with definable fibrils that were hard to define could be found in several samples, decellularized and controls, in particular concerning the small diameter fibrils, and therefore these samples did not reach the maximal score points (Fig. 4).
Cytocompatibility
Hematoxylin and eosin and DAPI staining demonstrated successful seeding of the constructs in both groups (Cryo-Triton and Cryo-SDS). At day 7, cells were mainly attached to the scaffold surface, with only few areas where cells penetrated the scaffold in both groups. By day 14, the cell layer on the scaffold surface had grown thicker, and especially in the Cryo-Triton group, there were more areas where the cells displayed migration into the scaffolds (Fig. 5).
FIG. 5.

Respresentative images of decellularized superficial digital flexor tendon scaffolds that had been reseeded with mesenchymal stromal cells and stained with (A) hematoxylin and eosin and (B) DAPI at day 7 or day 14 after seeding. d: day. Scale bars=100 μm. Color images available online at www.liebertpub.com/tec
LIVE/DEAD staining of the constructs showed that most regions of the surface were populated with vital MSC, which displayed an elongated morphology and were oriented along the collagen fibers (Fig. 5). However, in both groups, there were also regions where predominantly dead cells were found, or regions where the overall cell density was very low. As the cell distribution on the scaffolds was not homogeneous, quantification of seeding success was only performed on the basis of MR imaging, which allowed evaluation of each construct in whole.
Prussian blue staining and fluorescence-based visualization of the SPIO/rhodamine-labeled MSC in monolayer culture demonstrated successful labeling of the cells. Prussian blue staining of the seeded scaffolds at day 7 and 14 further confirmed that the SPIO particles persisted in the cells during the observation period (Fig. 6).
FIG. 6.

(A) Confirmation of successful cell labeling with superparamagnetic iron oxide particles and rhodamine: (i) Fluorescence-based visualization of the intracellular rhodamine in monolayer culture with DAPI counterstaining of the nuclei; (ii) Prussian blue staining of the intracellular iron oxide particles in monolayer culture; (iii) Prussian blue staining of the superficial digital flexor tendon slices at day 7 and 14 after seeding. All scale bars=100 μm. (B) Corresponding T1 weighted (T1w) and T2* weighted (T2*w) magnetic resonance images of seeded and nonseeded control samples at day 1 after seeding. (C) Dot plot displaying the respective calculated magnetic resonance imaging (MRI) seeding indices (n=3). Color images available online at www.liebertpub.com/tec
MR images of the nonseeded control scaffolds displayed a higher median signal intensity than images of the seeded constructs, due to the hypointense areas on the scaffold surface, which were evident in seeded samples of both decellularization groups (Fig. 6). These hypointense areas were more distinct in the T2*w than in the T1w sequence and thus can be interpreted as susceptibility artefacts generated by the SPIO-labeled MSC.
In the Cryo-Triton group, MR seeding indices increased from day 1 to day 14, indicating continuing distribution of the labeled MSC in the scaffold. In the Cryo-SDS group, seeding indices were higher than in the Cryo-Triton group at day 1 and 7, but then ceased to increase further until day 14 (Fig. 6).
Discussion
In the present study, we demonstrated that the use of freeze-thaw cycles enhances decellularization effectiveness in large tendons. While not being effective alone, especially in terms of DNA removal,18 with 88.7 up to 100% of residual DNA left in large tendons (unpublished data), freeze-thaw cycles combined with the use of detergents proved to increase cell and DNA removal compared with detergent-based protocols alone.
Decellularization effectiveness was similarly favorable in the Cryo-Triton and the Cryo-SDS group. However, differences were found between the Triton and the SDS group, which also seemed to depend on the size of the tendon structure used. In the smaller-sized SDFT, histology results following the SDS protocol were almost as good as following the Cryo-Triton or the Cryo-SDS protocol. In the larger-sized DDFT, however, histology results following the SDS protocol were not satisfactory and similar to the results obtained following the Triton protocol.
Although cell removal following the Triton protocol was least efficient among all protocols compared in this study, Triton X-100 was not as inefficient as reported before,15 potentially due to the pretreatment with hypotonic solution or the longer incubation performed here. More according to the results of the present study, a 100% removal of anterior cruciate ligament cells was reported after use of a protocol including Triton X-100, SDS, and nucleases.17 However, these results, which tend to be slightly better than those obtained in the current study, were only obtained for the mid-portion of the ligament, whereas cell removal was not as efficient at the ligament–bone junctions,17 again indicating an effect of the scaffold size. Moreover, the protocol described there includes the combination of various chemicals and enzymes, requires a long overall incubation time of 6 days not including the subsequent washing steps,17 and was later shown to lead to extracellular matrix alterations that decrease cytocompatibility.22,23 In other studies that evaluated decellularization protocols for tendons or ligaments, cell removal was not quantitatively assessed15,16,18–21 and can therefore not be compared to the results obtained here.
DNA content of decellularized samples was more commonly quantified, significantly differing from the controls.18,19 Following decellularization based on hypotonic solution, trypsin and Triton X-100, a 67% reduction in DNA content was reported.16 Similar results were obtained in the present study following the Triton and the SDS protocol, with a roughly 60% reduction in DNA content, while even better results were obtained when using freeze-thaw cycles, leading to a roughly 80% reduction in DNA content.
Re-seeding of decellularized tendon scaffolds is challenging, as cells tend to populate the scaffold surface only.18,19,24,26 Promising approaches to enhance cell distribution between the collagen fibers have been published, aiming to enable penetration by scaffold surface scoring24 or by injecting a collagen gel prior to injecting the cells.26 In this study, however, for basic assessment of cytocompatibility, the cell suspension was applied onto the scaffold surface only. While cell culture on scaffolds decellularized by both the Cryo-Triton and the Cryo-SDS protocol was possible, our results indicate a wider cell distribution in scaffolds prepared according to the Cryo-Triton protocol, with more cells exhibiting migration between the collagen fibers, which is in accordance with others.23 This might be due to the fact that SDS alters the extracellular matrix composition more than Triton X-100,15,17,19,20,23 which potentially negatively influences cytocompatibility.22 Although in the current study, no considerable fibril alterations following decellularization were evident, it is possible that morphological changes in the periphery or changes in matrix composition on biochemical level could be detected by further assessment. Based on the results obtained so far, seeding of the scaffolds needs to be optimized and further quantified by techniques that allow a more exact evaluation of the cell distribution, such as histological assessment of the whole scaffold and high-resolution MRI.
The horse was chosen as donor animal not only because this allowed convenient recovery of large tendon samples, but also because extensive research on equine tendon anatomy, physiology, pathophysiology, and biomechanical properties has already been performed.30–33 Even more importantly, horses are considered ideal model animals for human tendinopathy, as conditions very similar to those found in humans naturally occur in horses, the equine SDFT being the equivalent to the human Achilles tendon.34
The option to obtain a cell-free, native tendon structure that in function and morphology closely resembles the human Achilles tendon holds great promise not only for current research use, but also for future transplantations. According to the results of the current study, the Cryo-Triton protocol investigated here is most suitable for decellularization of such large tendon structures, being highly effective in cell and DNA removal while maintaining cytocompatibility of the scaffold.
Acknowledgments
The authors acknowledge Prof. Dr. A. Bader (Center for Biotechnology and Biomedicine, University of Leipzig) and Prof. Dr. J. Seeger (Institute of Veterinary Anatomy, University of Leipzig) for kindly providing the laboratory facilities, and further C. Gittel and F. Paebst (Large Animal Clinic for Surgery, University of Leipzig) for providing their support. The work presented in this article was funded by the Mehl-Muelhens Foundation, the Dres. Jutta and Georg Bruns Foundation, and the German Federal Ministry of Education and Research (BMBF 1315883).
Disclosure Statement
No competing financial interests exist.
References
- 1.Siegel L., Vandenakker-Albanese C., and Siegel D.Anterior cruciate ligament injuries: anatomy, physiology, biomechanics, and management. Clin J Sport Med 22,349, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Riley G.Tendinopathy—from basic science to treatment. Nat Clin Pract Rheumatol 4,82, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Delince P., and Ghafil D.Anterior cruciate ligament tears: conservative or surgical treatment? A critical review of the literature. Knee Surg Sports Traumatol Arthrosc 20,48, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Maffulli N., Longo U.G., Loppini M., and Denaro V.Current treatment options for tendinopathy. Expert Opin Pharmacother 11,2177, 2010 [DOI] [PubMed] [Google Scholar]
- 5.Soroceanu A., Sidhwa F., Aarabi S., Kaufman A., and Glazebrook M.Surgical versus nonsurgical treatment of acute Achilles tendon rupture: a meta-analysis of randomized trials. J Bone Joint Surg Am 94,2136, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller B., Bowman K.F., Jr., and Bedi A.ACL graft healing and biologics. Clin Sports Med 32,93, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Sadoghi P., Rosso C., Valderrabano V., Leithner A., and Vavken P.The role of platelets in the treatment of Achilles tendon injuries. J Orthop Res 31,111, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Pascual-Garrido C., Rolon A., and Makino A.Treatment of chronic patellar tendinopathy with autologous bone marrow stem cells: a 5-year-followup. Stem Cells Int 2012,953510, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinello T., Bronzini I., Perazzi A., Testoni S., De Benedictis G.M., Negro A., Caporale G., Mascarello F., Iacopetti I., and Patruno M.Effects of in vivo applications of peripheral blood-derived mesenchymal stromal cells (PB-MSCs) and platlet-rich plasma (PRP) on experimentally injured deep digital flexor tendons of sheep. J Orthop Res 31,306, 2013 [DOI] [PubMed] [Google Scholar]
- 10.Kuo C.K., Marturano J.E., and Tuan R.S.Novel strategies in tendon and ligament tissue engineering: advanced biomaterials and regeneration motifs. Sports Med Arthrosc Rehabil Ther Technol 2,20, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longo U.G., Lamberti A., Petrillo S., Maffulli N., and Denaro V.Scaffolds in tendon tissue engineering. Stem Cells Int 2012,517165, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGuire D.A., and Hendricks S.D.Allograft tissue in ACL reconstruction. Sports Med Arthrosc 17,224, 2009 [DOI] [PubMed] [Google Scholar]
- 13.Raghavan S.S., Woon C.Y., Kraus A., Megerle K., Choi M.S., Pridgen B.C., Pham H., and Chang J.Human flexor tendon tissue engineering: decellularization of human flexor tendons reduces immunogenicity in vivo. Tissue Eng Part A 18,796, 2012 [DOI] [PubMed] [Google Scholar]
- 14.Yoshida R., Vavken P., and Murray M.M.Decellularization of bovine anterior cruciate ligament tissues minimizes immunogenic reactions to alpha-gal epitopes by human peripheral blood mononuclear cells. Knee 19,672, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cartmell J.S., and Dunn M.G.Effect of chemical treatments on tendon cellularity and mechanical properties. J Biomed Mater Res 49,134, 2000 [DOI] [PubMed] [Google Scholar]
- 16.Whitlock P.W., Seyler T.M., Parks G.D., Ornelles D.A., Smith T.L., Van Dyke M.E., and Poehling G.G.A novel process for optimizing musculoskeletal allograft tissue to improve safety, ultrastructural properties, and cell infiltration. J Bone Joint Surg Am 94,1458, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Woods T., and Gratzer P.F.Effectiveness of three extraction techniques in the development of a decellularized bone-anterior cruciate ligament-bone graft. Biomaterials 26,7339, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Ning L.J., Zhang Y., Chen X.H., Luo J.C., Li X.Q., Yang Z.M., and Qin T.W.Preparation and characterization of decellularized tendon slices for tendon tissue engineering. J Biomed Mater Res A 100,1448, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Vavken P., Joshi S., and Murray M.M.TRITON-X is most effective among three decellularization agents for ACL tissue engineering. J Orthop Res 27,1612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deeken C.R., White A.K., Bachman S.L., Ramshaw B.J., Cleveland D.S., Loy T.S., and Grant S.A.Method of preparing a decellularized porcine tendon using tributyl phosphate. J Biomed Mater Res B Appl Biomater 96,199, 2011 [DOI] [PubMed] [Google Scholar]
- 21.Omae H., Zhao C., Sun Y.L., An K.N., and Amadio P.C.Multilayer tendon slices seeded with bone marrow stromal cells: a novel composite for tendon engineering. J Orthop Res 27,937, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gratzer P.F., Harrison R.D., and Woods T.Matrix alteration and not residual sodium dodecyl sulfate cytotoxicity affects the cellular repopulation of a decellularized matrix. Tissue Eng 12,2975, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Harrison R.D., and Gratzer P.F.Effect of extraction protocols and epidermal growth factor on the cellular repopulation of decellularized anterior cruciate ligament allografts. J Biomed Mater Res A 75,841, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Woon C.Y., Farnebo S., Schmitt T., Kraus A., Megerle K., Pham H., Yan X., Gambhir S.S., and Chang J.Human flexor tendon tissue engineering: revitalization of biostatic allograft scaffolds. Tissue Eng Part A 18,2406, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Stewart A.A., Barrett J.G., Byron C.R., Yates A.C., Durgam S.S., Evans R.B., and Stewart M.C.Comparison of equine tendon-, muscle-, and bone marrow-derived cells cultured on tendon matrix. Am J Vet Res 70,750, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Martinello T., Bronzini I., Volpin A., Vindigni V., Maccatrozzo L., Caporale G., Bassetto F., and Patruno M.Successful recellularization of human tendon scaffolds using adipose-derived mesenchymal stem cells and collagen gel. J Tissue Eng Regen, 2012. [Epub ahead of print]; DOI: 10.1002/term.1557 [DOI] [PubMed] [Google Scholar]
- 27.Addicott B., Willman M., Rodriguez J., Padgett K., Han D., Berman D., Hare J.M., and Kenyon N.S.Mesenchymal stem cell labeling and in vitro MR characterization at 1.5 T of new SPIO contrast agent: Molday ION Rhodamine-B. Contrast Media Mol Imaging 6,7, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peh W.C., and Chan J.H.The magic angle phenomenon in tendons: effect of varying the MR echo time. Br J Radiol 71,31, 1998 [DOI] [PubMed] [Google Scholar]
- 29.Bernsen J.Dynamic tresholding of grey level images. Proc Intl Conf Patt Recog 1251, 1986 [Google Scholar]
- 30.Dowling B.A., Dart A.J., Hodgson D.R., and Smith R.K.Superficial digital flexor tendonitis in the horse. Equine Vet J 32,369, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Dowling B.A., and Dart A.J.Mechanical and functional properties of the equine superficial digital flexor tendon. Vet J 170,184, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Thorpe C.T., Clegg P.D., and Birch H.L.A review of tendon injury: Why is the equine superficial digital flexor tendon most at risk? Equine Vet J 42,174, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Smith R.K., and McIlwraith C.W.Consensus on equine tendon disease: building on the 2007 Havemeyer symposium. Equine Vet J 44,2, 2012 [DOI] [PubMed] [Google Scholar]
- 34.Patterson-Kane J.C., Becker D.L., and Rich T.The pathogenesis of tendon microdamage in athletes: the horse as a natural model for basic cellular research. J Comp Pathol 147,227, 2012 [DOI] [PubMed] [Google Scholar]