

Abstract
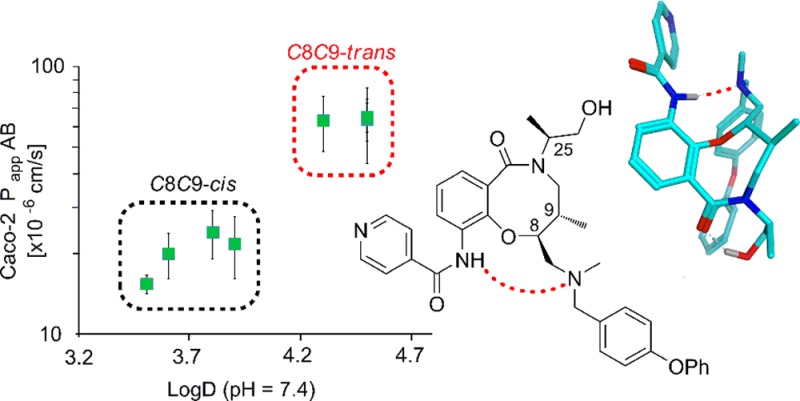
Profiling of eight stereoisomeric T. cruzi growth inhibitors revealed vastly different in vitro properties such as solubility, lipophilicity, pKa, and cell permeability for two sets of four stereoisomers. Using computational chemistry and NMR spectroscopy, we identified the formation of an intramolecular NH→NR3 hydrogen bond in the set of stereoisomers displaying lower solubility, higher lipophilicity, and higher cell permeability. The intramolecular hydrogen bond resulted in a significant pKa difference that accounts for the other structure–property relationships. Application of this knowledge could be of particular value to maintain the delicate balance of size, solubility, and lipophilicity required for cell penetration and oral administration for chemical probes or therapeutics with properties at, or beyond, Lipinski’s rule of 5.
Introduction
Lipinski’s “rule of 5”1,2 (RO5) has had a profound impact on the build-up of small molecule screening collections, as it delineates chemical space that is more likely to yield cell permeable compounds and allow oral administration. However, finding RO5 compliant ligands for targets with extended binding sites, e.g., proteases and peptidic GPCRs, as well as protein–protein interactions has turned out to be a major challenge. Instead, modulation of these targets often requires ligands substantially differentiated from traditional small molecule screening collections by having properties at the border of the RO5 or beyond (bRO5).3,4 Attention has therefore been focused on strategies for lead generation such as natural-product-derived5,6 approaches and diversity-oriented synthesis (DOS).7,8
It is uncertain how physicochemical properties apply to the cell permeability of natural-product- or DOS-derived compounds, especially when the compound’s properties are at, or beyond, the border of RO5.9−11 Understanding the key properties for design of pharmacodynamically active compounds in this space, which are also able to penetrate cells and modulate intracellular targets, is therefore a key challenge. Interestingly, extensive studies of the immunosuppressant cyclosporin A, a cyclic peptide with properties far outside RO5, have revealed it to be conformationally flexible which allows shielding of polarity through intramolecular hydrogen bonding.9,12 Most likely, this explains the unexpectedly high bioavailability of cyclosporin A, which consequently can be administered orally.13,14 Recent studies of cyclic tri- and hexapeptide model systems15−18 also showed that intramolecular hydrogen bonding may contribute to druglike membrane permeability. In a few cases intramolecular hydrogen bonding has also been found to improve the membrane permeability and oral bioavailability of traditional small molecule drug candidates with RO5 compliant properties.19−22
In order to capitalize on the opportunities for modulation of challenging targets, the Broad Institute has created a small molecule screening collection through a diversity-oriented synthesis (DOS) strategy.23 The DOS screening library consists of >100 000 novel, discrete compounds with a fraction of sp3 centers (Fsp3)24 and stereochemical diversity25−29 more consistent with those of natural products than traditional RO5 screening libraries.30 Furthermore, the DOS library is composed of a complete matrix of stereoisomers as a result of being assembled through a build/couple/pair strategy. This modular synthetic approach and complete coverage of stereoisomers provide a unique opportunity to systematically dissect the factors controlling cell permeability and oral bioavailability for natural product-like compounds with properties at or beyond RO5.
A high-throughput screening campaign with the Broad’s DOS screening library identified lactams 1–8 (Table 1) as novel growth inhibitors of the recombinant Tulahuen strain of T. cruzi, with potencies (IC50) for individual stereoisomers ranging from 1 nM to 0.5 μM.31 The key structural features of 1–8 include an eight-membered fused ring system, three stereocenters, and physicochemical properties at the border of the RO5 (Table 1). Medicinal chemistry efforts and structure–activity relationships have been described elsewhere.31 However, in the profiling of stereoisomers 1–8 the solubility was found to vary greatly depending on the stereochemical relationship of C8 and C9, with trans-C8,C9 diastereomers being 1–2 μM in phosphate buffered saline (PBS) and the cis-C8,C9 stereoisomers having an almost 100-fold higher solubility. Herein, we conduct detailed studies of 1–8 with regard to lipophilicity, pKa, and cell permeability to investigate this unexpected finding further. We observed major stereospecific differences in the experimental parameters and report our successful efforts to explain the contributing factors to these differences. Our findings suggest rational strategies for modulation of cell permeability for natural-product-like compounds that have properties at or beyond RO5.
Table 1. Structure and Solubility for the Eight Stereoisomeric T. cruzi Growth Inhibitors 1–8.
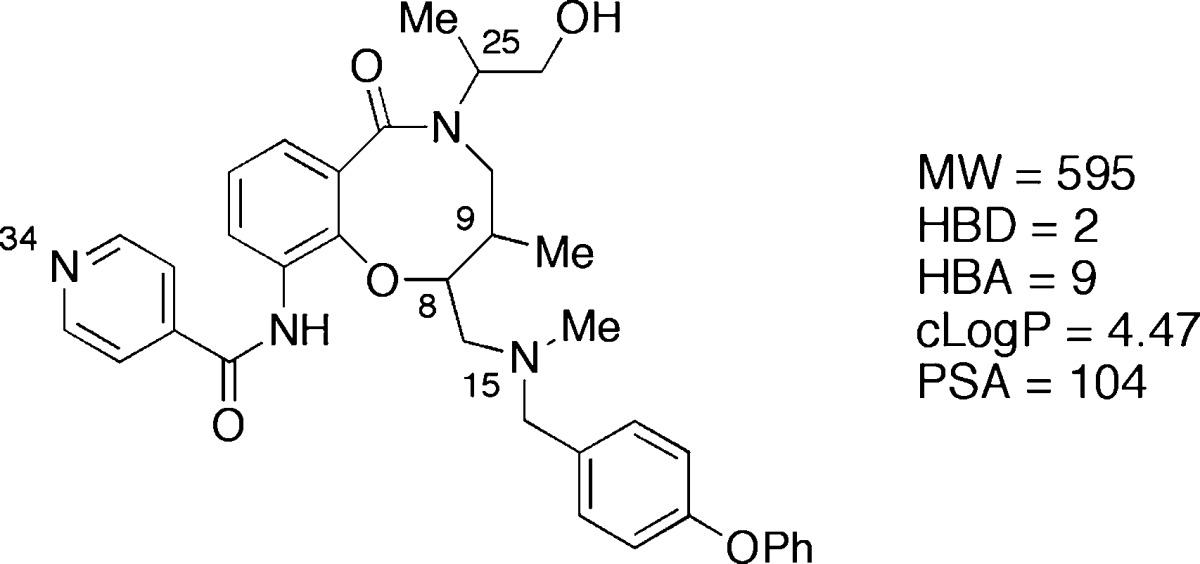
| compd | configuration (C8,C9,C25) | relative configuration (C8,C9) | solubility (μM)a |
|---|---|---|---|
| 1 | SSS | trans | 1 |
| 2 | RRS | trans | 1 |
| 3 | SSR | trans | 2 |
| 4 | RRR | trans | 1 |
| 5 | SRR | cis | 86 |
| 6 | RSS | cis | 93 |
| 7 | RSR | cis | 98 |
| 8 | SRS | cis | 87 |
Determined at pH 7.4 in PBS containing 1% DMSO.
Results
When the lipophilicities of 1–8 were measured, this again revealed that the compounds clustered into two groups depending on the stereochemical relationship of C8 and C9 (Figure 1a). Stereoisomers with a cis-C8,C9 relationship (5–8), which were of higher solubility, possessed log D values of 3.4–3.9, while the trans-C8,C9 stereoisomers (1–4) had log D values that were 0.4–1.1 units higher. In line with these findings, increased epithelial permeability through a Caco-2 cell monolayer was observed for the stereochemical cluster with higher log D, while a lower permeability was found for the low log D cluster (Figure 1b). The cellular permeability was determined both with a pH gradient from 6.5 on the apical side of the cellular monolayer to a pH of 7.4 on the basolateral side and with a pH of 7.4 on both sides of the monolayer. The former is a common model for uptake from the small intestine into the bloodstream, mimicking the upper intestinal pH gradient, whereas the latter reflects the transition from the bloodstream into most tissues.32 The trans-stereoisomers 1–4 did not show a dependency of permeability on pH. In contrast, cis-stereoisomers 5–8 demonstrated a significant increase in permeability with a pH of 7.4 on the apical side (orange diamonds), although the total values were still significantly lower than for 1–4. The ratio between the permeability from the basolateral to apical side of the cell monolayer and the permeability in the opposite direction (pH 7.4 on both sides), i.e., the efflux ratio, varied from 0.31 to 0.48 (Table SI 5). Thus, the cell permeabilities determined for the eight stereoisomers were not significantly influenced by efflux.
Figure 1.
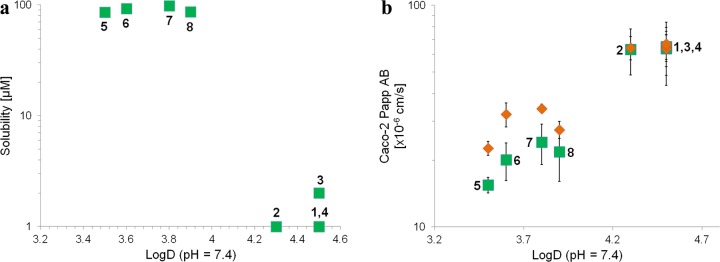
(a) Solubility versus lipophilicity (log D, pH 7.4) for compounds 1–8. (b) Permeability (Caco-2 Papp(AB)) versus lipophilicity (log D) for 1–8, determined with a pH gradient of 6.5 (apical) to 7.4 (basolateral) between the two sides (green squares for 1–8) and with a pH of 7.4 on both sides (orange diamonds). Standard deviations for n = 3–6 determinations are given as error bars.
On the basis of the pH dependencies in the permeability assay, we hypothesized that the differences in solubility, lipophilicity, and permeability could originate from differences in pKa between the stereochemical clusters. Determination of pKa for 1–8 by spectrophotometric titration indeed revealed a pKa difference of ∼1 unit for the tertiary amine, with 1–4 being less basic and consequently less soluble and more lipophilic than the more basic 5–8 (Table 2). This difference is most significant from a medicinal chemistry perspective because it determines the ionic state of the compounds at physiological pH values. The ionic state, in turn, will affect the cellular permeability, in both uptake from the intestine to blood and distribution from blood into tissues. A much smaller, but significant, difference (∼0.2 units) for the pyridine group was also observed between the two stereochemical clusters, but this does not influence compound properties at physiological pH.
Table 2. pKa Values for 1–8 Determined by Spectrophotometric Titrationa.
| compd | relative configuration (C8,C9) | pKa1 (N34, pyridine) | pKa2 (N15, 3′-amine) |
|---|---|---|---|
| 1 | trans | 2.76b | 6.08b |
| 2 | trans | 2.77b | 6.18c |
| 3 | trans | 2.76b | 6.14b |
| 4 | trans | 2.77b | 6.08b |
| 5 | cis | 2.98b | 7.07b |
| 6 | cis | 2.98b | 7.08b |
| 7 | cis | 2.98b | 7.16b |
| 8 | cis | 2.97c | 7.16b |
Three titrations for each of 1–8 (cf. structural formulas for 1–8 in Table 1).
SD = ±0.01.
SD = ±0.02.
After identification of two sets of compounds with identical calculated physicochemical properties and functional groups (Table 1) yet displaying vastly different in vitro properties, the underlying cause was concluded to be stereochemistry.33,34 It was assumed that the two clusters had major differences in intramolecular interactions and conformational preferences that led to the differences in pKa and in vitro properties. The pKa values reported here represent a thermodynamic process in which the ammonium ion (R3NH+) dissociates to the free amine (R3N:) and a proton, a reaction that is more endothermic for compounds with higher pKa values. Each pKa unit equates to a free energy difference of 1.36 kcal/mol at 298 K as defined by the Henderson–Hasselbalch equation and the relationship between the free energy difference and the equilibrium constant for the acid–base reaction (cf. Supporting Information).35 To investigate this process, diastereomers 1 and 8 were selected as representatives for the C8,C9-trans and C8,C9-cis stereochemical clusters, respectively. First, molecular mechanics calculations with a continuum solvation model36 (MMFFs/generalized Born solvation) were used to determine the difference in strain energy between the neutral and protonated forms of 1 and 8, respectively. Interestingly, the lowest energy conformations of the neutral forms of amines 1 and 8 were found to be rigid because of a strong intramolecular amide–amine hydrogen bond (Figure 2). However, the neutral form of cis-isomer 8 had a strain energy 2.6 kcal/mol higher than trans-isomer 1 (Table 3). For the more flexible ammonium ion forms, 8 was also higher in energy than 1 but only by 0.4 kcal/mol. Together the differences between the isomeric reactants and products in deprotonation of the ammonium ion add to a 2.2 kcal/mol difference, which would be equivalent to 1 having a pKa value 1.6 units lower than 8, assuming that the free energy difference is similar to the difference in strain energy. Quantum mechanics calculations with continuum solvation (B3LYP/6-31G* with the SM8 solvation model)37 provided the same conclusion but with a smaller energy difference between 1 and 8. The neutral cis-isomer 8 was 3.1 kcal/mol higher in free energy than the trans-isomer 1, while the cation form of 8 was higher in energy by 1.2 kcal/mol, leading to a total difference of 1.9 kcal/mol (Table 3). This difference between the calculated free energies of the two stereoisomers would result in a theoretical difference in pKa of 1.4 units, somewhat more consistent with the experimentally observed difference. Furthermore, use of a thermodynamic cycle that separates the gas phase deprotonation from solvation35 led to a predicted difference of 1.1 pKa units which, astonishingly, is identical to the experimental value (cf. Supporting Information for calculation).
Figure 2.

Comparison of the neutral state of diastereomers 1 and 8 showing Newman projections looking down the C10–C9 bond (left) and the C9–C8 bond (center) with gauche interactions highlighted with red arrows. The lowest energy conformations found by quantum mechanics (B3LYP/6-31G*) and molecular mechanics (MMFFs force field, GB solvation) are shown in color.
Table 3. Calculated Relative Energy Difference between the Minimum Energy Conformations of the Neutral Amine and the Protonated Form of Diastereomers 1 (C8,C9-Trans) and 8 (C8,C9-Cis).
| strain energy difference, MMFFs + GB (kcal/mol) | free energy (298 K), SM8//B3LYP/6-31G* (kcal/mol) | |
|---|---|---|
| Δ(8–1), neutral amine | 2.6 | 3.1 |
| Δ(8–1), ammonium ion | 0.4 | 1.2 |
| overall Δ(8–1) | 2.2 | 1.9 |
| predicted ΔpKa (8–1)a | 1.6 | 1.4 |
Experimental ΔpKa (8–1): 1.1 units.
The difference in deprotonation energy between 1 and 8 was thus predicted with excellent accuracy with the SM8 model; however, all methods did not yield the same accuracy. For instance, another continuum solvation model, the Poisson–Boltzmann finite element method (PBF),38,39 calculated a net difference in free energy of solvation that was over 5 kcal/mol higher, leading to a correspondingly larger predicted pKa difference. Importantly, the difference in magnitude of the solvation energy was in line with benchmark calculations that have shown SM8 to provide significantly better agreement with experimental solvation energies.37 All of the methods predicted trans-isomer 1 to be lower in energy than cis-isomer 8 and that this is predominantly driven by the difference in energy of the intramolecularly hydrogen bonded neutral amines.
Independent of whether molecular mechanics or quantum mechanics was used, the minimum energy conformation of the neutral form in both diastereomers had the same rigid core conformation containing an intramolecular hydrogen bond (Figure 2). This constrained conformation provides a model for explaining the large difference in energy between the neutral species of 1 and 8 and also of the difference in pKa. Newman projections along the C10–C9 and C9–C8 bonds highlight the larger number of gauche interactions in cis-isomer 8 versus trans-isomer 1. The energy difference between the protonated forms is lower because of the loss of the intramolecular hydrogen bond allowing for more conformational freedom for both compounds.
The results from the conformational calculations of compounds 1 and 8 were further investigated by NMR spectroscopy, with particular emphasis on the predicted intramolecular hydrogen bond. As 1 had a low solubility in aqueous solution, DMSO-d6 was used as a surrogate for a polar environment whereas CDCl3 was used to mimic the lipid environment of a cell membrane. Significant downfield shifts were displayed by N28-H of trans-isomer 1, compared to cis-isomer 8, both in DMSO-d6 and in CDCl3 (Figure 3a). For 1, the chemical shift of N28-H was almost identical in both solvents, whereas a significant upfield shift was found for 8 in CDCl3. The lack of solvent dependence of the shift of N28-H in 1, in combination with the downfield shift relative to 8, suggests that N28-H in 1 is involved in an intramolecular hydrogen bond in both solvents.40,41 In contrast, the solvent dependence found for 8 indicates that N28-H forms an intermolecular hydrogen bond to solvent in DMSO. In addition, the chemical shift of N28-H of compound 8 in DMSO-d6 has a greater temperature dependence than in compound 1 (Supporting Information spectrum SI 16), providing further support for the presence of an intramolecular hydrogen bond in 1 but not in 8.
Figure 3.
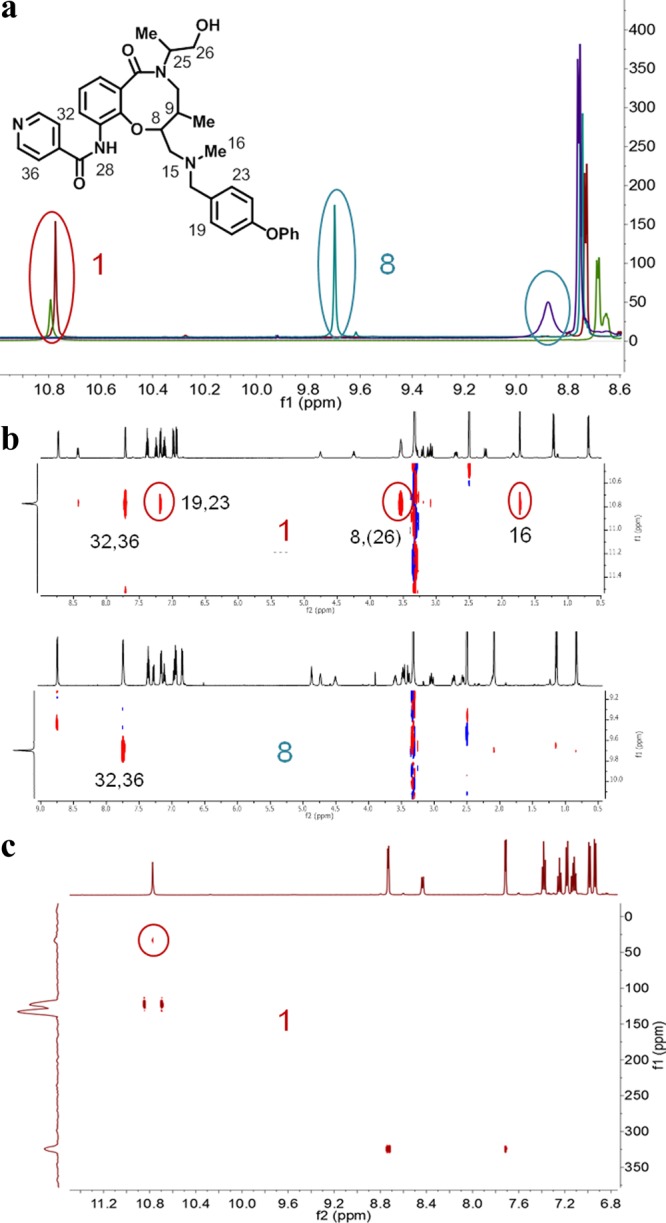
(a) Overlays of 1H NMR spectra of 1 (C8,C9-trans, red and green) and 8 (C8,C9-cis, blue and violet) in DMSO-d6 and CDCl3, respectively. The resonance of N28-H in 1 and 8 is marked with red and blue ovals in the overlays. (b) Parts of the ROESY spectra of 1 and 8 recorded in DMSO-d6 showing crosspeaks from N28-H. ROESY crosspeaks across the eight-membered hydrogen-bonded ring are marked with red ovals in the spectrum of 1. (c) 1H–15N HMBC of 1 in DMSO-d6. The crosspeak resulting from the coupling of N28-H and N15 is marked with a red circle.
To obtain information on intramolecular distances within 1 and 8, 2D-ROESY experiments were performed. For trans-isomer 1, several distinct rotating frame Overhauser effect (ROE) peaks support the presence of a hydrogen bond between N28-H and N15. Thus, strong ROE crosspeaks are observed between N28-H and C19,23-H, C8-H and C16-H3, revealing that N28-H is in proximity to the tertiary amine region of 1 (Figure 3b). These ROEs are not observed for the cis-isomer 8, which only displays one strong ROE correlation from N28-H to the adjacent C36,32-H. Finally, a correlation between N28-H and N15 was observed in a natural abundance 1H–15N heteronuclear multiple bond correlation (HMBC) spectrum of 1 (Figure 3c) but could not be detected for 8. This through bond scalar coupling definitively proves the existence of a hydrogen bond between these atoms and also indicates that it has a high stability.41 Thus, chemical shift differences, including solvent and temperature dependencies, together with distance information from the ROESY experiments and finally information from 1H–15N HMBC spectroscopy all verified the presence of an intramolecular hydrogen bond in compound 1 but not in 8. As DMSO-d6, which cannot act as a hydrogen bond donor, was used in the NMR studies, it could be argued that the intramolecular hydrogen bond may not be formed in aqueous solutions. However, the intramolecular hydrogen bond is strong in DMSO-d6 as revealed by the finding that it exists also at temperatures around 338 K (Figure SI 16) and by the fact that it is detected as a cross peak in the H–N HMBC spectrum. As the formation of an intramoleccular hydrogen bond is also in line with the calculations and experimental findings, it is likely that it is also formed in aqueous solutions of trans-C8,C9 stereoisomers 1–4.
The presence of an intramolecular hydrogen bond in trans-isomers 1–4, and its absence in cis-isomers 5–8, thus provides a rationale for the difference in pKa of the tertiary amine in the two sets of stereoisomers. The pKa difference also explains the influence of pH on cell permeability in the Caco-2 assay (cf. Figure 1b). With pKa values close to 6.1, the degree of intramolecular hydrogen bonding and thus the cell permeability of trans-isomers 1–4 were not measurably influenced by a change in pH from 6.5 to 7.4 on the apical side of the cell monolayer. On the other hand, cis-stereoisomers 5–8, which have pKa ≈ 7.1, will become increasingly deprotonated at higher pH, leading to an increased likelihood of intramolecular hydrogen bonding which in turn results in a higher cell permeability. In a final effort to verify the influence of intramolecular hydrogen bonding on the properties of the two sets of stereoisomers, N-methylated derivatives of 1 and 8 that are unable to form an intramolecular hydrogen bond were synthesized (cf. Supporting Information). As could be expected, prevention of intramolecular hydrogen bonding gave an N-methylated derivative of 1 that had a lipophilicity and cell permeability within the ranges observed for the non-hydrogen-bonded stereoisomers 5–8 (Figure SI 1, Tables SI 1 and SI 4). The N-methylated derivative of 8 retained a lipophilicity and cell permeability close to that of 8, also in line with expectations.
Discussion
In summary, profiling of the complete matrix of stereoisomeric T. cruzi inhibitors 1–8 unexpectedly revealed a striking influence of stereochemistry on solubility, lipophilicity, and cell permeability. This effect on compound properties was traced to the relative stereochemistry at the two adjacent stereocenters at C8 and C9 in the eight-membered rings of 1–8. For stereoisomers 1–4, which had a trans-C8,C9 relationship, an intramolecular hydrogen bond was favored that reduced the basicity of the tertiary amine of the inhibitors while simultaneously shielding polarity from the surrounding environment. As a consequence, 1–4 were more lipophilic, less soluble, and had higher cell permeabilities than cis-C8,C9 stereoisomers 5–8, for which an intramolecular hydrogen bond was less favorable. In comparison, limited profiling of other DOS compounds that lacked the opportunity of forming an intramolecular hydrogen bond showed statistically insignificant variation between stereoisomers with regard to their lipophilicity, solubility, and cell permeability (Figure SI 2). Another unexpected finding was that the intramolecular hydrogen bond that influences the properties of 1–4 came via formation of an eight-membered ring. In contrast, the majority of intramolecular hydrogen bonds found in a recent exhaustive analysis of crystal structure databases involve formation of five- or six-membered rings.42 Our observations emphasize the importance of preparing and screening pure stereoisomers in chemical probe or drug discovery projects, since their physicochemical as well as pharmacokinetic and pharmacodynamic properties may be significantly different.
The results obtained for the stereoisomeric T. cruzi inhibitors point to the opportunity that intramolecular hydrogen bonding can be used to “hide” hydrogen bond donors and adjust pKa in design of druglike compounds with properties at or beyond the RO5. They also suggest that not only thermodynamically favored five- and six-membered rings42 but also intramolecular hydrogen bonding leading to formation of larger rings may be used in optimization of compound properties. It can be assumed that opportunities to adjust physicochemical properties, and subsequently cell permeability and oral bioavailability, will be of increasing importance as compound properties deviate further and further beyond the RO5. This is supported by the fact that intramolecular hydrogen bonding confers cell permeability and oral bioavailability to cyclosporin A, a cyclic undecapeptide,9,12 with properties far outside of the RO5. As revealed in this work and suggested by others,9 conformational calculations may be used for prediction of intramolecular hydrogen bonding, and they may therefore have value as prospective tools for design and optimization of bRO5 compounds. Computed Δ log Poct–tol values, i.e., differences in log P determined for partitioning between water and octanol or toluene, respectively, have also been suggested for prediction of intramolecular hydrogen bonding.43 In addition, a recent analysis42 of crystal structure databases gave a list of intramolecular hydrogen bonding motifs for five- to seven-membered hydrogen-bonded rings that can also be deployed in compound design or optimization.
Modulation of challenging targets with extended binding sites requires compound classes that reach into chemical property space near the limit of what is acceptable for cell permeability and oral bioavailability,3,4 i.e., into bRO5 space.9,10 Macrocycles constitute one example of compounds that predominantly reside in bRO5 chemical space and that also have demonstrated success in modulation of challenging targets.44 A recent comprehensive investigation of macrocyclic drugs and clinical candidates revealed that a significant number that are orally bioavailable had molecular weights, lipophilicities, and polar surface areas that were higher than for traditional oral small molecule drugs.45 However, it was discovered that macrocycles, just as small molecule drugs, may have no more than five hydrogen bond donors to allow for oral administration.45 This observation, in combination with the findings reported herein, further emphasizes that masking of hydrogen bond donors by logical incorporation of intramolecular hydrogen bonds may be of particular value in efforts to improve cell permeability and oral bioavailability of compounds at the border of, or beyond, RO5 chemical space.
Methods
Solubility
Solubility was determined in phosphate buffered saline (PBS), pH 7.4, containing 1% DMSO. Each compound was prepared at 100 mM by diluting 5 μL of a 10 mM DMSO stock in triplicate with 495 μL of both 100% DMSO and PBS with 1% DMSO. Compounds were allowed to equilibrate at room temperature with a 750 rpm vortex shake for 18 h. StirStix were included in each preparation to lessen compound aggregation. After equilibration, samples were analyzed by UPLC–MS (Waters, Milford, MA) with compounds detected by SIR detection on a single quadrupole mass spectrometer. The DMSO samples were used to create a two-point calibration curve to which the response in PBS was fitted.
log D
The method for measurement of log D was based on the traditional shake flask technique, using UPLC with quantitative mass spectrometry (MS) to measure the relative octanol and aqueous concentrations. 1-Octanol (HPLC grade, ≥99%, Sigma-Aldrich) and a 10 mM phosphate buffer [Na2HPO4·2H2O (p.a. grade, Merck) and NaH2PO4·H2O (p.a. grade, Merck)] were used. Equal parts of buffer and 1-octanol were vigorously mixed in a separation funnel three times (at least 15 min between each mixing) to saturate the solutions. The mixture was left overnight to separate the upper octanol phase from the lower buffer phase before being used in the experiments. Compounds were assayed in pools of eight, and four dilutions of buffer and octanol samples were analyzed and evaluated for log D calculation. All liquid transfers were performed with a Beckman Biomek FX robot, and samples were analyzed on a fast-scanning triple quadropole mass spectrometer (Waters Micromass TQD with MassLynx 4.1) coupled to a Waters Acquity Ultra Performance LC using a Acquity UPLC HSS T3 1.8 μm, 2.1 mm × 50 mm or Acquity UPLC BEH C18 1.7 μm, 2.1 mm × 50 mm column.
Caco AB Permeability
Transepithelial transport of the compounds through a Caco-2 monolayer was determined in an automated fashion using a Tecan Freedom EVO 200 equipped with a Te-MO 96 and a TEER Station. Caco-2 cells were grown in DMEM supplemented with 10% FCS for 15–19 days on Costar 24-well cell culture cluster plates (polycarbonate membrane, 0.4 μm pore size). Chamber volumes were 288 and 950 μL on the apical and basolateral sides of the cell monolayers, respectively, and all incubations were performed with prewarmed buffers in a shaking incubator at 480 rpm and 37 °C. Prior to assay, cells were washed with HBSS supplemented with 25 mM HEPES (HBSS-HEPES), pH 7.4, to remove the culture medium. After 15 min equilibration time the transepithelial electrical resistance (TEER) was determined to assess acceptance of the cell plates into the assay. A second measurement and a lucifer yellow leakage determination was carried out after performing all the transport experiments to monitor integrity of the cell monolayers throughout the study.
Permeability in the absorptive direction (A–B, apical-to-basolateral) was studied over 120 min at 10 μM test compound. The compound solutions were freshly prepared from DMSO stock solutions diluted into HBSS supplemented with 25 mM MES, pH 6.5, or into HBSS-HEPES, pH 7.4; the final solvent concentration was always 1%. Samples from the donor side (2 μL) were drawn immediately after addition of test compound and after 45 and 120 min. The donor samples were diluted 1:100 with 198 μL in HBSS. From the receiver compartment an amount of 200 μL is withdrawn after 45 and 120 min and replaced with fresh HBSS-HEPES, pH 7.4. Upon completion of the study all samples were quenched with 67 μL of acetonitrile and analyzed subsequently using UPLC/MS/MS (see above). The permeability was determined as the appearance rate of compound on the receiver side, in relation to donor concentration, according to the following equation:
where dQ/[(dt)(T)] is the slope of the permeation profile across the Caco-2 cell monolayers, A is the surface area of the Transwell insert (0.33 cm2), and D is the concentration on the donor side. Following the permeability experiments, recovery from the donor and receiver compartments was calculated from the following equation:
where D0 and Dend are the donor sample concentrations at the beginning and last time points, respectively, Vd is the donor side volume (0.288 mL for A–B and 0.95 mL for B–A), Rend is the receiver sample concentration at the last time point, and Vr is the receiver volume (0.95 mL for A–B and 0.285 mL for B–A).
All measurements were performed in duplicate, and only minor standard deviations ((0.08–1.14) × 10–6 cm/s) and on average moderate to good mass recovery values (82%) were observed.
Caco ABBA Efflux
For the Caco ABBA measurements the cells were grown as described above and the same robotic system (Tecan Freedom EVO 200 equipped with a Te-MO 96 and a TEER station) was used. Permeabilities were determined in both A–B and B–A (basolateral-to-apical) directions, and identical buffers, HBSS-HEPES, pH7.4, were used on both sides of the cell monolayer, irrespective of transport direction. The test concentration was again 10 μM for each test compound, and incubations were performed in duplicate. During incubation the Transwell plates were placed in a shaking incubator at 480 rpm and 37 °C. Sampling in the A–B direction was at time points 0 and 60 min from the donor compartment (A, 2 μL, 1:100 dilution in HBSS) and 200 μL from the receiver side (B) after 60 min. Sampling in the B–A direction was at time points 0 and 30 min from the donor compartment (B, 1 μL, 1:100 dilution in HBSS) and 100 μL from the receiver side (A) after 30 min. Permeabilities were calculated as described above, and the efflux ratio was determined from the permeability in B–A and A–B directions.
where Papp(B–A) indicates the apparent permeability coefficient in the basolateral to apical direction and where Papp(A–B) indicates the apparent permeability coefficient in the apical to basolateral direction.
pKa
The pKa values of the compounds were determined with a GLpKa instrument, equipped with a D-PAS (dip probe absorption spectroscopy) lamp from Sirius Analytical Ltd. A 10 mM standard DMSO solution of each compound was made, 2–5 μL of which was added to 25 μL of phosphate buffer. The phosphate buffer was added because the amount of sample used is not enough to act as its own buffering system while performing the titration. The GLpKa instrument then added a predetermined volume of ionic-strength-adjusted (ISA) water (1.5 mL). A pH-metric titration from low to high pH was performed. During the titration, the GLpKa instrument collected a UV–vis spectrum by using the D-PAS technique to establish a titration curve. The electrode was calibrated using a blank titration from pH 1.8 to pH 12.0 before every individual determination. The measurements were performed under argon to minimize the effect of dissolved CO2. Precipitation was continuously monitored at 500 nm, which was apparent for the more insoluble analogues. The temperature was controlled throughout the experiment at 25 ± 1 °C.
Computational Methods
The calculated log P (cLogP) was generated using the ACD/Labs PhysChem Batch Predictor with the shipped database.46 The 2D polar surface area (PSA) was calculated in MOE 2013.47 Conformations for the tertiary amine and the ammonium conjugate acid were generated using the LowModeMD method in MOE 2012,48 using the MMFFs force field and the generalized Born solvation model.36 Unique conformers were defined using heavy atom rmsd criterion of 0.25 Å. These were then clustered using the conformer hierarchical clustering using Canvas, version 1.5, with atomic rmsd of heavy atoms plus −OH and −SH to yield 10 clusters. The lowest energy conformers within 2 kcal/mol, as well as the lowest energy conformer in each of the clusters, were kept for optimization using quantum mechanics in Jaguar 8.1.49 The optimization used a medium sized basis set (6-31G*) with the B3LYP hybrid density functional method50−52 in conjunction with the Poisson–Boltzmann finite element method38,39 for solvent reaction field optimization and aqueous solvation energy prediction. The geometries and energy ranking of the resulting conformers were similar for both methods. The SM8 and PBF solvent models were used to calculate the free energy of hydration for the gas-phase minimized geometries.
NMR Spectroscopy
A Bruker Avance 600 MHz system equipped with a 5 mm TCI (cryo) probe or a Bruker Avance III 500 MHz system equipped with a 5 mm RT BBFO plus probe was used to analyze the compounds in DMSO-d6 (Sigma Aldrich, 99.9% deuteration) or CDCl3 (Sigma Aldrich, 99.9% deuteration) at 298 K. For assignment typically 1–2 mg of substance was dissolved in 140 μL of DMSO-d6 and transferred to a 2.5 mm Bruker MATCH tube. 1H and 13C resonances were assigned using standard 1D and 2D Bruker pulse sequences (TopSpin, version 2.1). H–H COSY,53 C–H HSQC,54 and C–H HMBC54 experiments were performed to determine 1H, 13C connectivities. H–H ROESY55 was used for intramolecular distance information, and an H–N HMBC54 experiment was utilized to obtain indirect 15N chemical shift information and connectivities. 1H and 13C chemical shifts were referenced relative to residual solvent signals set to 2.50 and 39.5 ppm (DMSO) and to 7.27 and 77.0 ppm (CDCl3). 15N was indirectly referenced in TopSpin via the 1H, 15N gyromagnetic ratios. Temperature dependence studies were made on the same DMSO-d6 samples and on samples of similar concentration in CDCl3.
Acknowledgments
This work was funded by AstraZeneca R&D Mölndal, Sweden, the Carl Trygger Foundation, the Swedish Research Council (Grants 9478 and 21386), and the NIGMS-sponsored Center of Excellence in Chemical Methodology and Library Development (Broad Institute CMLD; Grant P50 GM069721). We thank Dr. Stephen Johnston and Sara Andersson for determining the solubility and pKa, respectively, of compounds 1–8, and Jan Holmgren for assistance with log D measurements. We also thank Dr. Joanne Kotz for critical reading of this manuscript and Drs. David Lahr and Carol Mulrooney for helpful discussions pertaining to the computational experiments.
Glossary
Abbreviations Used
- bRO5
beyond Lipinski’s rule of 5
- DOS
diversity-oriented synthesis
- Fsp3
fraction sp3
- SD
standard deviation
Supporting Information Available
Additional information on the conformational and relative pKa calculations; tables and figures with solubility, log D, and cell permeability data; NMR data for compounds 1 and 8; and procedures for synthesis of the N-methylated derivatives of 1 and 8. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
¶ M.F.: Tri-Institutional Therapeutics Discovery Institute Inc., New York, NY, U.S.
Author Present Address
∀ M.P.: Ecole Polytechnique Fédérale de Lausanne, Switzerland.
Author Contributions
Δ B.O. and P.M. contributed equally.
Δ The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- Lipinski’s rule of 5 states that compounds with a molecular weight of >500, >5 hydrogen bond donors, >10 hydrogen bond acceptors, and a calculated lipophilicity (cLogP) of >5 are more likely to have poor cell permeability and oral absorption.
- Vieth M.; Sutherland J. J. Dependence of molecular properties on proteomic family for marketed oral drugs. J. Med. Chem. 2006, 49, 3451–3453. [DOI] [PubMed] [Google Scholar]
- Paolini G. V.; Shapland R. H. B.; van Hoorn W. P.; Mason J. S.; Hopkins A. L. Global mapping of pharmacological space. Nat. Biotechnol. 2006, 24, 805–815. [DOI] [PubMed] [Google Scholar]
- Newman D. J. Natural products as leads to potential drugs: an old process or the new hope for drug discovery?. J. Med. Chem. 2008, 51, 2589–2599. [DOI] [PubMed] [Google Scholar]
- Li J. W.-H.; Vederas J. C. Drug discovery and natural products: end of an era or an endless frontier?. Science 2009, 325, 161–165. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [DOI] [PubMed] [Google Scholar]
- O’ Connor C. J.; Beckmann H. S. G.; Spring D. R. Diversity-oriented synthesis: producing chemical tools for dissecting biology. Chem. Soc. Rev. 2012, 41, 4444–4456. [DOI] [PubMed] [Google Scholar]
- Alex A.; Millan D. S.; Perez M.; Wakenhut F.; Whitlock G. A. Intramolecular hydrogen bonding to improve membrane permeability and absorption in beyond rule of five chemical space. Med. Chem. Commun. 2011, 2, 669–674. [Google Scholar]
- Terrett N. Drugs in middle space. Med. Chem. Commun. 2013, 4, 474–475. [Google Scholar]
- There is no clear and agreed and accepted definition of bRo5 or middle space. Lipinski pointed out that violation of one of his empirical rules still allowed compounds to have acceptable cell permeability and oral absorption. Therefore, a simple definition of bRo5 or middle space could be for compounds found in this chemical space to have ≥2 violations of Ro5.
- El Tayar N.; Mark A. E.; Vallat P.; Brunne R. M.; Testa B.; van Gunsteren W. F. Solvent-dependent conformation and hydrogen-bonding capacity of cyclosporin A: evidence from partition coefficients and molecular dynamics simulations. J. Med. Chem. 1993, 36, 3757–3764. [DOI] [PubMed] [Google Scholar]
- Ptachcinski R. J.; Burckart G. J.; Venkataramanan R. Cyclosporine. Drug Intell. Clin. Pharm. 1985, 19, 90–100. [DOI] [PubMed] [Google Scholar]
- Fahr A. Cyclosporin clinical pharmacokinetics. Clin. Pharmacokinet. 1993, 24, 472–495. [DOI] [PubMed] [Google Scholar]
- Rezai T.; Bock J. E.; Zhou M. V.; Kalyanaraman C.; Lokey R. S.; Jacobsen M. P. Conformational flexibility, internal hydrogen bonding, and passive membrane permeability: successful in silico prediction of the relative permeabilities of cyclic peptides. J. Am. Chem. Soc. 2006, 128, 14073–14080. [DOI] [PubMed] [Google Scholar]
- Chen J.; Rong F.; Shan B.; Chen Y.; Li Y.; Yu H.; Chen L.; Kuang T.; Li S.; Chen Y.; Duc J.; Ai C.; Li J.; Li X.; Shi C.; Jiang Z.; Long Y.; Gao Q.; Wang Z.; Xu K.; Ran X.; Yi H.; Zhao D.; Qiao H.; Shen J.; Liu B.; Liu C.; Wub K.; Geng X.; Tan J.; McLeod D.; Frost H.; Bai G.; Goetz G.; Federico J. III; Whitney-Pickett C.; Troutman M.; Noe M. C.; Guimaraes C.; Piotrowski D. W.; Magee T. V. Synthesis of 12-membered macrocyclic templates and library analogs for PPI. Tetrahedron Lett. 2013, 54, 3298–3301. [Google Scholar]
- White T. R.; Renzelman C. M.; Rand A. C.; Rezai T.; McEwen C. M.; Gelev V. M.; Turner R. A.; Linington R. G.; Leung S. S. F.; Kalgutkar A. S.; Bauman J. N.; Zhang Y.; Liras S.; Price D. A.; Mathiowetz A. M.; Jacobsen M. P.; Lokey R. S. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand A. C.; Leung S. S. F.; Eng H.; Rotter C. J.; Sharma R.; Kalgutkar A. S.; Zhang Y.; Varma M. V.; Farley K. A.; Khunte B.; Limberakis C.; Price D. A.; Liras S.; Mathiowetz A. M.; Jacobson M. P.; Lokey R. S. Optimizing PK properties of cyclic peptides: the effect of side chain substitutions on permeability and clearance. Med. Chem. Commun. 2012, 3, 1282–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood V. A.; Field M. J.; Horwell D. C.; Julien-Larose C.; Lewthwaite R. A.; McCleary S.; Pritchard M. C.; Raphy J.; Singh L. Utilization of an intramolecular hydrogen bond to increase the CNS penetration of an NK1 receptor antagonist. J. Med. Chem. 2001, 44, 2276–2285. [DOI] [PubMed] [Google Scholar]
- Ettorre A.; D’Andrea P.; Mauro S.; Porcelloni M.; Rossi C.; Altamura M.; Catalioto R. M.; Giuliani S.; Maggi C. A.; Fattori D. hNK2 receptor antagonists. The use of intramolecular hydrogen bonding to increase solubility and membrane permeability. Bioorg. Med. Chem. Lett. 2011, 21, 1807–1809. [DOI] [PubMed] [Google Scholar]
- Wu C.; Decker E. R.; Blok N.; Li J.; Bourgoyne A. R.; Bui H.; Keller K. M.; Knowles V.; Li W.; Stavros F. D.; Holland G. W.; Brock T. A.; Dixon R. A. F. Acyl substitution at the ortho position of anilides enhances oral bioavailability of thiophene sulfonamides: TBC3214, an ETA selective endothelin antagonist1. J. Med. Chem. 2001, 44, 1211–1216. [DOI] [PubMed] [Google Scholar]
- Sasaki S.; Cho N.; Nara Y.; Harada M.; Endo S.; Suzuki M.; Furuya S.; Fujino M. Discovery of a thieno[2,3-d]pyrimidine-2,4-dione bearing a p-methoxyureidophenyl moiety at the 6-position: a highly potent and orally bioavailable non-peptide antagonist for the human luteinizing hormone-releasing hormone receptor. J. Med. Chem. 2003, 46, 113–124. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L. Molecular diversity by design. Nature 2009, 457, 153–154. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- Gerard B.; Lee M. D. IV; Dandapani S.; Duvall J. R.; Fitzgerald M. E.; Kesavan S.; Lowe J. T.; Marié J.-C.; Pandya B. A.; Suh B.-C.; O’Shea M. W.; Dombrowski M.; Hamann D.; Lemercier B.; Murillo T.; Akella L. B.; Foley M. A.; Marcaurelle L. A. Synthesis of stereochemically and skeletally diverse fused ring systems from functionalized C-glycosides. J. Org. Chem. 2013, 78, 5160–5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J. T.; Lee M. D. IV; Akella L. B.; Davoine E.; Donckele E. J.; Durak L.; Duvall J. R.; Gerard B.; Holson E. B.; Joliton A.; Kesavan S.; Lemercier B. C.; Liu H.; Marié J.-C.; Mulrooney C. A.; Muncipinto G.; Welzel-O’Shea M.; Panko L. M.; Rowley A.; Suh B.-C.; Thomas M.; Wagner F. F.; Wei J.; Foley M. A.; Marcaurelle L. A. Synthesis and profiling of a diverse collection of azetidine-based scaffolds for the development of CNS-focused lead-like libraries. J. Org. Chem. 2012, 77, 7187–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald M. E.; Mulrooney C. A.; Duvall J. R.; Wei J.; Suh B.-C.; Akella L. B.; Vrcic A.; Marcaurelle L. A. Build/couple/pair strategy for the synthesis of stereochemically diverse macrolactams via head-to-tail cyclization. ACS Comb. Sci. 2012, 14, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcaurelle L. A.; Comer E.; Dandapani S.; Duvall J. R.; Gerard B.; Kesavan S.; Lee M. D. IV; Liu H.; Lowe J. T.; Marie J.-C.; Mulrooney C. A.; Pandya B. A.; Rowley A.; Ryba T. D.; Suh B.-C.; Wei J.; Young D. W.; Akella L. B.; Ross N. T.; Zhang Y.-L.; Fass D. M.; Reis S. A.; Zhao W.-N.; Haggarty S. J.; Palmer M.; Foley M. A. An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: discovery of macrocyclic histone deacetylase inhibitors. J. Am. Chem. Soc. 2010, 132, 16962–16976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comer E.; Liu H.; Joliton A.; Clabaut A.; Johnson C.; Akella L. B.; Marcaurelle L. A. Fragment-based domain shuffling approach for the synthesis of pyran-based macrocycles. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6751–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roughley S. D.; Jordan A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- Dandapani S.; Germain A. R.; Jewett I.; LeQuemen S.; Marie J.-C.; Muncipinto G.; Duvall J. R.; Carmody L. C.; Perez J. R.; Engel J. C.; Gut J.; Keller D.; de Siqueira Neto J. L.; McKerrow J. H.; Kaiser M.; Rodriguez A.; Palmer M. A. J.; Foley M.; Schreiber S. L.; Munoz B. Diversity-oriented synthesis yields a new drug lead for treatment of Chagas disease. ACS Med. Chem. Lett. 2014, 5, 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhoff S.; Ungell A.-L.; Zamora I.; Artursson P. pH-Dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: implications for drug–drug interactions. Pharm. Res. 2003, 20, 1141–1148. [DOI] [PubMed] [Google Scholar]
- Beckett A. H.; Dwuma-Badu D. The influence of stereochemistry on pKa, rate of quaternization and partition coefficients of corynantheidine-type alkaloids. J. Pharm. Pharmacol. 1969, 21Suppl.162S–168S. [DOI] [PubMed] [Google Scholar]
- Rupp M.; Körner R.; Tetko I. V. Predicting the pKa of small molecules. Comb. Chem. High Throughput Screening 2011, 14, 307–327. [DOI] [PubMed] [Google Scholar]
- Toth A. M.; Liptak M. D.; Phillips D. L.; Shields G. C. Accurate relative pKa calculations for carboxylic acids using complete basis set and Gaussian-n models combined with continuum solvation methods. J. Chem. Phys. 2001, 114, 4595–4606. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; MMFF V. I. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Olson R. M.; Kelly C. P.; Cramer C. J.; Truhlar D. G. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J. Chem. Theory Comput. 2007, 3, 2011–2033. [DOI] [PubMed] [Google Scholar]
- Tannor D. J.; Marten B.; Murphy R.; Friesner R. A.; Sitkoff D.; Nicholls A.; Ringnalda M.; Goddard W. A. III; Honig B. Accurate first principles calculation of molecular charge distributions and solvation energies from ab initio quantum mechanics and continuum dielectric theory. J. Am. Chem. Soc. 1994, 116, 11875–11882. [Google Scholar]
- Marten B.; Kim K.; Cortis C.; Friesner R. A.; Murphy R. B.; Ringnalda M. N.; Sitkoff D.; Honig B. New model for calculation of solvation free energies: correction of self-consistent reaction field continuum dielectric theory for short-range hydrogen-bonding effects. J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar]
- Wagner G.; Pardi A.; Wuthrich K. Hydrogen bond length and 1H NMR chemical shifts in proteins. J. Am. Chem. Soc. 1983, 105, 5948–5949. [Google Scholar]
- Jansma A.; Zhang Q.; Li B.; Ding Q.; Uno T.; Bursulaya B.; Liu Y.; Furet P.; Gray N. S.; Geierstanger B. H. Verification of a designed intramolecular hydrogen bond in a drug scaffold by nuclear magnetic resonance spectroscopy. J. Med. Chem. 2007, 50, 5875–5877. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Mohr P.; Stahl M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. [DOI] [PubMed] [Google Scholar]
- Shalaeva M.; Caron G.; Abramov Y. A.; O’Connell T. N.; Plummer M. S.; Yalamanchi G.; Farley K. A.; Goetz G. H.; Philippe L.; Shapiro M. J. Integrating intramolecular hydrogen bonding (IMHB) considerations in drug discovery using ΔlogP as a tool. J. Med. Chem. 2013, 56, 4870–4879. [DOI] [PubMed] [Google Scholar]
- Driggers E. M.; Hale S. P.; Lee J.; Terrett N. F. The exploration of macrocycles for drug discovery—an underexploited structural class. Nat. Rev. Drug Discovery 2008, 7, 608–624. [DOI] [PubMed] [Google Scholar]
- Giordanetto F.; Kihlberg J. Macrocyclic drugs and clinical candidates: What can medicinal chemists learn from their properties?. J. Med. Chem. 2014, 57, 278–295. [DOI] [PubMed] [Google Scholar]
- PhysChem Batch Predictor, version 9.04; ACDLabs: Toronto, Canada, 2005. [Google Scholar]
- MOE, version 2013.08; Chemical Computing Group: Montreal, Canada, 2013. [Google Scholar]
- Molecular Operating Environment (MOE), version 2012.10; Chemical Computing Group Inc. (1010 Sherbooke St. West, Suite No. 910, Montreal, QC, Canada, H3A 2R7: ), 2012. [Google Scholar]
- Jaguar, version 8.5; Schrödinger, LLC: New York, NY, 2013. [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- Vosko S. H.; Wilk L.; Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar]
- von Kienlin M.; Moonen C. T. W.; van der Toorn A.; van Zijl P. C. M. Rapid recording of solvent-suppressed 2D COSY spectra with inherent quadrature detection using pulsed field gradients. J. Magn. Reson. 1991, 93, 423–429. [Google Scholar]
- Willker W.; Leibfritz D.; Kerssebaum R.; Bermel W. Gradient selection in inverse heteronuclear correlation spectroscopy. Magn. Reson. Chem. 1993, 31, 287–292. [Google Scholar]
- Marion D. Rotating frame nuclear overhauser effect: a practical tool for the 1H NMR study of peptides in solution. FEBS Lett. 1985, 192, 99–103. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.