

Summary
Severe sepsis remains a poorly understood systemic inflammatory condition with high mortality rates and limited therapeutic options in addition to organ support measures. Here we show that the clinically approved group of anthracyclines acts therapeutically at a low dose regimen to confer robust protection against severe sepsis in mice. This salutary effect is strictly dependent on the activation of DNA damage response and autophagy pathways in the lung, as demonstrated by deletion of the ataxia telangiectasia mutated (Atm) or the autophagy-related protein 7 (Atg7) specifically in this organ. The protective effect of anthracyclines occurs irrespectively of pathogen burden, conferring disease tolerance to severe sepsis. These findings demonstrate that DNA damage responses, including the ATM and Fancony Anemia pathways, are important modulators of immune responses and might be exploited to confer protection to inflammation-driven conditions, including severe sepsis.
Keywords: Sepsis, ATM, Autophagy, Anthracyclines
INTRODUCTION
Sepsis is a life-threatening condition that arises as a systemic inflammatory response to an infection (Bone et al., 1992; Levy et al., 2003). It includes a continuum of clinical severity ranging from systemic inflammatory response syndrome (SIRS), sepsis, severe sepsis to septic shock (Suffredini and Munford, 2011). It is the leading cause of death in intensive care units and the third cause of overall hospital mortality (Angus and Wax, 2001; Ulloa and Tracey, 2005). In spite of substantial improvement in diagnosis and support measures, the global annual mortality rate is ~28% (Hotchkiss and Karl, 2003), ranging from less than 10% in SIRS to up to 70% in septic shock (Angus and Wax, 2001; Annane et al., 2003). The pathophysiology of sepsis remains poorly understood. As a result, the basic elements of treatment – early antibiotics, prompt control of the source of infection and organ support - have not changed substantially in the last fifty years, and attempts to translate basic research results into effective new interventions have been met with limited or no success (Suffredini and Munford, 2011). In the same period, the incidence of sepsis and its economic burden has increased by 1% each year (Martin et al., 2003; Ulloa and Tracey, 2005), indicating the urgent need for novel therapeutic options.
Inflammation is a response to harmful stimuli that limits tissue damage and aims at restoring homeostasis (Medzhitov, 2008). Pathogen-associated molecular patterns (PAMPs) on microorganisms and damage-associated molecular patterns (DAMPs) originating from dying cells are sensed by the host through germline-encoded pattern recognition receptors (PRRs) that recognize conserved signature structures in non-self and self (Janeway and Medzhitov, 2002). These sensors are present in both professional (including neutrophils, macrophages and dendritic cells) and non-professional immune cells and their activation initiates intracellular signaling cascades leading to the transcriptional expression of inflammatory mediators, such as cytokines and chemokines. Inflammation needs to be effectively terminated after removal of the original trigger for repair of damaged tissue to occur. In the susceptible host, overproduction of inflammatory mediators or an exaggerated response to their presence can lead to septic shock, tissue destruction or permanent loss of function (Takeuchi and Akira, 2010).
There are two evolutionarily conserved defense strategies against infection that can limit host disease severity. One relies on reducing pathogen load, i.e. resistance to infection, while the other provides host tissue damage control, limiting disease severity irrespectively of pathogen load, i.e. tolerance to infection (Raberg et al., 2009; Schneider and Ayres, 2008). As demonstrated originally for plants and thereafter in Drosophila, tolerance to infection also operates in mammals, as revealed for Plasmodium (Raberg et al., 2007; Seixas et al., 2009) and polymicrobial infections in severe sepsis (Larsen et al., 2010).
Here we show in an experimental mouse model that anthracyclines confer strong protection against sepsis by increasing disease tolerance to infection, that is, acting irrespectively of pathogen burden. We further show that ATM (ataxia telangiectasia mutated) kinase and the induction of autophagy are strictly required for the in vivo protection against sepsis. These molecular pathways provide strong damage control in tissues, specifically in the lung.
RESULTS
Anthracyclines confer strong protection against severe sepsis
In an in vitro chemical screen using ~2320 compounds, we identified several lead candidates capable of inhibiting inflammatory cytokine production in response to E. coli challenge by the THP-1 macrophage line (Figure S1a and Supplemental Table S1). This inhibitory effect was dissociated from cytotoxicity of the compounds tested on THP-1 cells (Figure S1b). Among these, we found 3 representatives of the anthracycline family of chemotherapeutic agents namely epirubicin, doxorubicin and daunorubicin, and validated their inhibitory activity on cytokine production (Figure S1c).
We then used the cecal ligation and puncture (CLP) mouse model of experimental sepsis to investigate the in vivo effects of epirubicin (Rittirsch et al., 2009). In CLP, sepsis results from a polymicrobial infection of abdominal origin, leading to bacteremia and a systemic inflammatory response (Rittirsch et al., 2009). We adjusted CLP severity to a high-grade sepsis, where at least 80% of C57BL/6 mice succumbed within 48 h after the initial procedure. Under these conditions, epirubicin administered i.p. at the time of CLP and again 24 h later in a total of 1.2μg/g of body weight reproducibly and significantly (p<0.001) increased the survival of C57BL/6 mice subjected to CLP by nearly 80%, without the use of antibiotics (Figure 1a). A similar protective effect was observed in epirubicin-treated animals with the same dose and schedule but administered i.v. (Figure S1d). This appeared to be a general property of the anthracycline family because other representative members of this family of drugs identified in the initial chemical screen conferred a similar degree of protection against CLP (Figure 1b). The protective effect of anthracyclines was not dependent on the mouse strain as outbread NMRI mice were similarly protected by epirubicin (Figure 1c). Epirubicin was equally effective against another clinically relevant pathogen causing sepsis, K. pneumoniae administered intranasally (Figure 1d), arguing that epirubicin can be effective in the treatment of sepsis of different origins in addition to peritoneal sepsis. Mice previously subjected to CLP and treated with epirubicin were not immunocompromised as they could clear a secondary intranasal viral infection similarly to control mice (Figure 1e). Taken together, these results indicate that low doses of the anthracycline family of chemotherapeutic agents confer strong protection against severe sepsis, without causing host immunosuppression.
Figure 1. Epirubicin affords protection against severe sepsis.

(a) Survival of C57BL/6 wild-type animals subjected to CLP treated with carrier (PBS) or epirubicin (Epi) (0.6μg/g body weight) at the time of procedure and 24 hours later. (b) Survival of C57BL/6 wild-type animals subjected to CLP treated with carrier (PBS), epirubicin (Epi), doxorubicin (Doxo) or daunorubicin (Dauno). Treatment schedule and doses as in (a). (c) Survival of NMRI mice subjected to CLP and treated with carrier (PBS) or epirubicin (Epi) as in (a). (d) Survival of C57BL/6 wild-type animals following intranasal inoculation of Klebsiella pneumoniae and treated with carrier (PBS) or epirubicin (Epi) as in (a). (e) Quantification of infectious viral MuHV-4 particles in lung of C57BL/6 wild-type animals previously subjected to mock CLP (S), mock CLP treated with epirubicin (S+E) or CLP treated with epirubicin (C+E). Epirubicin treatment dose and schedule as in (a). Mice were intranasally inoculated with 1000 PFU of MuHV-4 on day 3 post CLP and viral particles quantified by plaque assay at days 6 and 12 post viral infection. Each circle represents individual animals and horizontal lines indicate arithmetic means ± SEM from two independent assays. The dashed horizontal line represents the limit of detection of the assay. ns, not significant; *P<0.05; **P<0.01; ***P<0.001 (log-rank (Mantel-Cox) test for (a) to (d) and Mann-Whitney test for (e)). See also Figure S1 and Table S1.
Epirubicin acts therapeutically to promote disease tolerance to severe sepsis
We found that in epirubicin-treated mice subjected to CLP the bacterial load in blood and target organs of sepsis, e.g., lung, liver, kidney and spleen 24 h post-CLP did not differ from that of untreated controls (Figure 2a). While at 48 h post-CLP we noticed a trend towards a lower bacterial load in the target organs of epirubicin-treated animals, the differences were not statistically significant, even if most untreated control animals die between 24 and 48 h after the CLP procedure. These results raised the possibility that the protective effect of epirubicin in vivo is related to disease tolerance without directly affecting the pathogen burden(Medzhitov et al., 2012). This idea was supported by the observation that the serum concentrations of several markers of tissue damage such as LDH (lung and general cellular damage), CK (muscle), ALT (liver) and urea (kidney) were substantially reduced to almost basal levels in epirubicin-treated mice, 24 h after CLP, compared to untreated mice (Figure 2b). In addition, we observed a substantial reduction in the levels of inflammatory mediators including TNF, IL-1β, IL-6 and HMGB1 compared to non-treated CLP mice (Figure 2c). We have also observed improvement of histological lesions in the lung, liver and kidney after CLP by treatment with epirubicin (Figure S2). To explore this further in the absence of bacteria, we found that the drug protected C57BL/6 mice from lethal septic shock caused by lipopolysaccharide (LPS, endotoxin) (Figure 2d).
Figure 2. Epirubicin promotes disease tolerance to severe sepsis.

(a) Polymicrobial load (CFUs) in blood, lung, liver, kidney and spleen, at indicated time points, of C57BL/6 animals undergoing CLP and treated with PBS (C+P) or epirubicin (C+E) (0.6μg/g body weight) at the time of procedure and 24 hours later. Each circle represents individual animals. Horizontal lines indicate arithmetic means ± SEM. (b) and (c) Epirubicin counteracts tissue damage and inflammation associated with CLP as assessed by (b) LDH, CK, ALT, urea and (c) TNF, IL-1β, IL-6 and HMGB1 plasma concentrations in C57BL/6 wild-type animals 24 hours after mock CLP (S) (n=2) or CLP followed by treatment with PBS (C+P) (n=5) or epirubicin (C+E) (n=7) as in (a). Results shown represent arithmetic means ± SEM from duplicate (b) or triplicate (c) readings per animal. (d) Survival of C57BL/6 wild-type animals following lethal LPS injection and treatment with carrier (PBS) or epirubicin (Epi) as in (a). (e) Survival of C57BL/6 wild-type animals subjected to CLP treated with carrier (PBS), meropenem (40μg/g body weight/day) or epirubicin (Epi) as in (a). (f) CFUs in blood, at indicated time, of C57BL/6 animals undergoing mock CLP (S) or CLP followed by treatement with PBS (C+P), epirubicin (C+E) or meropenem (C+M) as in (a). Each circle represents individual animals. Horizontal lines indicate arithmetic means ± SEM. (g) IL-1β, TNF and HMGB1 plasma concentrations in C57BL/6 wild-type animals 24 hours after CLP followed by treatment with PBS (C+P) (n=4), epirubicin (C+E) (n=5) or meropenem (C+M) (n=5) as in (c). ns, not significant; *P<0.05; **P<0.01; ***P<0.001 (log-rank (Mantel-Cox) test for (d) and (e), Mann-Whitney test for (a) and (f), and unpaired t test for (b), (c) and (g)). See also Figure S2.
Large spectrum antibiotics such as meropenem are very effective at lowering bacteremia and are standard drugs used in sepsis (Russell, 2006). We tested the efficacy of meropenem in CLP in comparison to epirubicin and found that while meropenem delayed the death rate of CLP-subjected mice, it did not prevent mortality (Figure 2e), in spite of a strong impact on bacterial burden (Figure 2f). This was in sharp contrast to the action of epirubicin, that did not interfere with bacteremia (Figure 2f) but prevented CLP-induced mortality (Figure 2e), again arguing for a role of epirubicin in conferring disease tolerance against severe sepsis (Larsen et al., 2010; Medzhitov et al., 2012).
Both epirubicin and meropenem decreased the amounts of IL-1β, TNF and HMGB1 in the serum of mice subjected to CLP (Figure 2g). This indicates that whereas decreased circulating levels of inflammatory mediators may contribute to confer protection against severe sepsis, inhibition of IL-1β, TNF and HMGB1 is not sufficient per se to explain the protective effect of epirubicin, which is in accordance with what is observed for other therapeutic approaches in the clinical setting (Hotchkiss and Karl, 2003). Taken together these data suggest that epirubicin acts through an additional alternative mechanism to cytokine inhibition to confer tolerance to sepsis.
Epirubicin protection against sepsis is mediated by ATM
Next, in order to explore the molecular mechanism behind the protective effects of anthracyclines, we used our in vitro assay system to perform a short hairpin RNA (shRNA)-based screen in THP-1 cells, focusing on kinases and phosphatases and using IL-1β and TNF secretion as assay readouts. While our in vivo results suggested the possibility that anthracyclines ameliorate the lethal effects of sepsis by a mechanism affecting tissue tolerance, we reasoned that our in vitro assay would be useful for the identification of candidate pathways mediating the anthracycline effects. We found several negative regulators of IL-1β productionin response to E. coli challenge, including the genes encoding Ataxia Telangiectasia Mutated (ATM), Checkpoint Kinase 1 (CHEK1) and Ataxia Telangiectasia and Rad3 Related (ATR) (Figure S3 and Supplemental Table S2). These findings suggest that DNA damage response (DDR) components are negative regulators of IL-1β secretion. Using a phospho-specific antibody against the activated form of ATM, we found that although E. coli alone was a poor, but reproducible ATM activator (Figure S3), epirubicin alone or in combination with E. coli triggered a robust ATM activation (Figure S3). This was confirmed using immunoblotting (Figure S3).
ATM is a master regulator of the DDR (Ciccia and Elledge, 2010) and is known to be activated by anthracyclines and other DNA damaging agents (Siu et al., 2004). Therefore we used ATM-deficient mice to test the contribution of the DDR to the protective effect of anthracyclines against severe sepsis. ATM-deficient (Atm−/−) mice were not protected by epirubicin against CLP and died with similar kinetics to those of wild-type (Atm+/+) animals that were treated with PBS alone (Figure 3a). We conclude that ATM expression is necessary to mediate the protective effect of epirubicin in sepsis. In striking contrast to wild-type mice (Figures 2b and c), in the absence of ATM, epirubicin no longer normalized the serologic markers of organ lesion (Figure 3b) or decreased the levels of inflammatory mediators (Figure 3c). However, in mice subjected to CLP and treated with etoposide (after in vivo titration to find the best and most effective dose), an agent known to cause DNA double strand breaks and to activate ATM-dependent pathways (Montecucco and Biamonti, 2007), mortality induced by CLP was only partially rescued (Figure 3d), suggesting that ATM is necessary but not sufficient for the protection conferred by anthracyclines against sepsis.
Figure 3. The protection afforded by epirubicin against severe sepsis is mediated by ATM.

(a) Survival of Atm+/+ and Atm−/− C57BL/6 animals subjected to CLP and treated with PBS or epirubicin (Epi) with same schedule and dose as in fig 1. (b) LDH, CK, ALT, urea and (c) TNF, IL-1β and IL-6 plasma concentrations in Atm−/− C57BL/6 animals 24 hours after mock CLP (S) (n=2) or CLP followed by treatment with PBS (C+P) (n=8) or epirubicin (C+E) (n=8) as in (a). Results shown represent arithmetic means ± SEM from triplicate readings per animal. (d) Survival of PBS-, etoposide (Eto)-, and epirubicin (Epi)-treated wild-type C57BL/6 animals undergoing CLP. Etoposide dose was 2μg/g body weight. Treatment schedule as in (a). (e) FANCD2 and Ub-FANCD2 protein levels by immunoblotting in THP-1 cells following E. coli challenge after a pre-incubation (1 hour) with carrier, epirubicin or KU-55933 as indicated. (f) Survival of Fancd2+/+ and Fancd2−/− animals subjected to CLP and treated with PBS or epirubicin (Epi) with same schedule and dose as in (a). (g) Representative sections of γH2AX staining and percentage of γH2AX+ cells per field (right panel) in lungs isolated 1 hour after mice were subjected to whole body γ irradiation (4 Gy). Results shown represent arithmetic means ± SD from 3 fields. (h) Survival of C57BL/6 wild-type animals subjected to CLP following whole body γ irradiation (4Gy) or treated with carrier (PBS) or epirubicin (Epi) as in (a). ns, not significant; *P<0.05; **P<0.01; ***P<0.001 (log-rank (Mantel-Cox) test for (a), (d), (f) and (h) and unpaired t test for (b), (c) and (g)). See also Figure S3 and Table S2.
In addition to double strand breaks (repaired in an ATM-dependent manner), anthracyclines also cause DNA interstrand cross-links, a DNA lesion known to be repaired by the Fanconi Anemia (FA) pathway (Ciccia and Elledge, 2010). Interestingly, FA patients were reported to spontaneously overproduce TNF (Briot et al., 2008; Vanderwerf et al., 2009), possibly because the FA protein FANCD2can directly inhibit TNF promoter activity (Matsushita et al., 2011). In THP-1 cells, we observed that FANCD2 is activated in an ATM-independent manner upon epirubicin treatment, as shown by its mono-ubiquitination (Figure 3e). These findings support the independence of signaling events initiated by the generation of DNA double strand breaks and DNA interstrand cross-links. We examined the contribution of this pathway for epirubicin protection of CLP and found that Fancd2−/− mice were slightly but significantly (p<0.05) impaired for the protective effects (Figure 3f).
These results suggest that activation of DDR is protective against sepsis. To further test this hypothesis we have used whole body sub-lethal γ-irradiation. We found a significant increase in the number of cells with γH2AX-positive foci (p<0.001), a surrogate marker of ATM activation (Ciccia and Elledge, 2010), in the lungs of whole body sub-lethal γ-irradiated mice as compared to controls (figure 3g). Mice subjected to CLP that were irradiated showed a significant increased survival (p<0.001) as compared to non-irradiated mice (Figure 3h). We conclude that the protective phenotype induced by epirubicin is dependent on the activation of multiple pathways downstream of a DDR. The activation of the ATM pathway is the main contributor, but the full protection requires the activation of additional DDR pathways, including the FA pathway.
The protective effect of epirubicin is dependent on the autophagy pathway
Although it is possible that the dominant ATM-mediated protection against sepsis might rely on ROS scavenging (Cosentino et al., 2010), on the induction of apoptosis of inflammatory cells (Garrison et al., 2011), on the preservation of genomic stability (Westbrook and Schiestl, 2010), or on the biogenesis of anti-inflammatory microRNAs such as miR-146a (Zhang et al., 2011), we found no significant contribution for any of these processes (Figure S4). We, therefore, explored a possible role for autophagy in this process, given that ATM is a negative regulator of mTOR, which is itself, an inhibitor of autophagy (Alexander et al., 2010a; Alexander et al., 2010b). Using autophagy-defective (Lc3b−/−) mice, we found that the autophagy pathway is required for the in vivo effect of epirubicin (Figure 4a). Similarly to Atm−/− mice (Figure 3b and c), epirubicin was not able to decrease the serologic markers associated with organ lesion (Figure 4b) or to normalize cytokine levels in autophagy-defective mice (Figure 4c).
Figure 4. The ATM-dependent protection of epirubicin against severe sepsis relies on the induction of autophagy.

(a) Survival of Lc3b+/+ and Lc3b−/− animals subjected to CLP and treated with PBS or epirubicin (Epi) with same schedule and dose as in fig 1. (b) LDH, CK, ALT, urea and (c) TNF, IL-1β and IL-6 plasma concentrations in Lc3b−/− animals 24 hours after mock CLP (S) (n=2) or CLP followed by treatment with PBS (C+P) (n=4) or epirubicin (C+E) (n=7) as in (2b). Results shown represent arithmetic means ± SEM from triplicate readings per animal. ns, not significant; *P<0.05; **P<0.01; ***P<0.001 (log-rank (Mantel-Cox) test for (a), unpaired t test for (b) and (c)). See also Figure S4.
We then used LC3b-GFP mice to study the contribution of the autophagy pathway in the protection conferred by epirubicin. While FACS analysis shows that CLP alone induces LC3b aggregation in different splenocyte populations, namely monocytes and neutrophils, epirubicin treatment did not increase the autophagy pathway in these critical players in sepsis (Figure 5a). We then tested the impact of epirubicin on the survival of a conditional depletion of Atg7 specifically in neutrophils and monocytes upon CLP, using Atg7loxP/loxPLysMCre GFP-LC3b animals. Strikingly, these animals were equally protected by epirubicin as compared to control mice (Figure 5b), suggesting that the autophagy pathway is not required in the myeloid compartment for the protective effects of epirubicin against sepsis.
Figure 5. The protective effect of epirubicin is dependent on the activation of ATM and the autophagy pathway in the lung.

(a) GFP expression in blood monocytes and neutrophils, isolated from transgenic LC3b-GFP animals, 24 hours after mice were subjected to mock CLP (S) or CLP followed by treatment with PBS (C+P) or epirubicin (C+E) (0.6μg/g body weight) at the time of procedure and 24 hours later. Each circle represents individual animals. Horizontal lines indicate arithmetic means ± SEM. (b) Survival of Atg7loxP/loxP and Atg7loxP/loxP LysMcre/cre mice subjected to CLP and treated with PBS or epirubicin (Epi) with same schedule and dose as in (a). (c) LC3B-I and LC3B-II protein levels by immunoblotting using a specific antibody against LC3B in lung, liver and kidney, isolated at the indicated times, of naïve C57BL/6 animals (C) or mice subjected to mock CLP (S) or CLP followed by treatment with PBS (C+P) or epirubicin (C+E) as in (a). (d) Representative sections of LC3b staining in lungs, isolated at the indicated times, of mice subjected to CLP followed by treatment with PBS (C+P) or epirubicin (C+E) as in (a). (e) Survival of wild-type (B6) and Atg7loxP/loxP animals subjected to CLP and treated with PBS or epirubicin (Epi) with same schedule and dose as in (b) 5 days after inhalation of adenoviral vector encoding Cre (AdCre). (f) LDH, CK, ALT and urea plasma concentrations in wild-type (B6 AdCre) and Atg7loxP/loxP AdCre animals 24 hours after mock CLP (S) (n=2 for B6 AdCre) or CLP followed by treatment with PBS (C+P) (n=5 for B6 AdCre and n=2 for Atg7loxP/loxP AdCre) or epirubicin (C+E) (n=6 for B6 AdCre and n=3 for Atg7loxP/loxP AdCre) as in (a). (g) Survival of wild-type (B6) animals subjected to CLP 4 days after inhalation of adenoviral vector encoding GFP (AdGFP) or Atg7 (AdATG7). (h) Representative sections of γH2AX staining and percentage of γH2AX+ cells per field (right panel) in lungs, isolated 6 hours after mice were subjected to CLP followed by treatment with PBS (C+P) or epirubicin (C+E) as in (a). Results shown represent arithmetic means ± SD from 10 fields. (i) Survival of wild-type (B6) and AtmloxP/loxP animals subjected to CLP and treated with PBS or epirubicin (Epi) with same schedule and dose as in (b) 5 days after inhalation of AdCre. (j) Survival of C57BL/6 wild-type animals subjected to CLP treated with carrier (PBS), etoposide (40μg/g body weight/day) or epirubicin (Epi) (0.6μg/g body weight) intranasally, at the time of procedure and 24 hours later. ns, not significant; *P<0.05; **P<0.01; ***P<0.001 (log-rank (Mantel-Cox) test for (b), (e), (g), (i) and (j), Mann-Whitney test for (a), and unpaired t-test for (f) and (h) (right panel)). See also Figure S5.
Autophagy can be effectively monitored by the conversion and immobilization of LC3 (Kabeya et al., 2000). Because the autophagy pathway was not required in the myeloid compartment for protection against sepsis by epirubicin, we then looked at target organs of sepsis (lung, liver and kidney) using immunobloting to identify lipidation of LC3b as indicative of activation of the autophagy pathway. We found that epirubicin specifically induced lipidation of LC3b in the lung at 6 h, but not in the liver or kidney (Figure 5c). Although LC3 was transiently lipidated after CLP in the liver at 6 and 24 h as previously reported (Chien et al., 2011), levels of LC3 were not altered by epirubicin treatment (Figure 5c). We have further confirmed that autophagy was induced in the lung as shown by the increase of LC3b positive vesicles in lung sections at 6 h and 24 h comparing epirubicin treated and non-treated mice (Figure 5d).
We then deleted Atg7 specifically in the lung (Figure S5), using an adenovirus-expressing CRE (Adcre) to intranasally infect Atg7loxP/loxP mice (Komatsu et al., 2005). When subjected to CLP, these mice were no longer protected from CLP by epirubicin treatment (Figure 5e). In contrast to control mice, in Atg7loxP/loxPAd cre mice the treatment with epirubicin does not improve the levels of circulating markers of organ lesion (Figure 5f). Accordingly, we observed a significant protective effect in survival after overexpression of ATG7 in the lung using adenovirus (Figure 5g).
By assessing the levels of γH2AX in the lungs of control or epirubicin-treated CLP-subjected mice, we found a significant increase in the number of cells with γH2AX-positive foci in lungs of epirubicin-treated mice (Figure 5h). To test whether ATM activation was also required in the lung, we used AtmloxP/loxP mice and Adcre to delete ATM specifically in the lung. Upon Adcre-mediated ATM deletion in the lung, mice were no longer protected against sepsis by treatment with epirubicin (Figure 5i). We therefore conclude that the protective effect of epirubicin in sepsis is, at least in part, dependent on the activation of ATM and of autophagy in target organs, namely the lung. These conclusions are further supported by intranasal delivery of epirubicin or etoposide to the lung (Figure 5j), because the protective effects as measured by survival are similar to the i.p. administration of those drugs (Figure 5j).
Epirubicin has a 24 h therapeutic window to protect against sepsis
Finally, we studied the therapeutic window of epirubicin in mice. When given alone, epirubicin conferred strong protection at the time of the procedure or until 3 h after the initiation of CLP (Figure 6a). When administered only 6 h after CLP, epirubicin quickly lost its protective effect (Figure 6a). However, if given in combination with meropenem, even when this antibiotic is only administered 12 h after CLP, low dose epirubicin conferred complete protection until at least 24 h after the initial procedure (Figure 6b and 6c). These results suggest that anthracyclines can be used not only to prevent sepsis, but also that they can act therapeutically when their administration is combined with a large spectrum antibiotic.
Figure 6. Epirubicin confers protection against severe sepsis in a therapeutic manner.
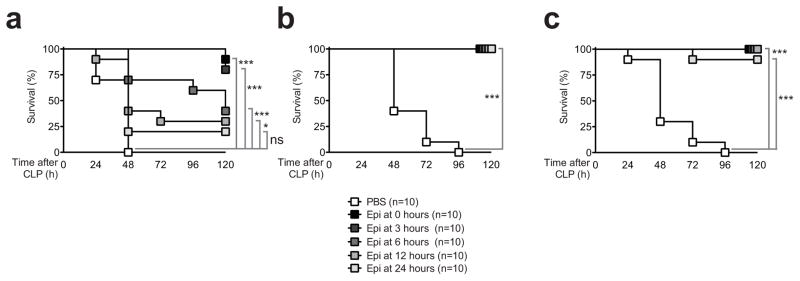
(a) Survival of C57BL/6 wild-type animals subjected to CLP treated with PBS or epirubicin (same dose as in Figure 1) at indicated times in the absence of meropenem; (b) with administration of meropenem (40μg/g body weight/day) starting at the time of the procedure or (c) with meropenem treatment starting 12 hours after CLP. ns, not significant; *P<0.05; **P<0.01; ***P<0.001 (log-rank (Mantel-Cox)).
Discussion
Here we report that epirubicin, and more generally the group of anthracyclines, are very effective at conferring protection against severe sepsis in mice, even when used up to 24 h after the onset of infection. This therapeutic window is likely to be sufficient to make these drugs good candidates as useful therapeutic options in the clinic to reduce the mortality of sepsis in most patients that are either in the hospital or seek medical attention within the first few hours of symptoms initiation.
Although we began our investigation of the use of anthracyclines in sepsis by virtue of their effects in inhibiting inflammatory cytokine expression in myeloid cells in vitro, our studies have identified a mode of protection that seems to be much stronger and perhaps completely independent of such effects, and rather manifests at the level of DNA damage response and autophagy-induced protection in the lung. Thus, our findings uncover an unexpected role for these pathways in tissue (lung) tolerance to the pathological consequences of infection. These findings are especially relevant given that agents discovered in studies over the last few years targeting various pro-inflammatory cytokines have had limited success in humans. Our studies suggest a critical role for protecting host tissues thereby conferring protection against sepsis. Recent studies have highlighted the role of tissue tolerance to infection as an important aspect of host pathology (Medzhitov et al., 2012).
Interestingly, the protective effect of epirubicin seems to act irrespectively of the host pathogen burden, revealing that it confers disease tolerance to polymicrobial infection (Larsen et al., 2010; Raberg et al., 2009; Schneider and Ayres, 2008). This finding reveals that pharmacologic agents that provide tissue damage control can limit disease severity irrespectively of pathogen load and represent a promising therapeutic strategy against sepsis. Moreover, based on our identification of ATM as a major mediator of epirubicin effects, we propose that this protein and other components of the DNA damage response machinery constitute novel regulators of tolerance, without affecting pathogen resistance mechanisms.
Recent reports make our findings counter-intuitive as doxorubicin and daunorubicin have been shown to induce acute inflammation when injected in the abdomen where they induce cytokine secretion (Krysko et al., 2011; Sauter et al., 2011). However, the concentrations of anthracyclines utilized in these studies were more than 10-fold higher than those used here. By using lower concentrations we may reduce the cytotoxicity of these drugs and the resulting release of pro-inflammatory DAMPs by dying cells and reveal the additional pharmacological effects mediated by the surviving target cells. Interestingly, fluoroquinolones that are bacterial type II topoisomerase inhibitors, as opposed to anthracyclines, which are eukaryotic type II topoisomerase inhibitors, were reported to have immunomodulatory effects (Dalhoff and Shalit, 2003) when used in supra-therapeutic concentrations. Fluoroquinolones have been shown to protect against LPS model of septic shock (Khan et al., 2000). While the molecular mechanisms that explain these effects have not been elucidated, it has been proposed that higher doses of fluoroquinolones can inhibit mammalian topoisomerase type II enzymes in addition to their bacterial targets (Dalhoff and Shalit, 2003), an effect that can be achieved with very low doses of anthracyclines.
The induction of autophagy is a common response to many forms of cellular stress, including DNA damage (Mizushima and Komatsu, 2011). The rationale for testing the role of autophagy was based on the knowledge that ATM is a negative regulator of mTOR and that mTOR is a negative regulator of autophagy(Alexander et al., 2010a; Alexander et al., 2010b). We therefore reasoned that if epirubicin activates ATM (as we have confirmed), then it is possible that it induces autophagy. This line of investigation made sense because several reports(Chien et al., 2011; Nakahira et al., 2010) have suggested a protective role for autophagy in sepsis. To probe the contribution of autophagy for the protective phenotype conferred by epirubicin, we have used two different genetic deletions in the autophagy pathway (LC3b and ATG7) and in both cases the protection normally conferred by epirubicin is lost. This is considered one of the best and most compelling ways to test the contribution of autophagy and the direction of autophagic flow (Klionsky et al., 2012).
Chien et al. have previously found that autophagy is transiently induced in the rat liver after CLP (Chien et al., 2011), and later made similar observations in the rat kidney (Hsiao et al., 2012). Chien et al. have speculated that the transient induction of autophagy in these organs could be required for protection and its decline at a later stage could contribute to the functional failure in liver during polymicrobial sepsis. After we found autophagy induction by epirubicin, we thought that one reason to explain anthracycline protection against CLP could be the sustained activation of autophagy in the liver and/or kidney preventing its decline as observed by Chien et al. Instead we found that both in the liver and in the kidney, treatment with epirubicin did not change this pattern, but instead transiently induced autophagy in the lung, a response that was not present in mice subjected to CLP in the absence of epirubicin treatment. We also report that epirubicin is highly protective when delivered directly to the lung and that overexpression of ATG7 specifically in the lung improves survival to CLP. Together, these observations strongly suggest that lung protection is critical and likely dominant because it prevents failure of additional organs, which makes our findings all the more relevant as the lung is the organ that often shows the first signs of dysfunction in septic patients and drives the failure of other target organs particularly the kidney and later the liver (Hotchkiss and Karl, 2003).
Nakahira et al. demonstrate that depletion of the autophagic proteins LC3b and beclin 1 enhanced the activation of caspase-1 and secretion of IL-1β and IL-18 (Nakahira et al., 2010). While we also observe inhibition of IL-1β secretion in vitro and in vivo by treatment with epirubicin, this finding is not dependent on the decreased activation of caspase-1 mediated by autophagy because even low concentrations of epirubicin lead to higher levels of active caspase-1 (Chora et al., data not shown) but this event is overshadowed by a strong inhibition of IL-1β, as well as most of pro-inflammatory mediators that are NF-kB dependent, at the transcriptional level. We have now identified the mechanism: epirubicin targets the N-terminal region of p65, inhibiting transcription by blocking the DNA-binding ability of NF-kB (Chora et al., data not shown). The Nakahira et al. report also suggested to us that severe sepsis could cause DNA lesions capable of activating the inflammasome leading to chronic inflammation and that the induction of autophagy could block the inflammasome and prevent excessive inflammation. To address this possibility, we have used comet assays to look at different types of DNA damage in response to bacterial challenge in the presence and absence of epirubicin. We did find that E. coli alone triggers a small but measurable increase of DNA single strand breaks (Neves-Costa et al., unpublished). However the presence of epirubicin does not decrease, rather it increases DNA damage as compared to E. coli alone. Therefore, epirubicin does not decrease the generation of DNA damaged species that can activate the inflammasome. We conclude that while the Nakahira et al. work clearly shows that autophagic proteins regulate NALP3-dependent inflammation by preserving mitochondrial integrity, the autophagy protection conferred by epirubicin to CLP does not depend on the mechanisms demonstrated in the Nakahira et al. paper, specifically the negative regulation of IL-1β secretion by autophagy.
Interestingly, the protective phenotype of epirubicin is strikingly similar to that of RIPK3-deficient mice (Duprez et al., 2011), suggesting that epirubicin-mediated, ATM-dependent, autophagy induction can possibly prevent TNF-driven necroptosis in such key organs in sepsis pathology as the lung. In fact, there have been recent works that support the role of autophagy in the inhibition of necroptosis (Bray et al., 2012; Degenhardt et al., 2006; Lu and Walsh, 2012; Shen and Codogno, 2012), which could be achieved by targeting key necroptosis signaling components (such as RIPK1 and RIPK3) for degradation. It is also possible that autophagy protects against severe sepsis because its activation increases the degradation of pro-inflammatory mediators with an important role in sepsis, like HMGB1 (Li et al., 2011). In addition, increased effective autophagy can be beneficial in sepsis due to its critical role in the removal of damaged mitochondria in an ATM-dependent manner(Valentin-Vega and Kastan, 2012). The molecular mechanisms at the basis of epirubicin-induced protection in sepsis by autophagy are certainly an interesting topic for future studies.
Experimental Procedures
Animal Model and Anthracycline Treatment
Animal care and experimental procedures were conducted in accordance with Portuguese and US guidelines and regulations after approval by the respective local committees (Instituto de Medicina Molecular and Instituto Gulbenkian de Ciência). All mice used were 8–12 weeks old. Mice were bred and maintained under specific pathogen-free (SPF) conditions. C57BL/6 and C57BL/6 Atm−/− mi ce were obtained from the Instituto Gulbenkian de Ciência (a kind gift from Dr. Vasco Barreto). C57BL/6 Nrf2−/− mice were provided originally from the RIKEN BioResource Center (Koyadai, Tsukuba, Ibaraki, Japan) and subsequently by the Instituto Gulbenkian de Ciência. LC3b−/− (B6129PF2/J background) and NMRI mice were purchased from Jackson and Charles River laboratories, respectively. miR-146-deficient mice were generated in the Baltimore’s laboratory (Boldin et al., 2011). Fancd2−/− mice were generated by the Grompe laboratory (Houghtaling et al., 2003). Atg7loxP/loxP were generated by Masaaki Komatsu and obtained from the Green laboratory. AtmloxP/loxP mice were generated and obtained from the F.W. Alt’s laboratory. CLP was performed as described previously (Rittirsch et al., 2009). The endotoxemia model was performed by injecting intraperitoneally (i.p.) a single dose of 50 μg/g body weight of LPS (from E. coli serotype 026:B6; Sigma-Aldrich). Pulmonary monostrain infections were carried out as described previously (Weber et al., 2011), using intranasal injection of Klebsiella pneumoniae (ATCC13803) at 8x107 colony-forming units (CFU). Epirubicin (Sigma-Aldrich), doxorubicin (Sigma-Aldrich), daunorubicin (Sigma-Aldrich) were dissolved in PBS, etoposide (Sigma-Aldrich) was dissolved in DMSO, aliquoted and stored at −80°C. Meropenem (AstraZeneca, Lisbon, Portugal). Epirubicin and daunorubicin (0.6μg/g body weight), doxorubicin (0.5μg/g body weight), etoposide (2μg/g body weight) were injected intraperitoneally at 0 and 24 h following CLP. Meropenem (20μg/g body weight b.i.d.) was injected i.p. for 5 consecutive days.
Colony-Forming Units Assay
Blood samples from septic or mock CLP mice were collected by cardiac puncture at indicated times after surgery. Mice were subsequentially perfused in toto with 10mL ice cold PBS and spleen, liver and kidneys were surgically removed and homogenized in 5mL of sterile PBS. Serial dilutions of blood and tissue homogenates were immediately plated on Trypticase Soy Agar II plates supplemented with 5% Sheep Blood. CFUs were counted after a 12 h incubation at 37 C.
Serology and Cytokine Measurement
Plasma from blood samples obtained 24 h post-CLP was collected after centrifugation. LDH, CK, ALT and urea levels were measured using the BioAssay Systems kits (BioAssay Systems, California) according to company’s protocol. Levels of TNF, IL-1β and IL-6 were measured using the murine ELISA kits (R&D Systems, Minneapolis) according to company’s protocol. Levels of HMGB1 were assessed using a murine ELISA kit (Shino Test Corporation, Tokyo) according to company’s protocol.
Histology
Mice were euthanized, perfused in toto with 10mL ice cold PBS and lungs and livers were surgically removed. Livers were placed in 10% phosphate buffered formalin for 24 h after which were embedded in paraffin. Sections were subsequently incubated with a primary antibody reactive to HMGB1 (Abcam, Cambridge, UK) followed by incubation with biotinylated secondary antibody and then with biotinylated horseradish peroxidase. Staining was developed by addition of diaminobenzidine (DAB) substrate (Vector Labs, Burlingame, CA) and counterstained with hematoxylin. Lungs were embedded in Tissue-Tek OCT (Sakura, Alphen aan den Rijn, Netherlands), and snap-frozen in liquid nitrogen. Lung sections (7 μm) were fixed in 1% paraformaldehyde in PBS for 2 min, followed by methanol at −20°C for 10 min and then in acetone for 2 min. Detection of LC3b and histone γH2AX was performed by incubating sections overnight at 4°C with rabbit polyclonal antibodies specific for, respectively, LC3b (L7543, Sigma Aldrich, USA) and γH2AX (phosphoS139) (ab2893; Abcam, Cambridge, UK); incubation with a secondary DyLight 488-coupled antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) was for 1 h at room temperature. Sections were counterstained with DAPI (0.5 μg/mL) to visualize DNA and mounted in Vectashield (Vector Laboratories Inc., Burlingame, CA) before confocal microscopy. Samples were examined with a Zeiss LSM 510 META laser scanning confocal microscope (Carl Zeiss, Jena, Germany). The acquired images were analyzed using a MATLAB (Mathworks; Natick, MA) routine developed in-house to perform automatic threshold segmentation and enumeration of individual cell nuclei stained with DAPI.
In vivo Viral Infection and Viral Titer Assay
Murid herpesvirus-4 infection and viral particle quantification was performed as previously described (Marques et al., 2008). Briefly, mice were intranasally inoculated with 1000 PFU of MuHV-4 strain 68 in 20 μLof PBS under light isoflurane anaesthesia. At 6 and 12 days post-infection, lungs were removed and homogenized in 5mLof Glasgow’s modified Eagle’s medium (GMEM). Infectious virus titers in freeze-thawed lung homogenates were determined by serial diluted suspension assay using Baby hamster kidney cells (BHK-21) cultured in GMEM supplemented with 10% fetal bovine serum, 10% tryptose phosphate broth, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (GMEM). Plates were incubated for four days, fixed with 10% formal saline and counterstained with toluidine blue. Viral plaques were counted with a plate microscope. Cre-adenovirus were obtained from the University of Iowa, prepared as a calcium-phosphate coprecipitate and incubated for 20 min at room temperature. Atg7loxP/loxP and ATMloxP/loxP were subjected to light isoflurane anesthesia and allowed to inhale 125μLof virus at a concentration of 2.5 × 107PFU. Additionally, wild -type C57BL/6 mice were included as controls. Mice were allowed to rest for 5 days after inhalation after which were subjected to CLP.
Stainings and Flow Cytometry
Peritoneal infiltrating leukocytes from either wild-type or LC3b-GFP transgenic animals were obtained 24 h post CLP by lavage with 5 mL of sterile ice-cold PBS, washed and blocked with mouse Ab anti-FcγIII/II (clone 93) receptor diluted in PBS containing 2% FCS (v/v) for 20 min at 4°C. Surface markers were detected by incubating for 30 min at 4°C with mouse Ab anti-CD4 (clone GK1.5), -CD8 (clone 53–6.7), -CD19 (clone 6D5), -Ly-6G (clone 1A8) (all Biolegend) and -neutrophil monoclonal antibody (clone 7/4) (Abcam, Cambridge, UK). Dead cells were excluded by co-staining with propidium iodide. Total cell number was determined by flow cytometry using a fixed number of latex beads (Beckman Coulter, CA, USA) co-acquired with a pre-established volume of the cellular suspension. For phospho-ATM intracellular staining, stimulated THP-1 cells were washed and fixed with ice-cold methanol. Mouse Ab anti-phosphoATM pS1981, clone 10H11.E12 (IgG1k) (Rockland, MA, USA) was incubated for 60 min at room temperature followed by an incubation of secondary Ab conjugated with Alexa 488 (Molecular Probes, CA, USA). Fluorescence was measured by flow cytometry, and data analyzed using FlowJo software.
Immunoblotting
Mouse phospho-ATM (4526, Cell Signaling, Danvers, MA, 1:1000 dilution), rabbit total ATM (2873, Cell Signaling, Danvers, MA, 1:1000 dilution), rabbit LC3b (Sigma, 1:1000 dilution) and the rabbit Fancd2 (Novus Biologicals, CO, USA; 1:1000 dilution) Ab were used overnight at 4°C. Primary Ab were detected using peroxidase conjugated secondary Ab (1h; RT) and developed with SuperSignal chemiluminescent detection kit (Pierce, Carcavelos, Portugal).
Supplementary Material
Highlights.
Anthracyclines confer strong protection against severe sepsis.
Anthracyclines act therapeutically by promoting disease tolerance to severe sepsis.
DDR and autophagy are required in the lung for anthracycline-induced protection.
ATM and FA pathways are required for protection.
Acknowledgments
We are grateful to Vasco Barreto for Atm−/− and Frederick Alt for AtmloxP/loxP mice. We thank Mario Ramirez for bacterial strains to probe epirubicin protection in different models of sepsis. L.F.M. receives support from FLAD and FCT (grants PTDC/SAU-IMU/110303/2009, PTDC/SAU-MII/100780/2008, and PTDC/SAU-IMU/110303/2009), A.C. receives support from FCT (PTDC/SAU-IMU/110303/2009), J.F. receives support from a Gulbenkian grant (96526/2009) and P.P. is an FCT fellow (SFRH/BD/45502/2008).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010a;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander A, Kim J, Walker CL. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy. 2010b;6 doi: 10.4161/auto.6.5.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus DC, Wax RS. Epidemiology of sepsis: an update. Crit Care Med. 2001;29:S109–116. doi: 10.1097/00003246-200107001-00035. [DOI] [PubMed] [Google Scholar]
- Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current epidemiology of septic shock: the CUB-Rea Network. Am J Respir Crit Care Med. 2003;168:165–172. doi: 10.1164/rccm.2201087. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med. 2011;208:1189–1201. doi: 10.1084/jem.20101823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone RC, Sibbald WJ, Sprung CL. The ACCP-SCCM consensus conference on sepsis and organ failure. Chest. 1992;101:1481–1483. doi: 10.1378/chest.101.6.1481. [DOI] [PubMed] [Google Scholar]
- Bray K, Mathew R, Lau A, Kamphorst JJ, Fan J, Chen J, Chen HY, Ghavami A, Stein M, DiPaola RS, et al. Autophagy suppresses RIP kinase-dependent necrosis enabling survival to mTOR inhibition. PLoS One. 2012;7:e41831. doi: 10.1371/journal.pone.0041831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briot D, Mace-Aime G, Subra F, Rosselli F. Aberrant activation of stress-response pathways leads to TNF-alpha oversecretion in Fanconi anemia. Blood. 2008;111:1913–1923. doi: 10.1182/blood-2007-07-099218. [DOI] [PubMed] [Google Scholar]
- Chien WS, Chen YH, Chiang PC, Hsiao HW, Chuang SM, Lue SI, Hsu C. Suppression of autophagy in rat liver at late stage of polymicrobial sepsis. Shock. 2011;35:506–511. doi: 10.1097/SHK.0b013e31820b2f05. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2010;30:546–555. doi: 10.1038/emboj.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalhoff A, Shalit I. Immunomodulatory effects of quinolones. Lancet Infect Dis. 2003;3:359–371. doi: 10.1016/s1473-3099(03)00658-3. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Garrison SP, Thornton JA, Hacker H, Webby R, Rehg JE, Parganas E, Zambetti GP, Tuomanen EI. The p53-target gene puma drives neutrophil-mediated protection against lethal bacterial sepsis. PLoS Pathog. 2011;6:e1001240. doi: 10.1371/journal.ppat.1001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003;17:2021–2035. doi: 10.1101/gad.1103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao HW, Tsai KL, Wang LF, Chen YH, Chiang PC, Chuang SM, Hsu C. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 2012;37:289–296. doi: 10.1097/SHK.0b013e318240b52a. [DOI] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Slifer TR, Araujo FG, Suzuki Y, Remington JS. Protection against lipopolysaccharide-induced death by fluoroquinolones. Antimicrob Agents Chemother. 2000;44:3169–3173. doi: 10.1128/aac.44.11.3169-3173.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Kaczmarek A, Krysko O, Heyndrickx L, Woznicki J, Bogaert P, Cauwels A, Takahashi N, Magez S, Bachert C, Vandenabeele P. TLR-2 and TLR-9 are sensors of apoptosis in a mouse model of doxorubicin-induced acute inflammation. Cell Death Differ. 2011;18:1316–1325. doi: 10.1038/cdd.2011.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, Bonaparte D, Cavalcante MM, Chora A, Ferreira A, et al. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med. 2010;2:51ra71. doi: 10.1126/scitranslmed.3001118. [DOI] [PubMed] [Google Scholar]
- Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- Li W, Zhu S, Li J, Assa A, Jundoria A, Xu J, Fan S, Eissa NT, Tracey KJ, Sama AE, Wang H. EGCG stimulates autophagy and reduces cytoplasmic HMGB1 levels in endotoxin-stimulated macrophages. Biochem Pharmacol. 2011;81:1152–1163. doi: 10.1016/j.bcp.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JV, Walsh CM. Programmed necrosis and autophagy in immune function. Immunol Rev. 2012;249:205–217. doi: 10.1111/j.1600-065X.2012.01147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques S, Alenquer M, Stevenson PG, Simas JP. A single CD8+ T cell epitope sets the long-term latent load of a murid herpesvirus. PLoS Pathog. 2008;4:e1000177. doi: 10.1371/journal.ppat.1000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- Matsushita N, Endo Y, Sato K, Kurumizaka H, Yamashita T, Takata M, Yanagi S. Direct inhibition of TNF-alpha promoter activity by Fanconi anemia protein FANCD2. PLoS One. 2011;6:e23324. doi: 10.1371/journal.pone.0023324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Montecucco A, Biamonti G. Cellular response to etoposide treatment. Cancer Lett. 2007;252:9–18. doi: 10.1016/j.canlet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2010;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raberg L, Graham AL, Read AF. Decomposing health: tolerance and resistance to parasites in animals. Philos Trans R Soc Lond B Biol Sci. 2009;364:37–49. doi: 10.1098/rstb.2008.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raberg L, Sim D, Read AF. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science. 2007;318:812–814. doi: 10.1126/science.1148526. [DOI] [PubMed] [Google Scholar]
- Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JA. Management of sepsis. N Engl J Med. 2006;355:1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- Sauter KA, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1beta through activation of the NLRP3 inflammasome. Cancer Biol Ther. 2011;11:1008–1016. doi: 10.4161/cbt.11.12.15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol. 2008;8:889–895. doi: 10.1038/nri2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A, Soares MP. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci U S A. 2009;106:15837–15842. doi: 10.1073/pnas.0903419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Codogno P. Autophagy is a survival force via suppression of necrotic cell death. Exp Cell Res. 2012;318:1304–1308. doi: 10.1016/j.yexcr.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Siu WY, Lau A, Arooz T, Chow JP, Ho HT, Poon RY. Topoisomerase poisons differentially activate DNA damage checkpoints through ataxia-telangiectasia mutated-dependent and -independent mechanisms. Mol Cancer Ther. 2004;3:621–632. [PubMed] [Google Scholar]
- Suffredini AF, Munford RS. Novel therapies for septic shock over the past 4 decades. JAMA. 2011;306:194–199. doi: 10.1001/jama.2011.909. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Ulloa L, Tracey KJ. The “cytokine profile”: a code for sepsis. Trends Mol Med. 2005;11:56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Valentin-Vega YA, Kastan MB. A new role for ATM: regulating mitochondrial function and mitophagy. Autophagy. 2012;8:840–841. doi: 10.4161/auto.19693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderwerf SM, Svahn J, Olson S, Rathbun RK, Harrington C, Yates J, Keeble W, Anderson DC, Anur P, Pereira NF, et al. TLR8-dependent TNF-(alpha) overexpression in Fanconi anemia group C cells. Blood. 2009;114:5290–5298. doi: 10.1182/blood-2009-05-222414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber SE, Tian H, Pirofski LA. CD8+ cells enhance resistance to pulmonary serotype 3 Streptococcus pneumoniae infection in mice. J Immunol. 2011;186:432–442. doi: 10.4049/jimmunol.1001963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook AM, Schiestl RH. Atm-deficient mice exhibit increased sensitivity to dextran sulfate sodium-induced colitis characterized by elevated DNA damage and persistent immune activation. Cancer Res. 2010;70:1875–1884. doi: 10.1158/0008-5472.CAN-09-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wan G, Berger FG, He X, Lu X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol Cell. 2011;41:371–383. doi: 10.1016/j.molcel.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.