

Abstract
Homogenization by bead beating is a fast and efficient way to release DNA, RNA, proteins, and metabolites from budding yeast cells, which are notoriously hard to disrupt. Here we describe the use of a bead mill homogenizer for the extraction of proteins into buffers optimized to maintain the functions, interactions and post-translational modifications of proteins. Logarithmically growing cells expressing the protein of interest are grown in a liquid growth media of choice. The growth media may be supplemented with reagents to induce protein expression from inducible promoters (e.g. galactose), synchronize cell cycle stage (e.g. nocodazole), or inhibit proteasome function (e.g. MG132). Cells are then pelleted and resuspended in a suitable buffer containing protease and/or phosphatase inhibitors and are either processed immediately or frozen in liquid nitrogen for later use. Homogenization is accomplished by six cycles of 20 sec bead-beating (5.5 m/sec), each followed by one minute incubation on ice. The resulting homogenate is cleared by centrifugation and small particulates can be removed by filtration. The resulting cleared whole cell extract (WCE) is precipitated using 20% TCA for direct analysis of total proteins by SDS-PAGE followed by Western blotting. Extracts are also suitable for affinity purification of specific proteins, the detection of post-translational modifications, or the analysis of co-purifying proteins. As is the case for most protein purification protocols, some enzymes and proteins may require unique conditions or buffer compositions for their purification and others may be unstable or insoluble under the conditions stated. In the latter case, the protocol presented may provide a useful starting point to empirically determine the best bead-beating strategy for protein extraction and purification. We show the extraction and purification of an epitope-tagged SUMO E3 ligase, Siz1, a cell cycle regulated protein that becomes both sumoylated and phosphorylated, as well as a SUMO-targeted ubiquitin ligase subunit, Slx5.
Keywords: Basic Protocol, Issue 80, Life Sciences (General), budding yeast, protein extracts, bead beating, sumo, Ubiquitin, post-translational modifications, 6xHis affinity tag
Introduction
The awesome power of yeast genetics is legendary, but the preparation and analysis of native proteins from budding yeast, Saccharomyces cerevisiae, is often fraught with problems. The latter is due to the considerable mechanical strength and elasticity of the yeast cell wall1. Different means have been described for the enzymatic, chemical, mechanical, and pressure-based disruption of yeast cells to obtain whole-cell protein extract 2-6. These techniques vary widely in their efficacy to yield cell-representative, native protein extracts that can be used for subsequent analyses or purification steps. For example, the yeast cell wall can be removed with lytic enzymes (e.g. zymolyase) and resulting spheroblasts can be disrupted by shearing, detergents, or osmotic lysis to release proteins. This approach has been successfully employed as the starting point for many subcellular fractionations but it requires lengthy incubations that are not compatible with the stability of some proteins7.
Proprietary yeast lysis reagents (such as detergents) for the chemical extraction of proteins of yeast cells are commercially available but the efficacy of these reagents in protein extraction and their effect on subsequent biochemical characterization of proteins is not always clear8. High pressure homogenizers, often referred to as French presses, effectively break yeast cells by first subjecting them to high pressure and then extruding them through a small opening in a pressure cell. This technique produces high quality extracts but the equipment is very expensive and may not be suitable for small quantities of cells or multiple samples9. Therefore, mechanical disruption of yeast cells in a bead mill is often the method of choice for native yeast protein preparations10. This technique involves mechanical disruption of the yeast cell wall with acid-washed glass beads, which can be conducted with a variety of shakers, vortexers or bead mills. Notably, this method can be used to simultaneously process multiple smaller samples (1 ml of cells or less). Many different beads or bead mill disruption matrices are now commercially available to disrupt almost any kind of cell type in 2 ml tubes. Considering the other techniques and equipment, a bead mill has the added advantage that the disruption of yeast cells occurs very fast, which helps to preserve post-translational modifications such as sumoylation, especially when the appropriate buffers with protease and/or proteasome inhibitors are utilized and the temperature of extracts is controlled.
This protocol focuses on the fast, effective, and reliable extraction of endogenous and over-expressed proteins under gentle conditions with the ultimate goal to preserve protein function, interactions, and post-translational modifications. Growth media, cell lysis buffer compositions, and bead mill settings are optimized to maintain protein interactions and post-translational modifications such as sumoylation and ubiquitylation.
Protocol
Purification of 6xHIS-tagged proteins expressed in budding yeast cells under native conditions
1. Growth of Yeast Cells and Induction of Protein Expression
(Modified from 2)
OPTIONAL: Use logarithmically growing yeast cultures expressing protein of interest instead of the galactose-induced cultures described below.
Transform cells of a Gal+ yeast strain with a plasmid encoding a galactose-inducible 6xHIS-tagged protein of choice. For example, see reagents list.
Inoculate transformants in 5 ml of appropriate selective media (e.g. SD-uracil) containing 2% sucrose. Incubate at 30 °C O/N, rotating.
Dilute O/N culture so that the optical density at 600 nm measures 0.3 (OD600 = 0.3) in 33 ml of selective media with 2% sucrose. Grow at 30 °C, shaking (Δ150 rpm).
When the culture has reached OD600 = 0.8-1.5, induce expression of the desired protein by adding 17 ml of 3x YEP with 6% galactose (Recipe in Table 1), for a final concentration of 1x YEP with 2% galactose. Total culture volume is now 50 ml. Incubate, shaking, at 30 °C for an additional 5-6 hr. Note: the culture volume can be varied. In step 1.3, dilute into two-thirds of the desired final volume. In step 1.4, add one-third the final volume of 3x YEP/6% galactose.
Measure the OD600 of the induced culture and centrifuge Δ150-200 OD600 units of cells for 5 min at 5,000 rpm at 4 °C.
Resuspend cell pellet with 1 ml ice-cold 1x PBS with 1x protease inhibitor cocktail and transfer to a 2 ml screw cap tube.
Centrifuge cells for 1 min at 15,000 rpm at 4 °C. Decant supernatant.
Snap freeze cell pellet in liquid nitrogen, followed by immediate cell lysis or storage at -80 °C until further use.
2. Homogenization of Yeast Cells and Extraction of Proteins
To the frozen cell pellet from the previous step, add 200 µl of acid-washed glass beads and 500 µl of ice-cold lysis buffer (Recipe in Table 1 or use cell lysis buffer of choice).
Briefly pipette up and down. It is not required to fully resuspend the cell pellet. Keep tubes on ice at all times. Note: The end of the pipette tip may be cut off before pipetting up and down.
At 4 °C, place the tube(s) with cells into the bead mill, balance, lock, and run the machine as per manufacturer's instructions.
Run the bead beater containing the tube(s) for 20 sec at 5.5 m/sec, then place on slushy ice for 1 min. Repeat 6x in total.
Clear the extracted proteins by centrifugation for 15 min at 15,000 rpm at 4 °C. OPTIONAL: remove small particulates by centrifugation through a SpinX filter.
- Prepare a sample of the whole cell extract (WCE) to confirm presence of the protein of interest by Western Blot:
- Add WCE corresponding with 2 OD600 units of cells (e.g. 5 µl of cleared extract if 200 OD600 units of cells were harvested) to 800 µl 20% trichloroacetic acid (TCA). Vortex to mix.
- Centrifuge for 2.5 min at 15,000 rpm at 4 °C. Decant the supernatant, but be careful to retain the pellet.
- Add 800 µl of 2% TCA to the pellet and vortex the tube, followed by centrifugation for 2.5 min at 15,000 rpm at 4 °C. Decant the supernatant, but be careful to retain the pellet.
- Add 100 µl of TCA Sample Buffer (Recipe in Table 1), vortex to dissolve pellet. Note: If the sample buffer becomes acidic (turns yellow) after addition to the pellet, add aliquots of Δ10 µl Tris base [1M] until the sample is blue again.
- Incubate in a 100 °C heat block for 2-5 min.
- Vortex again to fully dissolve if remnants of pellet are still present. Pellets prepared by this method are notoriously difficult to fully dissolve. It may take Δ10 min of vortexing to completely dissolve pellets.
- Store sample at -80 °C until further use.
Snap freeze aliquots of remaining cleared WCE in liquid nitrogen and store at -80 °C until further use.
3. Batch Purification of Proteins from Yeast Cell Homogenates
Note: This purification method was optimized for purification of 6xHIS-tagged proteins on Co2+ metal affinity resin.
- Resin Equilibration
- For a sample of cleared WCE prepared from Δ20-40 OD600 units of cells, add 50-100 µl of affinity resin to a microcentrifuge tube. Proteins extracted from cells that did not express the desired protein or a nonspecific resin, such as amylose, may be used as controls for nonspecific binding.
- Wash resin 5x with 1 ml of wash buffer: invert top-over-bottom until resin is resuspended, and then spin for 1 min at 5,000 rpm at 4 °C. Aspirate the supernatant. Note: if performing extraction and purification on the same day, resin equilibration can be performed prior to extraction.
- Protein Binding for Affinity Purification
- Add 100-200 µl of cleared lysate (Δ20-40 OD600 units) to 50-100 µl washed beads, and bring the total volume to 1 ml with lysis buffer.
- Incubate with nutation or rocking at 4 °C for 2-5 hr.
- Spin for 1 min at 5,000 rpm at 4 °C.
- If desired, save a sample of the remaining supernatant for subsequent analysis of unbound proteins. TCA precipitate as detailed above for the WCE (step 2.6).
- Wash resin with bound proteins 5x with 1 ml of wash buffer, followed by a spin for 1 min at 5,000 rpm at 4 °C. Keep samples cold during washes.
- Elution of Bound Proteins
- Add 150 µl elution buffer to resin, nutate at 4 °C for 5 min, spin for 1 min at 5,000 rpm at 4 °C and save the supernatant in a new tube. OPTIONAL: Repeat 2x and pool elutions.
- Prepare eluted sample for Western blot: To 25 µl of eluted proteins, add 25 µl 2x lithium dodecyl sulfate (LDS) sample buffer with 2 µl β-mercaptoethanol (BME) and incubate in a 100 °C heat block for 2 min. Note: Standard 2X Laemmli sample buffer may also be used.
- Snap freeze excess eluted protein in liquid nitrogen. OPTIONAL: strip remaining proteins from resin with an equal volume of 2x LDS sample buffer at 65 °C for 5 min, remove the LDS-eluted proteins to a new tube, then add 2 µl BME to the eluted proteins, not to the resin. This order is to prevent removal of any ions or antibodies that are conjugated to the beads.
- Store samples at -80 °C until further use.
- Western Blot and probe with appropriate antibodies to visualize proteins.
- Load 10-20 µl (Δ1-3 OD600 units) of each sample and 3-10 µl of a protein ladder in an SDS-PAGE gel of choice. Gels routinely used for this protocol include 4-12% Bis-Tris and 8% Tris-Glycine.
- Run gel at 200 V for 50 min.
- Transfer proteins from gel to a PVDF membrane by semi-dry transfer at 19 V for 20-30 min (recipe in Table 1).
- Block membrane in 4% milk/1x Tris buffered saline-TWEEN (TBST) for 1 hr at RT or O/N at 4 °C (recipe in Table 1).
- Incubate membrane with primary antibody to the epitope-tagged protein of interest in 4% milk/1x TBST for 1-3 hr at RT or O/N at 4 °C.
- Wash membrane 3x for 5 min each with 1x TBST.
- Incubate membrane with appropriate secondary horseradish peroxidase (HRP) -conjugated antibody for 1-3 hr at RT.
- Wash membrane 3x for 15 min each in a large volume of 1x TBST.
- Cover membrane with ECL substrate and wrap in saran wrap.
- Expose membrane to film and develop to visualize proteins.
Representative Results
Our representative results reveal that the described bead-beating and protein extraction protocol is useful for the reproducible preparation of proteins for a variety of downstream applications (summarized in Table 2). SDS-PAGE followed by Coomassie staining of WCEs show that a wide range of proteins (~12 to >250 kDa) can be reproducibly extracted from yeast cells (Figure 1A). Discrete bands over a range of molecular weights are indicative of high quality protein extracts. The quality of extracts can be confirmed by western blotting for specific epitope-tagged proteins. Shown is a representative result for the galactose-overexpressed, V5-tagged SUMO ligase Siz1 which migrates at Δ120kDa (Figure 1B). Generally, partially degraded proteins would run as multiple fragments below the expected weight, but here only full length protein is observed. Additionally, high molecular weight adducts of a given protein may indicate post-translational modifications. For example, we show galactose-overexpressed sumoylated Siz1Δ440 and full length Siz1 (Figure 1C and Fig 1D). Modifications can also be preserved on endogenously expressed Siz1 (Figure 1E).
We provide representative results that two nuclear enriched proteins, Slx5 and Siz1Δ440, were successfully purified from our WCEs (Figures 2A, 2B, and 2C). Notably, we show that a 6xHIS-tagged protein (Figure 2A; Slx5) can be affinity purified from our WCEs both under native and denaturing conditions. In contrast, Slx5-GST (Figure 2B) and Siz1Δ440-HA (Figure 2C) lack a 6xHIS epitope but under native conditions still interact with the metal-affinity resin in an imidazole-sensitive manner. We propose that Siz1, and to a lesser extent Slx5, are able to bind the metal-affinity resin via their metal-coordinating RING domain. While, to our knowledge, this particular phenomenon has not yet been reported, a similar situation has been described in which cholera toxin B subunit binds Ni2+ affinity resin in a manner mediated by its natural histidine residues11. Importantly, this observation suggests that proteins in the WCE are, at least in part, natively folded.
Especially for the study of protein function it is important to show that protein complexes remain intact in whole cell extracts. Our representative data reveal that Siz1Δ440, Slx5, and Pgk1 (a cytosolic protein that serves as a loading control) were present in WCEs. Siz1Δ440 was purified from these extracts based on its inherent ability to bind to metal affinity resins (compare Figure 2C). When Slx5 and Siz1 were co-expressed in yeast cells, Slx5 levels in the eluates were increased, raising the possibility that Slx5 may interact with Siz1 (Figure 3).
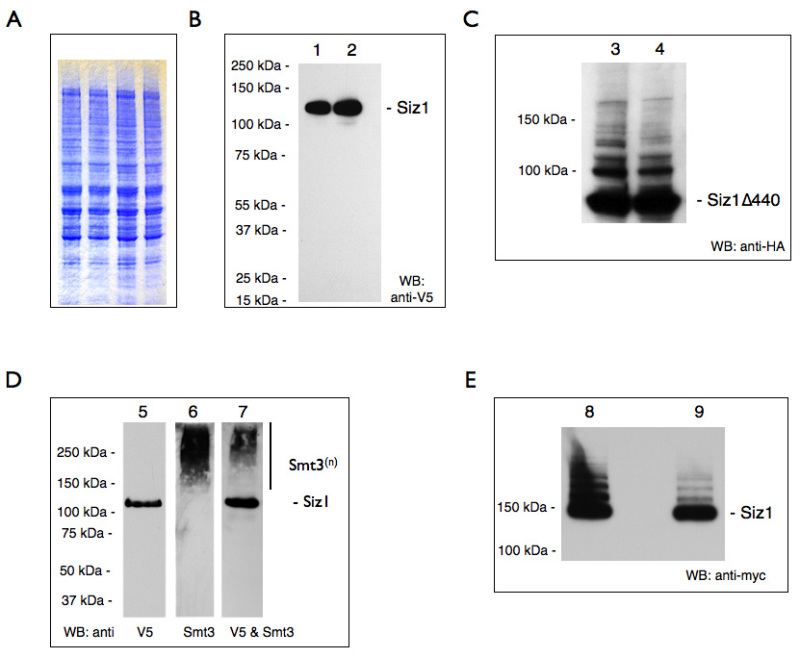 Figure 1. Presence of intact and sumoylated proteins in yeast cell lysate after extraction. Unless otherwise stated whole cell extracts were prepared as described in this protocol. Samples were separated by SDS-PAGE and after Western blotting were probed with antibodies to either the HA, V5, or myc epitope tags. Yeast strains are compiled in Table 3. WB: western blot. (A) Representative samples of TCA- precipitated WCE (from YOK 2514 (two lanes on the left) and YOK 2592 (two lanes on the right), (Δ0.2 OD/lane)) were visualized by staining with a gel stain (see Materials section). (B) Lane 1 & 2: homogenates of yeast strain YOK 2510 and YOK 2512 containing galactose-overexpressed Siz1-V5/6xHIS. Note the absence of partially degraded protein fragments. For unknown reasons, this particular sample does not reveal sumoylated adducts when probed with an anti-V5 antibody. However, sumoylation can be detected with an anti SUMO (anti-Smt3) antibody as shown in Fig. 1D. (C) Lane 3 & 4: homogenates of yeast strain YOK 2508 and YOK 2509 containing galactose-overexpressed Siz1Δ440-HA. (D) Lane 5, 6 & 7: galactose-overexpressed Siz1-V5/6xHIS was purified using Co2+ metal affinity resin, eluted with 200 mM imidazole, and probed with anti-V5 (lane 5), anti-Smt3 (lane 6). Lane 7 is a reprobe of lane 6 with the anti-V5 antibody. and shows both Siz1 and sumoylated Siz1 adducts (Smt3n). (E) Lane 8: JD52 wildtype cells containing endogenous levels of myc-tagged Siz1 (YOK 2397). Cleared lysate prepared using a mammalian lysis buffer. Lane 9: Lysate supernatant after a 2 hr incubation with anti-V5 agarose. Note sumoylated adducts of Siz1. Click here to view larger image.
Figure 1. Presence of intact and sumoylated proteins in yeast cell lysate after extraction. Unless otherwise stated whole cell extracts were prepared as described in this protocol. Samples were separated by SDS-PAGE and after Western blotting were probed with antibodies to either the HA, V5, or myc epitope tags. Yeast strains are compiled in Table 3. WB: western blot. (A) Representative samples of TCA- precipitated WCE (from YOK 2514 (two lanes on the left) and YOK 2592 (two lanes on the right), (Δ0.2 OD/lane)) were visualized by staining with a gel stain (see Materials section). (B) Lane 1 & 2: homogenates of yeast strain YOK 2510 and YOK 2512 containing galactose-overexpressed Siz1-V5/6xHIS. Note the absence of partially degraded protein fragments. For unknown reasons, this particular sample does not reveal sumoylated adducts when probed with an anti-V5 antibody. However, sumoylation can be detected with an anti SUMO (anti-Smt3) antibody as shown in Fig. 1D. (C) Lane 3 & 4: homogenates of yeast strain YOK 2508 and YOK 2509 containing galactose-overexpressed Siz1Δ440-HA. (D) Lane 5, 6 & 7: galactose-overexpressed Siz1-V5/6xHIS was purified using Co2+ metal affinity resin, eluted with 200 mM imidazole, and probed with anti-V5 (lane 5), anti-Smt3 (lane 6). Lane 7 is a reprobe of lane 6 with the anti-V5 antibody. and shows both Siz1 and sumoylated Siz1 adducts (Smt3n). (E) Lane 8: JD52 wildtype cells containing endogenous levels of myc-tagged Siz1 (YOK 2397). Cleared lysate prepared using a mammalian lysis buffer. Lane 9: Lysate supernatant after a 2 hr incubation with anti-V5 agarose. Note sumoylated adducts of Siz1. Click here to view larger image.
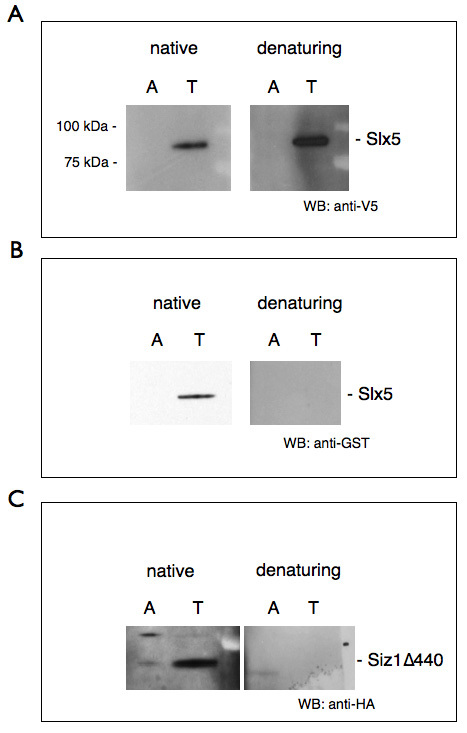 Figure 2. Purification of over-expressed RING-domain containing proteins with TALON Metal Affinity resin. Galactose induction, protein extraction and purification were performed as described in this protocol. Purification was performed under native conditions and denaturing conditions, for which guanidinium hydrochloride was added to the sample to 6 M before incubation with resin. Lysate was rotated with both amylose control resin (A) and TALON Metal Affinity resin (T). Proteins were eluted with 200 mM imidazole in elution buffer. Samples were separated by SDS-PAGE and Western blotted (WB) with an antibody to the appropriate epitope tag. A) Galactose-induced Slx5-V5/6xHIS (YOK 2096) was purified with TALON resin under both native and denaturing conditions B) Galactose-induced Slx5-GST (YOK 2071) was purified with TALON resin in native, but not denaturing conditions. Presumably, the metal coordinating RING domain interacts with TALON resin when properly folded. C) Galactose-induced Siz1Δ440-HA (YOK 2353) was also purified with TALON resin in native, but not denaturing conditions. These proteins do not contain 6xHIS tags, but each does contain a RING domain, which naturally coordinates Zn2+ ions and may confer the ability to intrinsically bind TALON Metal Affinity resin when properly folded. Click here to view larger image.
Figure 2. Purification of over-expressed RING-domain containing proteins with TALON Metal Affinity resin. Galactose induction, protein extraction and purification were performed as described in this protocol. Purification was performed under native conditions and denaturing conditions, for which guanidinium hydrochloride was added to the sample to 6 M before incubation with resin. Lysate was rotated with both amylose control resin (A) and TALON Metal Affinity resin (T). Proteins were eluted with 200 mM imidazole in elution buffer. Samples were separated by SDS-PAGE and Western blotted (WB) with an antibody to the appropriate epitope tag. A) Galactose-induced Slx5-V5/6xHIS (YOK 2096) was purified with TALON resin under both native and denaturing conditions B) Galactose-induced Slx5-GST (YOK 2071) was purified with TALON resin in native, but not denaturing conditions. Presumably, the metal coordinating RING domain interacts with TALON resin when properly folded. C) Galactose-induced Siz1Δ440-HA (YOK 2353) was also purified with TALON resin in native, but not denaturing conditions. These proteins do not contain 6xHIS tags, but each does contain a RING domain, which naturally coordinates Zn2+ ions and may confer the ability to intrinsically bind TALON Metal Affinity resin when properly folded. Click here to view larger image.
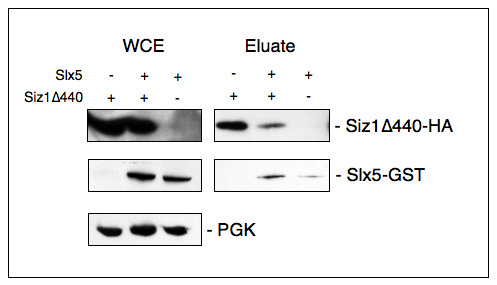 Figure 3. Co-purification of over-expressed proteins on TALON Metal Affinity resin. Slx5-GST and Siz1Δ440-HA were expressed by themselves or in combination (+/-) in JD52 cells (strains YOKs 2353, 2402, 2224 respectively). Proteins were extracted as described (WCE) and lysates were nutated with TALON Metal Affinity resin (see Figure 2). Proteins were eluted from the resin with 200 mM imidazole in elution buffer (Eluate) and prepared in LDS sample buffer. Proteins were separated by SDS-PAGE and after Western blotting were probed with an antibody to the HA tag or the GST tag as appropriate. Equal loading of proteins is confirmed with PGK as a loading control. Note that Slx5 purification is enhanced in the presence of Siz1Δ440. Click here to view larger image.
Figure 3. Co-purification of over-expressed proteins on TALON Metal Affinity resin. Slx5-GST and Siz1Δ440-HA were expressed by themselves or in combination (+/-) in JD52 cells (strains YOKs 2353, 2402, 2224 respectively). Proteins were extracted as described (WCE) and lysates were nutated with TALON Metal Affinity resin (see Figure 2). Proteins were eluted from the resin with 200 mM imidazole in elution buffer (Eluate) and prepared in LDS sample buffer. Proteins were separated by SDS-PAGE and after Western blotting were probed with an antibody to the HA tag or the GST tag as appropriate. Equal loading of proteins is confirmed with PGK as a loading control. Note that Slx5 purification is enhanced in the presence of Siz1Δ440. Click here to view larger image.
| Solution | Components | Comments |
|---|---|---|
| 3x YEP | 30 g Yeast Extract 60 g Peptone in 700 ml of ddH2O | Mix ingredients and autoclave to sterilize. Add filter sterilized galactose to 6% to individual aliquots of 3x YEP when ready for use |
| TCA Sample Buffer | 15% glycerol 80 mM Tris Base (non-pH'd) 3.5% SDS bromophenol blue "to taste." Bring volume to 25 ml with ddH2O | Before use: add 40 µl of β-mercaptoethanol (BME) to 1 ml TCA Sample Buffer |
| Wash Buffer | 50 mM HEPES pH 7.3 200 mM NaCl 1% Triton X-100 20 mM imidazole | Imidazole used for Ni2+ or Co2+ metal affinity purification only |
| Lysis Buffer | 50 mM HEPES pH 7.3 200 mM NaCl 1% Triton X-100 10 mM imidazole 1x protease inhibitor cocktail | Add protease inhibitor cocktail immediately before use. Imidazole used for Ni2+ or Co2+ metal affinity purification only. OPTIONAL: 25 mM N-ethylmaleimide (cysteine protease inhibitor, to preserve sumoylation), 1 mM sodium orthovanadate (protein tyrosine phosphatase inhibitor), and/or other empirically determined protease inhibitors based on specific area of study |
| Elution Buffer | 50 mM HEPES pH 7.3 200 mM NaCl 200 mM imidazole | Imidazole used to compete for histidine binding sites in Ni2+ or Co2+ metal affinity purification only. Other methods of elution may be used for different affinity resins. |
| Semi Dry Transfer Buffer (10x) | In 1 L of ddH2O: 58 g Tris 29.3 g Glycine 18.75 ml 20% SDS | To make 1x Semi Dry Transfer Buffer: mix 100 ml 10x Semi Dry Transfer Buffer with 200 ml Methanol and 700 ml ddH2Os |
| Tris Buffered Saline (TBS, 10x) | 50 ml 1 M Tris-HCl, pH 8.0 150 ml 5 M NaCl 300 ml ddH2O | To make 1x TBST, mix 100 ml 10x TBS with 900 ml ddH2O and add 1 ml of TWEEN-20 |
Table 1. Solutions and Buffers.
| Application | Amount of Cells | Next Steps |
|---|---|---|
| Cell Lysis | 150-200 ODs of harvested cell pellet | Bead beating as described in step 2 |
| Western Blot of WCE | 2 ODs of lysed cells prepared | Prepare for Western blot by TCA precipitation as described in step 2.6. Δ0.3 OD units (Δ15 µl) run out on gel |
| Affinity Purification and/or Pulldown | 30 ODs of lysed cells | Bind, elute, and prepare proteins as described in steps 3.2 and 3.3 |
| Western Blot of Purified Proteins | Approximately 1-2 ODs (if prepared as in step 3.3) | Western blot as described in step 3.4 |
Table 2. Summary of Applications for this Protocol.
| Name | Relevant Genotype or Parent Strain | Plasmid(s) or Cassette insertion | Reference |
|---|---|---|---|
| YOK 2062 | MATa ura3-52 his3-Δ200 leu2-3,112 trp1-Δ63 lys2-801 | Dohmen et al., 1995 | |
| YOK 2071 | JD52 | GAL1/10-GST-Slx5 (BOK 629, OpenBiosystems Yeast GST collection YSC4515-202484078) | this study |
| YOK 2096 | JD52 | pYES2.1-GAL-Slx5-V5/His6-TOPO (BOK 390) | this study |
| YOK 2224 | JD52 | GAL1/10-GST-Slx5 (BOK 629, OpenBiosystems Yeast GST collection YSC4515-202484078); pRS425 (BOK 343) | this study |
| YOK 2353 | JD52 | pAG425-GAL1-ccdB-Siz1Δ440-HA (BOK 795) | this study |
| YOK 2397 | JD52 | Siz1-13xmyc/HIS5 (endogenously tagged) | this study |
| YOK 2402 | JD52 | GAL1/10-GST-Slx5(BOK 629, OpenBiosystems Yeast GST collection YSC4515-202484078); pAG425-GAL1-ccdB-Siz1Δ440-HA (BOK 795) | this study |
| YOK 2501 | MHY3765, alpha mating type, ura3-52, lys2-801, trp1-Δ63, his3-Δ200, leu2-Δ1 | ubc4Δ::HIS3; ubc6Δ::TRP1; mat-alpha2Δ::kanMX | Xie et al., 2010 |
| YOK 2508 | ubc4Δ::HIS3; ubc6Δ::TRP1; mat-alpha2Δ::kanMX (YOK 2501) | pAG425-GAL1-ccdB-Siz1Δ440-HA (BOK 795) | this study |
| YOK 2509 | ubc4Δ::HIS3; ubc6Δ::TRP1; mat-alpha2Δ::kanMX (YOK 2501) | GAL1/10-GST-Slx5 (BOK 629, OpenBiosystems Yeast GST collection YSC4515-202484078); pAG425-GAL1-ccdB-Siz1Δ440-HA (BOK 795) | this study |
| YOK 2510 | ubc4Δ::HIS3; ubc6Δ::TRP1; mat-alpha2Δ::kanMX (YOK 2501) | pYES2.1-GAL-Siz1-V5/His6-TOPO (BOK 898); pRS425 (BOK 343) | this study |
| YOK 2512 | ubc4Δ::HIS3; ubc6Δ::TRP1; mat-alpha2Δ::kanMX (YOK 2501) | Slx5-Protein A (BOK 762); pYES2.1-GAL-Siz1-V5/His6-TOPO (BOK 898) | this study |
| YOK 2514 | Siz1-13xmyc/HIS5 (YOK 2397) | msn5Δ::hygromycin | this study |
| YOK 2592 | msn5Δ::hygromycin in JD52 (YOK 2514) | slx5Δ::kanMX4 | this study |
| YOK 2721 | JD52 | pYES2.1-GAL-Siz1-V5/His6-TOPO (BOK 898) | this study |
Table 3. Yeast Strains.
Discussion
This protocol focuses on the preparation of intact, native, and post-translationally modified proteins from budding yeast cells for down-stream applications. Before attempting this protocol, it is critical to determine if the protein of interest can be readily detected in protein extracts prepared under denaturing conditions12. If polyclonal antibodies are not available it may be advantageous to epitope tag the protein of interest so that the fusion protein can be detected on Western blots. In the present protocol, we tagged SIZ1 with the HA, 6xHIS-V5, or myc epitope tags and this strategy allowed us to clearly identify the protein and its sumoylated forms on Western blots (Figure 1). We had less success detecting GFP-tagged proteins, possibly because high-affinity anti-GFP antibodies are hard to obtain (data not shown). Another critical issue concerns the expression level of the protein of interest. Levels of individual proteins vary widely in budding yeast cells4 and some proteins have to be overexpressed so they can be detected and/or purified. Galactose-inducible vectors are available for the high-level expression of the yeast gene of interest2. In the case of Siz1, we were able to detect the full length or truncated protein when expressed from chromosomally-tagged ORFs or when expressed under control of the strong inducible galactose promotor from a plasmid (Figure 1). The use of truncations may be useful in structure/function analyses or when the full length molecule is difficult to express or unstable. While galactose-induced overexpression enhances protein detection and purification, it has clear limitations when studying the physiological relevance of potential cell-cycle specific post-translational modifications and interactions. For example, sumoylation of Siz1 is most prevalent at G2/M stage of the cell cycle, and modifications or interactions of the overexpressed protein may have to be confirmed by other experimental means. Finally, the addition of specific protease, phosphatase, and/or proteasome inhibitors is an important consideration for the success of this protocol. Generally, the best results are obtained by adding these inhibitors before cells are frozen and into extraction and dilution buffers. The addition of EDTA, a metal ion chelator sometimes present in protease inhibitor cocktails, is not advisable as it may interfere with subsequent metal affinity purifications.
Protein extracts obtained by bead-beating are well suited as starting material for the purification of individual proteins. Specifically, we show that overexpressed Slx5 carrying a 6xHIS affinity tag can be purified from WCEs using a metal affinity resin (Figure 2A). To preserve post-translational modifications and reduce protein degradation, 6xHIS-tagged proteins are often purified under denaturing conditions5. However, there are distinct advantages associated with the purification of proteins under native conditions. For example, these proteins may co-purify with binding partners or may be used in subsequent in vitro assays13,14. In the case of Siz1 and Slx5 we serendipitously determined that both proteins exhibit intrinsic affinity for the metal affinity resin, independent of the 6xHIS tag, and this observation suggested to us that both proteins assumed native confirmations. Unfortunately, proper folding and biological activity of purified proteins has to be determined on a case to case basis. To this end, the placement of affinity and epitope tags at either the amino or carboxy terminus, may greatly affect the activity or stability of fusion protein and is an important consideration in planning a protein purification strategy.
Future work will focus on how to enhance post-translational modifications during purification, for example through the use of protease deficient yeast strains, different protease inhibitors, or other affinity tags. We predict that this protocol in its current or buffer-optimized form is applicable for the scalable extraction, purification and subsequent study of many budding yeast proteins.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank all members of the Kerscher lab for their support. We thank Mark Hochstrasser for yeast strain MHY3765. This work was supported by NSF grant 1051970 (to OK) and a Howard Hughes Medical Institute Undergraduate Travel grant and Monroe Scholars Program Grant (to ES).
References
- Klis FM, Boorsma A, De Groot PWJ. Cell wall construction inSaccharomyces cerevisiae. Yeast. 2006;23(3):185–202. doi: 10.1002/yea.1349. [DOI] [PubMed] [Google Scholar]
- Gelperin DM, White MA, et al. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev. 2005;19(23):2816–2826. doi: 10.1101/gad.1362105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caserta E, Haemig HAH, et al. In Vivo and In Vitro Analyses of Regulation of the Pheromone-Responsive prgQ Promoter by the PrgX Pheromone Receptor Protein. J. Bacteriol. 2012;194(13):3386–3394. doi: 10.1128/JB.00364-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh W-K, et al. Global analysis of protein expression in yeast. Nature. 2003;425(6959):737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Hannich JT, Lewis A, et al. Defining the SUMO-modified proteome by multiple approaches in Saccharomyces cerevisiae. J. Biol. Chem. 2005;280(6):4102–4110. doi: 10.1074/jbc.M413209200. [DOI] [PubMed] [Google Scholar]
- Xie Y, Rubenstein EM, Matt T, Hochstrasser M. SUMO-independent in vivo activity of a SUMO-targeted ubiquitin ligase toward a short-lived transcription factor. Genes Dev. 2010;24(9):893–903. doi: 10.1101/gad.1906510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum GG, Böhni PCP, Schatz GG. Import of proteins into mitochondria. Cytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J. Biol. Chem. 1982;257(21):13028–13033. [PubMed] [Google Scholar]
- Deutscher MP. Guide to protein purification. 1990.
- Puig O, Caspary F, et al. The Tandem Affinity Purification (TAP) Method: A General Procedure of Protein Complex Purification. Methods. 2001;24(3):218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Conzelmann A, Riezman H, Desponds C, Bron C. A major 125-kd membrane glycoprotein of Saccharomyces cerevisiae is attached to the lipid bilayer through an inositol-containing phospholipid. EMBO J. 1988;7(7):2233. doi: 10.1002/j.1460-2075.1988.tb03063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dertzbaugh MTM, Cox LML. The affinity of cholera toxin for Ni2+ ion. Protein Eng. 1998;11(7):577–581. doi: 10.1093/protein/11.7.577. [DOI] [PubMed] [Google Scholar]
- Hautbergue GG, Goguel VV. The yeast C-type cyclin Ctk2p is phosphorylated and rapidly degraded by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 1999;19(4):2527–2534. doi: 10.1128/mcb.19.4.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore ZC, Donaher M, Matson BC, Murphy H, Westerbeck JW, Kerscher O. Sumo-dependent substrate targeting of the SUMO protease Ulp1. BMC Biol. 2011;9:74. doi: 10.1186/1741-7007-9-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Kerscher O, Kroetz MB, McConchie HF, Sung P, Hochstrasser M. The yeast Hex3.Slx8 heterodimer is a ubiquitin ligase stimulated by substrate sumoylation. J. Biol. Chem. 2007;282(47):34176–34184. doi: 10.1074/jbc.M706025200. [DOI] [PubMed] [Google Scholar]