

Abstract
In nature, bacteria rarely exist in isolation; they are instead surrounded by a diverse array of other microorganisms that alter the local environment by secreting metabolites. These metabolites have the potential to modulate the physiology and differentiation of their microbial neighbors and are likely important factors in the establishment and maintenance of complex microbial communities. We have developed a fluorescence-based coculture screen to identify such chemically mediated microbial interactions. The screen involves combining a fluorescent transcriptional reporter strain with environmental microbes on solid media and allowing the colonies to grow in coculture. The fluorescent transcriptional reporter is designed so that the chosen bacterial strain fluoresces when it is expressing a particular phenotype of interest (i.e. biofilm formation, sporulation, virulence factor production, etc.) Screening is performed under growth conditions where this phenotype is not expressed (and therefore the reporter strain is typically nonfluorescent). When an environmental microbe secretes a metabolite that activates this phenotype, it diffuses through the agar and activates the fluorescent reporter construct. This allows the inducing-metabolite-producing microbe to be detected: they are the nonfluorescent colonies most proximal to the fluorescent colonies. Thus, this screen allows the identification of environmental microbes that produce diffusible metabolites that activate a particular physiological response in a reporter strain. This publication discusses how to: a) select appropriate coculture screening conditions, b) prepare the reporter and environmental microbes for screening, c) perform the coculture screen, d) isolate putative inducing organisms, and e) confirm their activity in a secondary screen. We developed this method to screen for soil organisms that activate biofilm matrix-production in Bacillus subtilis; however, we also discuss considerations for applying this approach to other genetically tractable bacteria.
Keywords: Microbiology, Issue 80, High-Throughput Screening Assays, Genes, Reporter, Microbial Interactions, Soil Microbiology, Coculture, microbial interactions, screen, fluorescent transcriptional reporters, Bacillus subtilis
Introduction
We are interested in understanding how the metabolites that bacteria secrete affect the physiology and development of neighboring microbes. Many metabolites have been characterized for their bioactive effects on other microbes. Two well-described examples include antibiotics, which inhibit the growth of other microbes, and quorum sensing molecules, which alter the global gene expression of other microbes. However, bacteria produce many other small molecule natural products that have no known bioactivities1. We hypothesize that bacteria have evolved and preserved the ability to produce some of these metabolites because they allow them to modulate the cellular physiology of their microbial neighbors in the complex microbial communities within which most bacteria exist.
Bacillus subtilis cell types
We have focused our studies on chemically mediated microbial interactions that involve Bacillus subtilis. This is not only because of its status as the Gram-positive model bacterium and the resultant genetic tools available for its manipulation, but also because of its ability to differentiate into characterized cell types. Examples include cells that are: swimming; producing the extracellular matrix that is required for robust biofilm formation; competent to take up DNA from the environment; and sporulating, among others2. Each of these cell types expresses a characteristic transcriptional regulon that makes them physiologically and/or physically distinct from their genetically identical siblings. Under many growth conditions, multiple cell types coexist as various subpopulations within a single colony of B. subtilis cells3. Although many species of bacteria may exhibit analogous cell type heterogeneity, this phenomenon has been particularly well studied in B. subtilis.
In particular, genes that are upregulated within each of these specific B. subtilis cell types have been identified. Identifying such upregulated genes is essential for the work described here because many of these microbial phenotypes of interest are difficult or impossible to observe directly. For instance, we cannot visually detect a trait such as swimming on solid (1.5%) agar plates, even though a subpopulation of B. subtilis cells produce flagella under those conditions3. Another example is biofilm matrix-production. Matrix production can be visualized by colony morphology (as it results in macroscopically wrinkly colonies), but only on certain growth medium, and only after multiple days of growth4. However, by knowing which genes are upregulated during differentiation, we can construct transcriptional reporters that act as markers for cellular differentiation into these cell types.
Reporter constructs
These fluorescent transcriptional reporters consist of the promoters for cell-type specific genes driving the production of a reporter gene, for instance a fluorescent protein. Examples include Phag-yfp (for swimming cells), PtapA-yfp (for biofilm matrix-producing cells), and PsspB-yfp (for sporulating cells), where Px indicates the promoter region for gene x. These reporter constructs are integrated into a neutral locus on the chromosome (Figure 1 and see below) so that the native regulation of the phenotype is left intact. However, now when a cell expresses this phenotype, it also expresses a fluorescent protein. This provides an easily visualized read-out of the activation of particular phenotypic behavior, allowing us to screen for microbes that activate this physiological response. Although such reporters are commonly used in microbiology, they have not been broadly applied in screens to identify metabolic interactions between microbes before this method was described5.
There are a number of important considerations in the design and construction of cell-type-specific reporter strains. We have utilized exclusively transcriptional fluorescent reporters, although other types of constructs are certainly possible. We discourage the use of translational fusions as markers for cell type differentiation in our screen, however, for two reasons: 1) the desire to leave the native cell-type-specific protein unperturbed, and 2) the recognition that a diffuse, cell-wide fluorescence will be easier to detect than localized puncta within cells (common with translational fusions).
Reporter gene selection
After deciding to use transcription as a read-out, the reporter gene must be selected (e.g. LacZ, fluorescence, or luciferase). LacZ has the advantage of needing the least specialized equipment to detect, but there is a much higher likelihood of false positives among environmental microbes. In our hands, the background level of Lac+ organisms among soil microbes was prohibitively high (>>10% of soil microbes were blue (Lac+) on X-gal plates; data not shown). It is possible that by titrating the concentration of X-gal in the medium, this could be optimized to allow the use of an X-gal reporter, although we did not attempt this. Luciferase provides high sensitivity of detection and is the most orthogonal reporter: there is almost no chance of environmental microbes being inherently luminescent. However, we found it difficult to identify instrumentation at our institution that allowed luminescence detection across entire Petri plates, as most were designed to scan only localized regions in multi-well plates. There might also be complications in visualizing luminescent colonies in a manner that also allowed the simultaneous physical isolation of inducing organisms. While using fiduciaries may have made this possible, we instead elected to use fluorescent transcriptional reporters, which were proven to work in B. subtilis, provided adequate sensitivity of detection and low false positive rates among soil organisms, and allowed to use of easily available instrumentation for both visualization and isolation procedures.
Fluorophore selection
The specific fluorophore selected will depend on your bacterial species, the agar growth medium you are using, and the particular fluorescence filter sets you have available. With our instrumentation, we found that both the B. subtilis colonies themselves and the agar they were grown on exhibited less background fluorescence when YFP (yellow fluorescent protein) filters were used, making that reporter superior to GFP (green fluorescent protein) in our hands. The codon usage of fluorescent proteins are frequently optimized for eukaryotes, making it important to select a fluorophore either known from the literature to work in your bacterial species, or to test it explicitly using a constitutive promoter. A large number of ever-evolving fluorescent protein variants are currently available6, which have been reviewed in a number of sources7,8, some of which explicitly provide guidance on choosing an appropriate fluorescent protein for your experiment9.
Promoter selection
The selection of a promoter will largely depend on your cell type or phenotype of interest. For organisms such as B. subtilis, some cell-type specific reporter genes have been established in the literature. For other bacterial strains, examining microarray or transcriptional data will be necessary to provide information about which genes are highly upregulated under the conditions where your cell type of interest is manifested. A recent study cataloged the transcription of B. subtilis under 104 different growth conditions using tiling microarrays10. This paper provides comprehensive information about which genes are highly upregulated under different conditions, which is invaluable for less-well-characterized phenotypes.
Rather than mapping precise promoter regions for every gene of interest, we typically simply use the sequence 200-500 bp upstream of the gene as the promoter. The exact sequence length depends on the genomic context: shorter regions are used when necessary to avoid including upstream coding regions from neighboring open reading frames.
Neutral loci and integration
How to maintain the reporter construct in your bacterial strain becomes the final question in designing a fluorescent transcriptional reporter strain. In bacteria, genes of interest are frequently maintained on plasmids using antibiotic selection. However, it may not be possible to use antibiotics during coculture without killing the environmental microbes. If plasmids are stably maintained in your bacterial species, it may be possible grow your bacteria containing a plasmid-borne reporter in the presence of antibiotics to prepare your reporter for screening, and then eliminate antibiotics during the coculture itself in the hope that the plasmid will be sufficiently maintained to allow for fluorescence. However, if plasmids are easily lost in your bacterium, or are lost under stress conditions, this will not be a viable option. In many cases, the best solution will be to integrate the reporter construct onto the bacterial chromosome, which allows stable maintenance of the reporter even in the absence of selection. In order for the integration to not disrupt the normal expression or regulation of your gene of interest, we recommend integrating into an ectopic site on the chromosome that can act as a "neutral locus." In B. subtilis these integration sites are genes that - when mutated - convey a phenotype in certain minimal media (allowing integrants to be identified without antibiotic selection), yet do not alter growth or sporulation rates in rich media, and include such genes as amyE , lacA, thrC , pyrD , gltA, and sacA (conveying the ability to utilize starch, β-galactosides, threonine, uracil, glutamate, and sucrose, respectively)11-13 .
While integration in these genes have been used reliably for many years in B. subtilis (particularly at amyE and lacA), similar knowledge may not be available for genes in many other bacterial species. The use of phage attachment sites are great alternatives for neutral chromosomal integration sites: many species-specific14-16, as well as general integration sites such as the Tn7 attachment site (attTn7) have been identified and utilized for gene insertions in many bacterial species17,18.
Environmental microbes
We use soil as a direct source of environmental microbes for our coculture screen. The soil contains a high diversity of microbes, and many of these organisms are rich source of natural products. By using liquid suspensions of soil placed directly onto plates with our fluorescent transcriptional reporter strain (without prior isolation of bacteria from the soil), we greatly simplify the experimental approach. The soil can either be used immediately after harvesting, or be frozen at -80 °C for future use. Immediate use has the advantage that a greater diversity of microbes can potentially be grown, including those that will not survive freezing well. It has the disadvantage that the concentration of cultivable soil organisms from these samples is unknown, increasing the number of screen plates that must be used. Delayed use has the advantage that the cfu/ml for each soil source can be determined in advance, allowing an optimized number of colonies to be grown on each screen plate. However, it requires that the soil organisms be capable of surviving freezing.
Note that diversifying the inducer pool being examined (i.e. the soil sources) appears to be more effective at identifying new interspecies interactions than in-depth screening on the same soil: greater phylogenetic diversity was observed in the hits identified in our matrix-induction screen as additional soil sources were examined rather than screening the same soil sources more thoroughly (E.A. Shank and R. Kolter, Harvard Medical School, unpublished results).
Overview
The approach we describe here is straightforward in terms of its technical requirements. It involves: 1) constructing a fluorescent transcriptional reporter in B. subtilis or another bacterial species of interest, 2) identifying conditions under which this reporter is not activated, 3) preparing aliquots of this reporter strain and organisms to be screened (in our case soil, but other sources could be utilized instead), 4) mixing these two sets of microbes on solid media, 5) identifying and isolating putative inducing organisms, and 6) confirming that these organisms do indeed activate this phenotype in a secondary screen. Once identified, these organisms and their metabolites provide us with chemical tools to modulate bacterial behavior, to study bacterial physiology and microbial interactions, and to potentially act as novel scaffolds for future therapeutic compounds.
Protocol
1. Select a Reporter Gene and Construct a Fluorescent Transcriptional Reporter
For B. subtilis:
See the JoVE article in reference 19 for a protocol describing the construction of fluorescent transcriptional reporters in Bacillus subtilis.
For other bacterial species:
Identify a gene that is upregulated during the physiological response of interest. This can be based on existing literature or transcriptional analysis of the microbe under particular conditions.
Construct a fluorescent transcriptional reporter for this gene to act as a proxy for the change in phenotype. This construct should include the promoter of this upregulated gene driving the production of an appropriate fluorescent protein (see Figure 1).
Integrate this construct into a neutral locus on the chromosome. This ensures that native regulation of the gene of interest is not disrupted, and avoids the need for plasmid selection mechanisms (e.g. antibiotics) that might interfere with the growth of environmental microbes.
2. Determine Coculture Conditions
For B. subtilis PtapA-yfp reporter:
Use 0.1x LB, Lennox (1 g tryptone, 0.5 g yeast extract, 0.5 g NaCl per liter) medium for this reporter, since B. subtilis matrix-production is minimal on Luria Broth20. This medium allows B. subtilis colonies to grow to submillimeter but observable size, while allowing diverse taxa from soil to grow5.
Include 100 mM MOPS buffer to minimize potential pH changes.
For other bacterial species:
Use published transcriptional data or empirically test various culture conditions to identify one where the microbe grows but the activation of the fluorescent reporter is negligible (to allow its activation to be detected when the reporter strain is grown in coculture with inducing microbes.)
Use a medium with low nutrient content (relative to traditionally rich microbiological media) when screening environmental microbes from oligotrophic environments (such as the soil), since many oligotrophic bacteria do not grow when presented with high nutrient conditions21. A low nutrient medium also reduces colony size, increasing the throughput of the screen.
Select a medium with low background fluorescence and good optical clarity.
Optimize growth temperature to allow both the reporter strain and environmental microbes to grow simultaneously.
Consider the addition of a buffering agent. The use of buffer in the plates will reduce the possibility of detecting pH-mediated changes in physiology22, unless such interactions are of interest.
3. Prepare Reporter Aliquots
For B. subtilis PtapA-yfp reporter:
Streak reporter strain from -80 °C frozen stock onto a fresh LB plate using a sterile toothpick or applicator stick.
Grow overnight at 30 °C.
- Perform serial dilutions in liquid culture to minimize background fluorescence arising from growth on a solid medium:
- Inoculate a 5 ml liquid culture of LB and grow with shaking at 37 °C.
- When the culture reaches an OD600 ~0.6, dilute into 5 ml fresh LB to an OD600 of 0.02.
- Grow at 37 °C again with shaking until cultures reaches OD600 ~0.6.
- Repeat serial growth dilutions a total of 3x.
- Let final serial dilution culture grow to OD600 ~0.4.
Add glycerol to 15-20%.
Aliquot 50-200 μl into 0.5 ml microfuge tubes and freeze at -80 °C.
Make equivalent aliquots for the nonfluorescent parent strain of the reporter (they will be required during secondary screening).
For other bacterial species:
Use cells that have a low background fluorescence (i.e. are grown under conditions where there is little expression from the promoter used in the reporter).
Make and freeze aliquots containing known cfu/ml (colony forming units per ml) of the reporter strain so that an appropriate number of colonies can be grown on each coculture screen plate (see section above for details).
Make equivalent aliquots for the nonfluorescent parent strain (they will be required during secondary screening).
4. Obtain Soil Samples
Collect soil into sterile conical tubes or sterile bags using a spatula, discarding the top 0.5 cm of exposed surface soil.
Add sterile saline (0.85% NaCl) at a ratio of 10 ml per 1 g of soil to make a soil slurry.
- Select a method to dislodge bacteria from soil particles: a) immediate use of fresh sample or b) delayed use after freezing of sample.
- For immediate use, vortex slurry for 1 min.
- For delayed use, blend soil slurry in blender for three 1 min cycles, placing the blender jar on ice for 1 min rests between blending cycles.
Let soil slurry settle for ~1 min.
Move upper aqueous layer to a fresh tube.
Add glycerol to a final concentration of 15-20%.
Aliquot 50-200 μl into 0.5 ml microfuge tubes and freeze at -80 °C.
5. Determine cfu/ml of Frozen Reporter And Soil Aliquots
Thaw a frozen soil and reporter aliquot. Soil microbes can be thawed on ice. Because B. subtilis lyses at 4 °C, those aliquots should be thawed quickly at RT to minimize the time spent at low temperature.
Make two replicate serial dilutions (to 10-8) in 0.1x LB or other isotonic buffer.
Plate 5 μl spots of each serial dilution onto agar plates of the same medium that will be used for coculture screening.
Grow at RT (or the temperature that will be using for screening).
The next day, count the number of colonies within each spot and calculate cfu/ml of each of the frozen aliquots.
6. Confirm Aliquot Concentrations for Spread Screen Plates
Thaw a frozen aliquot as in step 5.1.
Dilute to 1 x 105, 2.5 x 105, 5 x 105, 1 x 106, and 2.5 x 107 cfu/ml.
Add 50 μl spot of each dilution to center of individual plates. These should yield plates with the calculated optimum of 25,000 colonies per plate, as well as 2- and 5- fold more and less, allowing the best actual dilution to be determined.
Add approximately 20 sterile glass (3 mm) beads by gently tapping them onto the plate. Beads provide a more even distribution of colonies across the plate than a bent glass spreader does.
Spread cells by keeping plates on the benchtop and shaking them back and forth, rotating them as you work, until the liquid has been absorbed. Do not continue shake the beads once the plate is dry; otherwise it will begin to kill the bacteria.
Flip plates over and discard beads into waste beaker containing ethanol.
Let plates grow for the time of your assay (e.g. 24 hr) at correct temperature for your assay (e.g. 24 °C/RT).
Using a dissecting stereoscope, count the number of colonies in two or more fields of view.
Calculate the number of colonies per area, and determine how many colonies are on each plate.
Adjust future dilutions as necessary. The actual number of colonies is not as important as having the same number on each comparable screen plate.
7. Prepare Coculture Plates
Thaw reporter aliquot (and soil aliquot if frozen) as in step 5.1.
- Dilute reporter (to concentrations optimized in section 6) in 0.1x LB or other low nutrient, isotonic solution.
- For frozen soil: Dilute to concentration optimized in section 6 in 0.1x LB or other low nutrient, isotonic solution.
- For fresh soil: Make dilutions of fresh soil slurry, based on prediction that the cfu/ml of the soil slurry could range from 10-4 to 10-9 cfu/ml.
Spot 50 μl of soil and reporter dilutions onto center of coculture screen plate. Also, plate soil alone and reporter alone as controls.
Spread using glass beads as described in step 6.4-6.6.
If there are known growth conditions that activate your fluorescent reporter, streak or plate your reporter under these conditions and grow to use as a positive control during screening.
Incubate at 24 °C for 24 - 28 hr (or as appropriate for your reporter/assay)
8. Screen CoCulture Plates for Fluorescence
Using brightfield illumination, focus your stereoscope so that the colonies are sharp.
- Look at the soil-only plates to determine whether your soil sample has autofluorescent colonies. If so, this soil may result in a high rate of false positives (with a concomitant increase in secondary screening).
- Use a high ratio of reporter:soil in your coculture plates to increase the chance that an inducer will be surrounded by multiple reporter colonies, reducing the chance they will be detected as false-positives (Figure 2).
- Use a different fluorescent protein (one that emits in a different channel).
- Determine the assay timing. For most new reporters, the timing of potential induction is unknown, and thus must be determined empirically.
- Begin screening for fluorescence as soon as colonies become clearly visible with the dissecting stereoscope (magnification ~30X) and continue examining the plates periodically until growth has ceased and/or background fluorescence becomes too high.
- Once the time window of potential induction is determined for a particular reporter, it should be similar for all coculture plates containing that reporter, simplifying the monitoring of the screen plates. For the B. subtilis PtapA-yfp reporter, the appropriate time to examine the plates is between 24-28 hr after plating the cells; for the B. subtilis PsspB-yfp reporter, it is between 26-32 hr of growth.
- Maximize your fluorescence sensitivity:
- Ensure your fluorescence dissecting scope is in a dark room or surrounded by blackout curtains. Induction will likely be less intense than a constitutively produced FP and requires greater sensitivity to detect.
- Allow time for the fluorescence lamp to stabilize and your eyes to adjust to the darkness before attempting to detect fluorescence from your coculture plates (at least 1-2 min).
Using a positive control (if you have one), ensure that the magnification you are using allows you to detect fluorescence. The magnification is typically best if your field of view is approximately 30-50x your typical colony diameter (200-400X).
Turn off the bright light and open the shutter for your fluorescence.
- After your eyes have adjusted to the darkness, slowly move the plate back and forth across your field of view, looking for bright spots.
- Start from the top of the plate and use a zigzag pattern to move the plate side-to-side as you move towards the bottom of the plate.
- Practice moving the plate in brightfield to get a sense for how slowly to move to plate, and to be sure that you are covering the entire surface area.
- Move the plate slowly enough that the colonies do not become blurry. It is better to oversample the surface rather than miss areas.
- After one full sweep, turn the plate 90° and repeat. Human eyes are remarkably good at detecting even faint fluorescence through this method.
If you detect fluorescence, stop moving the plate and go back and find the fluorescent area.
Turning the brightfield on slowly, determine whether the fluorescence is associated with a bacterial colony (and not autofluorescent soil detritus or media components). If so, the nonfluorescent colonies proximal to the fluorescent colony are putative inducing organisms.
9. Isolate Putative Inducing Organisms
- Once induced (fluorescent) colonies have been identified, isolate those colonies secreting the inducing compounds.
- If a high enough concentration of reporter colonies are growing on the plate (>0.5:1 reporter:soil cfu), the putative inducing colonies will be surrounded by multiple fluorescent colonies (again, see Figure 2).
- In cases where the complexity of the coculture growth makes it ambiguous which colony is the inducer, isolate multiple potential inducing colonies for subsequent testing in the secondary screen.
- Localize the colonies you want to pick in the center of the field of view.
- If they are close to the edge of the plate, rotate the plate so that the lip of the plate is away from your dominant hand (i.e. if you are right-handed, put the lip of the plate on the left). This allows you to approach the colonies at a low angle, which improves the accuracy of your picking.
Place fresh plates to streak the putative inducing organisms on to nearby, as well as a waste beaker to discard used tips into.
Leaving the fluorescence lamp on, turn the bright light slowly on, so that you will be able to identify (by shape and position), the fluorescent colony and the surrounding putative inducing organisms. You may need to go back and forth with the light a few times to be able to identify the colonies you want to pick when there is no fluorescence and only bright light.
Use a glass rod (200 mm long x 5 mm diameter) to pick up a sterile, round 200 μl gel-loading tip and hold it like a pencil. This is the picking tool you will use to isolate individual colonies.
Rest your outer palm against the stage to stabilize it on the work surface. Place your other hand on the inner, thumb side of your hand to stabilize your picking tool.
If necessary, flip again between fluorescence and brightfield views to identify the colonies you want to pick.
Keeping the pipette tip above the plate surface, move it into your field of view and center it above the colony you would like to pick. The tip will be out of focus.
Using the outer edge of your hand (which is resting on the microscope stage or work surface), pivot the pipette tip slowly down to the colony to be picked. Touch it very lightly, trying to minimize how many other colonies the tip contacts.
Without rotating the picking tool, streak onto a section of a fresh plate. Spread with a soft touch to avoid gouging the agar. Because the number of cells transferred by this method is quite small (the colonies are much smaller than typically manipulated), a single continuous streak will result in isolated colonies.
Repeat this process with other putative inducing organisms.
Incubate plates at 24 °C (or the temperature of your assay).
10. Streak Putative Inducing Organisms to Obtain Isolated Single Colonies
- Using a stereoscope, determine - based on colony structure or morphology - whether there are different colony types contained within each of your putative picked inducing organisms.
- Look at the colonies using a stage where you can illuminate the colonies from both above as well as from below to detect potential colony differences.
Restreak each different morphotype onto a fresh plate and incubate until grown.
Restreak one more time and let grow. If different morphotypes persist, continue to restreak to purity.
11. Retest Putative Inducing Organisms in Secondary Screen
Retest all of the putative inducing organisms in a secondary screen to determine which are activating the fluorescent transcriptional reporter. The secondary screen consists of a lawn of microcolonies of the fluorescent transcriptional reporter strain, along with control plates, onto which putative inducing organisms are patched or spotted.
- Set up three identical plates: one containing a microcolony lawn of the fluorescent transcriptional reporter strain (at the same concentration as was used during coculture screening); one containing a microcolony lawn of the wild-type parent strain without a fluorescence reporter; and one containing no lawn.
- Mark the top of the back of each plate to determine the plate orientation.
- Add positional markers for the patches/spots; up to ten putative inducing organisms can be tested on each set of plates (Figure 3).
- Thaw lawn aliquots (reporter and nonfluorescent parent strain) as in step 5.1.
- Spread 50 μl of a 5 x 105 cfu/ml dilution onto lawn plates (or other optimized dilutions) using sterile beads as in step 6.4.
- Let plates dry.
- Patch or spot putative inducing organisms:
- Select patching if an easier and faster approach is desired, and it is acceptable to have a less precise number of cells deposited. To patch:
- Touch a sterile toothpick to the colony to test - do not pick up all of the cells.
- Patch (make a small streak) onto the blank plate.
- Repeat patch with fresh toothpick onto reporter plate.
- Repeat patch with fresh toothpick onto control plate.
- Select spotting if a quantitative and reproducible approach is desired. Spotting allows the number of deposited cells to be normalized (see reference 5 for complete details), and permits the relative potency of the different inducing organisms to be compared. To spot:
- Resuspend putative inducing organisms in 1 ml of liquid media in a sterile plastic cuvette.
- Take the OD600 of the resuspensions.
- Using the formula X = 250 ÷ (OD600 - 0.5), add the volume X for each resuspension to 500 μl of liquid medium to get a solution with an OD600 of 0.5. This method simplifies the required pipetting steps when performing multiple dilutions to normalize OD's because you can use the same volume (500 μl) for all of your dilutants.
- Spot 1 μl of each OD-normalized resuspension to each of the three plates.
Let grow at 24 °C for 24-28 hr (or as appropriate for your reporter/assay).
Use the fluorescent dissecting microscope to identify putative inducing organisms that activate your fluorescent reporter strain but not your parental control strain. These isolates are your positive hits - the environmental microbes that secrete compounds that induce your phenotype of interest.
Representative Results
This screen was used to identify soil organisms secreting compounds that alter the physiology of B. subtilis. The results described here focus on the matrix-producing cell type of B. subtilis, which produces the protein and exopolysaccharide that are required for robust biofilm formation in this bacterium. We selected the promoter of the tapA-sipW-tasA operon for our fluorescent reporter construct (PtapA-yfp). This operon encodes the protein structural component of the matrix and is upregulated during biofilm matrix production23. Our matrix reporter (Figure 1) was constructed as previously described19.
Previous work has shown that B. subtilis produces matrix in response to the self-produced quorum-sensing-like molecule surfactin, as well as purified metabolites produced by other soil bacteria20. We were interested in expanding these studies to investigate more broadly which soil microbes make metabolites capable of inducing matrix production in B. subtilis. We elected to use dilute LB for growth, since this medium was already known to lead to poor matrix production20, providing us with a growth condition where our reporter strain was nonfluorescent. We then optimized the number of colonies appropriate for screening under these growth conditions. In order to optimize each screen plate, it is necessary to determine how many colonies grow from the frozen soil and reporter aliquots and what the appropriate concentration of colonies and nutrient conditions are. Ideally we want each coculture plate to contain an equivalent number of soil and reporter colonies (i.e. a 1:1 ratio of reporter:soil) and to be closely spaced, individual colonies. This high ratio of reporter colonies increases the likelihood that an inducer will activate multiple surrounding reporter colonies. Having multiple activated inducer colonies surrounding a putative inducer colony increases confidence in pinpointing the actual inducing organism (Figure 2). The nutrient content controls the extent of growth/colony formation while the dilution of the inoculum determines whether the resulting colonies are appropriately dispersed. On a standard 10 cm diameter Petri plate with low nutrient medium, we found that approximately 25,000 colonies total per plate (50 μl of a 5 x 105 cfu/ml dilution) provided the best separation of B. subtilis colonies on 0.1x LB MOPS medium (Figure 4).
Although the calculated cfu/ml from the serial dilutions provides an approximate concentration of bacteria in the aliquots, it is necessary to ensure that the concentration of the resulting colonies is appropriate when an entire plate is spread with cells. The calculated cfu/plate and actual cfu/plate are not always identical (Figure 4). Plating colony lawns of equivalent concentrations is important to allow different reporter strains to be compared (otherwise, differences in nutrient availability may alter their physiological state and interfere with the results).
After preparing aliquots of the reporter and soil, we mixed them on coculture screen plates and examined them for fluorescence using a stereoscope (Figure 5). We also plated controls that were only inoculated with either soil or the B. subtilis PtapA-yfp reporter strain. B. subtilis produces biofilm matrix (fluorescence) in response to numerous microbes from the soil as seen by the fluorescent colonies in the coculture image in Figure 5. For the soils we examined, we had a high hit rates for the PtapA-yfp reporter. As described in reference 5, between 12-67% of the isolates (from six different soil samples) had the ability to induce fluorescence in the PtapA-yfp reporter strain. This is in contrast to our unpublished results from analogous screens using the sporulation (PsspB-yfp) and competence (PcomG-yfp) reporters. After extensive screening (>200,000 colonies for each reporter), only two organisms were identified that induce sporulation, while none were identified that induce competence. Thus, the hit rates for different cell types are be highly variable and may be difficult to predict in advance.
We then picked individual putative inducing colonies. The colonies on the coculture screen plates are quite small on the low-nutrient medium we recommend (submillimeter diameter). Nevertheless, it is possible to accurately pick and isolate very small colonies by hand (Figure 6) from within a complicated coculture screen plate. The manual method that we use is simple and requires neither specialized tools nor flame sterilization. These putative inducer colonies are then restruck to isolation. Because the coculture plates are crowded with colonies, it is not unusual - even with very careful picking technique - to have more than one organism growing from each putative inducer sample. Careful examination should allow isolation of morphologically distinct colonies. All putative inducing organisms are then tested in a secondary screen. Positive and negative results from both the patch and spot method are shown in Figure 3. Considering their dense growth, our ability to physically collect inducing colonies from the coculture plates was quite good, with approximately 50% of the colonies examined in our secondary screen being true positives. Additional results from this screen as well as follow-up work emerging from it have been previously described5.
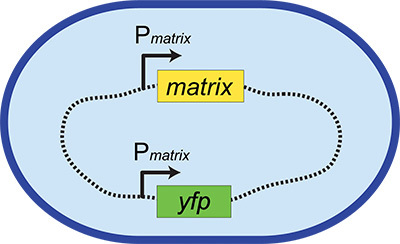 Figure 1. Fluorescent transcriptional reporter construct. The blue oval represents a bacterial cell and the dashed line represents its chromosome. This example shows a fluorescent transcriptional reporter for the production of matrix. The native locus remains intact (Pmatrix-matrix, where "P" and the arrow indicates the promoter region), while the reporter construct (Pmatrix-yfp) is inserted elsewhere in the chromosome in a neutral locus.
Figure 1. Fluorescent transcriptional reporter construct. The blue oval represents a bacterial cell and the dashed line represents its chromosome. This example shows a fluorescent transcriptional reporter for the production of matrix. The native locus remains intact (Pmatrix-matrix, where "P" and the arrow indicates the promoter region), while the reporter construct (Pmatrix-yfp) is inserted elsewhere in the chromosome in a neutral locus.
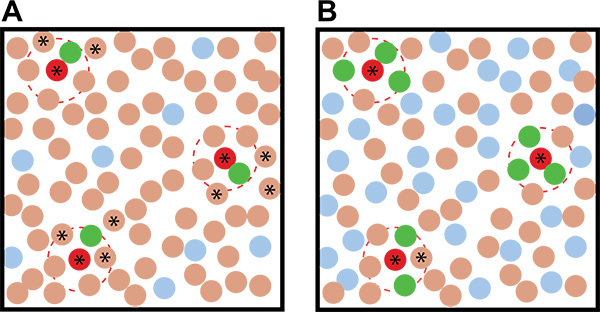 Figure 2. Idealized examples of coculture screening results with different ratios of the reporter:environmental microbes. A) Using a low reporter:environmental microbe ratio leads to more ambiguity in identifying putative inducing organisms than when B) a high reporter:environmental microbe ratio is used. The brown circles represent soil organisms, the red circles represent inducing soil organisms, the blue circles represent uninduced reporter colonies, and the green colonies represent induced reporter colonies. The dashed red lines indicate the action radius of the inducing metabolite. Stars indicate nonfluorescent colonies that - based on their proximity to the fluorescent colonies - are putative inducing organisms and should be picked and retested in the secondary screen.
Figure 2. Idealized examples of coculture screening results with different ratios of the reporter:environmental microbes. A) Using a low reporter:environmental microbe ratio leads to more ambiguity in identifying putative inducing organisms than when B) a high reporter:environmental microbe ratio is used. The brown circles represent soil organisms, the red circles represent inducing soil organisms, the blue circles represent uninduced reporter colonies, and the green colonies represent induced reporter colonies. The dashed red lines indicate the action radius of the inducing metabolite. Stars indicate nonfluorescent colonies that - based on their proximity to the fluorescent colonies - are putative inducing organisms and should be picked and retested in the secondary screen.
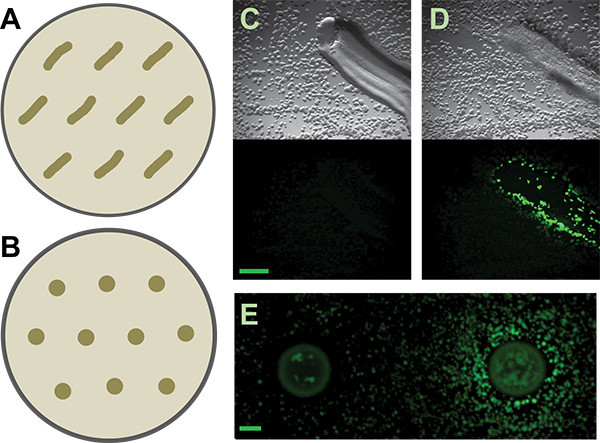 Figure 3. Secondary screen. A and B) Schematics of how to distribute patched or spotted isolates on secondary screen plates, respectively, for the B. subtilis matrix reporter. More generous spacing may be required for other reporters or inducing isolates, depending on the diffusibility of their active metabolites. C and D) Representative results from patched soil isolates that are negative and positive, respectively, for inducing the B. subtilis PtapA-yfp- reporter. Top panels are the brightfield images; lower panels are the fluorescence images. Scale bar is 1 mm. E) Negative and positive results from spotted soil isolates for the same reporter. Scale bar is 2 mm.
Figure 3. Secondary screen. A and B) Schematics of how to distribute patched or spotted isolates on secondary screen plates, respectively, for the B. subtilis matrix reporter. More generous spacing may be required for other reporters or inducing isolates, depending on the diffusibility of their active metabolites. C and D) Representative results from patched soil isolates that are negative and positive, respectively, for inducing the B. subtilis PtapA-yfp- reporter. Top panels are the brightfield images; lower panels are the fluorescence images. Scale bar is 1 mm. E) Negative and positive results from spotted soil isolates for the same reporter. Scale bar is 2 mm.
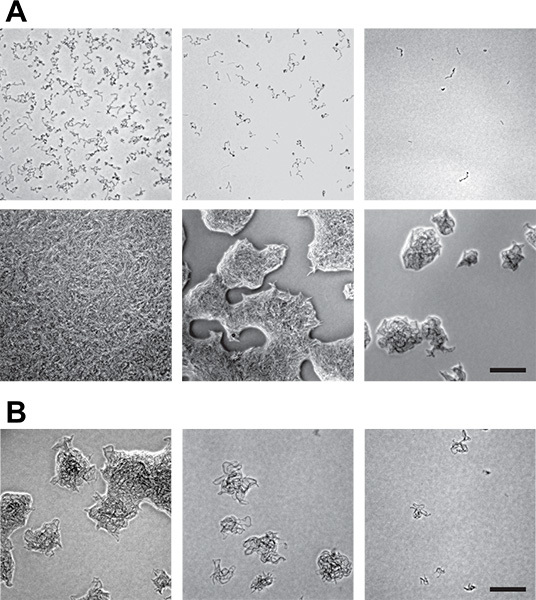 Figure 4. Determination of microcolony concentration. The distribution and size of your colonies will depend both on nutrient and cell concentrations. A) Differences in growth of B. subtilis on 0.01x LB (upper row) versus 0.08x LB (lower row). Cells on 0.01x LB do not form into microcolonies, while those on 0.08x LB do. (Note that for our screens we increased the nutrient levels slightly from those shown here: from 0.08x LB to 0.1x LB.) These images are from 1 μl spots of sequential 1:5 dilutions at known cfu/ml. Extrapolating from these concentrations, to get similar distributions of colonies across a 10 cm Petri plate would require plating (from left): 3,200,000; 640,000; and 128,000 cfu total per plate. However, spotting results in uneven distribution of cells (they are concentrated at the spot edges) compared to spreading cells over the entire plate. Thus, once a nutrient concentration is selected, it is important to examine plates spread with a variety of concentrations. Scale bar is 0.1 mm. B) These panels show the results of spreading (from left) 50,000; 25,000; and 5,000 total cfu per plate on 0.08x LB plates. From these images, we selected 25,000 as our target number of cfu/plate. Scale bar is 0.1 mm.
Figure 4. Determination of microcolony concentration. The distribution and size of your colonies will depend both on nutrient and cell concentrations. A) Differences in growth of B. subtilis on 0.01x LB (upper row) versus 0.08x LB (lower row). Cells on 0.01x LB do not form into microcolonies, while those on 0.08x LB do. (Note that for our screens we increased the nutrient levels slightly from those shown here: from 0.08x LB to 0.1x LB.) These images are from 1 μl spots of sequential 1:5 dilutions at known cfu/ml. Extrapolating from these concentrations, to get similar distributions of colonies across a 10 cm Petri plate would require plating (from left): 3,200,000; 640,000; and 128,000 cfu total per plate. However, spotting results in uneven distribution of cells (they are concentrated at the spot edges) compared to spreading cells over the entire plate. Thus, once a nutrient concentration is selected, it is important to examine plates spread with a variety of concentrations. Scale bar is 0.1 mm. B) These panels show the results of spreading (from left) 50,000; 25,000; and 5,000 total cfu per plate on 0.08x LB plates. From these images, we selected 25,000 as our target number of cfu/plate. Scale bar is 0.1 mm.
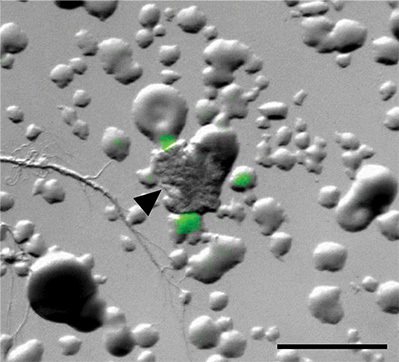 Figure 5. Coculture of B. subtilis PtapA-yfpmixed with soil organisms. Overlay of brightfield and fluorescence image from a coculture screen plate containing the B. subtilis PtapA-yfp matrix reporter mixed with soil organisms. Arrowhead indicates putative inducer surrounded by fluorescence reporter microcolonies. Scale bar is 1 mm.
Figure 5. Coculture of B. subtilis PtapA-yfpmixed with soil organisms. Overlay of brightfield and fluorescence image from a coculture screen plate containing the B. subtilis PtapA-yfp matrix reporter mixed with soil organisms. Arrowhead indicates putative inducer surrounded by fluorescence reporter microcolonies. Scale bar is 1 mm.
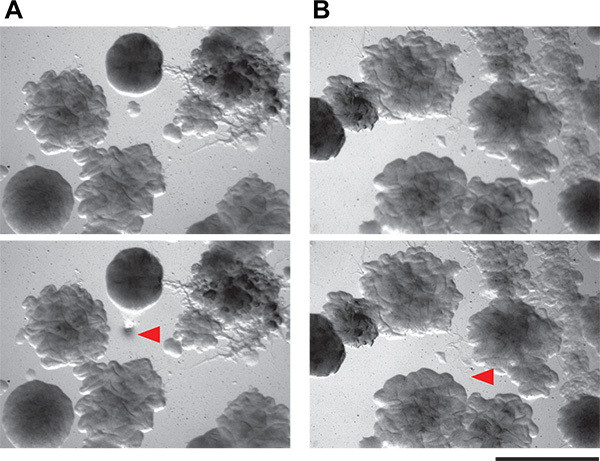 Figure 6. Demonstration of feasibility of isolating tiny bacterial colonies from coculture plates. A and B) These panels show two fields of view of agar plates containing complex microbial communities from soil. Colonies as small as 0.1 mm can be isolated using the picking technique described here. Top panels are the field of view before colony picking, and bottom panel are the same fields of view after colony picking. Red arrowheads indicate where cells have been removed.
Figure 6. Demonstration of feasibility of isolating tiny bacterial colonies from coculture plates. A and B) These panels show two fields of view of agar plates containing complex microbial communities from soil. Colonies as small as 0.1 mm can be isolated using the picking technique described here. Top panels are the field of view before colony picking, and bottom panel are the same fields of view after colony picking. Red arrowheads indicate where cells have been removed.
Discussion
One of the inherent limitations of this protocol is that it relies on the cultivability of microbial organisms. As has been well documented24, most microbial life on the planet cannot (yet) be grown under the culturing conditions explored to date. Thus, a huge number of interactions between microbial species that are occurring in natural settings will go undetected using this approach. However, since our desire is to not only identify the existence of such interactions, but then also study the mechanisms and molecules involved in mediating them, the ability to cultivate these microbes is a necessity. Even within cultivable species, this area has been poorly explored, making the approach described here a valuable contribution as a method to identify chemically mediated interactions between microbes. Additionally, although this protocol has been optimized to screen for matrix-induction of Bacillus subtilis, it can theoretically be applied to any transcriptional fluorescent reporter in any other bacterial species.
Another related limitation of this approach is that this screen (by definition) requires coculture. In natural environments, microbes with different growth rates may still coexist in spatial proximity while exploiting different environmental niches. Such microbial interactions would go undetected by our coculture screen, however, which will only allow the growth of environmental microbes with nutrient requirements and growth rates similar to those of the reporter species. Modifications that would separate the growth of the potential inducing organisms from the growth of the reporter strain are certainly possible. We also anticipated that the hyphal growth of fungi - common in soil - might cause difficulties in the co-culture screen. While the short timescale of our screen with B. subtilis meant that few fungi were detected, adding antifungal compounds to the growth medium could minimize this concern.
The ability to select an appropriate phenotype and gene for the fluorescent reporter construct should not be difficult, considering the wealth of sequencing and transcriptional data either already available or easily obtainable for many bacterial species. However, one difficulty with the approach described here is the need to identify growth conditions that minimize the background fluorescence of your reporter strain, allowing detection of fluorescence induction. The identification of these conditions must often be done empirically, although transcriptional data can assist this search (for instance the tiling microarray data available for growth of B. subtilis permits identification of conditions where genes of interest are poorly expressed10). For some reporters this empirical search may be challenging, in part because the expression of many bacterial phenotypes is heterogeneous. In other words, it is rare to find conditions in which no cells within the population are expressing Phenotype X. Thus, depending on the number of cells within that subpopulation and the strength of gene expression, it may be difficult to identify conditions that provide sufficiently low background fluorescence to allow induction to be detected. An alternative to this empirical search for ideal screening conditions may be to "tune" the expression levels of the reporter using directed mutagenesis. By altering the promoter region and/or ribosomal binding site of the reporter construct, the background fluorescence levels could be decreased. This could expand the usefulness of this screen by allowing even genes with some constitutive activation to be examined for induction.
Once inducing organisms have been identified and confirmed in a secondary screen, they can be phylogenetically identified by sequencing their 16S rRNA gene. It is also possible to quantify the extent of fluorescence using OD600-normalized spot in the secondary screen5. This can provide information about which members of the community produce compounds that affect your reporter strain and to what extent. Consequently, this can lead to hypotheses about which microbial interactions may be occurring in natural settings and the ability to explore the potential coevolution of these producing and responding organisms. Other future directions include elucidating the structure of the secreted molecule itself, determining the mechanism(s) by which the responding organism senses this compound, and using it as a chemical tool to modulate bacterial phenotypes.
Even with the considerations outlined above, the method described here is a significant contribution. It avoids the labor involved in assembling a library of environmental microbes, but allows their physical separation and isolation by using solid media. The strength of this coculture screen is that it provides a conceptually and technically straightforward method to screen through thousands of microbial species to identify those that secrete bioactive compounds of interest while being applicable to many bacterial species and phenotypes.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The author thanks Roberto Kolter (Harvard Medical School) for his invaluable advice and assistance during the development of this coculture screen. She also thanks Matthew Powers for reading the manuscript for clarity, and Chia-yi Cheng for assistance with obtaining Figure 6.
References
- Berdy J. Bioactive microbial metabolites. J. Antibiot. 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- Lopez D, Vlamakis H, Kolter R. Generation of multiple cell types in Bacillus subtilis. FEMS Microbiol. Rev. 2009;33:152–163. doi: 10.1111/j.1574-6976.2008.00148.x. [DOI] [PubMed] [Google Scholar]
- Vlamakis H, Aguilar C, Losick R, Kolter R. Control of cell fate by the formation of an architecturally complex bacterial community. Genes Dev. 2008;22:945–953. doi: 10.1101/gad.1645008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, Gonzalez-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. Fruiting body formation by Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11621–11626. doi: 10.1073/pnas.191384198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shank EA, et al. Interspecies interactions that result in Bacillus subtilis forming biofilms are mediated mainly by members of its own genus. Proc. Natl. Acad. Sci. U.S.A. 2011;108:1236–1243. doi: 10.1073/pnas.1103630108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piston DW, Patterson GH, Lippincott-Schwartz J, Claxton NS, Davidson MW. Introduction to Fluorescent Proteins. 2013.
- Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010;90:1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Patterson GH, Davidson MW. Advances in fluorescent protein technology. J. Cell Sci. 2007;120:4247–4260. doi: 10.1242/jcs.005801. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat. Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Nicolas P, et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012;335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- Middleton R, Hofmeister A. New shuttle vectors for ectopic insertion of genes into Bacillus subtilis. Plasmid. 2004;51:238–245. doi: 10.1016/j.plasmid.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Shimotsu H, Henner DJ. Construction of a single-copy integration vector and its use in analysis of regulation of the trp operon of Bacillus subtilis. Gene. 1986;43:85–94. doi: 10.1016/0378-1119(86)90011-9. [DOI] [PubMed] [Google Scholar]
- Guerout-Fleury AM, Frandsen N, Stragier P. Plasmids for ectopic integration in Bacillus subtilis. Gene. 1996;180:57–61. doi: 10.1016/s0378-1119(96)00404-0. [DOI] [PubMed] [Google Scholar]
- Semsey S, Blaha B, Koles K, Orosz L, Papp PP. Site-specific integrative elements of rhizobiophage 16-3 can integrate into proline tRNA (CGG) genes in different bacterial genera. J. Bacteriol. 2002;184:177–182. doi: 10.1128/JB.184.1.177-182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier E, et al. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 2004;70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Kim YW, Chang HI. Construction of an integration-proficient vector based on the site-specific recombination mechanism of enterococcal temperate phage phiFC1. J. Bacteriol. 2002;184:1859–1864. doi: 10.1128/JB.184.7.1859-1864.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KH, Schweizer HP. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc. 2006;1:153–161. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- Craig NL. Tn7: a target site-specific transposon. Mol. Microbiol. 1991;5:2569–2573. doi: 10.1111/j.1365-2958.1991.tb01964.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Betancur JC, Yepes A, Schneider J, Lopez D. Single-cell analysis of Bacillus subtilis biofilms using fluorescence microscopy and flow cytometry. J. Vis. Exp. 2012. p. e3796. [DOI] [PMC free article] [PubMed]
- Lopez D, Fischbach MA, Chu F, Losick R, Kolter R. Structurally diverse natural products that cause potassium leakage trigger multicellularity in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 2009;106:280–285. doi: 10.1073/pnas.0810940106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartoukian SR, Palmer RM, Wade WG. Strategies for culture of 'unculturable' bacteria. FEMS Microbiol. Lett. 2010;309:1–7. doi: 10.1111/j.1574-6968.2010.02000.x. [DOI] [PubMed] [Google Scholar]
- Romano JD, Kolter R. Pseudomonas-Saccharomyces interactions: influence of fungal metabolism on bacterial physiology and survival. J. Bacteriol. 2005;187:940–948. doi: 10.1128/JB.187.3.940-948.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda SS, Chu F, Kearns DB, Losick R, Kolter R. A major protein component of the Bacillus subtilis biofilm matrix. Mol. Microbiol. 2006;59:1229–1238. doi: 10.1111/j.1365-2958.2005.05020.x. [DOI] [PubMed] [Google Scholar]
- Zengler K, et al. Cultivating the uncultured. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15681–15686. doi: 10.1073/pnas.252630999. [DOI] [PMC free article] [PubMed] [Google Scholar]