

Abstract
The NF-κB pathway transcriptionally controls a large set of target genes that play important roles in cell survival, inflammation, and immune responses. While many studies showed anti-tumorigenic and pro-survival role of NF-κB in cancer cells, recent findings postulate that NF-κB participates in a senescence-associated cytokine response, thereby suggesting a tumor restraining role of NF-κB. In this review, we discuss implications of the NF-κB signaling pathway in cancer. Particularly, we emphasize the connection of NF-κB with cellular senescence as a response to chemotherapy, and furthermore, present examples how distinct oncogenic network contexts surrounding NF-κB produce fundamentally different treatment outcomes in aggressive B-cell lymphomas as an example.
Keywords: cancer, chemotherapy, NF-κB, senescence
INTRODUCTION
The nuclear factor κB (NF-κB) is a transcription factor complex composed of homo- and heterodimers of five members of the Rel family including RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105), NF-κB2 (p52/p100). More than 25 years after it was first described as a nuclear protein binding to the kappa immunoglobulin light chain enhancer in B cells (Sen and Baltimore, 1986), NF-κB is now known to control a complex signaling network via a long list of target genes in response to a variety of cellular stimuli. However, probably due to the pathway array surrounding NF-κB, ascribing common functions to NF-κB in general biological scenarios has been difficult. In tumor development, NF-κB presents with Janus-like features that may have pro- and anti-tumorigenic implications. Similar opposing functionalities may apply to NF-κB’s role in cancer therapy, thereby making thorough analysis of NF-κB network a prerequisite for targeted treatment strategies related to NF-κB.
NF-κB SIGNALING
Of the five NF-κB family members in mammalian cells, p105 and p100 are precursors, and, after post-translational modification and cleavage, become p50 and p52, respectively. They share a conserved Rel homology domain (RHD), which is responsible for binding to target DNA sequence, homo/heterodimerization, and the interaction with the inhibitor protein IκB. With each other, NF-κB family members form homo- or heterodimers, and in the absence of any stimulation (inactive state), also with IκB. Activation of the NF-κB signaling cascade can be divided into two pathways depending on how the active homo-/heterodimers are produced. In the classical (or canonical) pathway, activation is achieved by releasing IκB from the inactive complex via IκB kinase (IKK)-mediated phosphorylation followed by ubiquitin-mediated proteasomal degradation of IκB. The free NF-κB dimers (mainly p50/p65 and p50/c-Rel) trans-locate to nucleus and activate target gene transcription. In the alternative (non-canonical) pathway, the activation of the NF-κB pathway is achieved by inducing post-translational processing of p100 to the p52 subunit. Here, IKK (mainly IKKα) phosphorylates p100, leading to polyubiquitination and proteasomal processing to p52, which forms the transcriptionally active p52/RelB dimer (Bonizzi and Karin, 2004; Hayden and Ghosh, 2004).
A wide range of stimuli, including inflammatory cytokines, bacterial and viral products, are known to activate the NF-κB pathway resulting in transcriptionally active NF-κB complexes as described above. The active complexes translocate into the nucleus and bind to discrete DNA sequences in promoters and enhancers of target genes to activate transcription. There are many NF-κB target genes, orchestrating a plethora of biological functions, including anti-apoptosis, cell adhesion, cellular stress responses, inflammation, and immunity (Hayden and Ghosh, 2008).
CONTROVERSIAL ROLES OF NF-κB IN CANCER DEVELOPMENT
NF-κB activities are frequently deregulated in cancer, representing one of the most important signaling cascades in transformed cells with mainly proto-oncogenic impact (Perkins, 2007). Constitutive activation of NF-κB has been observed in different kinds of cancer, including lymphoma, leukemia, breast, colon, liver, pancreas, prostate, and ovarian cancers. Moreover, activation of NF-κB has often been linked to recurrence, poor survival, tumor progression, aggressiveness, and chemoresistance (Arkan and Greten, 2011; Basseres and Baldwin, 2006; Prasad et al., 2010). However, there are also studies that found NF-κB transcription factors or upstream activators rather to act as tumor suppressors. NF-κB controls the expression of a large set of target genes, including pro-survival and pro-inflammatory transcripts, but the actual role of NF-κB as a tumor suppressor or rather a tumor promoter, as well as it’s potential interference with treatment outcome is controversial (Ben-Neriah and Karin, 2011).
Pro-oncogenic NF-κB
Since NF-κB is normally involved in B-cell maturation and activation, deregulation of the NF-κB pathway is a prominent feature of hematological malignancies. Mutations in genes encoding NF-κB subunits, IκB proteins, or upstream regulators were identified in a variety of hematological malignancies (Compagno et al., 2009; Franzoso et al., 1992; Neri et al., 1991). There are now a number of lymphoid malignancies known, where constitutive NF-κB has been implicated as an essential oncogenic lesion. Mutations of multiple genes in receptor complexes (e.g. CD79-ITAM and MyD88), signaling complexes (e.g. TNFAIP3 [A20], CARD11-BCL10-MALT1 [CBM]), and the core signaling complexes (e.g. IKK, c-Rel) cause deregulation of NF-κB pathway in human lymphomas (Ngo et al., 2011; Rosenwald et al., 2002; Sun et al., 2004; Zhou et al., 2004). A good example of NF-κB’s essential role in cancer development can be found in diffuse large B-cell lymphoma (DLBCL) which can be divided into at least two subtypes according to their gene-expression profiling: the activated B-cell-like (ABC) subtype and the germinal-center B-cell-like (GCB) subtype. The main signature of the ABC subtype is the constitutive activation of the NF-κB pathway, which is rarely hyperactivated in GCB-DLBCL. Activating mutations in CARD11 (which encodes Carma-1) have been detected in ABC DLBCL, generating constitutively active CARD11 that associates with the Bcl-10-MALT1 complex (CBM complex; mediates NF-κB signaling between antigen receptor and IKK) without antigenic stimulation, leading to persistent activation of NF-κB (Lenz et al., 2008a; Ngo et al., 2006; Staudt, 2010). A20 is a negative regulator of the NF-κB pathway, as it prevents excessive activation of NF-κB in response to a variety of external stimuli (Heyninck and Beyaert, 2005; Wertz et al., 2004). It is frequently inactivated by truncating mutations and/or deletions in B-cell lymphomas, further adding to the theme that uncontrolled signaling of NF-κB is involved in the pathogenesis of B-cell malignancies. In multiple myeloma, another lymphoid neoplasia associated with NF-κB activation, no mutations in NF-κB or IκB encoding genes have been unveiled so far but extensive genetic analysis of primary tumors and multiple myeloma cell lines discovered a number of mutations in genes encoding upstream signaling molecules that lead to stabilization and accumulation of the NF-κB inducing kinase (NIK) in non-canonical pathway (Annunziata et al., 2007; Keats et al., 2007).
Oncogenic mutations in NF-κB signal mediators appear to be rare in solid tumors. However, oncogenic signaling, for example from activated Ras, may enhance NF-κB activity in the absence of mutations. Moreover, inflammation-associated NF-κB-driven cytokine production promotes carcinogenesis in a non cell- autonomous fashion (Ben and Karin, 2011; Staudt, 2010). Accordingly, mouse models demonstrated an oncogenic role for NF-κB in the development of lymphomas and solid tumors (Basseres et al., 2010; Calado et al., 2010; Yang et al., 2010).
In addition, the number of tumors with activated nuclear NF-κB is much larger than the fraction of malignancies with confirmed mutations in NF-κB-, IκB-, or typical upstream signal mediator-encoding genes. Such observations led to the proposal that some of the NF-κB activation seen in cancer is due to crosstalk from other deregulated pathways that fuel into the NF-κB cascade or is the result of external stimuli such as exposure to inflammatory cytokines in the tumor microenvironment. Production of the cytokines by immune cells that activate the NF-κB pathway in pre-malignant cells to induce genes that stimulate cell proliferation and survival is a major tumor-promoting mechanism (Grivennikov et al., 2010). For example, TNFα and IL-1 secreted by the environmental cells were shown to act on premalignant cells where they activate NF-κB. NF-κB activation further induces expression of genes involved in blockade of apoptosis, promotion of proliferation and angiogenesis, mechanisms collectively contributing to malignant conversion (Popivanova et al., 2008; Tu et al., 2008).
Anti-oncogenic NF-κB
Although NF-κB transcription factors have an oncogenic role in cancer development and confer drug resistance in cancer therapy in some settings, other studies found NF-κB transcription factors or upstream activators rather to act as tumor suppressors, thereby underscoring the complexity and potential context dependency of NF-κB network-mediated effector functions in both tumor development and therapy.
Contributing to its anti-cancer property, NF-κB has been shown to mediate apoptosis in a variety of cell types (Ryan et al., 2000; Wang et al., 1998). For instance, RelA and c-Rel exert proapoptotic function in T cells, B cells, fibroblasts, neuronal cells, and HeLa cells (Kaltschmidt et al., 2000; Kasibhatla et al., 1999; Martin et al., 2009; Schneider et al., 1999; Sheehy and Schlissel, 1999). There are also hints that NF-κB may modulate the apoptotic response depending on the developmental stage of the immune cells. For example, overexpression of RelA caused a cell-cycle arrest that is followed by apoptosis in the pro-B cell line 220–8, whereas overexpression of RelA in the WEHI 231 immature B-cell line or in the mature B-cell line M12 did not induce apoptosis (Sheehy and Schlissel, 1999).
Importantly, genetically defined mouse models supported the view that NF-κB transcription factors or upstream activators possess tumor suppressor functions. First evidence directly linking NF-κB to tumor suppression came from experiments using epidermal cells. Functional blockade of NF-κB in epidermal cells resulted in severe hyperplasia of the skin in transgenic mice expressing dominant negative IκBα mutant, which was reversible upon overexpression of active RelA and p50 subunits of NF-κB, suggesting a tumor-suppressive effect of NF-κB (Seitz et al., 1998). In a diethylnitrosamine-induced hepatocellular carcinoma (HCC) mouse model, hepatocyte-specific ablation of IKKβ strongly enhanced the development of HCC (Maeda et al., 2005). In other studies, hepatocyte-specific ablation of IKKγ (NEMO) or TAK1 (TGF-β-activated kinase 1), both required for the activation of IKK and NF-κB, resulted in spontaneous liver damage, hepatocyte death, and interestingly, release of factors leading to liver fibrosis and development of HCC (Inokuchi et al., 2010; Luedde et al., 2007). In the Eμ-myc transgenic mouse lymphoma model, in which oncogene Myc is overexpressed and drives B-cell lymphoma, NF-κB2 loss accelerated tumor development by impairing Myc’s apoptotic response, adding an example for a tumor suppressive function of NF-κB via its non-canonical pathway (Keller et al., 2010).
NF-κB IN CANCER TREATMENT RESPONSE: ANTI-APOPTOSIS
Given the well-established overlap between failsafe barriers such as apoptosis and senescence in tumor development and therapy, the NF-κB pathway has also been linked to chemoresistance in cancer treatment. Activation of NF-κB occurs in response to DNA damage, the mode by which most conventional chemotherapeutic agents exert their anti-cancer activity. Among the NF-κB target genes activated by DNA damage are bcl-2 (Catz and Johnson, 2001), bcl-xL (Tamatani et al., 1999), COX-2 (Yamamoto et al., 1995), cyclin D1 (Guttridge et al., 1999), survivin (Zhu et al., 2001), and XIAP (Stehlik et al., 1998), which are closely involved in anti-apoptotic, pro-survival functions of NF-κB, thereby operating as candidate mediators of chemoresistance in a wide variety of tumor cells.
Since the downstream targets of NF-κB are involved in apoptosis inhibition and may thereby block the action of many forms of chemotherapy (Baldwin, 2001), it may also contribute to the poor response of the ABC DLBCL to chemotherapy (Alizadeh et al., 2000; Lenz et al., 2008b; Rosenwald et al., 2002). This hypothesis is supported by several in vitro experiments. Introduction of the NF-κB super repressor (a non-degradable mutant of IκBα) resulted in rapid apoptosis and cell-cycle arrest, selectively in ABC-DLBCL cells (Davis et al., 2001). GCB-DLBCL cells were not affected in the same condition, indicating that the constitutive NF-κB activity is specifically required for the survival and proliferation of ABC-DLBCL cells. Strictly speaking, such result shows only the lymphoma’s dependency on hyper-active NF-κB, not the contribution of it to chemoresistance. However, it is non-disputable that NF-kB as the “Achilles’ heel” of ABC DLBCL is a good therapeutic target. Likewise, the IKKβ inhibitors PS-1145 and MLX105 were selectively toxic for ABC-DLBCL cells but not for GCB DLBCL cells. Introduction of an estrogen-inducible RelA fusion protein into ABC-DLBCL restored NF-κB activity even in the presence of IKK inhibition. The active form of RelA also stopped apoptosis induction by the kinase inhibitors PS-1145 and MLX105, demonstrating that the NF-κB inhibition is directly responsible for tumor cell death (Lam et al., 2005).
NF-κB IN CANCER TREATMENT RESPONSE: CELLULAR SENESCENCE
Premature cellular senescence is a terminal cell-cycle arrest that can be induced by oncogenic activation or chemotherapy, involving DNA damage response (DDR) signaling in both settings (Kuilman et al., 2010; Schmitt, 2007). It has been demonstrated that oncogene-induced senescence (OIS) imposes a critical barrier to tumor formation in vivo (Braig et al., 2005). Moreover, cellular senescence is increasingly recognized as a clinically relevant response to anti-cancer treatment, because it can be executed in response to therapeutic drugs and irradiation (Chang et al., 1999). Indeed, therapy-induced senescence (TIS) is detectable in resected human tumor material after neoadjuvant chemotherapy (te Poele et al., 2002). Importantly, genetically defined mouse tumor models were instrumental to demonstrate that TIS indeed extends survival of the tumor-bearing mice after chemotherapy (Schmitt et al., 2002).
Interestingly, recent publications postulated that senescence-associated and predominantly NF-κB-driven cytokines, collectively termed “senescence-associated secretory phenotype (SASP)”, may reinforce the senescent cell-cycle arrest (Acosta et al., 2008; Coppe et al., 2008; Kuilmann et al., 2008; Rovillain et al., 2011). These - on first sight - counter-intuitive, double-edged functions of NF-κB to promote both chemoresistance via anti-apoptosis and chemosensitivity via cellular senescence caught our attention to initiate an investigation to genetically determine the role of NF-κB in response to chemotherapy, with a special emphasis on TIS. By utilizing the Eμ-myc transgenic mouse lymphoma model, we first showed in vivo that TIS presents with and depends on active NF-κB signaling. To build a genetically modifiable model system reminiscent of the distinguishable NF-κB activities in ABC- vs. GCB-DLBCL, we classified primary Eμ-myc lymphomas by their NF-κB activity at diagnosis, i.e. prior to any therapy, and found “NF-κB high” (provocatively named “ABC-like”) lymphomas to be apoptosis-defective and chemoresistant due to their strongly elevated expression levels of the NF-κB target bcl2. Notably, NF-κB high Myc-lymphomas failed to enter senescence in response to therapy, thereby further contributing to their drug insensitivity, although the underlying senescence-compromising mechanism is not entirely clear. Moreover, mice that relapsed upon chemotherapy had lymphomas with significantly higher NF-κB activity at diagnosis as compared to those achieving cure after a single application of cyclophosphamide, a common chemotherapeutic agent widely used in clinical oncology. Accordingly, we designated “NF-κB low” mouse lymphomas “GCB-like”. Subsequently, we tested these “NF-κB low” lymphomas for their ability to enter TIS: after blocking apoptosis via retroviral introduction of bcl2, we observed more senescence in response to therapy if we genetically increased NF-κB levels. Parallel to mouse work, transcriptome and clinical data from DLBCL patients were also analyzed. Importantly - unlike ABC-DLBCL - almost half of the GCB-DLBCL are known to present with high Bcl2 levels that are independent of NF-κB activity, because they result from a t(14;18) translocation, which constitutively drives Bcl2 expression from an immunoglobulin heavy chain promoter (Huang et al., 2002; Iqbal et al., 2004), a scenario we recapitulated by retroviral, hence, NF-κB-independent Bcl2 expression in “GCB-like” mouse lymphomas. Given the particularly TIS-prone condition in „GCB-like“ mouse lymphomas with high Bcl2 and high NF-κB levels, we re-applied these determinants to a 233-DLBCL patient transcriptome data set with known subsequent clinical courses under standard immunochemotherapy. Indeed, patients with Bcl2-high and NF-κB-high GCB-DLBCL at diagnosis were identified as a clinically relevant subcohort that achieved a significantly longer (P < 0.005) progression-free survival than patients with Bcl2-high GCB-DLBCL but low NF-κB activity. Hence, functional investigations in mice identified an array of novel stratifiers of response that unveiled the clinically unexpected finding of a patient subgroup with a particularly good prognosis despite high-level NF-κB activity. All together, these data underscore the opposing roles NF-κB plays in cancer treatment, dependent on the cellular context, which accounts for fundamentally different clinical outcomes even in the same cancer entity.
NF-κB IN CANCER TREATMENT RESPONSE: ONCOGENIC NETWORKS
The data described above highlight how oncogenic networks and interdependencies, wired up during tumor development, may regulate the actual functions and even opposing roles of NF-κB in subsequent responses to therapy. NF-κB and Bcl2 are generally considered as indicators of aggressive tumor biology and poor outcome, but our mechanistic analyses in a genetically tractable mouse lymphoma model unveiled a setting in which these moieties contribute to superior outcome. Our approach demonstrates that functional understanding of oncogenic networks - and NF-κB/Bcl2 forms the most simple model of such a “two-factor network” with a linear connection in one (ABC-type), but an independent role of both factors in the other setting (GCB-type) - is required to properly utilize molecular lesions as biomarkers or even therapeutic targets (Fig. 2).
Fig. 2.
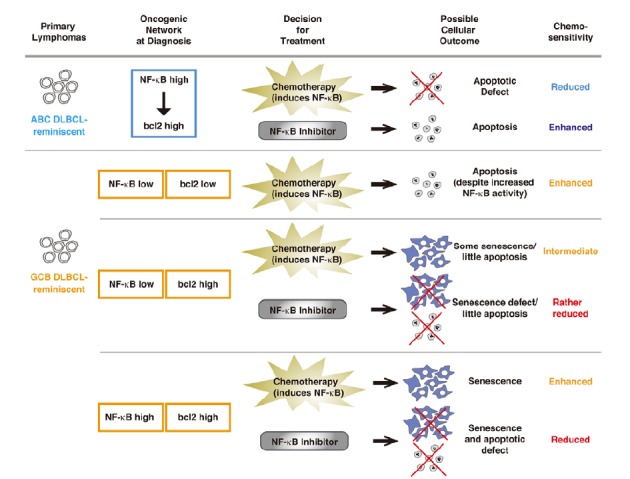
A model of how oncogenic network influences cancer treatment outcome. How two oncogenic moieties (NF-κB and bcl2) are connected in a network at diagnosis - a linear pathway in which NF-κB drives bcl2 expression reminiscent of ABC DLBCL, or in independent parallel pathways reminiscent of GCB DLBCL - influences the cellular outcome and sensitivity to treatment using conventional chemotherapy and/or novel NF-κB inhibitor.
NF-κB IN CANCER TREATMENT RESPONSE: SASP AND TUMOR MICTROENVIRONMENT
The SASP is composed of pro-inflammatory cytokines such as IL-1α, IL-1β, IL-6, IL-8, bFGF, TGF-β (in some settings), GMCSF, as well as inflammation-related chemokines such as CXCL-1/-2/-3/-5/-7, MIP-1α, or MCP-1 (a.k.a. CCL2). It also contains factors with, at least in some contexts, anti-proliferative activity such as IGFBPs or PAI-1, as well as factors like the MMPs that remodel the extracellular matrix (Acosta et al., 2008; Kortlever et al., 2006; Kuilman and Peeper, 2009). Moreover, in an extend view on the SASP program as a senescence-associated pro-inflammatory signaling array, this phenotype is not restricted to secreted factors, but also includes membrane-bound cell surface molecules serving as ligands and receptors, for instance TNF receptors, CXCR2 or the IL-6R, thereby creating potential autocrine loops (e.g. CXCL-1/-5/-7 or IL-8 with CXCR2) or even, as reported, intra-cellular short cuts (e.g. IL-6 and the IL-6R), and self-amplifying cascades (e.g. NF-κB signaling via TNF-R).
Therefore, the expression of surface-presented receptors and ligands and the secretion of a plethora of factors by senescent cancer cells may have complex, growth-inhibitory or -promoting effects on adjacent tumor and surrounding bystander cells. In particular, factors secreted by senescent cells can also act on macrophages, neutrophils, and NK cells, thereby promoting immune responses that, on one hand, may ultimately lead to the clearance of senescent tumor cells (Xue et al., 2007), but, on the other hand, might also create a microenvironment that fosters tumor progression (Coppé et al., 2010). Given our own observation that macrophage-derived TGFb evokes lymphoma cell senescence in a non-cell-autonomous fashion (Reimann et al., 2010), SASP-activated macrophages are likely to contribute to TIS in vivo, or, in turn, NF-κB inactivation in lymphoma cells might result in an inability to launch TIS because of the disrupted SASP/macrophage link. Thus, the outcome of anticancer therapy is not only determined by a quantitative effect on cancer cells forced to irreversibly exit the cell cycle but may also depend on novel capabilities acquired by senescent cells that can impact on their malignant and non-malignant neighbors in different ways. With further elucidation of these complex interdependencies that regulate tumor cell survival, senescence, and immune clearance, we expect non-genotoxic senescence-inducing agents to become a very promising perspective to be further exploited in cancer treatment.
NR-κB AS CANCER TREATMENT TARGET: A CAUTIOUS NOTE
Chemotherapy is still the most important treatment method for many types of cancer. The possible outcomes of chemotherapeutic treatments reach from necrosis, apoptosis, mitotic catastrophe, and autophagy to cellular senescence. Most of the chemotherapeutic agents used in the clinic are assumed to exert their anti-tumor effect through the induction of apoptosis. Accordingly, cancer cells with apoptotic defects would exhibit chemoresistance. As an example, ABC-subtype DLBCL present with an inferior prognosis after chemotherapy, and are characterized by constitutive activation of the NF-κB pathway, which appears to confer treatment resistance via an apoptotic block. In contrast, another subtype of DLBCL with lower NF-κB activity (i.e. the GCB subtype) is associated with a much better response to chemotherapy. In vitro analyses demonstrated that inhibition of NF-κB can sensitize ABC- but not GCB-DLBCL to apoptotic cell death; therefore, the hyperactive NF-κB pathway is central to the pathogenesis of ABC DLBCL and serves as a potential treatment target selectively in this subgroup. Many trials using specific NF-κB inhibitors or more broad range of proteasome inhibitors, which suppress NF-κB activation by preventing the degradation of IκB, are ongoing (Lim et al., 2012; clinicaltrialsfeeds.org). However, such treatment should be applied with caution. Because, in addition to the concerns of acquired resistance and impaired immunological functions of patients after repeated treatments, it may not be even clear who can benefit from NF-κB inhibition, considering the significant role NF-κB plays in cellular senescence. Coming back to the example of DLBCL, cross-species analysis of mouse lymphoma model and patient data revealed a patient cohort of GCB DLBCL, which seems not to be affected by NF-κB activation at first sight, with high Bcl2 level, actually benefit from high NF-κB activity (Fig. 2). Here, cellular senescence induced by chemotherapy and mediated by NF-κB is a key factor for the better treatment outcome. This contrasting impact of NF-κB in the same cancer entity should alert us to the consequence of (simply) adding NF-κB antagonists in cancer treatment regimens. Even more, the SASP factors, many of which controlled by NF-κB, may also contribute to treatment outcome not only by affecting tumor cells themselves, but also by modifying the tumor microenvironment in non-cell-autonomous ways. Considering that the simple oncogenic network of only two factors (NF-κB and bcl2) already create multiple scenarios for possible treatment responses (Fig. 2), one can easily imagine how complicated it can be to predict cancer treatment outcomes in individuals with still unknown factors, emphasizing the importance of understanding the network of genes for the development of personalized medicine designed for individual patient.
Fig. 1.
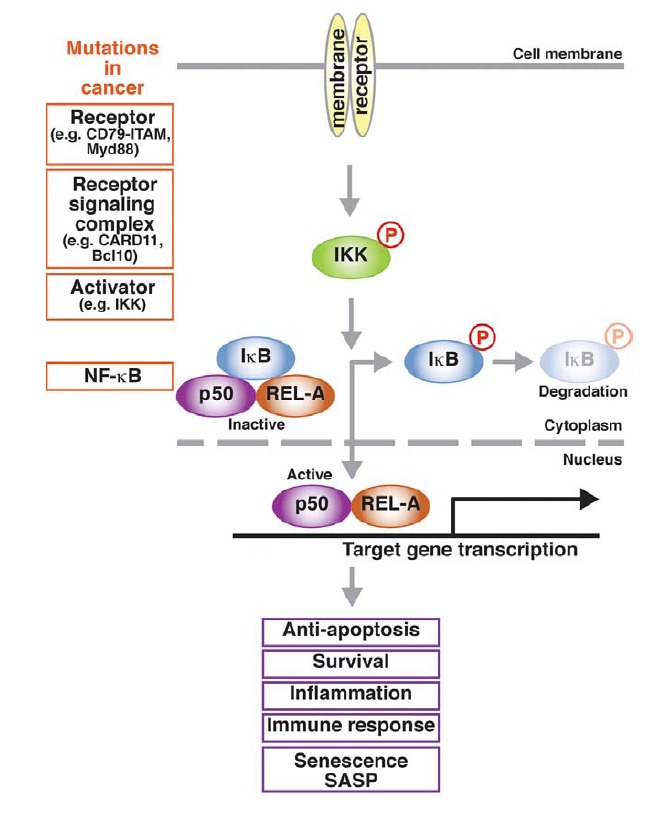
NF-κB signaling pathway and cancer related mutations. Only canonical (classical) pathway is shown. Extracellular stimuli transmitted though receptor complexes activate IKK via phosphorylation. Consequently, IKK phosphorylates IκB, leading to its dissociation from inactive NF-κB complex and ubiquitin-mediated proteasomal degradation. Freed from IκB, active NF-κB dimer translocates into the nucleus and induces the transcription of various target genes involved in multiple cellular pathways. Cancer-relevant mutations, indicated in orange boxes, can occur in all steps of NF-κB signaling cascade.
Acknowledgments
We would like to thank members of the C. A. Schmitt lab for critical discussions.
REFERENCES
- Acosta J.C., O’Loghlen A., Banito A., Guijarro M.V., Augert A., Raguz S., Fumagalli M., Da Costa M., Brown C., Popov N., et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Alizadeh A.A., Eisen M.B., Davis R.E., Ma C., Lossos I.S., Rosenwald A., Boldrick J.C., Sabet H., Tran T., Yu X., et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Annunziata C.M., Davis R.E., Demchenko Y., Bellamy W., Gabrea A., Zhan F., Lenz G., Hanamura I., Wright G., Xiao W., et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkan M.C., Greten F.R. IKK- and NF-κB-mediated functions in carcinogenesis. Curr. Top. Microbiol. Immunol. 2011;349:159–169. doi: 10.1007/82_2010_97. [DOI] [PubMed] [Google Scholar]
- Baldwin A.S. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J. Clin. Invest. 2001;107:241–246. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basseres D.S., Baldwin A.S. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- Basseres D.S., Ebbs A., Levantini E., Baldwin A.S. Requirement of the NF-κB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537–354. doi: 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Neriah Y., Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- Bonizzi G., Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Braig M., Lee S., Loddenkemper C., Rudolph C., Peters A.H., Schlegelberger B., Stein H., Dörken B., Jenuwein T., Schmitt C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- Calado D.P., Zhang B., Srinivasan L., Sasaki Y., Seagal J., Unitt C., Rodig S., Kutok J., Tarakhovsky A., Schmidt-Supprian M., et al. Constitutive canonical NF-κB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. 2010;18:580–589. doi: 10.1016/j.ccr.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catz S.D., Johnson J.L. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001;20:7342–7351. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- Chang B.D., Broude E.V., Dokmanovic M., Zhu H., Ruth A., Xuan Y., Kandel ES., Lausch E., Christov K., Roninson I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59:3761–3767. [PubMed] [Google Scholar]
- Compagno M., Lim W.K., Grunn A., Nandula S.V., Brahmachary M., Shen Q., Bertoni F., Ponzoni M., Scandurra M., Califano A., et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J.P., Patil C.K., Rodier F., Sun Y., Muñoz D.P., Goldstein J., Nelson P.S., Desprez P.Y., Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J.P., Desprez P.Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.E., Brown K.D., Siebenlist U., Staudt L.M. Constitutive nuclear factor kappa B activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoso G., Bours V., Park S., Tomita-Yamaguchi M., Kelly K., Siebenlist U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-κB-mediated inhibition. Nature. 1992;359:339–342. doi: 10.1038/359339a0. [DOI] [PubMed] [Google Scholar]
- Guttridge D.C., Albanese C., Reuther J.Y., Pestell R.G., Baldwin A.S., Jr. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov S., Greten F.R., Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden M.S., Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Hayden M.S., Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Heyninck K., Beyaert R. A20 inhibits NF-κB activation by dual ubiquitin-editing functions. Trends Biochem. Sci. 2005;30:1–4. doi: 10.1016/j.tibs.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Huang J.Z., Sanger W.G., Greiner T.C., Staudt L.M., Weisenburger D.D., Pickering D.L., Lynch J.C., Armitage J.O., Warnke R.A., Alizadeh A.A., et al. The t(14;18) defines a unique subset of diffuse large B-cell lymphoma with a germinal center B-cell gene expression profile. Blood. 2002;99:2285–2290. doi: 10.1182/blood.v99.7.2285. [DOI] [PubMed] [Google Scholar]
- Inokuchi S., Aoyama T., Miura K., Osterreicher C.H., Kodama Y., Miyai K., Akira S., Brenner D.A., Seki E. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc. Natl. Acad. Sci USA. 2010;107:844–849. doi: 10.1073/pnas.0909781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal J., Sanger W.G., Horsman D.E., Rosenwald A., Pickering D.L., Dave B., Dave S., Xiao L., Cao K., Zhu Q., et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol. 2004;165:159–166. doi: 10.1016/s0002-9440(10)63284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B., Kaltschmidt C., Hofmann T.G., Hehner S.P., Dröge W., Schmitz M.L. The pro- or anti-apoptotic function of NF-kappaB is determined by the nature of the apoptotic stimulus. Eur. J. Biochem. 2000;267:3828–3835. doi: 10.1046/j.1432-1327.2000.01421.x. [DOI] [PubMed] [Google Scholar]
- Kasibhatla S., Genestier L., Green D.R. Regulation of fas-ligand expression during activation-induced cell death in T lymphocytes via nuclear factor kappaB. J. Biol. Chem. 1999;274:987–992. doi: 10.1074/jbc.274.2.987. [DOI] [PubMed] [Google Scholar]
- Keats J.J., Fonseca R., Chesi M., Schop R., Baker A., Chng W.J., Van Wier S., Tiedemann R., Shi C.X., Sebag M., et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller U., Huber J., Nilsson J.A., Fallahi M., Hall M.A., Peschel C., Cleveland J.L. Myc suppression of Nfkb2 accelerates lymphomagenesis. BMC Cancer. 2010;10:348. doi: 10.1186/1471-2407-10-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever R.M., Higgins P.J., Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006;8:877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T., Peeper D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- Kuilman T., Michaloglou C., Vredeveld L.C., Douma S., van Doorn R., Desmet C.J., Aarden L.A., Mooi W.J., Peeper D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- Kuilman T., Michaloglou C., Mooi W.J., Peeper D.S. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam L.T., Davis R.E., Pierce J., Hepperle M., Xu Y., Hottelet M., Nong Y., Wen D., Adams J., Dang L., et al. Small molecule inhibitors of IkappaB kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin. Cancer Res. 2005;11:28–40. [PubMed] [Google Scholar]
- Lenz G., Davis R.E., Ngo V.N., Lam L., George T.C., Wright G. W., Dave S.S., Zhao H., Xu W., Rosenwald A., et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008a;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- Lenz G., Wright G., Dave S.S., Xiao W., Powell J., Zhao H., Xu W., Tan B., Goldschmidt N., Iqbal J., et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008b;359:2313–232. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K.H., Yang Y., Staudt L.M. Pathogenetic importance and therapeutic implications of NF-κB in lymphoid malignancies. Immunol. Rev. 2012;246:359–378. doi: 10.1111/j.1600-065X.2012.01105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luedde T., Beraza N., Kotsikoris V., van Loo G., Nenci A., De Vos R., Roskams T., Trautwein C., Pasparakis M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11:119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Maeda S., Kamata H., Luo J.L., Leffert H., Karin M. IKK couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Martin A.G., Trama J., Crighton D., Ryan K.M., Fearnhead H.O. Activation of p73 and induction of Noxa by DNA damage requires NF-kappa B. Aging. 2009;1:335–349. doi: 10.18632/aging.100026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri A., Chang C.C., Lombardi L., Salina M., Corradini P., Maiolo AT., Chaganti R.S., Dalla-Favera R. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-κB p50. Cell. 1991;67:1075–1087. doi: 10.1016/0092-8674(91)90285-7. [DOI] [PubMed] [Google Scholar]
- Ngo V.N., Davis R.E., Lamy L., Yu X., Zhao H., Lenz G., Lam L.T., Dave S., Yang L., Powell J., et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- Ngo V.N., Young R.M., Schmitz R., Jhavar S., Xiao W., Lim K.H., Kohlhammer H., Xu W., Yang Y., Zhao H., et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins N.D. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Popivanova B.K., Kitamura K., Wu Y., Kondo T., Kagaya T., Kaneko S., Oshima M., Fujii C., Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 2008;118:560–570. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad S., Ravindran J., Aggarwal B.B. NF-kappaB and cancer: how intimate is this relationship. Mol. Cell. Biochem. 2010;336:25–37. doi: 10.1007/s11010-009-0267-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann M., Lee S., Loddenkemper C., Dörr J.R., Tabor V., Aichele P., Stein H., Dörken B., Jenuwein T., Schmitt C. A. Tumor stroma-derived TGF-beta limits myc-driven lymphomagenesis via Suv39h1-dependent senescence. Cancer Cell. 2010;17:262–272. doi: 10.1016/j.ccr.2009.12.043. [DOI] [PubMed] [Google Scholar]
- Rosenwald A., Wright G., Chan W.C., Connors J.M., Campo E., Fisher R.I., Gascoyne R.D., Muller-Hermelink H.K., Smeland E.B., Giltnane J.M., et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002;346:1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- Rovillain E., Mansfield L., Caetano C., Alvarez-Fernandez M., Caballero O.L., Medema R.H., Hummerich H., Jat P.S. Activation of nuclear factor-kappa B signalling promotes cellular senescence. Oncogene. 2011;30:2356–2366. doi: 10.1038/onc.2010.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K.M., Ernst M.K., Rice N.R., Vousden K.H. Role of NF-kappaB in p53-mediated programmed cell death. Nature. 2000;404:892–897. doi: 10.1038/35009130. [DOI] [PubMed] [Google Scholar]
- Schmitt C.A. Cellular senescence and cancer treatment. Biochim. Biophys Acta. 2007;1775:5–20. doi: 10.1016/j.bbcan.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Schmitt C.A., Fridman J.S., Yang M., Lee S., Baranov E., Hoffman R.M., Lowe S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]
- Schneider A., Martin-Villalba A., Weih F., Vogel J., Wirth T., Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Seitz C.S., Lin Q., Deng H., Khavari P.A. Alterations in NF-κB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-κB. Proc. Natl. Acad. Sci USA. 1998;95:2307–2312. doi: 10.1073/pnas.95.5.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R., Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–716. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- Sheehy A.M., Schlissel M.S. Overexpression of RelA causes G1 arrest and apoptosis in a Pro-B cell line. J. Biol. Chem. 1999;274:8708–8716. doi: 10.1074/jbc.274.13.8708. [DOI] [PubMed] [Google Scholar]
- Staudt L.M. Oncogenic activation of NF-κB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehlik C., de Martin R., Kumabashiri I., Schmid J.A., Binder B.R., Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J. Exp. Med. 1998;188:211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Deng L., Ea C.K., Xia Z.P., Chen Z.J. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- Tamatani M., Che Y.H., Matsuzaki H., Ogawa S., Okado H., Miyake S., Mizuno T., Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J. Biol. Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- te Poele R.H., Okorokov A.L., Jardine L., Cummings J., Joel S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–1883. [PubMed] [Google Scholar]
- Tu S., Bhagat G., Cui G., Takaishi S., Kurt-Jones E.A., Rickman B., Betz K.S., Penz-Oesterreicher M., Bjorkdahl O., Fox J.G., et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.Y., Mayo M.W., Korneluk R.G., Goeddel D.V., Baldwin A.S., Jr NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wertz I.E., O’Rourke K.M., Zhou H., Eby M., Aravind L., Seshagiri S., Wu P., Wiesmann C., Baker R., Boone D.L., et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Xue W., Zender L., Miething C., Dickins R.A., Hernando E., Krizhanovsky V., Cordon-Cardo C., Lowe S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K., Arakawa T., Ueda N., Yamamoto S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J. Biol. Chem. 1995;270:31315–31320. doi: 10.1074/jbc.270.52.31315. [DOI] [PubMed] [Google Scholar]
- Yang J., Splittgerber R., Yull F.E., Kantrow S., Ayers G.D., Karin M., Richmond A. Conditional ablation of Ikkβ inhibits melanoma tumor development in mice. J. Clin. Invest. 2010;120:2563–2574. doi: 10.1172/JCI42358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H., Wertz I., O’Rourke K., Ultsch M., Seshagiri S., Eby M., Xiao W., Dixit V.M. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature. 2004;427:167–171. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]
- Zhu L., Fukuda S., Cordis G., Das D.K., Maulik N. Anti-apoptotic protein surviving plays a significant role in tubular morphogenesis of human coronary arteriolar endothelial cells by hypoxic preconditioning. FEBS Lett. 2001;508:369–374. doi: 10.1016/s0014-5793(01)03084-8. [DOI] [PubMed] [Google Scholar]