

Abstract
ClC-1 is a member of a large family of voltage-gated chloride channels, abundantly expressed in human skeletal muscle. Mutations in ClC-1 are associated with myotonia congenita (MC) and result in loss of regulation of membrane excitability in skeletal muscle. We studied the electrophysiological characteristics of six mutants found among Korean MC patients, using patch clamp methods in HEK293 cells. Here, we found that the autosomal dominant mutants S189C and P480S displayed reduced chloride conductances compared to WT. Autosomal recessive mutant M128I did not show a typical rapid deactivation of Cl− currents. While sporadic mutant G523D displayed sustained activation of Cl− currents in the whole cell traces, the other sporadic mutants, M373L and M609K, demonstrated rapid deactivations. V1/2 of these mutants was shifted to more depolarizing potentials. In order to identify potential effects on gating processes, slow and fast gating was analyzed for each mutant. We show that slow gating of the mutants tends to be shifted toward more positive potentials in comparison to WT. Collectively, these six mutants found among Korean patients demonstrated modifications of channel gating behaviors and reduced chloride conductances that likely contribute to the physiologic changes of MC.
Keywords: chloride channel, ClC-1, myotonia congenital, skeletal muscle
INTRODUCTION
Myotonia congenita (MC) is the genetic disorder characterized by muscle stiffness resulting from prolonged action potentials in skeletal muscle after relaxation (Accardi and Puschi, 2000; Dutka et al., 2000; Fahlke et al., 1996; Fialho et al., 2007). Two types of myotonia are recognized: the autosomal dominant disease called myotonia congenita (Thomsen’s disease, OMIM 160800) (George et al., 1993) and the autosomal recessive disease called generalized myotonia (Becker’s disease, OMIM 255700) (Hsiao et al., 2010; Jentsch et al., 2010; Koch et al., 1992; 1993). Both conditions display similar clinical features, but can be distinguished based on severity and inheritance patterns (Koch et al., 1992; 1993). For example, it is commonly reported that the recessive pattern of the disease is more severe than the dominant pattern (Koch et al., 1992; 1993; Lossin and George, 2008). However, the causative factors of either pattern of disease have yet to be uncovered (Lossin and George, 2008; Tang and Chen, 2011). It has been known that mutations in the gene encoding the skeletal muscle Cl− channel, ClC-1 are a major contributor to the pathogenesis of MC (Lossin and George, 2008; Tang and Chen, 2011).
ClC-1 is one of a subfamily of voltage-gated anion channels, and is distributed mainly to T-tubules and sarcolemmal membranes in skeletal muscle (Bryant et al., 1971; Dulhunty, 1979; Fahlke et al., 1997a; Heiny et al., 1990). ClC-1 plays a significant role in stabilization of the membrane potential at the resting level after action potential firing from influx of sodium ions through voltage-gated sodium channels (Bryant et al., 1971; Dulhunty, 1979; Fahlke et al., 1997a). Structurally ClC-1 protein forms a homodimeric double-barrel and consists of 18 domains and two tandem cystathionine-β-synthase (CBS) domains located in the intracellular c- terminal region (Markovic and Dutzler, 2007; Meyer and Dutzler, 2006). The CBS domain is one of the key structures observed in the eukaryotic ClC family. Two CBS domains are located in the cytoplasmic c-terminal and are essential to ClC channel function (Dutzler et al., 2002; Markovic and Dutzler, 2007; Meyer and Dutzler, 2006).
The channel gating of ClC-1 involves two identical protopores which function independently (Duzler, 2006; Dutzler et al., 2002; Mailänder et al., 1996). Due to its distinctive dimeric structure, the gating mechanism of ClC-1 shows two distinct patterns based on voltage stimulation: “slow gating” and “fast gating” (Accardi and Puschi, 2000; Dutzler, 2006; Fahlke et al., 1997b). Slow gating is known to regulate both identical pores simultaneously (Accardi and Puschi, 2000; Dutzler, 2006). On the contrary, when two protopores function separately at the same time in the dimeric channel complex, it is called fast gating (Accardi and Puschi, 2000; Aromataris et al., 2001; Dutzler, 2006). Six hClC-1 mutations among Korean patients with myotonia congenita, M128I, S189C, M373L, P480S, G523D, and M609K, and their clinical characteristics were previously reported (Moon et al., 2009). Among these six mutations, characteristics of S189C had been described previously, but the gating function remained uncharacterized (Fahlke et al., 2001; Wu et al., 2002). The remaining five mutations were first described in this report. Here, we analyze the physiological consequences of these six mutations though electrophysiological studies.
MATERIALS AND METHODS
Antibodies and reagents
The following primary antibodies, anti-GFP (sc-8334) and GAPDH (sc-20357) were used (Santa Cruz Biotechnology). The following secondary antibody, normal rabbit (sc-2027) was used (Santa Cruz Biotechnology).
Cell culture and transient transfection
HEK293 cells (American Type Culture Collection, USA) were incubated in Dulbecco’s modified Eagle’s medium (Hyclone), supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 ug/ml streptomycin according to the supplier’s recommendations. hClC-1 and mutant channels were transiently expressed using fugene 6 transfection reagent (Promega), based on manufacturer’s guide. To mimic heterozygous effect of autosomal dominant pattern, mutants were co-expressed in 1:1 ratio with WT, using fugene 6 transfection reagent (Promega) and incubated in 37°C. All experiments were conducted after 24–48 h of transfection.
Construction of DNA plasmids
pEGFP-N1 (Clontech) was substituted for the mammalian expression vector construct pRc/CMV (Invitrogen) containing hClC-1. The restriction enzyme sites were XhoΙ and EcoRΙ. We utilized site-directed mutagenesis with QuikChange XL Site-Directed Mutagenesis Kit.
Western blot
Transfected cells were washed with the phosphate buffer saline (PBS) containing (in mM/L): NaCl (136), KCl (2.5), KH2PO4 (1.5), Na2HPO4 (6.5), adjusted with NaOH to pH 7.4, then solubilized in the radioimmunoprecipitation assay (RIPA) buffer containing (in mM/L): Tris-Cl (50), NaCl (150), 1% Triton X-100 (1%), 0.1% sodium deoxycholate, 0.1% SDS, EDTA (1), a protease inhibitor cocktail (Calbiochem) and phosphatase inhibitor (Calbiochem), adjusted with NaOH to pH 7.4. Solubilized cell lysates were separated by SDS/PAGE and transferred to nitrocellulose membrane (BioRad). Proteins were previously blocked in 5% nonfat milk for 1 h, and incubated with primary antibodies, GFP and GAPDH overnight at 4°C. After incubation with horseradish peroxidase coupled secondary antibody for 1 h at room temperature, bound antibodies were detected using ECL detection reagent (Thermo Scientific).
Electrophysiology
Electrophysiology experiments were carried out as described below. Standard whole-cell patch-clamp recordings were conducted using Axopatch 200B amplifier (Axon Instruments) and digitized with the Digidata 1440A (Molecular Devices). Data were analyzed off-line by using pCLAMP 10.3 software (Molecular Devices). Pipettes were pulled from borosilicate glass (Harvard), and had resistances of 2.2–4.0 MΩ. Contribution of leak current was negligible under our conditions, and no correction for leakage current was used. The extracellular solution containing (in mM/L): NaCl (140), KCl (4), CaCl2 (2), MgCl2 (1), HEPES (5), adjusted with NaOH to pH 7.4 was used. 134 mM of Cl− concentration intracellular solution were composed of (in mM/L): NaCl (130), MgCl2 (2), EGTA (5), HEPES (10), adjusted with NaOH to pH 7.4. In the low-chloride internal solution, NaCl was substituted by Na-glutamate (120). For pharmacological test, 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS) was dissolved in dimethylsulfoxide (DMSO), and added to the extra-cellular solution at 50 μM and 500 μM concentration.
The 12 voltage pulses for the experiment were applied from −165 mV to 75 mV in 20 mV increments. Each voltage pulse was given with a 160 ms duration. For S189C, the voltage pulses were added up to 195 mV to obtain a complete Boltzmann fitting. Holding potential was fixed at 0 mV, and tail pulse was given at −125 mV for 20 ms duration in the voltage pulse protocol. For the low chloride experiment, holding potential was fixed at −85 mV.
Open probability of slow gating was determined from a step pulse protocol similar to those for overall Po, with the addition of a short (500μs) prepulse to 170 mV between a series of step pulses and −125 mV tail pulse, to maximize Po of fast gating (Accardi and Puschi, 2000).
Data analysis
Data analysis was conducted using Origin 8 (OriginLab) and Clampfit (Axon Instruments). Overall apparent open probability (Po) for WT and six mutant channels was calculated from the tail currents after voltage pulses ranging from −164 mV to 75 or 195 mV.
| (1) |
Data points gained by the equation (1) were fitted with a Boltzmann distribution with an offset to gain Po curves.
| (2) |
where Pmin is an offset, or a minimum Po at the most negative potential, V is the membrane potential, V1/2 is the half-maximal activation potential, and κ is the slope factor.
For separation of slow and fast gating,
| (3) |
where Pso and Pfo indicate the open probability of slow and fast gating at Vp. N is the number of channels expressed at −120 mV. Here, when prepulse at +170 mV is given after a series of voltage steps, which can be assumed that the open probability of fast gating is ∼1. In this state, the slow gate is not substantially affected. Therefore, the equation,
| (4) |
is established. When Pso (Vp) was obtained, using the equation (3),
| (5) |
can be drawn. Data obtained using the equation (5) were plotted.
Peak current density was calculated for comparison of data across cells of different sizes. It was obtained from peak current (pA) from each recording divided by cell capacitance (pF).
The time course of current activation of S189C was examined by fitting the currents obtained in 134 mM [Cl−]i between 35 mV and 115 mV in 20 mV increments.
where τ is the time constant of current activation, and A, I0, I (t) are the amplitudes, the offset, and the normalized currents, respectively.
Statistical analysis
Experimental values were analyzed for mean and s.e.m. The quantitative figures in this work represent the mean ± s.e.m. Data sets were statistically evaluated utilizing an unpaired t-test. Differences in the mean values of P < 0.05 were considered significant unless indicated otherwise.
RESULTS
Channel characteristics of WT hClC-1
Whole cell patch-clamp recordings from HEK 293 cells expressing WT hClC-1 were performed in both 134 mM [Cl−]i and 4 mM [Cl−]i to mimic physiological condition. WT hClC-1 demonstrated an instantaneous activation at the tested voltage ranges and a typical rapid deactivation of Cl− currents over time at the hyperpolarized regions after instantaneous increase under 134 mM [Cl−]i. (Fig. 1Aa). Under physiological condition (4 mM), WT hClC-1 generated instantaneous currents and slow activation of Cl− currents on a typical time course (Fig. 1Ab). To exclude the possibility of endogenous channels influencing the chloride currents, mock transfection test was performed with HEK cells expressing pEGFP-N1 vector alone under 134 mM [Cl−]i, and no currents were observed. (Fig. 1Ac). Instantaneous and steady-state current densities (pA/pF) were obtained from the representative current traces under both conditions to observe the current-voltage relationships. For instantaneous currents, the inward rectifications were observed in 134 mM [Cl−]i (Fig. 1Ad). For steady-state currents, the current-voltage relationship showed an outward rectification under physiological condition (4 mM) (Fig. 1Ae). Current-voltage relationships of WT hClC-1 under 4 mM [Cl−]i revealed that reversal (equilibrium) potentials were shifted to −60 mV (instantaneous currents) and −45 mV (steady-state currents), respectively. The reversal potential was shifted to lesser degree than expected. The discrepancy may be due to permeability when glutamate used as a substitute for internal Cl−.
Fig. 1.
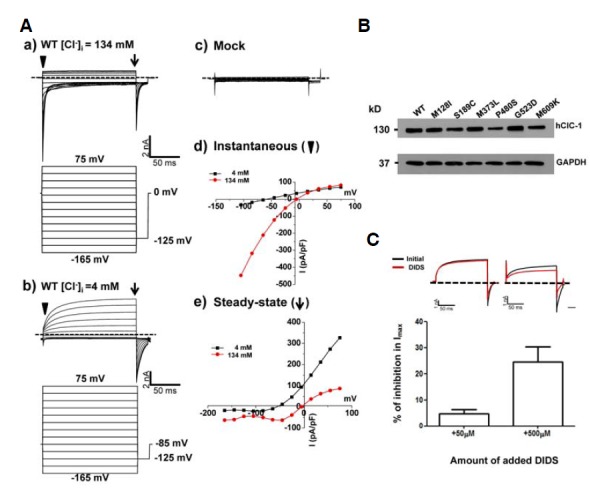
Channel characteristics and expression of WT hClC-1 in HEK293 cells. (A) a) Representative whole cell current traces for WT hClC-1 and voltage protocol for hClC-1 channel activity under 134 mM [Cl−]i in HEK293 cell expressing hClC-1. For voltage protocol, each holding potential and tail pulse was fixed at 0 mV and −125 mV. A series of voltage step pulses was applied from −165 mV to +75 mV in 20 mV steps for WT or mock transfection test. b) Representative whole cell current traces for WT hClC-1 and voltage protocol for hClC-1 channel activity under 4 mM [Cl−]i in HEK293 cell expressing hClC-1. For voltage protocol, each holding potential and tail pulse was fixed at −85 mV and −125 mV. A series of voltage step pulses was applied from −165 mV to +75 mV in 20 mV steps. c) Mock transfection test in HEK 293 cells. pEGFP-N1 vector was transiently expressed in HEK293 cells and the same voltage pulses were applied as WT. d) The instantaneous currents (where an inverted triangle indicates) were measured under both 134 mM [Cl−]i and 4 mM [Cl−]i at the beginning of test voltage pulses, normalized with respect to cell capacitance (pA/pF). e) Steady-state currents (where an arrow indicates) were measured under both 134 mM [Cl−]i and 4 mM [Cl−]i at the end of test voltage pulses, normalized with respect to cell capacitance (pA/pF). Dashed lines indicate zero-current level. (B) Western blotting analysis of the protein expression of hClC-1 and mutants. 24 h after transfection the total cell lysates were examined. GAPDH was used as a loading control. (C) Inhibition of hClC-1 WT by extracellular application of 50 μM and 500 μM DIDS. hClC-1 was transiently expressed in HEK293 cells and representative whole cell current traces were elicited by the voltage protocol which applied at 75 mV under 4 mM [Cl−]i. Each holding potential and tail pulse was fixed at −85 mV and −125 mV. Dashed lines indicate zero current line (top). Currents were measured initially (black), after steady-state inhibition in 50 μM and 500 μM DIDS for 30 s (red). Reduced maximal currents obtained at 75 mV under 50 μM (n = 5) and 500 μM (n = 5) DIDS treatment compared to initial currents were indicated in the bar graph (bottom).
Western blotting analysis was conducted to evaluate protein expression. Expression of WT and mutant proteins was confirmed in HEK 293 cells over expressing GFP-tagged WT hClC-1 and mutant hClC-1 proteins (Fig. 1B). Chloride channel expression was also evaluated using the general chloride channel blocker, 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS), as previously described (Matulef et al., 2008). When extracellular 50 μM DIDS was applied, Cl− currents were only minimally blocked (5%). However, application of extracellular 500 μM DIDS caused Cl− currents to be inhibited up to 25% compared to initial currents, and this result was similar to that previously reported (Matulef et al., 2008) (Fig. 1C). Taken together, these results suggest that WT hClC-1 and the mutants were properly expressed, and the characteristics of WT hClC-1 were confirmed.
Channel characteristics of autosomal dominant mutants, S189C and P480S
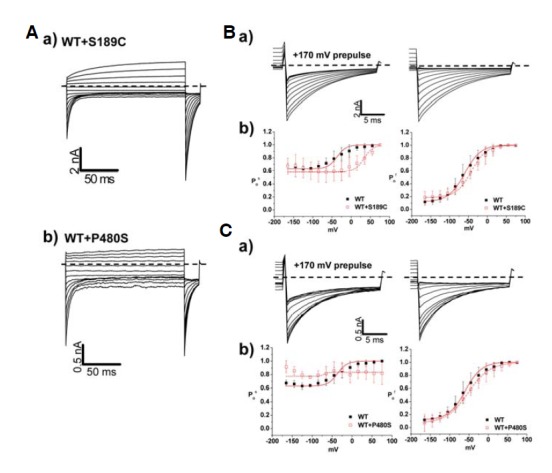
Whole cell patch-clamp recording from HEK293 cells expressing WT CIC-1 showed typical voltage-dependent Cl− currents with V1/2 of −54 ± 3 mV (Fig. 2Ac, Table 2). Surprisingly, the channel activation of the S189C mutant showed more than a 100 mV shift of V1/2 value toward depolarizing voltage (58 ± 4 mV) with shallower voltage-dependency (Fig. 2Ac and Table 2). In order to clarify the type of gating process involved in the phenotype of S189C mutant, whole cell recordings were performed using the “umbrella protocol” described earlier (Accardi and Pusch, 2000). V1/2 values of WT for “slow” gating and “fast” gating were −36 ± 17 mV and −60 ± 10 mV, respectively (Table 3), well matched with the previously reported values (Accardi and Pusch, 2000). For S189C mutant, V1/2 of both gating processes was shifted toward more depolarizing potentials relative to WT (Fig. 3Bb). In particular, the V1/2 of slow gating was remarkably shifted by 86 mV, implying that altered “slow” gating process is mainly responsible for the functional consequence of the S189C mutation (Table 3). This right shifted voltage-dependency also resulted in remarkably decreased Cl− current density (∼5% of WT channel) in the range of physiological membrane potential (Table 1). One interesting feature of S189C is slow activation as compared to instantaneous activation of WT hClC-1. The activations at the depolarized regions were voltage dependent (Fig. 4 and Table 5).
Fig. 2.
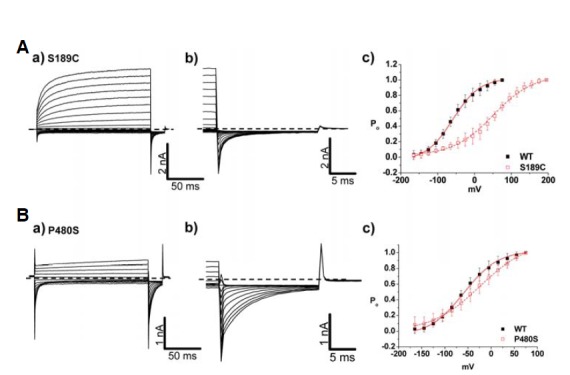
Channel characteristics of autosomal dominant mutants, S189C and P480S. (A, B) a) Whole cell current traces for S189C and P480S. For voltage protocol, a series of voltage step pulses was applied from −165 mV to 195 mV (S189C) and 75 mV (P480S). Each holding potential and tail pulse was fixed at 0 mV and −125 mV. b) Enlargement of whole cell current traces elicited during tail pulse for S189C and P480S. c) The voltage dependence of activation of WT, S189C, and P480S, calculated from tail currents measured at −125 mV. Closed black squares and opened red squares represent WT (n = 10) and mutants, S189C (n =10), P480S (n = 14), respectively. The averaged data were fit with a Boltzmann distribution, and V1/2 parameters are reported in Table 2. Dashed lines indicate zero-current level.
Table 2.
Botlzmann parameters of the activation curves of currents obtained from WT and six mutants
| Mutant | Inheritance | V1/2 (mV) | κ (mV) | Total N |
|---|---|---|---|---|
| WT | Normal | −54 ± 3 | 27 ± 3 | 10 |
| S189C | A.D | 58 ± 4 | 59 ±14 | 10 |
| P480S | A.D | −33 ± 6 | 48 ± 5 | 14 |
| M128I | A.R | 56 ± 2 | 29 ± 12 | 12 |
| M373L | Sporadic | −36 ± 3 | 27 ± 1 | 6 |
| G523D | Sporadic | - | - | 12 |
| M609K | Sporadic | −50 ± 4 | 25 ± 2 | 7 |
indicates that no value was calculated to fit with Boltzmann distribution.
Table 3.
Boltzmann parameters of the activation curves for fast and slow gating analysis results from WT and six mutants
| Mutant | Inheritance | V 1/2 (mV) | κ (mV) | Total N | ||
|---|---|---|---|---|---|---|
|
| ||||||
| Fast | Slow | Fast | Slow | |||
| WT | Normal | −60 ± 10 | −36 ± 17 | 20 ± 2 | 18 ± 2 | 10 |
| S189C | A.D | 5 ± 14 | 50 ± 5 | 52 ± 5 | 10 ± 1 | 13 |
| P480S | A.D | −61 ± 5 | −19 ± 15 | 28 ± 2 | 15 ± 2 | 6 |
| M128I♦ | A.R | 0 ± 8 | −100 ± 6 | 45 ± 8 | 49 ± 6 | 10 |
| M373L♦ | Sporadic | −52 ± 6 | −126 ± 10 | 20 ± 2 | 18 ± 1 | 5 |
| G523D♦ | Sporadic | −91 ± 11 | - | 63 ± 7 | - | 10 |
| M609K♦ | Sporadic | −60 ± 1 | - | 18 ± 1 | - | 8 |
indicates that the mutant showed reversed pattern of activation curve for slow gating.
indicates that no value was calculated to fit with Boltzmann distribution.
Fig. 3.
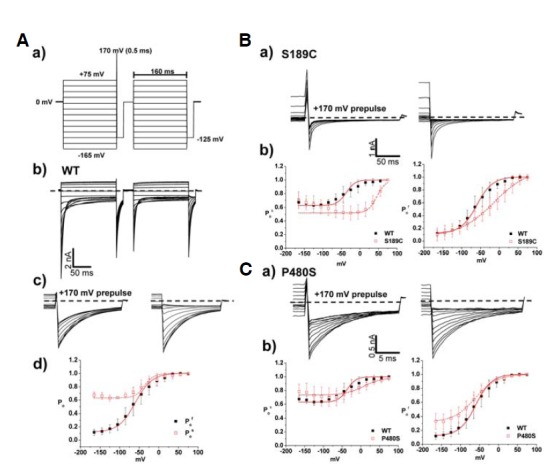
Analysis of fast and slow gating in WT, S189C, and P480S. (A) a) Voltage pulse protocol for fast and slow gating analysis. Holding potential and tail pulse were fixed at 0 mV and −125 mV. First series of voltage pulses was given from −165 mV to +75 mV during 160 ms, and prepulse at +170 mV during 0.5 ms was followed. Second series of voltage pulses was followed after previous pulse, and it is identical to the first voltage pulse protocol except prepulse. b) Whole cell current traces of hClC-1 WT elicited from the voltage pulse protocol for fast and slow gating analysis. c) Whole cell tail current traces at −125 mV. d) The voltage dependence of activation of WT for slow and fast gating analysis, calculated from tail currents measured at −125 mV. Black closed squares and red opened squares represent relative open probability of fast (n = 10) and slow (n = 10) gating, respectively. The averaged data were fit with a Boltzmann distribution, and V1/2 parameters are reported in Table 3. Dashed lines indicate zero-current level. (B, C) a) Whole cell current traces at tail pulse, −125 mV in fast and slow gating analysis of S189C and P480S. Prepulse at 170 mV was indicated (left). b) Comparison of slow gating channel activation curves (left) and fast gating channel activation curves (right) in WT (n = 10), S189C (n = 13), and P480S (n = 6) Dashed lines indicate zero-current level.
Table 1.
Peak Current Density of hClC-1 and six mutants
| Mutant | Inheritance | Peak Current Density (pA/pF) | Total N |
|---|---|---|---|
| WT | Normal | −498 ± 86.3 | 10 |
| S189C | A.D | −28.8 ± 15.2*** | 10 |
| P480S | A.D | −117.8 ± 16.3*** | 14 |
| M128I | A.R | −138.5 ± 12.4*** | 12 |
| M373L | Sporadic | −654.5 ± 222.0 n.s | 6 |
| G523D | Sporadic | −452.2 ± 57.0 n.s | 12 |
| M609K | Sporadic | −155.3 ± 68.6 * | 7 |
*P < 0.05
***P < 0.001 n.s represent not significant.
Each A.D and A.R indicate autosomal dominant and autosomal recessive pattern.
Fig. 4.
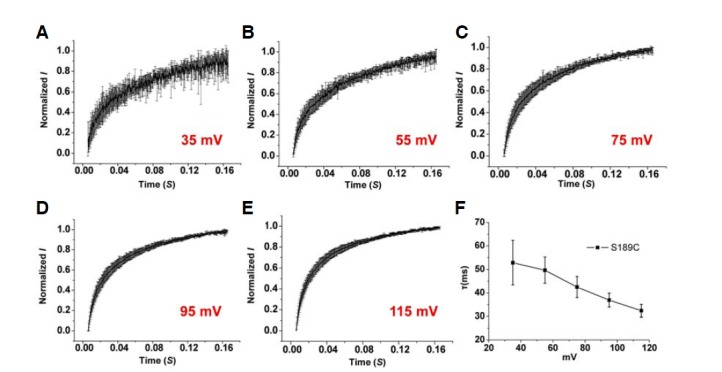
Kinetics of slow activation for autosomal dominant mutant, S189C in 134 mM [Cl−]i. (A–E) Time course of S189C as determined by whole cell current recording with 134 mM [Cl−]i in HEK293 cells. The averaged time dependence of current amplitude from each voltage pulse from 35 mV to 115 mV in 20 mV steps was shown (n = 5). Currents were normalized to maximal values obtained from each voltage pulse. (F) The fit parameters of a time constant.
Table 5.
Slow activation kinetics of S189C
| Voltage pulse (mV) | Mean of τ (ms) | Total N |
|---|---|---|
| 115 | 32.4 ± 3 | 5 |
| 95 | 36.9 ± 3 | 5 |
| 75 | 42.5 ± 5 | 5 |
| 55 | 49.7 ± 6 | 5 |
| 35 | 52.9 ± 10 | 5 |
Interestingly, P480S showed largely unaffected characteristics of both slow and fast gating processes as compared to WT (Fig. 2Bc and Fig. 3Cb). Though it is difficult to explain the abnormal phenotype of P480S mutation given this WT-like voltage-dependency, the P480S mutation might lead to reduced channel activity in the plasma membrane. The decreased Cl− conductance (∼26% of WT channel) from P480S could support this possiblity (Table 1).
Co-transfection experiment for two AD mutants
Illustrating the heterozygous form of autosomal dominant mutants, co-expression experiments were conducted in order to recapitulate heterozygous effects. Interestingly, when co-expressed with WT in HEK293 cells, S189C showed both inwardly and outwardly rectifying currents in whole cell traces (Fig. 5A). In the channel activation of co-expressed WT with S189C, V1/2 of slow and fast gating were 45 ± 5 mV and −37 ± 8 mV, respectively (Table 4). For S189C co-expressed with WT, slow gating process was significantly shifted more than 100 mV, suggesting that slow gating still governs the function of the channel. Co-expression tests of P480S also suggest that altered slow gating plays a key role in the functional consequence of the channel. V1/2 of co-expressed P480S in slow gating clearly showed less voltage dependency relative to WT whereas fast gating was largely unaffected (−50 ± 5 mV) (Fig. 5Cb). Interestingly, compared to V1/2 of S189C and P480S, V1/2 of both AD mutants co-expressed with WT displayed less shifting toward depolarizing potentials (Tables 3 and 4). These results show that 1) co-expression experiments with WT mimic dominant negative effects, and 2) the slow gating process largely contributes to the function of the channel.
Fig. 5.
Co-transfection test for auto-somal dominant mutants, S189C and P480S. (A) Whole cell current traces for co-expressing WT and S189C (a) or WT and P480S (b) in HEK293 cell. For voltage protocol, a series of voltage step pulses was applied from −165 mV to 75 mV. Each holding potential and tail pulse was fixed at 0 mV and −125 mV. Dashed lines indicate zero-current level. (B, C) a) Enlargement of whole cell current traces elicited at tail pulse, −125 mV for co-expressing WT and S189C (B) or WT and P480S (C) in HEK293 cell. Dashed lines indicate zero current level. b) The voltage dependence of activation of WT, WT co-expressed mutants, calculated from tail currents measured at −125 mV. Closed black squares and opened red squares represent WT (n = 10) and mutants, WT and S189C (n = 5), WT and P480S (n = 7), respectively. The averaged data were fit with a Boltzmann distribution, and V1/2 parameters are reported in Table 3.
Table 4.
Boltzmann parameters of the activation curves for fast and slow gating analysis obtained from WT and co-transfection test for autosomal dominant mutants
| Mutant | Inheritance | V 1/2 (mV) | κ (mV) | Total N | ||
|---|---|---|---|---|---|---|
|
| ||||||
| Fast | Slow | Fast | Slow | |||
| WT | Normal | −60 ± 10 | −38 ± 17 | 26 ± 2 | 18 ± 2 | 10 |
| S189C | A.D | −37 ± 8 | 45 ± 5 | 28 ± 4 | 20 ± 1 | 5 |
| P480S | A.D | −50 ± 5 | - | 27 ± 3 | - | 7 |
indicates that no value was calculated to fit with Boltzmann distribution.
Unique behaviors of autosomal recessive mutant channel
M128I is the only autosomal recessive mutant among these six mutants. Surprisingly, M128I did not show the typical deactivation of Cl− current on a time course (Fig. 6Aa). In addition, the channel activation of M128I was consistent with open state of M128I (Fig. 6Ac), indicating drastically right shifted V1/2 toward more depolarizing potentials by 100 mV (56 ± 2 mV) (Table 2). In slow gating analysis, however, M128I displayed reversed voltage-dependency (Fig. 6Bb). V1/2 of fast gating in M128I was shifted to more depolarizing potentials by 30 mV relative to WT (Table 3). Collectively, this modified voltage-dependency in both gating is responsible for nearly 72% reduction in Cl− current density (−138.5 ± 12.4 pA/pF) relative to WT (Table 1). The remarkably reduced Cl− current density and the right shifted V1/2 in fast gating can still explain the pathological output of M128I mutation in MC patient.
Fig. 6.
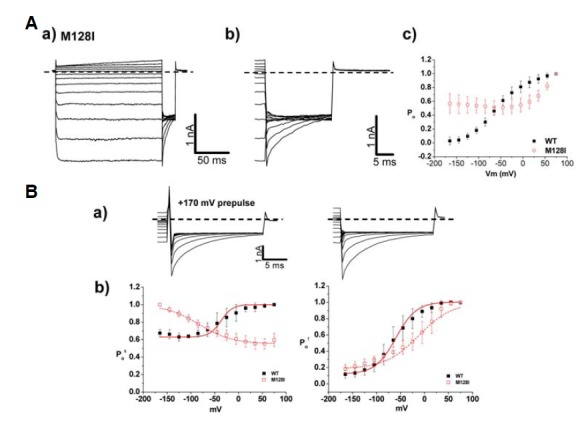
Channel characteristics of autosomal recessive mutant, M128I A. (A) a) Whole cell current traces for M128I. For voltage protocol, a series of voltage step pulses was applied from −165 mV to 75 mV. Each holding potential and tail pulse was fixed at 0 mV and −125 mV. b) Enlargement of whole cell current traces elicited during tail pulse for M128I. c) The voltage dependence of activation of M128I, calculated from tail currents measured at −125 mV. Closed black squares and opened red squares represent WT (n=10) and M128I (n=12), respectively. The averaged data were fit with a Boltzmann distribution, and V1/2 parameters are reported in Table 2. Dashed lines indicate zero-current level. (B) a) Characteristics of fast and slow gating in M128I. Whole cell current traces with prepulse, 170 mV (left) at −125 mV were indicated. Dashed lines indicate zero current level. b) Comparison of slow gating channel activation curves (left) and fast gating channel activation curves (right) of WT and M128I. Black closed squares and red opened squares represent (n=10) and M128I (n=10), respectively. The averaged data were fit with a Boltzmann distribution, and V1/2 parameters are reported in Table 3.
Various characteristics of sporadic mutants
In addition to the inherited mutants, some mutants are not associated with a familial history. M373L, G523D, and M609K are sporadic mutants, and whole cell patch-clamp recording from HEK 293 cells described the characteristics of these three mutants. M373L and M609K showed similar behaviors as WT, illustrating rapid activation and slow deactivation of Cl− currents on a typical time course (Figs. 7A and 7C). However, deactivation of current was completely absent in G523D (Fig. 7Ba). The reversed channel activation was observed in G523D whereas M373L and M609K showed WT-like characteristics (Fig. 7Bc). Interestingly, all sporadic mutants commonly revealed the reversed voltage dependency in slow gating process (Fig. 8Ab, Bb, and Cb). V1/2 values of M373L and M609K for fast gating were −36 ± 3 mV and −50 ± 4 mV, respectively, suggesting there was no significant effect on fast gating process in the two mutants (Table 3). However, M609K resulted in a decreased Cl− conductance (∼30% of WT channel), suggesting destabilization of the membrane potential.
Fig. 7.
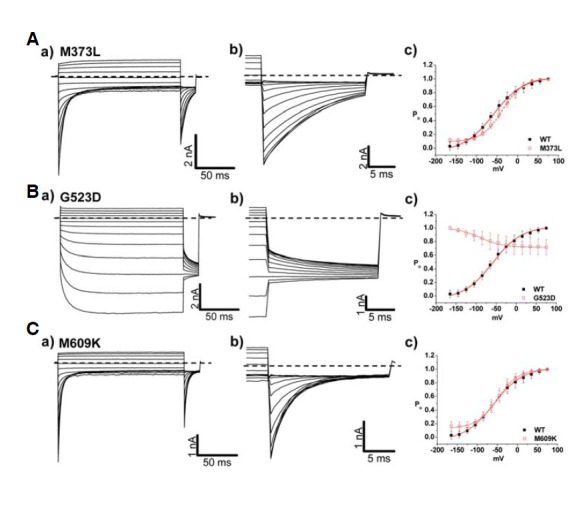
Analysis of channel characteristics of sporadic mutants, M373L, G523D, and M609K. (A–C) a) Whole cell current traces for M373L (A), G523D (B), and M609K (C) in HEK 293 cells under 134 mM [Cl−]i. For voltage protocol, a series of voltage step pulses was applied from −165 mV to 75 mV. Each holding potential and tail pulse was fixed at 0 mV and −125 mV. b) Enlargement of whole cell current traces elicited during tail pulse for M373L, G523D, and M609K. Dashed line indicates zero current level. c) The voltage dependence of activation of M373L, G523D, and M609K, calculated from tail currents measured at −125 mV. Closed black squares and opened red squares represent WT (n = 10) and mutants, M373L (n = 6), G523D (n = 12), M609K (n = 7), respectively. The averaged data were fit with a Boltzmann distribution, and V1/2 parameters are reported in Table 2.
Fig. 8.
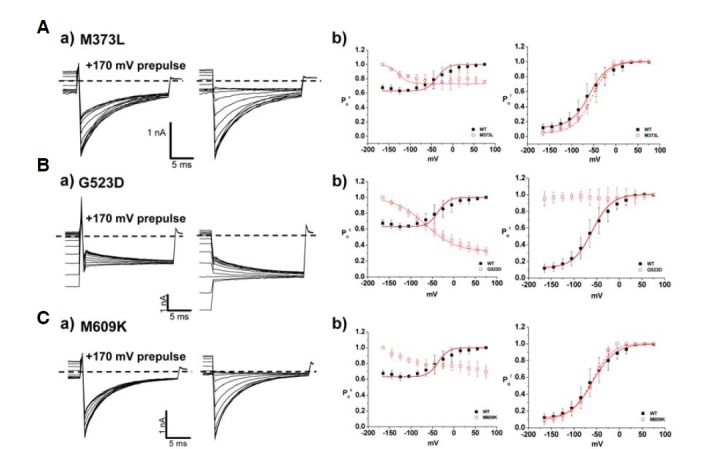
Analysis of fast and slow gating in sporadic mutants. (A–C) a) Characteristics of fast and slow gating in M373L (A), G523D (B), and M609K (C). Whole cell current traces with prepulse, 170 mV (left) at −125 mV were indicated. Dashed lines indicate zero current level. b) Comparison of slow gating channel activation curves (left) and fast gating channel activation curves (right) of WT and sporadic mutants. Black closed squares and red opened squares represent WT (n = 10) and mutants, M373L (n = 5), G523D (n = 10), M609K (n = 8), respectively. The averaged data were fit with a Boltzmann distribution, and V1/2 parameters are reported in Table 3.
Interestingly, G523D showed mainly characteristics in fast gating when compared to the WT channel (Fig. 8Bb). The typical voltage dependency in the channel activation was totally abrogated, and fitting with Boltzmann distributions was impossible. It remains to be determined how G523D contributes to pathological changes while showing consistent open state channel activation and adequate Cl− conductance in the range of physiological membrane potentials (−452.2 ± 52.0 pA/pF). However, the reversed voltage-dependency in slow gating process still supports the abnormal consequences of the G523D mutant channel.
DISCUSSION
We report the channel gating properties of six mutants found among Korean patients. The clinical presentation and inheritance associated with these mutants underscore the importance of their channel function on physiology.
We identified the functional consequences of the autosomal dominant mutants through the whole cell patch-clamp approach. Firstly, S189C demonstrated not only outwardly rectifying chloride currents but also modified channel gating in both slow and fast gating modalities. Co-expressed S189C with WT showed both inwardly and outwardly rectifying currents as well as shifted V1/2 to more depolarizing potentials. The S189C mutation is located in the linker region between domains C and D of ClC-1, which is known for the GSGIP region, a well-conserved domain among different species, functioning as the Cl− selectivity filter (Dutzler et al., 2002). Helices D, F, N, and R participate in the formation of the chloride conducting pore of a ClC monomer (Dutzler et al., 2002). The Cl− selectivity may be destabilized due to the mutation that occurs at this position. Taking into consideration both the importance of conserved residues and significant alteration that occurred in channel gating, we hypothesize that mutant S189C specifically plays a key role in slow gating of ClC-1 channel, causing outwardly rectifying currents. At depolarizing potentials, S189C activated with time constants that were drastically increased compared to WT which cannot be analyzed due to simultaneous and rapid deactivation of current. The slow activation observed in S189C might partially cause the repolarization of the action potential, leading to myotonic runs. In contrast, mutant P480S sustained whole cell traces that are similar to WT. P480S showed an appreciable right shift in slow gating whereas fast gating was not significantly altered. Co-expression of both S189C and P480S s resulted in a shift to more depolarizing potentials, particularly in slow gating, although less pronounced than that observed for solely mutant expressed channels. Collectively, these data suggest that a dominant negative effect is seen for both mutants, affecting slow gating in particular.
M128I resulted in significant changes not only in the whole cell current but also in gating. Although current was not deactivated on a time course as in WT, the whole cell density was reduced approximately 70%, which could explain the clinical abnormalities of MC found in the patient. Furthermore, both fast and slow gating of M128I was significantly modified. Taken together, we hypothesize that significant alteration in channel function as well as reduction in chloride conductance contributes to the clinical presentation of autosomal recessive MC. It is also interesting that M128V was previously reported to cause autosomal dominant type of MC (Grunnet et al., 2003), suggesting that the specific amino acid position mutated is not a major factor in determining the inheritance pattern. M128V resulted in a slow deactivation of outwardly rectifying current in the whole cell trace whereas it showed overlapping behavior with that of M128I, including a reduction in chloride current and right shifted V1/2 (Grunnet et al., 2003). Thus, we conclude that substitution of isoleucine instead of valine in domain B might abolish a typical slow deactivation of Cl− current in ClC-1. Further studies regarding structures of amino acid were not conducted in this study, but it reinforces the importance of this helix part of gating function (Grunnet et al., 2003).
In the sporadic mutants, a reduced chloride conductance in both G523D and M609K was observed. Slow gating of M373L, G523D, and M609K was commonly activated at negative potentials, which is the opposite behavior to that of WT. In addition, G523D demonstrated abnormal function in fast gating. Although the property of G523D was not investigated further in this study, it is plausible that the mutant is deeply involved in both slow and fast gating mechanisms. M609K is the only mutation in the present study that is located in the CBS1 domain (Supplementary Fig. 1). This site could be crucial in regulation of chloride conductance and slow gating in that channel activation of fast gating in M609K was similar to WT. M373L, on the other hand, simply resulted in the modified channel activation in slow gating, although it produced sufficient chloride conductance at physiologically relevant potentials. It remains puzzling how increased Cl− conductance induces myotonia. Recently, Richman et al. (2012) showed that the G233S mutant results in a near constitutively open channel by rendering the fast gate of the channel mostly open, while the common gating appears to be relatively normal (Richman et al., 2012). The functional characterizations of the mutant appear to be consistent with the structural role of the G233 residue according to the high-resolution structures of the homologous bacterial ClC molecules (Richman et al., 2012). This “gain-of-function” mutation for the molecular function of ClC-1 paradoxically appears to result in a reduction in membrane stability leading to myotonia. Because an increase in Cl− conductance in response to membrane depolarization is essential to repolarize the membrane potential to the resting state, a defect in the dynamics of the response to a voltage change as occurs in the G233S mutant may contribute to the over-excitability of the membrane and the clinical myotonia, in contrary to the previous studies (Richman et al., 2012). In this regard, the effect of the G523D mutation can be considered to be similar to those caused by the loss-of-function mutations of CLCN1 that result in reduced conductance at positive membrane potentials.
The two patients with P480S or G523D mutations responded well to mexiletine (Moon et al., 2009). The common findings between these two mutations are 1) high open probability of both slow and fast gating at the hyperpolarized membrane potentials and 2) low open probability of both slow and fast gating at the depolarized membrane potentials; although both effects are more severe with the G523D mutation. The prolonged repolarization, hyperexcitability, and spontaneous action potentials would be expected if there was a reduction in the resting muscle chloride conductance through ClC-1 (gClC-1). Our findings suggest that prolonged repolarization due to decreased Po of slow gating at the depolarized regions of ClC-1 channels is important for MC and decreasing Na+ current by mexiletine improved the myotonia symptoms. Decreased Cl− conductance was not a common factor for the response to treatment with mexiletine, a Na+ channel blocker causing a reduction in Na+ current amplitude.
The response to treatment is poor for M609K and M128I but excellent for P480S and G523D (Moon et al., 2009). The main physiologic difference is the voltage sensitivity (κ) of ClC-1 channel. The lower voltage sensitivity of ClC-1 mutants P480S and G523D appears to be important for the response to treatment. A similar finding is observed in mutant S189C (κ of 59 mV). However, the mutation is associated with a fair response to treatment in our previous study (patient 6 versus patient 9) (Moon et al., 2009). Although both patients have the same mutation, the symptoms and signs such as muscle hypertrophy, transient weakness, cold sensitivity, CK level, and EMG findings, were different (Moon et al., 2009).
The clinical severity of MC may be graded by Jøegensen (Colding-Jørgense, 2005). The severity grade was 5 (pronounced myotonia, transient weakness, and dystrophic features) for M609K and 2 [mild (and/or fluctuating) symptoms] for the M128I mutation (Moon et al., 2009). Both mutants commonly displayed reduced Cl− conductances. The open probability of slow gating is similar, but the open probability of fast gating is quite different between two mutants. These results suggest that right shift of the voltage dependence of fast gating is important for the clinical severity if other parameters such as decreased Cl− conductance and changes in slow gating (a reversed relationship between Po and voltage) are similar.
In conclusion, we identified and characterized six novel mutants found in ClC-1 among Korean patients who suffer from MC. The electrophysiological property as well as analysis of slow and fast gating is expected to provide a sufficient basis for investigation of ClC-1 channels in both physiological and pathological terms.
Acknowledgments
We appreciate Dr. Alfred L. George from Vanderbilt University, Nashiville, TN, USA for kindly providing the hCLCN-1 gene, Hyun-Ho Lim from Brandeis University, MA, USA for helpful comments and supplementary figure, and Han Sung Lee from University of California San Francisco for manuscript edits. This study was supported by grants from the National Research Foundation of Korea funded by the Korean government (MEST) (2008-2005948, 2010-0019472 and 2012R1A2A1A0 1003073). K. Ha, S. Y. Kim, and C. Hong were supported by the graduate program of Brain Korea 21 project from the Ministry of Education, Science and Technology (MEST).
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Accardi A., Pusch M. Fast and slow gating relaxations in the muscle chloride channel CLC-1. J. Gen. Physiol. 2000;116:433–444. doi: 10.1085/jgp.116.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aromataris E.C., Rychkov G.Y., Bennetts B., Hughes B.P., Bretag A.H., Roberts M.L. Fast and slow gating of CLC-1: differential effects of 2-(4-chlorophenoxy) propionic acid and dominant negative mutations. Mol. Pharmacol. 2001;60:200–208. doi: 10.1124/mol.60.1.200. [DOI] [PubMed] [Google Scholar]
- Bryant S.H., Morales-Aguilera A. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J. Physiol. 1971;219:367–383. doi: 10.1113/jphysiol.1971.sp009667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colding-Jørgensen E. Phenotypic variability in myotonia congenita. Muscle Nerve. 2002;32:19–34. doi: 10.1002/mus.20295. [DOI] [PubMed] [Google Scholar]
- Dulhunty A.F. Distribution of potassium and chloride permeability over the surface and T-tubule membranes of mammalian skeletal muscle. J. Membr. Biol. 1979;45:293–310. doi: 10.1007/BF01869290. [DOI] [PubMed] [Google Scholar]
- Dutka T.L., Murphy R.M., Stephenson D.G., Lamb G.D. Chloride conductance in the transverse tubular system of rat skeletal muscle fibres: importance inexcitation-contraction coupling and fatigue. J. Physiol. 2000;586:875–887. doi: 10.1113/jphysiol.2007.144667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R. The clc family of chloride channels and transporters. Curr. Opin. Struct. Biol. 2006;4:439–446. doi: 10.1016/j.sbi.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Dutzler R., Campbell E.B., Cadene M., Chait B.T., MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Fahlke C., Rosenbohm A., Mitrovic N., George A.L, Jr, Rüdel R. Mechanism of voltage-dependent gating in skeletal muscle chloride channels. Biophys. J. 1996;71:695–706. doi: 10.1016/S0006-3495(96)79269-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke C., Beck C.L., George A.L., Jr A mutation in autosomal dominant myotonia congenita affects pore properties of the muscle chloride channel. Proc. Natl. Acad. Sci USA. 1997a;94:2729–2734. doi: 10.1073/pnas.94.6.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke C., Knittle T., Gurnett C.A., Campbell K.P., George A.L., Jr. Subunit stoichiometry of human muscle chloride channels. J. Gen. Physiol. 1997b;109:93–104. doi: 10.1085/jgp.109.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke C., Desai R.R., Gillani N., George A.L., Jr. Residues lining the inner pore vestibule of human muscle chloride channels. J. Biol. Chem. 2001;276:1759–1765. doi: 10.1074/jbc.M007649200. [DOI] [PubMed] [Google Scholar]
- Fialho D., Schorge S., Pucovska. U., Davies N.P., Labrum R., Haworth A., Stanley E., Sud R., Wakeling W., Davis M.B., et al. Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain. 2007;130:3265–3274. doi: 10.1093/brain/awm248. [DOI] [PubMed] [Google Scholar]
- George A.L, Jr., Crackower M.A., Abdalla J.A., Hudson A.J., Ebers G.C. Molecular basis of Thomsen’s disease (autosomal dominant myotonia congenita) Nat. Genet. 1993;3:305–310. doi: 10.1038/ng0493-305. [DOI] [PubMed] [Google Scholar]
- Grunnet M., Jespersen T., Colding-Jørgensen E., Schwartz M., Klaerke D.A., Vissing J., Olesen S.P., Dunø M. Characterization of two new dominant ClC-1 channel mutations associated with myotonia. Muscle Nerve. 2003;28:722–732. doi: 10.1002/mus.10501. [DOI] [PubMed] [Google Scholar]
- Heiny J.A., Valle J.R., Bryant S.H. Optical evidence for a chloride conductance in the T-system of frog skeletal muscle. Pflugers Arch. 1990;416:288–295. doi: 10.1007/BF00392065. [DOI] [PubMed] [Google Scholar]
- Hsiao K.M., Huang R.Y., Tang P.H., Lin M.J. Functional study of CLC-1 mutants expressed in Xenopus oocytes reveals that a C-terminal region Thr891-Ser892-Thr893 is responsible for the effects of protein kinase C activator. Cell. Physiol. Biochem. 2010;25:687–694. doi: 10.1159/000315088. [DOI] [PubMed] [Google Scholar]
- Jentsch T.J., Lorenz C., Pusch M., Steinmeyer K. Myotonias due to CLC-1 chloride channel mutations. Soc. Gen. Physiol. 2010;50:149–159. [PubMed] [Google Scholar]
- Koch M.C., Steinmeyer K., Lorenz C., Ricker K., Wolf F., Otto M., Zoll B., Lehmann-Horn F., Grzeschik K.H., Jentsch T.J. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992;257:797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- Koch M.C., Ricker K., Otto M., Wolf F., Zoll B., Lorenz C., Steinmeyer K., Jentsch T.J. Evidence for genetic homogeneity in autosomal recessive generalised myotonia (Becker) J. Med. Genet. 1993;30:914–917. doi: 10.1136/jmg.30.11.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossin C., George A.L., Jr. Myotonia congenita. Adv. Genet. 2008;63:25–55. doi: 10.1016/S0065-2660(08)01002-X. [DOI] [PubMed] [Google Scholar]
- Mailänder V., Heine R., Deymeer F., Lehmann-Horn F. Novel muscle chloride channel mutations and their effects on heterozygous carriers. Am. J. Hum. Genet. 1996;58:317–324. [PMC free article] [PubMed] [Google Scholar]
- Markovic S., Dutzler R. The structure of the cytoplasmic domain of the chloride channel ClC-Ka reveals a conserved interaction interface. Structure. 2007;15:715–725. doi: 10.1016/j.str.2007.04.013. [DOI] [PubMed] [Google Scholar]
- Meyer S., Dutzler R. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure. 2006;14:299–307. doi: 10.1016/j.str.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Moon I.S., Kim H.S., Shin J.H., Park Y.E., Park K.H., Shin Y.B., Bae J.S., Choi Y.C., Kim D.S. Novel CLCN1 mutations and clinical features of Korean patients with myotonia congenita. J. Korean Med. Sci. 2009;24:1038–44. doi: 10.3346/jkms.2009.24.6.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulef K., Howery A.E., Kobertz W.R., Bois J.D., Maduke M. Discovery of potent CLC chloride channel inhibitors. ACS Chem. Biol. 2008;3:419–428. doi: 10.1021/cb800083a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman D.P., Yu Y., Lee T.T., Tseng P.Y., Yu W.P., Maselli R.A., Tang C.Y., Chen T.Y. Dominantly inherited myotonia congenita resulting from a mutation that increases open probability of the muscle chloride channel. CLC-1 Neuromol. Med. 2012;14:328–337. doi: 10.1007/s12017-012-8190-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C.Y., Chen T.Y. Physiology and pathophysiology of CLC-1: mechanisms of a chloride channel disease, myotonia. J. Biomed. Biotechnol. 2011;2011:685328. doi: 10.1155/2011/685328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F.F., Ryan A., Devaney J., Warnstedt M., Korade-Mirnics Z., Poser B., Escriva M.J., Pegoraro E., Yee A.S., Felice K.J., et al. Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain. 2002;125:2392–2407. doi: 10.1093/brain/awf246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.