

Abstract
A mammalian cell renovates itself by autophagy, a process through which cellular components are recycled to produce energy and maintain homeostasis. Recently, the abundance of gap junction proteins was shown to be regulated by autophagy during starvation conditions, suggesting that transmembrane proteins are also regulated by autophagy. Transient receptor potential vanilloid type 1 (TRPV1), an ion channel localized to the plasma membrane and endoplasmic reticulum (ER), is a sensory transducer that is activated by a wide variety of exogenous and endogenous physical and chemical stimuli. Intriguingly, the abundance of cellular TRPV1 can change dynamically under pathological conditions. However, the mechanisms by which the protein levels of TRPV1 are regulated have not yet been explored. Therefore, we investigated the mechanisms of TRPV1 recycling using HeLa cells constitutively expressing TRPV1. Endogenous TRPV1 was degraded in starvation conditions; this degradation was blocked by chloroquine (CLQ), 3MA, or downregulation of Atg7. Interestingly, a glucocorticoid (cortisol) was capable of inducing autophagy in HeLa cells. Cortisol increased cellular conversion of LC3-I to LC-3II, leading autophagy and resulting in TRPV1 degradation, which was similarly inhibited by treatment with CLQ, 3MA, or downregulation of Atg7. Furthermore, cortisol treatment induced the colocalization of GFP-LC3 with endogenous TRPV1. Cumulatively, these observations provide evidence that degradation of TRPV1 is mediated by autophagy, and that this pathway can be enhanced by cortisol.
Keywords: autophagy, glucocorticoid (cortisol), protein degradation, TRPV1
INTRODUCTION
Transient receptor potential vanilloid type 1 (TRPV1) is a ligand-gated cation channel, composed of six transmembrane (TM) domains, harboring a short pore-forming region between the fifth and sixth TM domains (Kedei et al., 2001). TRPV1 is a sensory transducer, and can be activated by a wide variety of exogenous and endogenous physical and chemical stimuli, such as heat, capsaicin, protons, and several inflammatory mediators (Caterina et al., 1997; Clapham, 2003; Hwang et al., 2000; Ross, 2003; Szallasi and Blumberg, 2007). The main functions of TRPV1 are to regulate body temperature and sensations such as scalding heat, pain and pungency (Caterina et al., 1997; Clapham, 2003; Szallasi and Blumberg, 2007). Intriguingly, the functions of TRPV1 are known to change dynamically in response to sensitization or desensitization under pathological conditions. For example, TRPV1 can be sensitized by bradykinin through PKC activation in inflammation (Sugiura et al., 2002). In addition, the sensitization and desensitization of TRPV1 can be controlled by its phosphorylation status, which is modulated by CaMKII and calcineurin (Jung et al., 2004). However, the mechanisms by which the expression levels of endogenous TRPV1 are regulated in neuropathic pain models remain as yet undefined.
In mammalian cells, proteins are subjected to degradation in two major organelles: proteasomes and lysosomes. Proteasomes specifically degrade ubiquitinated substrates; in contrast, lysosomes degrade cellular materials from various pathways called autophagy. Three main types of autophagy pathways exist: macroautophagy (the major type), microautophagy, and chaperone-mediated autophagy. In mammalian cells, autophagy is a complex process involving many different proteins, including autophagy-related proteins (Atgs), which regulate autophagosome formation [(Mizushima and Komatsu, 2011; Settembre et al., 2013) and references therein]. Autophagy is induced when cells are exposed to deleterious conditions, such as starvation or endoplasmic reticulum (ER) stress. Connexin, a gap junction protein, was recently demonstrated to be internalized upon exposure to stress and to traffic to autophagosomes, which ultimately degrade proteins (Bejarano et al., 2012; Fong et al., 2012; Lichtenstein et al., 2011). This finding suggests that transmembrane proteins can be regulated by autophagy. Interestingly, autophagy can also be induced by a glucocorticoid (cortisol), which is commonly used to treat noninfectious inflammatory diseases (Jia et al., 2011; Molitoris et al., 2011; Xia et al., 2010). In addition, cortisol induces numerous changes in gene expression, resulting in the inhibition of mammalian target of rapamycin (mTOR) and the induction of autophagy, accompanied by the accumulation of autophagosomes (Molitoris et al., 2011; Xia et al., 2010).
In this study, we studied the relationship between the degradation of TRPV1 and autophagy. We determined TRPV1 was degraded in two pathways, the starvation- and cortisol-induced autophagy pathways. Starvation or cortisol treatment in HeLa cells induced conversion of LC3-I to LC3-II, which resulted in the induction of autophagy. As a result, TRPV1 was degraded; this degradation was reduced by treatment with anti-autophagy inhibitors or by genetic silencing of Atg7. Overall, our studies identify a link between cortisol treatment and autophagy, and identify autophagy as a central player in TRPV1 regulation.
MATERIALS AND METHODS
Cell culture, plasmid and siRNA transfection, cell lysis
HeLa cells were incubated in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C with 5% CO2. Transient transfections were performed with a plasmid encoding a green fluorescent protein (GFP)-tagged version of LC3 (pGFP-LC3) (Pyo et al., 2005) and siRNAs by Lipofectamine 2000 (Invitrogen) and Oligofectamine (Invitrogen), respectively. Whole cell lysates were obtained by lysing cells in a hypotonic solution containing 10 mM Tris-HCl (pH 7.5), 10 mM NaCl, 10 mM EDTA, and 0.5% Triton X-100.
For starvation, HeLa cells were grown to approximately 40–50% confluence. One day later, cells were washed twice and then incubated in Hank’s Balanced Salt Solution (HBSS; Invitrogen) for the indicated time. When specified, cells were treated with 10 mM 3-methyladenine (3MA, Sigma), 100 μM chloroquine (CLQ, Sigma), or 5 μM MG132 (Agscientific) during starvation for 8 h. For cortisol incubations, HeLa cells were seeded one day before treatment and, when specified, cells were then incubated with various concentrations of cortisol (0–1.0 μM) for 24 h, or with 1 μM cortisol for various periods of time (corresponding to Figs. 4A and 4B).
Fig. 4.
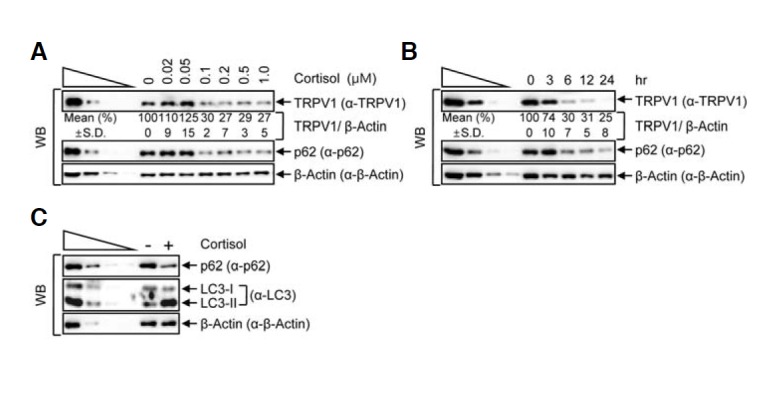
Cortisol treatment reduces the level of cellular TRPV1. (A, B) HeLa cells were treated with various cortisol concentrations ranging from 0 to 1 μM for 24 h (A) or 1 μM of cortisol for various periods of time (B). Western blotting (WB) was performed with the specific antibodies. (C) HeLa cell lysates that were incubated in the presence or absence of 1 μM of cortisol for 24 h were analyzed by WB. The normalized level in the absence of cortisol treatment was set to 100 %. Mean values (%) and standard deviations (S.D.) were calculated from three independent experiments.
siRNA
An siRNA sequence targeting Atg7 [5′-r(AAAGACUCGAGUGU GUUGGUGUUAUdTdT)-3′] (Liu et al., 2009) was employed to silence the expression of cellular Atg7 in HeLa cells. Control siRNA was obtained from Bioneer (Ahn et al., 2013).
Semiquantitative RT-PCR (RT-sqPCR)
Trizol (Invitrogen) was used to extract RNA from cells. Reverse transcriptase reactions were performed with a random hexamer (Invitrogen) and reverse transcriptase (Invitrogen). To amplify cDNA, semiquantitative PCR was performed using GoTaq polymerase (Promega). Primers used to PCR-amplify GAPDH mRNA and Atg7 mRNA were: 5′- CAA GAT CAT CAG CAA TGC C-3′ (sense) and 5’-CTGTGGTCATGAGTCCTT CC-3′ (antisense), and 5′-GAAACCAAAGCAGCAAGG-3′ (sense) and 5′-CTTCTGGATGCTGCAAAACA-3′ (antisense), respectively (Ahn et al., 2013; Liu et al., 2009). PCR products were analyzed on 1% agarose gels containing ethidium bromide.
Western blotting (WB)
For immunodetection of specific proteins, proteins in total cell lysates were boiled in sample buffer containing SDS and β-mercaptoethanol. Equal amounts of protein were then resolved on 8–15% polyacrylamide gels and transferred to nitrocellulose membranes. For immunoblotting, the primary antibodies used included anti-TRPV1 (Novus), anti-p62 (Santa Cruz Biotechnology), anti-LC3 (Novus), anti-Atg7 (Santa Cruz Biotechnology) and anti-β-actin (Santa Cruz Biotechnology). Specific antibodies were used at 1:1000–1:3000 dilutions in a 0.1% Tween 20/TBS solution containing 5% non-fat milk, and incubated overnight at 4°C with rocking. Immunoreactive bands were visualized with ECL solution (Thermo).
Immunofluorescence
Cells were fixed in 3.7% formaldehyde/PBS for 20 min. Fixed cells were incubated with primary antibodies and then washed and permeabilized with 1× PBS/0.2% saponin. Next, cells were incubated with Alexa Fluor 546 goat anti-rabbit IgG secondary antibodies (Invitrogen). Immunofluorescence images were obtained on a DeltaVision microscope, using the factory settings for the appropriate excitation and emission wavelengths of Alexa Fluor 546 and GFP. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Vector).
RESULTS
Starvation-induced autophagy decreases the level of cellular TRPV1
HeLa cells, which constitutively express TRPV1, were used to determine if the cellular level of TRPV1 is reduced upon induction of autophagy, through a degradation pattern similar to that observed with the transmembrane protein, connexin (Goswami and Hucho, 2007; Takahata et al., 1999). The level of TRPV1 was significantly reduced to approximately 25% of the normal level after 8 h starvation. Moreover, the level of the autophagosomal marker, p62 (Mathew et al., 2009), gradually decreased during starvation, whereas the level of β-actin, a control protein, remained unchanged, suggesting that cellular TRPV1 was gradually degraded in starvation conditions (Fig. 1A). Expectedly, the level of the well-established autophagosome marker, LC3-II (Kabeya et al., 2000), increased dramatically and the level of LC3-I decreased, which indicates that starvation can induce autophagy in HeLa cells (Fig. 1B). Since lysosomes are the major organelles which degrade proteins in autophagy pathway, we used chloroquine (CLQ) to inhibit the formation of autolysosomes. Treatment with CLQ during starvation did not affect the abundance of TRPV1, whereas 8 h starvation in the absence of CLQ led to degradation of TRPV1, suggesting that the level of TRPV1 is regulated by autophagy (Fig. 2A). Lysosomal degradation inhibition was further verified by employing 3-methyladenine (3MA), an inhibitor which blocks autophagosome formation by inhibiting class III PI3-kinase (Wu et al., 2010). Treatment with 3MA during starvation did not lead to TRPV1 degradation, which also indicates that TRPV1 degradation under starvation is mediated by autophagy (Fig. 2B). However, protein degradation could alternatively be mediated by ubiquitination-mediated proteasomal degradation during starvation. To exclude the possibility that TRPV1 degradation was mediated by proteasomal lysis, the proteasomal inhibitor, MG132, was added to the cells during starvation (Fig. 2C). The level of TRPV1 in cells treated with MG132 during starvation was not stabilized to the level observed upon CLQ or 3MA treatment during starvation. This result suggests that the degradation of TRPV1 during starvation is specifically mediated by autophagy, not by ubiquitination-mediated protein degradation.
Fig. 1.
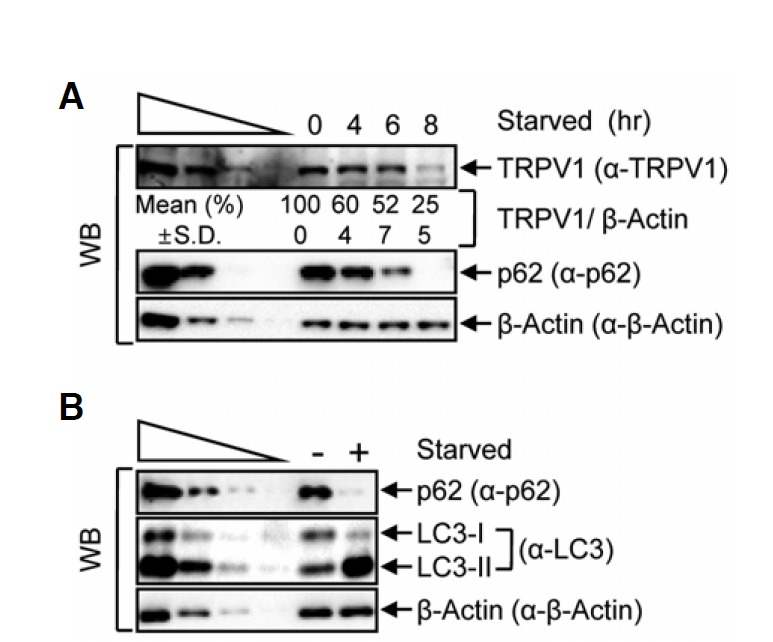
TRPV1 is degraded in starvation conditions. (A) HeLa cells were starved for 4, 6 or 8 h and cell lysates were analyzed by Western blotting (WB) with the specified antibodies, where β-actin served as a loading control. The four left lanes were three-fold dilutions of lysates and indicated that WB provides a semi-quantitative measurement of protein levels in the absence (−) or presence (+, 8 h) of starvation. (B) Levels of the cellular autophagy markers, p62 and LC3, in protein lysates from starved HeLa cell lysates were detected by WB. The normalized level without starvation (0 h) was set to 100 %. Mean values (%) and standard deviations (S.D.) were calculated from three independent experiments.
Fig. 2.
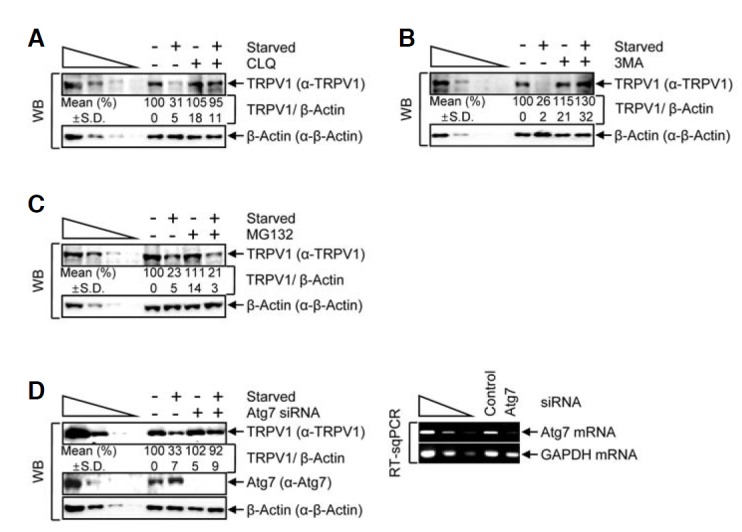
Starvation-mediated autophagy reduces the cellular level of TRPV1. (A–C) HeLa cells were treated with the following inhibitors: chloroquine (CLQ) (A), 3-methyladenine (3MA) (B) or a proteasome inhibitor, MG132 (C), in the presence or absence of starvation. Cell lysates were analyzed by WB. (D) HeLa cells were transiently transfected with either Atg7-targeting or negative control siRNA. After a 3-day incubation, cells were starved for 8 h. WB (left panel) and RT-sqPCR (right panel) were performed, where β-actin (WB) and GAPDH mRNA (RT-sqPCR) served as loading controls. The normalized level in DMSO treatment without starvation or control siRNA transfection without starvation was set to 100 %. Mean values (%) and standard deviations (S.D.) were calculated from three independent experiments.
To further test our hypothesis that TRPV1 degradation during starvation is mediated by autophagy, we used siRNA-mediated knockdown to silence the expression of Atg7, a protein which is essential for formation of the Atg12-Atg5 conjugate and maturation of the lysosomal membrane. We transiently transfected cells with either Atg7-targeting siRNA or a nonspecific control siRNA (Fig. 2D). Analysis of the levels of protein by WB (normalized to β-actin to control for variations in protein loading) demonstrated that Atg7-targeting siRNA reduced the level of Atg7 to less than 10% of that found in control siRNA-transfected cells (Fig. 2D, left panel). Additionally, Atg7 was also efficiently silenced on the mRNA level, where GAPDH mRNA served as an internal control (Fig. 2D, right panel). Upon siRNA-mediated silencing of Atg7, TRPV1 was not degraded during starvation. Altogether, these findings suggest that the level of TRPV1 is regulated by autophagy.
TRPV1 colocalizes with the autophagosomal marker, GFP-LC3
In autophagy, the target protein is expected to colocalize with LC3 while lysosomal degradation is inhibited. To test whether TRPV1 colocalizes with LC3, we transiently transfected HeLa cells with a plasmid expressing a GFP-tagged autophagy marker protein, GFP-LC3, and then incubated the transfected cells in starvation conditions (Figs. 3D–3F and 3J–3L) in the absence (Figs. 3A–3F) or the presence (Figs. 3G–3L) of CLQ. The immunofluorescence results showed that TRPV1 was mainly cytoplasmic, consistent with the previous observation that TRPV1 is localized to both the plasma membrane and ER membranes (Gallego-Sandin et al., 2009). When cells transiently expressing GFP-LC3 in normal conditions were not treated with CLQ, we did not observe any colocalization of GFP-LC3 with TRPV1 (Fig. 3C). When transiently transfected cells were starved, 16% of the GFP-LC3 transfected cells retained colocalization of TRPV1 with GFP-LC3 (Fig. 3F and right panel). In addition, 26% of the GFP-LC3 transfected cells with CLQ treatment without starvation also exhibited colocalization of GFP-LC3 with TRPV1 (Fig. 3I and right panel). Furthermore, when transiently transfected cells were starved and then treated with CLQ, 26% of the cells expressing GFP-LC3 exhibited colocalization with GFP-LC3 and TRPV1 (Fig. 3L and right panel). All these observations indicate that TRPV1 is targeted to degradation via autophagy, while TRPV1 colocalizes with GFP-LC3.
Fig. 3.
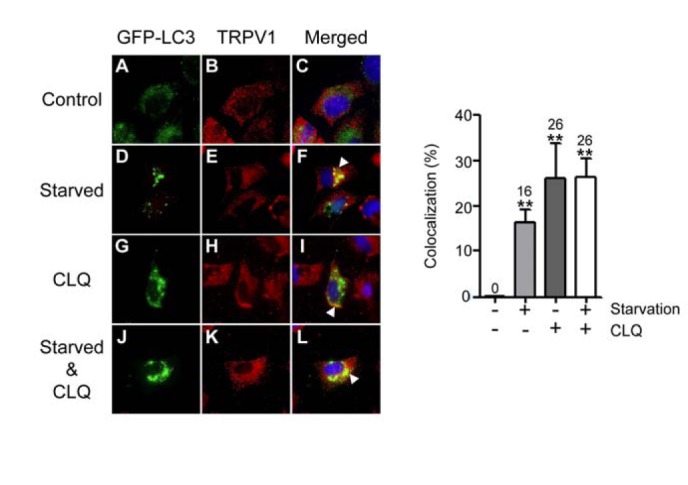
Starvation augments the colocalization of GFP-LC3 with TRPV1. (A–L) HeLa cells were transiently transfected with pGFP-LC3. After 2 days, cells were incubated in either the absence or presence of CLQ for 6 h in normal medium containing serum (A–C and G–I) or in HBBS to stimulate starvation (D–F and J–L). Control represents DMSO treatment instead of CLQ. Immunofluorescence was performed using rabbit anti-TRPV1 and Alexa Fluor 546 goat anti-rabbit IgG antibody for TRPV1 (red), GFP-LC3 (green) and DAPI for nuclei (blue). White arrow heads indicate the regions of colocalization of GFP-LC3 with TRPV1. (Right panel) Colocalization of GFPLC3 with TRPV1 in the left panel (C, F, I, and L) was quantified. Columns and error bars represent the mean values and standard deviations. The numbers above the error bars indicate the mean values. **P < 0.01 as determined by the unpaired Student’s t-test.
Cortisol reduces the level of cellular TRPV1 in an autophagy-dependent manner
Next, we investigated the level of TRPV1 in the context of a physiologically relevant inducer of autophagy. Many studies have reported that cortisol induces autophagy in osteocytes and T lymphocytes (Jia et al., 2011; Molitoris et al., 2011; Xia et al., 2010). To determine whether TRPV1 is degraded by cortisol-induced autophagy, we treated HeLa cells, which constitutively express functional cortisol receptors (Wallace and Cidlowski, 2001), with various concentrations of cortisol for 24 h (Fig. 4A). Surprisingly, the amount of TRPV1 when cells were treated with 1 μM of cortisol was reduced to approximately 27% of that in DMSO-treated control cells, while the level of an internal control, β-actin, remained unchanged. Furthermore, the level of p62 also gradually decreased depending on the concentration of cortisol (Fig. 4A). Interestingly, cortisol-mediated autophagy reduced the levels of both TRPV1 and p62 in a time-dependent manner. When cells were treated with 1 μM cortisol for various incubation periods, the relative amount of TRPV1 was gradually reduced, to approximately 25% that in the 0 h cortisol-treated cells (Fig. 4B). To confirm that cortisol is associated with autophagy, we observed the changes of LC3-I and -II by treatment of 1 μM cortisol for 24 h. As shown in Fig. 4C, cortisol treatment increased the level of LC3-II, but decreased the level of p62, similar to starvation-induced autophagy. These results imply that cortisol induces autophagy, and that this induction leads to the degradation of TRPV1.
Next, we examined whether cortisol reduces the level of cellular TRPV1 via an autophagy-dependent pathway. We employed the same approach that was used in the starvation-induced autophagy experiments. HeLa cells that had been incubated with 1 μM cortisol for 24 h were then treated with CLQ (Fig. 5A), 3MA (Fig. 5B) or MG132 (Fig. 5C). Similar to the starvation-induced autophagy results, the level of TRPV1 was stabilized when cells were treated with either CLQ or 3MA during the incubation with cortisol (Figs. 5A and 5B, respectively), while TRPV1 expression remained unchanged by CLQ or 3MA itself. However, MG132 did not have any effect on TRPV1 degradation in the context of cortisol treatment (Fig. 5C), suggesting that TRPV1 does not undergo proteasomal degradation in the presence of cortisol. We also employed siRNA-mediated silencing of Atg7 to verify that TRPV1 degradation was mediated by cortisol and an autophagy-dependent pathway. The level of Atg7 silencing was verified by WB and RT-sqPCR (Fig. 5D). As expected, these results indicated that the level of Atg7 was reduced to approximately 10% of that in control siRNA-transfected cells. Furthermore, cortisol treatment did not affect the siRNA-mediated knockdown of Atg7. Consistent with the results shown in Figs. 5A and 5B, siRNA-mediated silencing of Atg7 inhibited cortisol-induced degradation of TRPV1, compared with the significant degradation observed in control siRNA-transfected cells. These observations indicate that cortisol-induced degradation of TRPV1 is mediated by an autophagy-dependent pathway.
Fig. 5.

Cortisol induces autophagy and decreases the level of TRPV1. (A–C) HeLa cells were incubated in the presence or absence of 1 μM of cortisol for 24 h and then treated with CLQ (A), 3MA (B) or MG132 (C). WB was performed using the specified antibodies. (D) As in Fig. 2D, except that HeLa cells that were transiently transfected with siRNAs, Atg7-targeting or control were incubated with 1 μM of cortisol. Protein and gene expression levels were analyzed by WB (left panel) and RT-sqPCR (right panel), respectively. The normalized level in the absence of cortisol treatment was set to 100 %. Mean values (%) and standard deviations (S.D.) were calculated from three independent experiments.
To confirm the degradation of TRPV1 by cortisol-mediated autophagy, we examined the colocalizations of GFP-LC3 with TRPV1 upon cortisol treatment. Cells were transiently transfected with pGFP-LC3 and then treated either with or without cortisol for 24 h in the absence or presence of CLQ (Figs. 6A–6L). Immunofluorescence analysis revealed that 31% of GFP positive cells had colocalization of GFP-LC3 with TRPV1 during cortisol treatment (Fig. 6F and right panel). In addition, 35% and 42% of cells expressing GFP-LC3 had colocalization of GFP-LC3 with TRPV1 was observed with CLQ treatment in the absence or presence of cortisol, respectively (Figs. 6I and 6L, right panel). These findings provide insight into TRPV1 degradation via cortisol-induced autophagy.
Fig. 6.

Treatment with cortisol increases the extent of colocalization of GFP-LC3 and TRPV1. (A–L) As in Fig. 3, however, after transfection of pGFP-LC3, HeLa cells were incubated with 1 μM of cortisol for 24 h followed by CLQ or DMSO (control treatment) for 8 h. White arrow heads indicate regions of colocalization of GFP-LC3 with TRPV1. (Right panel) Colocalization of GFP-LC3 with TRPV1 in the left panel (C, F, I, and L) was quantified. Columns and error bars represent the mean values and standard deviations. The numbers above the error bars indicate the mean values. **P < 0.01 as determined by the unpaired Student’s t-test.
DISCUSSION
The main question addressed in this study was whether autophagy is involved in TRPV1 degradation. The major findings of this study include: (i) endogenous TRPV1 is degraded by starvation via autophagy (Figs. 1–3), (ii) cortisol enhances autophagy, thus resulting in TRPV1 degradation (Figs. 4 and 5), and (iii) cortisol induces colocalization of GFP-LC3 with TRPV1 (Fig. 6).
TRPV1 is found in the central nervous system, GI tract, keratinocytes, and sensory neurons. TRPV1 has multiple functions, including thermo-reception, and these functions may depend on the distribution of TRPV1 (Mezey et al., 2000; Szallasi and Blumberg, 2007). The functions of TRPV1 are regulated by phosphorylation, desensitization and phosphatidylinositol 4,5-bisphosphate (PIP2), a molecule that is involved not only in thermal pain but also in some types of pathological pain, such as inflammatory pain or neuropathic pain (Szallasi and Blumberg, 2007). In addition to the signaling pathways which regulate TRPV1 (phosphorylation and desensitization, etc.), the expression level of TRPV1 is also considered to play a pivotal role in pain hypersensitivity under chronic pain conditions. Current models suggest that the levels of TRPV1 increase in neuropathic pain conditions (Biggs et al., 2007; Fukuoka et al., 2002; Kanai et al., 2005) and in uninjured or spared neurons after nerve ligation/transection; in contrast, TRPV1 levels are thought to decrease in injured neurons (Hudson et al., 2001; Kim et al., 2008; Michael and Priestley, 1999). Thus, regulation of the protein levels of TRPV1 has been suggested to be a major determinant of TRPV1 function in pathological conditions. Also, sensitization of spinal TRPV1 by its upregulation was involved in the development and/or maintenance of mechanical allodynia (Kanai et al., 2005; Kim et al., 2012). However, no evidence for a suitable mechanism of TRPV1 recycling has yet been obtained, although changes in the levels of TRPV1 in nerves or the spinal cord have been observed after nerve injury, or in various pain models. In the present study, we found that TRPV1 is regulated via an autophagy-dependent pathway, and that this pathway is enhanced by cortisol. To our knowledge, this is the first evidence concerning TRPV1 degradation and recycling. Recent evidence has demonstrated that autophagy, not ubiquitination, can also regulate the cellular levels of two wild-type connexins, CX50 and CX43, whose functions can be rapidly modulated by degradation (Fong et al., 2012; Lichtenstein et al., 2011). We propose that TRPV1 is regulated via an autophagy-dependent pathway, in a manner similar to the involvement of autophagy in the proteolysis of connexins.
Recently, dexamethasone has been shown to increase the expression of a gene encoding a stress response protein (Dig2/RTP801/REDD1), which is known to contribute to the induction of autophagy by inhibiting mTOR signaling (Molitoris et al., 2011). Furthermore, glucocorticoids have been shown to induce autophagy in osteocytes (Xia et al., 2010). Consistent with these previous studies, we observed that cortisol induced autophagy in HeLa cells. We revealed that the mechanism of TRPV1 degradation involves autophagy under cortisol exposure conditions. Our results suggest a plausible recycling mechanism by which autophagy contributes to the degradation of TRPV1, thus regulating its expression level. The observation that TRPV1 degradation is also mediated by cortisol-induced autophagy suggests that autophagy could contribute to TRPV1-mediated pain responses under pathologic pain conditions.
Acknowledgments
We are grateful to Yong-Keun Jung for the pGFP-LC3 plasmid. This research was supported by the research fund of Hanyang University (HY-2011-N to J.H.) and the Basic Science Research Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (2012R1A1A2009458 to S.J.J.).
REFERENCES
- Ahn S., Kim J., Hwang J. CK2-mediated TEL2 phosphorylation augments nonsense-mediated mRNA decay (NMD) by increase of SMG1 stability. Biochim. Biophys Acta. 2013;1829:1047–1055. doi: 10.1016/j.bbagrm.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Bejarano E., Girao H., Yuste A., Patel B., Marques C., Spray D.C., Pereira P., Cuervo A.M. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol. Biol Cell. 2012;23:2156–2169. doi: 10.1091/mbc.E11-10-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs J.E., Yates J.M., Loescher A.R., Clayton N.M., Boissonade F.M., Robinson P.P. Changes in vanilloid receptor 1 (TRPV1) expression following lingual nerve injury. Eur. J Pain. 2007;11:192–201. doi: 10.1016/j.ejpain.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Caterina M.J., Schumacher M.A., Tominaga M., Rosen T.A., Levine J.D., Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Clapham D.E. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Fong J.T., Kells R.M., Gumpert A.M., Marzillier J.Y., Davidson M.W., Falk M.M. Internalized gap junctions are degraded by autophagy. Autophagy. 2012;8:794–811. doi: 10.4161/auto.19390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka T., Tokunaga A., Tachibana T., Dai Y., Yamanaka H., Noguchi K. VR1, but not P2X(3), increases in the spared L4 DRG in rats with L5 spinal nerve ligation. Pain. 2002;99:111–120. doi: 10.1016/s0304-3959(02)00067-2. [DOI] [PubMed] [Google Scholar]
- Gallego-Sandin S., Rodriguez-Garcia A., Alonso M.T., Garcia-Sancho J. The endoplasmic reticulum of dorsal root ganglion neurons contains functional TRPV1 channels. J. Biol. Chem. 2009;284:32591–32601. doi: 10.1074/jbc.M109.019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami C., Hucho T. TRPV1 expression-dependent initiation and regulation of filopodia. J. Neurochem. 2007;103:1319–1333. doi: 10.1111/j.1471-4159.2007.04846.x. [DOI] [PubMed] [Google Scholar]
- Hudson L.J., Bevan S., Wotherspoon G., Gentry C., Fox A., Winter J. VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur. J. Neurosci. 2001;13:2105–2114. doi: 10.1046/j.0953-816x.2001.01591.x. [DOI] [PubMed] [Google Scholar]
- Hwang S.W., Cho H., Kwak J., Lee S.Y., Kang C.J., Jung J., Cho S., Min K.H., Suh Y.G., Kim D., et al. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc. Natl. Acad. Sci USA. 2000;97:6155–6160. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J., Yao W., Guan M., Dai W., Shahnazari M., Kar R., Bonewald L., Jiang J.X., Lane N.E. Glucocorticoid dose determines osteocyte cell fate. FASEB J. 2011;25:3366–3376. doi: 10.1096/fj.11-182519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J., Shin J.S., Lee S.Y., Hwang S.W., Koo J., Cho H., Oh U. Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J. Biol. Chem. 2004;279:7048–7054. doi: 10.1074/jbc.M311448200. [DOI] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y., Nakazato E., Fujiuchi A., Hara T., Imai A. Involvement of an increased spinal TRPV1 sensitization through its up-regulation in mechanical allodynia of CCI rats. Neuropharmacology. 2005;49:977–984. doi: 10.1016/j.neuropharm.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Kedei N., Szabo T., Lile J.D., Treanor J.J., Olah Z., Iadarola M.J., Blumberg P.M. Analysis of the native quarternary structure of vanilloid receptor 1. J. Biol. Chem. 2001;276:28613–28619. doi: 10.1074/jbc.M103272200. [DOI] [PubMed] [Google Scholar]
- Kim H.Y., Park C.K., Cho I.H., Jung S.J., Kim J.S., Oh S.B. Differential changes in TRPV1 expression after trigeminal sensory nerve injury. J Pain. 2008;9:280–288. doi: 10.1016/j.jpain.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Kim Y.H., Back S.K., Davies A.J., Jeong H., Jo H.J., Chung G., Na H.S., Bae Y.C., Kim S.J., Kim J.S., et al. TRPV1 in GABAergic interneurons mediates neuropathic mechanical allodynia and disinhibition of the nociceptive circuitry in the spinal cord. Neuron. 2012;74:640–647. doi: 10.1016/j.neuron.2012.02.039. [DOI] [PubMed] [Google Scholar]
- Lichtenstein A., Minogue P.J., Beyer E.C., Berthoud V.M. Autophagy: a pathway that contributes to connexin degradation. J. Cell Sci. 2011;124:910–920. doi: 10.1242/jcs.073072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Wang P., Song W., Sun X. Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J. 2009;23:3383–3392. doi: 10.1096/fj.09-134296. [DOI] [PubMed] [Google Scholar]
- Mathew R., Karp C.M., Beaudoin B., Vuong N., Chen G., Chen H.Y., Bray K., Reddy A., Bhanot G., Gelinas C., et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezey E., Toth Z.E., Cortright D.N., Arzubi M.K., Krause J.E., Elde R., Guo A., Blumberg P.M., Szallasi A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc. Natl. Acad. Sci USA. 2000;97:3655–3660. doi: 10.1073/pnas.060496197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael G.J., Priestley J.V. Differential expression of the mRNA for the vanilloid receptor subtype 1 in cells of the adult rat dorsal root and nodose ganglia and its downregulation by axotomy. J. Neurosci. 1999;19:1844–1854. doi: 10.1523/JNEUROSCI.19-05-01844.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Molitoris J.K., McColl K.S., Swerdlow S., Matsuyama M., Lam M., Finkel T.H., Matsuyama S., Distelhorst C.W. Glucocorticoid elevation of dexamethasone-induced gene 2 (Dig2/RTP801/REDD1) protein mediates autophagy in lymphocytes. J. Biol. Chem. 2011;286:30181–30189. doi: 10.1074/jbc.M111.245423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo J.O., Jang M.H., Kwon Y.K., Lee H.J., Jun J.I., Woo H.N., Cho D.H., Choi B., Lee H., Kim J.H., et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J. Biol. Chem. 2005;280:20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- Ross R.A. Anandamide and vanilloid TRPV1 receptors. Br. J. Pharmacol. 2003;140:790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Fraldi A., Medina D.L., Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T., Tominaga M., Katsuya H., Mizumura K. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. J. Neurophysiol. 2002;88:544–548. doi: 10.1152/jn.2002.88.1.544. [DOI] [PubMed] [Google Scholar]
- Szallasi A., Blumberg P.M. In: Complex regulation of TRPV1 by vanilloids; in TRP ion channel function in sensory transduction and cellular signaling cascades. Liedtke W.B., Heller S., editors. Boca Raton (FL): 2007. [PubMed] [Google Scholar]
- Takahata K., Chen X., Monobe K., Tada M. Growth inhibition of capsaicin on HeLa cells is not mediated by intra-cellular calcium mobilization. Life Sci. 1999;64:PL165–171. doi: 10.1016/s0024-3205(99)00046-6. [DOI] [PubMed] [Google Scholar]
- Wallace A.D., Cidlowski J.A. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J. Biol. Chem. 2001;276:42714–42721. doi: 10.1074/jbc.M106033200. [DOI] [PubMed] [Google Scholar]
- Wu Y.T., Tan H.L., Shui G., Bauvy C., Huang Q., Wenk M.R., Ong C.N., Codogno P., Shen H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010;285:10850–10861. doi: 10.1074/jbc.M109.080796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., Kar R., Gluhak-Heinrich J., Yao W., Lane N.E., Bonewald L.F., Biswas S.K., Lo W.K., Jiang J.X. Glucocorticoid-induced autophagy in osteocytes. J. Bone Miner. Res. 2010;25:2479–2488. doi: 10.1002/jbmr.160. [DOI] [PMC free article] [PubMed] [Google Scholar]