

Abstract
Pulmonary arterial hypertension (PAH) is a complex disease characterized by elevated pulmonary arterial pressure, pulmonary vascular remodelling and occlusive pulmonary vascular lesions, leading to right heart failure. Evidence from recent epidemiological studies suggests the influence of gender on the development of PAH with an approximate female to male ratio of 4:1, depending on the underlying disease pathology. Overall, the therapeutic strategy for PAH remains suboptimal with poor survival rates observed in both genders. Endogenous sex hormones, in particular 17β oestradiol and its metabolites, have been implicated in the development of the disease; however, the influence of sex hormones on the underlying pathobiology remains controversial. Further understanding of the influence of sex hormones on the normal and diseased pulmonary circulation will be critical to our understanding the pathology of PAH and future therapeutic strategies. In this review, we will discuss the influence of sex hormones on the development of PAH and address recent controversies.
Linked Articles
This article is part of a themed section on Biological Sex and Cardiovascular Pharmacology. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2014.171.issue-3
Keywords: pulmonary arterial hypertension, sex hormones, oestrogen, testosterone, serotonin, vascular remodelling
Pulmonary arterial hypertension (PAH) is a progressive disease leading to right heart failure. Recent epidemiological data reports an increased incidence of PAH among females compared to males. For example, in the UK/Ireland and the USA, the percentage of female patients is 70 and 80% respectively (Badesch et al., 2010; Ling et al., 2012). There is mounting evidence to suggest that oestrogen and its metabolites may influence the pathogenesis of PAH. Here, we will examine the pathology, current therapies and the basis for gender differences in PAH, considering evidence gathered from both patient data and animal studies.
Pulmonary arterial hypertension
PAH is defined by vascular remodelling and complex vascular lesion formation arising from the accelerated proliferation of pulmonary endothelial, smooth muscle and fibroblast cells (Rabinovitch, 2008). Clinically, the disease is defined as a mean pulmonary artery pressure of >25 mmHg at rest or >30 mmHg during exercise. Symptoms are often non-specific, including fatigue, exertional dyspnoea, oedema and syncope and as a result diagnosis is frequently delayed until the disease is well established.
The main genetic defect associated with PAH is a mutation in the gene encoding bone morphogenetic protein receptor 2 (BMPR2). Germ line mutations in BMPR2 were originally identified in patients with heritable PAH (HPAH; Deng et al., 2000; Lane et al., 2000). In these families, the disease segregates in an autosomal dominant fashion, with markedly reduced penetrance, approximately 20 to 30%. In addition, up to 25% of patients with apparently sporadic idiopathic PAH (IPAH) have been found to harbour similar mutations (Thomson et al., 2000). A proportion of these mutation carriers are examples of HPAH where the condition has not manifested in relatives due to low penetrance, while others are examples of de novo mutation (Newman et al., 2001). The low penetrance of the disease among BMPR2 mutation carriers suggests other factors are important in the onset of clinical PAH and that at least one more genetic or environmental ‘hit’, in addition to a mutation in BMPR2, underlies established PAH. Indeed, a multitude of factors have been implicated in the pathobiology of the condition including endothelin-1 (ET-1; Giaid et al., 1993), phosphodiesterases (Murray et al., 2011), oestrogens (White et al., 2011a) and serotonin (Maclean and Dempsie, 2010). Heterozygous mutations in the activin receptor-like kinase 1 and endoglin have also been associated with PAH (Trembath et al., 2001; Chaouat et al., 2004). The plethora of agents highlighted as potential mediators of PAH is consistent with the complex and multifactorial nature of the disease pathology.
Currently pulmonary hypertension (PH) is classified by the World Health Organization (WHO) into five main categories subject to underlying causes (Table 1). PAH, the focus of this review is categorized as group 1 PH and subdivided into IPAH, HPAH, drug- or toxin-induced PAH or PAH associated with diseases such as connective tissue diseases, HIV infection, portal hypertension, congenital heart disease, schistosomiasis and chronic haemolytic anaemia (APAH). Despite the wide spectra of initiating factors, the disease arises as a result of pulmonary vasoconstriction, vascular remodelling and thrombosis (Rabinovitch, 2008).
Table 1.
Dana Point classification of pulmonary hypertension
| Dana Point clinical classification of pulmonary hypertension updated 2008 | |
|---|---|
| 1. PAH | |
| 1.1 Idiopathic PAH | |
| 1.2 Heritable | 1.2.1 BMPR2 |
| 1.2.2 ALK1, endoglin (with or without hereditary haemorrhagic telangiectasia) | |
| 1.2.3 Unknown | |
| 1.3 Drug and toxin induced | |
| 1.4 Associated with | 1.4.1 Connective tissue diseases |
| 1.4.2 HIV infection | |
| 1.4.3 Portal hypertension | |
| 1.4.4 Congenital heart diseases | |
| 1.4.5 Schistosomiasis | |
| 1.4.6 Chronic hemolytic anaemia | |
| 1.5 Persistent pulmonary hypertension of the newborn | |
| 1′ Pulmonary veno-occlusive disease (PVOD) and/or pulmonary capillary haemangiomatosis (PCH) | |
| 2. Pulmonary hypertension owing to left heart disease | |
| 3. Pulmonary hypertension owing to lung diseases and/or hypoxia | |
| 4. Chronic thromboembolic pulmonary hypertension (CTEPH) | |
| 5. Pulmonary hypertension with unclear multifactorial mechanisms | |
Table summarizing the updated 2008 Dana Point classification of pulmonary hypertension with main focus on category 1 PAH. Adapted from Simonneau et al. (2009).
Maintaining a vasodilated pulmonary circulation is essential for oxygenation of the blood. The pulmonary arterial endothelium is a source of vasodilators such as NO and prostacyclins. In PAH there is an imbalance in the production of vasodilatory and vasoactive agents in the pulmonary artery ultimately favouring vasoconstriction. Elevated levels of ET-1, a potent pulmonary vasoconstrictor and a decrease in vasodilators such as NO have been reported in patients with the disease (Giaid and Saleh, 1995; Cacoub et al., 1997). An imbalance in prostacyclin and thromboxane A2 (TXA2) levels in the favour of TXA2 also leads to vasoconstriction, thrombosis and platelet activation (Christman et al., 1992; Tuder et al., 1999).
Furthermore, in normal circumstances the diameter of the pulmonary artery wall is maintained by a balance between apoptosis and proliferation of the cells of the vessel wall. However in PAH proliferation is dominant, resulting in the muscularization of previously non-muscular pulmonary arterioles and a thickening of the medial layer of the vessel wall resulting in a narrowing of the lumen and the loss of small pre-capillary arteries (Rabinovitch, 2008). Ultimately, this vascular remodelling results in the formation of vascular lesions which eventually occlude the vessel. Plexiform lesions, comprised of a mass of disorganized vessels with proliferating endothelial cells (main component), smooth muscle cells, myofibroblasts and macrophages, also occur in PAH. These are angioproliferative lesions which arise from pre-existing pulmonary arteries and are often symptomatic of end-stage disease (Cool et al., 1999).
In summary, the combination of vasoconstriction and arterial remodelling that occurs during PAH results in vascular pruning (the obliteration of small resistance arteries) and increases pulmonary vascular resistance and pressure. Thus, the disease pathology results in an excessive burden on the right ventricle due to the increased workload required to compensate for the elevated downstream pressure and eventually results in right-sided heart failure. The extent of right heart dysfunction is often used as predictor of prognosis in PAH.
Epidemiology, sex differences and survival
As evident by recent epidemiological registries, the incidence of PAH varies from 1.1, 2.0 and 2.4 per million of the adult population per year in the UK/Ireland, USA and France respectively (Humbert et al., 2006; Frost et al., 2011; Ling et al., 2012). The reported incidence of IPAH in the USA and French registries of PAH is 46.2 and 39.2% respectively with APAH incidence also showing similarity at 50.7 and 52.7% respectively. The data documented from the UK and Ireland's PAH registries highlights different subcategory demographics with IPAH incidence at 92.9% and APAH at 1.7% (Ling et al., 2012).
In the REVEAL registry, it was found that 83.1% of patients studied fell into the age category group 19–64 years with 12.8 and 4.1% in the 65–74 years and 75+ years group respectively (Badesch et al., 2010). In this instance, the mean age of diagnosis appears similar between the sexes, 46 ± 19 years for men and 48 ± 17 years for women. However, it was documented that men present with significantly higher mean pulmonary artery pressure than women at baseline (Badesch et al., 2010).
There is a general consensus that female gender is a risk factor for PAH, with recent studies showing a female to male ratio of 4.3:1 among the total PAH group (Walker et al., 2006) and 4.1:1 in the IPAH subcategory (Badesch et al., 2010). This female prevalence is much stronger compared to a previous report from the NIH registry where the female to male ratio was reported as 1.7:1 (Rich et al., 1987). However, this ratio represented the total PAH population studied and was not further analysed into subcategories.
In some cohorts of patients, there has been considerable improvement in survival over the past two decades since the establishment of the NIH registry. This is likely to be due to changes in treatments and improved patient support strategies (Benza et al., 2012). However, the current 3-year survival for patients with PAH managed with state-of-the-art multiple drug therapy remains troubling at approximately 58% (Humbert et al., 2010a).
Although the incidence of PAH is higher in women than in men, the estimated 5-year survival rate from diagnosis in the REVEAL registry is ∼52% in men, compared to 62% in women (Shapiro et al., 2012). This suggests that either women respond better to treatment options or that female sex hormones are mediating protective effects. Interestingly, when age was accounted for, survival was only different between males and females after the age of 60 (Shapiro et al., 2012). This again highlights the importance of considering gender and age differences in both the diagnosis of PAH and importantly, also in the treatment.
Current therapies for PAH
The current therapeutic strategy for PAH is suboptimal. The majority of therapies alleviate the symptoms and subsequently improve the quality of life of patients, but fail to address the underlying disease pathology. In order to make progress in the management and treatment of the condition, therapies that drastically regenerate the lost distal pulmonary arteries and prevent the extensive pulmonary vascular remodelling observed in PAH are urgently required. At present, patients will inevitably succumb to the disease, or are considered for lung transplantation. Although an immense amount of research effort has advanced our understanding of this once poorly characterized disease, the exact mechanisms that underlie the disease pathology are still being clarified. PAH is multifactorial and complex in nature and individual variability may also suggest personalized treatment options in the future. Furthermore, given the increased frequency of PAH in females, therapies specifically targeted to women may be a potential therapeutic strategy.
As PAH is a multifactorial process with varying aetiologies, treatment options therefore depend on the PAH classification. The majority of patients receive supportive therapies including diuretics, anticoagulants, oxygen and digoxin (Galie et al., 2009b). Where possible therapies directed at the underlying cause of the PH are utilized. However, in WHO Group 1 PAH, the underlying cause of the disease is typically unknown, and therefore more advanced therapies which attempt to target the PAH are considered. If patients react positively to a vasoreactivity test (requiring right heart catheterization) calcium channel blockers are administered, and a certain percentage of patients will respond. The most commonly used are nifedipine and diltiazem (Galie et al., 2009b).
Prostacyclin is a potent vasodilator and inhibitor of platelet aggregation. Dysregulation of prostacyclin metabolic pathways have been reported in patients with PAH (Tuder et al., 1999), and as such, several prostacyclin analogues including epoprostenol, iloprost and treprostinil are used clinically in the treatment of PAH (Galie et al., 2009b). Although these drugs improve symptoms and exercise capacity, many require administration by intravenous infusion due to the short half-life of prostacyclin. There are serious adverse effects related to this delivery system, such as local site infection, catheter obstruction and in severe instances sepsis. Treprostinil is currently available in a subcutaneously administered form, while oral and aerosol forms of iloprost and treprostinil have now been developed (Olschewski, 2009; Channick et al., 2012; Tapson et al., 2012). Inhaled drugs, which target PAH, are an attractive possibility as this would potentially make them selective for the pulmonary circulation.
ET-1 is abundantly over-expressed in endothelial cells from patients with PH (Giaid et al., 1993) and stimulates proliferation of pulmonary artery smooth muscle cells (PASMC; Wort et al., 2001) and vasoconstriction. ET-1 receptor antagonists, bosentan, sitaxsentan and ambrisentan are currently approved for the treatment of PAH, preventing the aberrant activity of ET-1 observed in patients (Oudiz et al., 2009; Dwyer and Kilpatrick, 2011; Rubin, 2012). Precautions must also be taken when administering this class of compounds as they have been reported to display marked liver toxicity, and as a result, regular liver function test are required (McGoon et al., 2009).
Finally, inhibitors of PDE5 have also been licensed for the treatment of PAH, including sildenifil and tadalafil (Galie et al., 2009a). PDE5 inhibitors prevent the breakdown of cGMP and mediate vasorelaxation as well as having antiproliferative properties.
In instances where response to a single therapy is inadequate, combination therapy, utilizing more than one PAH-specific class of drug has become standard (Chen et al., 2009; Galie et al., 2009c). There are many questions regarding combination therapy including which combinations to use and whether a combination of drugs should be used initially or sequentially depending on response to the first drug.
Despite PAH being more frequently observed in females, a personalized medicine approach is not yet possible. Recently, it has been reported that women with PAH exhibit a greater clinical benefit from ET-1 receptor antagonists than men, as measured by a 44.1 metre improvement in the 6 min walk distance compared to 16.7 metre improvement in men (Gabler et al., 2012). As men often present with significantly higher mean pulmonary arterial pressure than women at baseline (Shapiro et al., 2012), this could be the reason as to why women respond better to treatment as their phenotype is less severe. There may also be differing circulating ET-1 levels between women and men with men having higher ET-1 compared to women (Miyauchi et al., 1992; Polderman et al., 1993). This heterogeneity in treatment response may reflect pathophysiological differences between sexes or distinct disease phenotypes.
PAH in females
Some studies have linked the use of the contraceptive pill and hormone replacement therapy to the increased incidence of PAH in females (Masi, 1976; Thorne et al., 2006; Sweeney and Voelkel, 2009). However, a clear link between hormonal-based therapies and PAH has not been established. Despite this, the increased frequency of PAH in women has given rise to the hypothesis that oestrogen may play a role in disease development and progression. This idea may not only suggest why more women appear to be affected by PAH but may also allow us to propose possible mechanisms involved in disease development.
Other risk factors associated with being female such as increased autoimmunity may also play a role in the development of PAH. For instance, PAH is a devastating complication in patients with the autoimmune diseases such as systemic sclerosis and systemic lupus erythematosus (Ruiz-Irastorza et al., 2012). These conditions are well documented to be more prevalent in females (Rubtsov et al., 2010). Moreover, female gender and autoimmune hepatitis are also associated with an increased risk for portopulmonary hypertension, a subcategory of PAH (Kawut et al., 2008).
Most studies, registries or clinical trials will report the effects of treatment on PAH indices and patient survival but not necessarily divide this into female and male cohorts. Retrospective analysis of this data would provide valuable information. However, some studies have reported gender differences in treatment response and in doing so these findings may eventually challenge the therapeutic decision made by clinicians. Further analysis of the longitudinal REVEAL study shows that women, in the particular cohort studied, have improved survival rates compared to men (Shapiro et al., 2012). However, this analysis does not declare which, if any, treatments were better than others in causing this improvement. The underlying factors responsible for this remarkable difference are unclear. The study does suggest that in certain cohorts there may be male/female differences in the pathology of PAH.
Although the incidence of PAH is greater in female patients, numerous studies have demonstrated better outcome in female animals using classical PAH models such as chronic hypoxia and monocrotaline-induced PAH (Rabinovitch et al., 1981; Umar et al., 2011). This coupled with the fact that oestrogen is a potent pulmonary vasodilator (Lahm et al., 2008) and that removal of female sex hormones by ovariectomy induces a more severe PAH phenotype (Ahn et al., 2003; Nadadur et al., 2012) has been coined the ‘oestrogen paradox’ of PAH (Umar et al., 2012). Research into why PAH occur more frequently in women has therefore been hampered by lack of appropriate models.
However, more recently, transgenic mouse models have been characterized that demonstrate female susceptibility. These include mice over-expressing the human serotonin transporter (SERT+ mice; White et al., 2011a), those that over-express the calcium-binding protein S100A4/Mts1 (Dempsie et al., 2011) and dexfenfluramine-treated mice (Dempsie et al., 2009). What all these models have in common is overactivity of the serotonin system. Indeed, synthesis of serotonin has also been associated with the development of the SUGEN/hypoxic model of PAH. Inhibition of the VEGF receptor by SUGEN combined with hypoxia (Su-Hx) generates a preclinical model of PAH that recapitulates human PAH more closely than previously characterized models (Gomez-Arroyo et al., 2012). This model is associated with increased expression of tryptophan hydroxylase 1 (Tph1), the rate-limiting enzyme in serotonin synthesis (Ciuclan et al., 2011, 2013). It has also recently been reported that unlike classical PAH models, female Su-Hx rats develop a more severe disease phenotype than males suggesting oestrogen may be a risk factor (Tofovic et al., 2012). However, this difference is not observed in the Su-Hx mouse (White et al., 2012). Models which demonstrate the development of PAH in females rather than males are already yielding important information and may give rise to novel therapeutic strategies (Dempsie et al., 2011; White et al., 2011a,b2011b).
Role of oestrogens in PAH
Oestrogens are steroid hormones, which in addition to their key role in the development of secondary sex characteristics, also play a role in memory, bone density and have been show to have cardiovascular effects. The three major oestrogens are oestrone, oestradiol and oestriol. Aromatase (CYP19A1), a member of the cytochrome P450 superfamily, synthesizes oestrogens through the aromatization of androgens, specifically testosterone and androstenedione (Figure 1). Oestrogen synthesis occurs mainly in the ovarian follicles and corpus luteum from where oestrogen is released into the circulation. Synthesis also occurs to a lesser extent in non-glandular tissues such as adipose tissue, liver, skin, muscle and brain (Simpson et al., 2005). MacLean et al., have recently demonstrated that CYP19A1 is expressed in the medial layer of pulmonary arteries from PAH patients, providing evidence of de novo synthesis (unpublished).
Figure 1.

Oestrogen metabolism in PAH. The circulating C19 precursors, testosterone and androstenedione are converted by CYP19A1 to 17β oestradiol and oestrone respectively. These metabolites then undergo hydroxylation at the C2, C4 or C16 positions by activity of CYP enzymes, including CYP1B1. While hydroxylation at C2 produces anti-proliferative metabolites, C16 hydroxylation induces proliferative metabolites. The CYPs are therefore key determinants of metabolite formation and can perturb the pathway towards an anti- or pro-proliferative state. Hydroxylated oestrogens can be further metabolized to the methoxyestrogens, which may also influence PASMC proliferation.
17β oestradiol, the main circulating premenopausal hormone, mediates protective effects in models of PAH. For example, 17β oestradiol attenuates the development of a PAH phenotype in both the chronic hypoxic model of PAH (Xu et al., 2010; Lahm et al., 2012) and in the inflammatory monocrotaline model of the disease (Farhat et al., 1993). These studies all used male rats; however, as females have much higher circulating levels of oestrogen with cyclical fluctuations, comparative studies using females would be of interest. However, there is evidence that oestrogen may also attenuate the development of PAH in female animals. These studies used monocrotaline or the chronic hypoxic model, which as discussed previously are more effective at inducing PAH in males (Resta et al., 2001; Nadadur et al., 2012; Yuan et al., 2012).
In contrast, there is evidence that oestrogen is a causative factor in novel models that demonstrate the development of PAH in females but not males. For example, the PAH disease phenotype in the SERT+ female mice can be prevented by ovariectomy and recovered by chronic administration of 17β oestradiol, indicating an essential function of this hormone in the development of a PAH phenotype (White et al., 2011a). These findings suggest that serotonin is facilitating and amplifying the effects of oestrogen in the pulmonary circulation. Indeed, it has been shown that oestrogen increases expression of Tph1, SERT and the 5-hydroxytryptamine 1B receptor in human PASMCs (White et al., 2011a).
Given that right ventricular dysfunction is the best predictor of prognosis in PAH and females are afflicted less severely than males with regard to a failing right ventricle, it is conceivable that oestrogen is cardioprotective. Indeed, in an animal model of right ventricular failure, oestrogen therapy is shown to completely reverse PH-induced right ventricular remodelling associated with right ventricular dysfunction (Lahm et al., 2012; Nadadur et al., 2012). Moreover, higher right ventricular ejection fraction and survival rates in females correlate with oestradiol levels (Kawut et al., 2008; Ventetuolo et al. 2011). Cardioprotective properties of oestradiol perhaps contribute to improved survival in female patients with PAH despite the higher female prevalence in the disease. In support of this, female Su-Hx rats display a significantly smaller increase in right ventricular mass compared to males even although they develop a more severe disease phenotype (Tofovic et al., 2012). Thus, the potential diverging effects of oestrogens in the cardiopulmonary unit merit further investigation before we consider treatment options. However, these findings highlight the importance of incorporating gender differences into basic research and may go some way to explaining the ‘oestrogen paradox’ whereby exogenous oestrogens appear protective in male animals.
Currently, two promoter single nucleotide polymorphisms in the gene coding for CYP19A1 that result in elevated oestrogen production have been associated with an increased risk of portopulmonary hypertension (Roberts et al., 2009). Furthermore, preliminary unpublished studies from MacLean et al., indicate that CYP19A1 is present in both smooth muscle cells and endothelial cells of the pulmonary vasculature. Locally synthesized oestrogen could therefore act in a paracrine fashion on smooth muscle cells and exert a more powerful influence than circulating oestrogens. Furthermore, expression of CYP19A1 is regulated, in part, by tissue-specific promoters and by alternative splicing mechanisms (Simpson et al., 2005). Further investigation into the specific mechanisms that regulate CYP19A1 expression in the lung will contribute to improved understanding of the role of CYP19A1 and oestrogen in the lung under both physiological and pathophysiological conditions.
Oestrogen receptors
Oestrogen activates three oestrogen receptors (ER), ERα, ERβ and G protein-coupled oestrogen receptor (GPER). ERα and ERβ mainly mediate the genomic effects of oestrogen; however, some non-genomic effects have been reported (Lahm et al., 2008). GPER signals rapidly through non-genomic mechanisms (Filardo et al., 2000). All three receptor types are present in the human pulmonary artery. In the hypoxic rat model, the beneficial effects of oestrogen on the pulmonary circulation are mediated by genomic ERs, ERα and ERβ (Lahm et al., 2012). Additionally, the ERβ agonist, diarylpropionitrile, has been shown to rescue severe monocrotaline-induced PAH to a similar extent as 17β oestradiol (Umar et al., 2011).
Transcript levels of ESR1, the gene encoding ERα are increased in both male and female PAH patients (Rajkumar et al., 2010), raising the novel paradigm of ER-positive PAH. This would certainly impact the way we consider individual patient treatment options. Recently, evidence has emerged that ERα has an evolutionary conserved binding site on the BMPR2 promoter and can thereby reduce its expression (Austin et al., 2009), giving evidence for a functional role of ERα in PAH. Furthermore, BMPR2 expression was found to be decreased in both lymphocytes from female patients and in whole lungs from female mice compared with their male counterparts. It is plausible that the increased endogenous oestrogen levels in females compared to males results in the decrease in BMPR2 expression observed and contributes to the increased incidence of PAH in females.
While evidence suggests that ERα may mediate the detrimental effects of oestrogens in PAH development, clinical data regarding ERβ in PAH is lacking. ERβ agonists have been shown to be protective in animal models of PAH (Umar et al., 2011). The phytoestrogen, genistein has much higher affinity for ERβ than ERα and reverses severe PAH in rats (Matori et al. 2012). However, lung dysfunction has been reported in ERβ knockout mice. Loss of ERβ in female mice leads to abnormal lung structures and a hypoxic environment, which ultimately contributes to ventricular hypertrophy (Morani et al. 2006).
Similarly, changes in the receptor expression profile of GPER in the pathogenesis of PAH are yet to be investigated. Detailed studies to determine the precise function of all three ERs in the healthy lung compared to PAH may yield vital information on the contribution of oestrogen signalling in the disease process. Furthermore, teasing out differences in changes in the receptor expression profile between males and females may prove useful in identifying underlying mechanisms responsible for the female predominance in PAH patients and contribute to more targeted therapies for the condition.
Oestrogen metabolites and PAH
Oestrogen metabolites and alterations in oestrogen metabolism have also been implicated in the pathobiology of PAH. 17β oestradiol is readily metabolized by cytochrome P450 (CYP) enzymes, which are abundantly expressed in the lung. CYPs catalyse the oxidation of 17β oestradiol, in the presence of NADPH and oxygen, to the 2-, 4- and 16α hydroxyestradiols. 17β oestradiol can also be inter-converted to oestrone by enzymatic activity of the 17β hydroxysteroid dehydrogenases, which can subsequently be converted to the 2-, 4- and the 16α hydroxyestrones (Figure 1). The 2- and 4-hydroxyestrogens can then undergo methylation by the activity of catechol O-methyltransferase (COMT) to the respective methoxyestrogens.
The compounds formed during this metabolic process can play differential roles in PAH. For instance, 2-hydroxyestradiol, its methylated metabolite, 2-methoxyestradiol and the synthetic analogue 2-ethoxyestradiol mediate protective effects in experimental models of PAH (Tofovic et al., 2008). In contrast, 16α hydroxyestrone induces significant proliferation of human PASMCs and administration of this oestrogen metabolite can induce PAH in mice (White et al., 2012). Furthermore, 16α hydroxyestrone may be actively involved in the pathogenesis of PAH, as indicated by increased urinary concentrations in experimental PAH (White et al., 2012). Importantly, female urinary 16α hydroxyestrone levels are also increased in patients with BMPR2-associated PAH (Austin et al., 2009). At present, the precise function and relationship between oestrogen and its metabolites in PAH is still largely unknown. A metabolic shift towards the formation of pro-proliferative metabolites by altered expression of enzymatic proteins could have potentially detrimental effects on the pulmonary circulation. For example, increased expression of the oestrogen-metabolizing enzyme, CYP4501B1 (cytochrome P450 1B1; CYP1B1), altering the metabolic pathway, is associated with the development and progression of PAH, potentially through the increased formation of 16α hydroxyestrone (White et al., 2012).
CYP1B1 metabolizes oestrogen predominantly to the 4-hydroxyestrogens and to a lesser extent to the 2- and 16α-hydroxyestrogens (Badawi et al., 2001; Lee et al., 2003). Under basal conditions, CYP1B1 expression is low. However CYP1B1 expression is up-regulated in animal models of PAH and critically, it is also consistently up-regulated in both IPAH and HPAH (White et al., 2012). In line with this, loss of function and inhibition of CYP1B1 is protective in preclinical models of PAH, giving evidence that CYP1B1 is involved in the pathogenesis of PAH (White et al., 2012). In this study, CYP1B1 was integral to the pathogenesis of PAH in both male and female animals, with no major gender differences reported, emphasizing the potential impact of locally synthesized oestrogens (White et al., 2012). Additionally, CYP1B1 is also involved in other metabolic pathways, such as metabolism of arachidonic acid, which has been implicated in PAH (Choudhary et al., 2004; Zhu and Ran, 2006). Thus, additional protective effects may be mediated by other pathways. Consistent with the hypothesis that serotonin may amplify the effects of oestrogen, serotonin has been found to increase CYP1B1 expression in human PASMCs and CYP1B1 expression is also increased in pulmonary arteries from female SERT+ mice (White et al., 2011b). These finding suggest serotonin may alter oestrogen metabolism and promote the formation of pro-proliferative oestrogen metabolites leading to the development of PAH.
In addition, oestrogen metabolites may interact and signal through the ERs. In a comprehensive analysis, the binding affinity of natural and synthetic oestrogen metabolites for the different receptor subtypes was studied. Interestingly, 17β oestradiol, 4-hydroxyestradiol and 4-hydroxyestrone and 2-hydroxyestradiol have an equal binding affinity for both ERα and ERβ receptors, whereas oestrone and 2-hydroxyestrone preferentially bind at ERα (Zhu et al., 2006). In contrast both 16α hydroxyestradiol and 16α hydroxyestrone have a higher binding affinity for ERβ (Zhu et al., 2006). Taken together, these findings suggest that modulating oestrogen metabolism, for example, by targeting CYP1B1 may be a novel therapeutic strategy for the treatment of PAH.
Androgens and PAH
Despite females developing PAH more frequently, male patients are consistently shown to have poorer survival even with treatment (Benza et al., 2010; Humbert et al., 2010a; 2010b). Androgens may therefore play a role in the gender differences observed in PAH. There is a paucity of data on male hormones in the pulmonary vasculature and the exact role of androgens in the physiology and pathophysiology in PAH remains uncertain.
Testosterone is the main secreted androgenic steroid. It is primarily biosynthesized in the testes and by the ovaries in females, although small amounts are also secreted from the adrenal cortex. The effects of testosterone are mediated via the androgen receptor (AR), which is located in a variety of tissues including vascular smooth muscle and endothelial cells (Dubey et al., 2002; Liu et al., 2003) and also in the lung (Mikkonen et al., 2010). Testosterone can activate the AR directly, or following its conversion by the enzyme 5α-reductase to the more potent androgen dihydrotestosterone (DHT; Figure 2). In general, testosterone plays a crucial role in the development, growth and function of the male reproductive tissues although it is becoming increasingly apparent that androgens also play a pivotal role in cardiovascular disease (Dubey et al., 2002).
Figure 2.

Schematic representation of androgen and oestradiol metabolism. Sex hormones are derived from cholesterol and converge on the circulating precursor DHEA and its sulphated form (DHEA-S). Both males and females possess the enzyme 17β-hydroxysteroid dehydrogenase (17β-HSD) that enables the conversion androgens to testosterone. The enzyme 5α-reductase then converts testosterone into the more potent metabolite DHT, which is subsequently deactivated by HSD enzymes. Testosterone can also be metabolized by the cytochrome P450 enzyme CYP19A1 to oestradiol which is further metabolized by HSD enzymes to oestrone and oestriol.
As previously mentioned, in PAH, right ventricular dysfunction is the most important prognostic factor and indicator of survival (D'Alonzo et al., 1991). ARs have been identified in both the right and left ventricle (Lizotte et al., 2009); however, to date, the left ventricle has been more extensively studied. Both testosterone and its primary metabolite, DHT, mediate genomic effects through the AR, although DHT is 10 times more potent in activation of the AR than testosterone itself (Liu et al., 2003). Furthermore, there is strong evidence that testosterone does indeed cause structural and morphological changes in human hearts (Achar et al., 2010) and both testosterone and DHT initiate cardiac hypertrophy (Hayward et al., 2001). In addition, metabolism of testosterone is significantly changed in the hypertrophic heart, with elevated levels of DHT described in human left ventricular hypertrophy (Thum and Borlak, 2002) and this is thought to exacerbate cardiac hypertrophy. As survival is worse in males, and survival is closely linked to right ventricle function, a correlation between testosterone and the right ventricle is proposed. The degree of right ventricular hypertrophy in rats exposed to high altitude is greater in castrated males treated with testosterone (Vander et al., 1978) and the effect of testosterone and hypoxia appear additive. Recently, Hemnes et al. provided the first evidence for testosterone-induced right ventricular fibrosis and increased myocyte size in a model of PAH. In this study, it was observed that there were no alterations in haemodynamic properties resulting from testosterone manipulation suggesting that the effects of testosterone are primarily involved in dysfunctional right ventricular (RV) hypertrophy (Hemnes et al., 2012). Therefore, following increased afterload inflicted by elevated pulmonary pressures in PAH, testosterone may be the underlying cause of some of the differences in survival between males and females.
Testosterone is also a potent vasodilator in isolated human pulmonary vasculature, an effect that is independent of gender (Smith et al., 2008; Rowell et al., 2009). It is in fact a more potent vasodilator than oestrogen in this vascular bed (English et al., 2001). Importantly, this action of testosterone is generally accepted to be a rapid, non-genomic effect and therefore is independent of the AR (Yue et al., 1995; Jones et al., 2002). The mechanism of vasodilation may be independent of the endothelium and NO (Yue et al., 1995; Jones et al., 2002) and due to inhibition of Ca2+ entry via voltage-gated calcium channels (Scragg et al., 2004; Hall et al., 2006). Acute and long-term vasodilation by testosterone is often suggested to be due to conversion to oestrogen. However, several studies have demonstrated that the vasodilator effect of testosterone is not inhibited by CYP19A1 inhibition or by ER antagonism (Teoh et al., 2000; Deenadayalu et al., 2001; Tep-areenan et al., 2002). Thus, the protective vascular effects of testosterone could provide rationale for a lower incidence of PAH in men. However, as PAH becomes established, the disease progress may be more exaggerated in men due to the negative effects of testosterone on the right ventricle leading to subsequent right ventricular failure. For therapeutic intervention in men, treatments reversing right ventricular remodelling may be more beneficial in prolonging survival compared to the currently available vasodilator options.
Another interesting androgen target, which is becoming increasingly studied in the setting of PAH, is dehydroepiandosterone (DHEA). DHEA is a naturally occurring steroid derived from the adrenal glands and is the most abundantly secreted circulating steroid. The sulphated ester, DHEA-S, serves as an inactive reservoir with conversion by sulfotransferases occurring in a wide range of tissues. Previously, age-related declines in serum DHEA and DHEA-S have been attributed to development of cardiovascular diseases (Barrett-Connor et al., 1986; Baulieu, 2002) and epidemiological and animal studies support a beneficial role for DHEA (Barrett-Connor and Goodman-Gruen, 1995; Williams, 2000). In addition, clear gender differences have been described in circulating levels of DHEA-S with levels twice as high in men as in women (Parker, 1999). DHEA has recently been described as having a multifunctional protective role in PAH. Pulmonary vasodilator properties of DHEA have been demonstrated in chronic hypoxic male rats and monocrotaline models associated with both opening of voltage-gated potassium channels (Farrukh et al., 1998; Gupte et al., 2002) and increased expression and function of pulmonary artery Ca2+-activated K+ channels (Bonnet et al., 2003; Hampl et al., 2003).
Moreover, antioxidant properties of DHEA have also been demonstrated in human PASMCs, decreasing proliferation and resistance to apoptosis by modulating mitochondrial functions (Dumas de la Roque et al., 2010). Effects of DHEA on proliferation and apoptosis appear directly mediated as they are independent of both AR and ERs (Williams et al., 2002; 2004; Oka et al., 2007; Bonnet et al., 2009). In line with this, DHEA reverses pulmonary arterial remodelling by various mechanistic pathways. Normalizing RhoA/ROCK activity in hypoxia was associated with prevention of vascular remodelling (Homma et al., 2007), decreased Src/STAT3 activation with restored BMPR2 (Paulin et al., 2011) and decreased accumulation of hypoxia-inducible factor α in PASMC during hypoxia (Dessouroux et al., 2008) contribute to decreased PASMC proliferation and remodelling by DHEA.
In summary, it is possible that the lower DHEA levels in women compared to men contributes to the higher frequency of PAH in women. The ability of DHEA to inhibit proliferation and induce significant vasodilation in pulmonary vasculature makes it an attractive drug target for treatment of PAH. Although there is yet no data on the clinical effect of DHEA treatment of PAH, DHEA has been shown to reverse PAH in chronic hypoxic and monocrotaline male rat models (Hampl et al., 2003; Homma et al., 2007; Oka et al., 2007; Dumas de la Roque et al., 2010). Interestingly, DHEA has also been found to act as a potent suppressor of CYP1B1 expression in cancer cells (Ciolino et al., 2003; Mikstacka et al., 2008). Data regarding the effect of DHEA in females are lacking. However, DHEA treatments in females should be approached with caution given that metabolism of DHEA to oestrogen may augment PAH (Dempsie et al., 2011; White et al., 2011a) and long-term exposure could promote hormone-dependent breast cancer.
Conclusion
Despite the female predominance in patients with PAH, no sex-based treatments are currently offered. Most issues specifically related to the care of women with PAH are related to birth control and pregnancy, as pregnancy in PAH carries a significant risk to maternal health (Weiss et al., 1998). Recently, it has been reported that women with PAH exhibit a greater clinical benefit from endothelin receptor antagonists than men (Gabler et al., 2012); this heterogeneity in treatment response may reflect underlying gender-specific pathophysiological differences in the development of PAH.
In PAH, sex hormones appear to influence the development and progression of the disease. The evidence suggests that oestradiol promotes cardioprotection while testosterone contributes to cardiac hypertrophy and consequently RV failure. Where other mediators of PAH such as serotonin are elevated, oestrogen may actually promote PASMC proliferation via accumulation of damaging oestrogen metabolites. The differential effects of male and female hormones in the pulmonary circulation highlight the advantages of incorporating comparative studies on males and female gender into both basic and clinical research.
Furthermore, drugs that target components of the oestrogen pathway such as CYP19A1 (i.e. anastrozole), CYP1B1 (2, 4, 3′, 5′-tetramethoxystilbene) and ERs such as selective oestradiol receptor modulators (tamoxifen) may prove to be novel therapeutic strategies for the treatment of PAH (Figure 3). As these drugs are already widely used in the treatment of cancer, this improves the translational potential of preclinical research.
Figure 3.
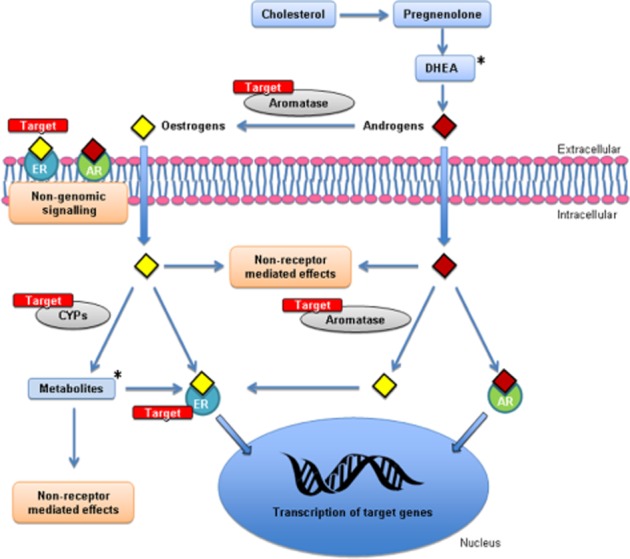
A schematic representation highlighting the potential therapeutic targets within sex hormone pathways in pulmonary arterial smooth muscle cells. *Metabolites of oestrogens and DHEA may also be useful in the treatment of PAH.
Acknowledgments
Supported by the British Heart Foundation and the Medical Research Council.
Glossary
- AR
androgen receptor
- BMPR2
bone morphogenetic protein receptor 2
- CYP19A1
aromatase
- CYP1B1
cytochrome P450 1B1
- DHEA
dehydroepiandosterone
- DHT
dihydrotestosterone
- ER
oestrogen receptor
- ERα
oestrogen receptor alpha
- ERβ
oestrogen receptor beta
- ET-1
endothelin-1
- GPER
G protein-coupled oestrogen receptor
- PAH
pulmonary arterial hypertension
- PASMC
pulmonary artery smooth muscle cell
- SERT
serotonin transporter
- Su-Hx
SUGEN hypoxic
- Tph1
tryptophan hydroxylase 1
Conflict of interest
None.
References
- Achar S, Rostamian A, Narayan SM. Cardiac and metabolic effects of anabolic-androgenic steroid abuse on lipids, blood pressure, left ventricular dimensions, and rhythm. Am J Cardiol. 2010;106:893–901. doi: 10.1016/j.amjcard.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn BH, Park HK, Cho HG, Lee HA, Lee YM, Yang EK, et al. Estrogen and enalapril attenuate the development of right ventricular hypertrophy induced by monocrotaline in ovariectomized rats. J Korean Med Sci. 2003;18:641–648. doi: 10.3346/jkms.2003.18.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin ED, Cogan JD, West JD, Hedges LK, Hamid R, Dawson EP, et al. Alterations in oestrogen metabolism: implications for higher penetrance of familial pulmonary arterial hypertension in females. Eur Respir. 34:1093–1099. doi: 10.1183/09031936.00010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin ED, Hamid R, Hemnes AR, Loyd JE, Blackwell T, Yu C, et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Biol Sex Differ. 2009-2012;3:6. doi: 10.1186/2042-6410-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badawi AF, Cavalieri EL, Rogan EG. Role of human cytochrome P450 1A1, 1A2, 1B1, and 3A4 in the 2-, 4-, and 16alpha-hydroxylation of 17beta-estradiol. Metabolism. 2001;50:1001–1003. doi: 10.1053/meta.2001.25592. [DOI] [PubMed] [Google Scholar]
- Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137:376–387. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- Barrett-Connor E, Goodman-Gruen D. The epidemiology of DHEAS and cardiovascular disease. Ann N Y Acad Sci. 1995;774:259–270. doi: 10.1111/j.1749-6632.1995.tb17386.x-i1. [DOI] [PubMed] [Google Scholar]
- Barrett-Connor E, Khaw KT, Yen SS. A prospective study of dehydroepiandrosterone sulfate, mortality, and cardiovascular disease. N Engl J Med. 1986;315:1519–1524. doi: 10.1056/NEJM198612113152405. [DOI] [PubMed] [Google Scholar]
- Baulieu EE. Androgens and aging men. Mol Cell Endocrinol. 2002;198:41–49. doi: 10.1016/s0303-7207(02)00367-2. [DOI] [PubMed] [Google Scholar]
- Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122:164–172. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142:448–456. doi: 10.1378/chest.11-1460. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Dumas-de-La-Roque E, Begueret H, Marthan R, Fayon M, Dos Santos P, et al. Dehydroepiandrosterone (DHEA) prevents and reverses chronic hypoxic pulmonary hypertension. Proc Natl Acad Sci U S A. 2003;100:9488–9493. doi: 10.1073/pnas.1633724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet S, Paulin R, Sutendra G, Dromparis P, Roy M, Watson KO, et al. Dehydroepiandrosterone reverses systemic vascular remodeling through the inhibition of the Akt/GSK3-{beta}/NFAT axis. Circulation. 2009;120:1231–1240. doi: 10.1161/CIRCULATIONAHA.109.848911. [DOI] [PubMed] [Google Scholar]
- Cacoub P, Dorent R, Nataf P, Carayon A, Riquet M, Noe E, et al. Endothelin-1 in the lungs of patients with pulmonary hypertension. Cardiovasc Res. 1997;33:196–200. doi: 10.1016/s0008-6363(96)00189-7. [DOI] [PubMed] [Google Scholar]
- Channick RN, Voswinckel R, Rubin LJ. Inhaled treprostinil: a therapeutic review. Drug Des Devel Ther. 2012;6:19–28. doi: 10.2147/DDDT.S19281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaouat A, Coulet F, Favre C, Simonneau G, Weitzenblum E, Soubrier F, et al. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax. 2004;59:446–448. doi: 10.1136/thx.2003.11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YF, Jowett S, Barton P, Malottki K, Hyde C, Gibbs JS, et al. Clinical and cost-effectiveness of epoprostenol, iloprost, bosentan, sitaxentan and sildenafil for pulmonary arterial hypertension within their licensed indications: a systematic review and economic evaluation. Health Technol Assess. 2009;13:1–320. doi: 10.3310/hta13490. [DOI] [PubMed] [Google Scholar]
- Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. Metabolism of retinoids and arachidonic acid by human and mouse cytochrome P450 1b1. Drug Metab Dispos. 2004;32:840–847. doi: 10.1124/dmd.32.8.840. [DOI] [PubMed] [Google Scholar]
- Christman BW, McPherson CD, Newman JH, King GA, Bernard GR, Groves BM, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70–75. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- Ciolino H, MacDonald C, Memon O, Dankwah M, Yeh GC. Dehydroepiandrosterone inhibits the expression of carcinogen-activating enzymes in vivo. Int J Cancer. 2003;105:321–325. doi: 10.1002/ijc.11075. [DOI] [PubMed] [Google Scholar]
- Ciuclan L, Bonneau O, Hussey M, Duggan N, Holmes AM, Good R, et al. A novel murine model of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:1171–1182. doi: 10.1164/rccm.201103-0412OC. [DOI] [PubMed] [Google Scholar]
- Ciuclan L, Hussey MJ, Burton V, Good R, Duggan N, Beach S, et al. Imatinib Attenuates Hypoxia-induced Pulmonary Arterial Hypertension Pathology via Reduction in 5-Hydroxytryptamine through Inhibition of Tryptophan Hydroxylase 1 Expression. Am J Respir Crit Care Med. 2013;187:78–89. doi: 10.1164/rccm.201206-1028OC. [DOI] [PubMed] [Google Scholar]
- Cool CD, Stewart JS, Werahera P, Miller GJ, Williams RL, Voelkel NF, et al. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol. 1999;155:411–419. doi: 10.1016/S0002-9440(10)65137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–349. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- Deenadayalu VP, White RE, Stallone JN, Gao X, Garcia AJ. Testosterone relaxes coronary arteries by opening the large-conductance, calcium-activated potassium channel. Am J Physiol Heart Circ Physiol. 2001;281:H1720–H1727. doi: 10.1152/ajpheart.2001.281.4.H1720. [DOI] [PubMed] [Google Scholar]
- Dempsie Y, MacRitchie NA, Morecroft I, Nilsen M, Loughlin L, MacLean MR. The effects of gender on the development of dexfenfluramine-induced pulmonary arterial hypertension in mice. Am J Respir Crit Care Med. 2009;179:A1808. [Google Scholar]
- Dempsie Y, Nilsen M, White K, Mair KM, Loughlin L, Ambartsumian N, et al. Development of pulmonary arterial hypertension in mice over-expressing S100A4/Mts1 is specific to females. Respir Res. 2011;12:159. doi: 10.1186/1465-9921-12-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessouroux A, Akwa Y, Baulieu EE. DHEA decreases HIF-1alpha accumulation under hypoxia in human pulmonary artery cells: potential role in the treatment of pulmonary arterial hypertension. J Steroid Biochem Mol Biol. 2008;109:81–89. doi: 10.1016/j.jsbmb.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Dubey RK, Oparil S, Imthurn B, Jackson EK. Sex hormones and hypertension. Cardiovasc Res. 2002;53:688–708. doi: 10.1016/s0008-6363(01)00527-2. [DOI] [PubMed] [Google Scholar]
- Dumas de la Roque E, Savineau JP, Bonnet S. Dehydroepiandrosterone: a new treatment for vascular remodeling diseases including pulmonary arterial hypertension. Pharmacol Ther. 2010;126:186–199. doi: 10.1016/j.pharmthera.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Dwyer N, Kilpatrick D. Bosentan for the treatment of adult pulmonary hypertension. Future Cardiol. 2011;7:19–37. doi: 10.2217/fca.10.114. [DOI] [PubMed] [Google Scholar]
- English KM, Jones RD, Jones TH, Morice AH, Channer KS. Gender differences in the vasomotor effects of different steroid hormones in rat pulmonary and coronary arteries. Horm Metab Res. 2001;33:645–652. doi: 10.1055/s-2001-18689. [DOI] [PubMed] [Google Scholar]
- Farhat MY, Chen MF, Bhatti T, Iqbal A, Cathapermal S, Ramwell PW. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br J Pharmacol. 1993;110:719–723. doi: 10.1111/j.1476-5381.1993.tb13871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrukh IS, Peng W, Orlinska U, Hoidal JR. Effect of dehydroepiandrosterone on hypoxic pulmonary vasoconstriction: a Ca(2+)-activated K(+)-channel opener. Am J Physiol. 1998;274:L186–L195. doi: 10.1152/ajplung.1998.274.2.L186. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- Frost AE, Badesch DB, Barst RJ, Benza RL, Elliott CG, Farber HW, et al. The changing picture of patients with pulmonary arterial hypertension in the United States: how REVEAL differs from historic and non-US Contemporary Registries. Chest. 2011;139:128–137. doi: 10.1378/chest.10-0075. [DOI] [PubMed] [Google Scholar]
- Gabler NB, French B, Strom BL, Liu Z, Palevsky HI, Taichman DB, et al. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest. 2012;141:20–26. doi: 10.1378/chest.11-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galie N, Brundage BH, Ghofrani HA, Oudiz RJ, Simonneau G, Safdar Z, et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009a;119:2894–2903. doi: 10.1161/CIRCULATIONAHA.108.839274. [DOI] [PubMed] [Google Scholar]
- Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT) Eur Heart J. 2009b;30:2493–2537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- Galie N, Negro L, Simonneau G. The use of combination therapy in pulmonary arterial hypertension: new developments. Eur Respir Rev. 2009c;18:148–153. doi: 10.1183/09059180.00003809. [DOI] [PubMed] [Google Scholar]
- Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–221. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–1739. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- Gomez-Arroyo J, Saleem SJ, Mizuno S, Syed AA, Bogaard HJ, Abbate A, et al. A brief overview of mouse models of pulmonary arterial hypertension: problems and prospects. Am J Physiol Lung Cell Mol Physiol. 2012;302:L977–L991. doi: 10.1152/ajplung.00362.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte SA, Li KX, Okada T, Sato K, Oka M. Inhibitors of pentose phosphate pathway cause vasodilation: involvement of voltage-gated potassium channels. J Pharmacol Exp Ther. 2002;301:299–305. doi: 10.1124/jpet.301.1.299. [DOI] [PubMed] [Google Scholar]
- Hall J, Jones RD, Jones TH, Channer KS, Peers C. Selective inhibition of L-type Ca2+ channels in A7r5 cells by physiological levels of testosterone. Endocrinology. 2006;147:2675–2680. doi: 10.1210/en.2005-1243. [DOI] [PubMed] [Google Scholar]
- Hampl V, Bibova J, Povysilova V, Herget J. Dehydroepiandrosterone sulphate reduces chronic hypoxic pulmonary hypertension in rats. Eur Respir J. 2003;21:862–865. doi: 10.1183/09031936.03.00084503. [DOI] [PubMed] [Google Scholar]
- Hayward CS, Webb CM, Collins P. Effect of sex hormones on cardiac mass. Lancet. 2001;357:1354–1356. doi: 10.1016/S0140-6736(00)04523-2. [DOI] [PubMed] [Google Scholar]
- Hemnes AR, Maynard KB, Champion HC, Gleaves L, Penner N, West J, et al. Testosterone negatively regulates right ventricular load stress responses in mice. Pulm Circ. 2012;2:352–358. doi: 10.4103/2045-8932.101647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma N, Nagaoka T, Morio Y, Ota H, Gebb SA, Karoor V, et al. Endothelin-1 and serotonin are involved in activation of RhoA/Rho kinase signaling in the chronically hypoxic hypertensive rat pulmonary circulation. J Cardiovasc Pharmacol. 2007;50:697–702. doi: 10.1097/FJC.0b013e3181593774. [DOI] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Respir Crit Care Med. 2006;173:1023–1030. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010a;122:156–163. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Yaici A, Montani D, O'Callaghan DS, Jais X, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010b;36:549–555. doi: 10.1183/09031936.00057010. [DOI] [PubMed] [Google Scholar]
- Jones RD, English KM, Pugh PJ, Morice AH, Jones TH, Channer KS. Pulmonary vasodilatory action of testosterone: evidence of a calcium antagonistic action. J Cardiovasc Pharmacol. 2002;39:814–823. doi: 10.1097/00005344-200206000-00006. [DOI] [PubMed] [Google Scholar]
- Kawut SM, Krowka MJ, Trotter JF, Roberts KE, Benza RL, Badesch DB, et al. Clinical risk factors for portopulmonary hypertension. Hepatology. 2008;48:196–203. doi: 10.1002/hep.22275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahm T, Crisostomo PR, Markel TA, Wang M, Wang Y, Weil B, et al. Exogenous estrogen rapidly attenuates pulmonary artery vasoreactivity and acute hypoxic pulmonary vasoconstriction. Shock. 2008;30:660–667. doi: 10.1097/SHK.0b013e31816f239f. [DOI] [PubMed] [Google Scholar]
- Lahm T, Albrecht M, Fisher AJ, Selej M, Patel NG, Brown JA, et al. 17beta-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am J Respir Crit Care Med. 2012;185:965–980. doi: 10.1164/rccm.201107-1293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, 3rd, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- Lee AJ, Cai MX, Thomas PE, Conney AH, Zhu BT. Characterization of the oxidative metabolites of 17beta-estradiol and estrone formed by 15 selectively expressed human cytochrome p450 isoforms. Endocrinology. 2003;144:3382–3398. doi: 10.1210/en.2003-0192. [DOI] [PubMed] [Google Scholar]
- Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med. 2012;186:790–796. doi: 10.1164/rccm.201203-0383OC. [DOI] [PubMed] [Google Scholar]
- Liu PY, Death AK, Handelsman DJ. Androgens and cardiovascular disease. Endocr Rev. 2003;24:313–340. doi: 10.1210/er.2003-0005. [DOI] [PubMed] [Google Scholar]
- Lizotte E, Grandy SA, Tremblay A, Allen BG, Fiset C. Expression, distribution and regulation of sex steroid hormone receptors in mouse heart. Cell Physiol Biochem. 2009;23:75–86. doi: 10.1159/000204096. [DOI] [PubMed] [Google Scholar]
- Maclean MR, Dempsie Y. The serotonin hypothesis of pulmonary hypertension revisited. Adv Exp Med Biol. 2010;661:309–322. doi: 10.1007/978-1-60761-500-2_20. [DOI] [PubMed] [Google Scholar]
- Masi AT. Editorial: pulmonary hypertension and oral contraceptive usage. Chest. 1976;69:451–453. doi: 10.1378/chest.69.4.451b. [DOI] [PubMed] [Google Scholar]
- Matori H, Umar S, Nadadur RD, Sharma S, Partow-Navid R, Afkhami M, et al. Genistein, a soy phytoestrogen, reverses severe pulmonary hypertension and prevents right heart failure in rats. Hypertension. 2012;60:425–430. doi: 10.1161/HYPERTENSIONAHA.112.191445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGoon MD, Frost AE, Oudiz RJ, Badesch DB, Galie N, Olschewski H, et al. Ambrisentan therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function test abnormalities. Chest. 2009;135:122–129. doi: 10.1378/chest.08-1028. [DOI] [PubMed] [Google Scholar]
- Mikkonen L, Pihlajamaa P, Sahu B, Zhang FP, Janne OA. Androgen receptor and androgen-dependent gene expression in lung. Mol Cell Endocrinol. 2010;317:14–24. doi: 10.1016/j.mce.2009.12.022. [DOI] [PubMed] [Google Scholar]
- Mikstacka R, Baer-Dubowska W, Wieczorek M, Sobiak S. Thiomethylstilbenes as inhibitors of CYP1A1, CYP1A2 and CYP1B1 activities. Mol Nutr Food Res. 2008;52(Suppl. 1):S77–S83. doi: 10.1002/mnfr.200700202. [DOI] [PubMed] [Google Scholar]
- Miyauchi T, Yanagisawa M, Iida K, Ajisaka R, Suzuki N, Fujino M, et al. Age- and sex-related variation of plasma endothelin-1 concentration in normal and hypertensive subjects. Am Heart J. 1992;123:1092–1093. doi: 10.1016/0002-8703(92)90734-d. [DOI] [PubMed] [Google Scholar]
- Morani A, Barros RP, Imamov O, Hultenby K, Arner A, Warner M, et al. Lung dysfunction causes systemic hypoxia in estrogen receptor beta knockout (ERbeta-/-) mice. Proc Natl Acad Sci U S A. 2006;103:7165–7169. doi: 10.1073/pnas.0602194103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray F, Maclean MR, Insel PA. Role of phosphodiesterases in adult-onset pulmonary arterial hypertension. Handb Exp Pharmacol. 2011;204:279–305. doi: 10.1007/978-3-642-17969-3_12. [DOI] [PubMed] [Google Scholar]
- Nadadur RD, Umar S, Wong G, Eghbali M, Iorga A, Matori H, et al. Reverse right ventricular structural and extracellular matrix remodeling by estrogen in severe pulmonary hypertension. J Appl Physiol. 2012;113:149–158. doi: 10.1152/japplphysiol.01349.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA, 3rd, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345:319–324. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, Golembeski SM, et al. Dehydroepiandrosterone upregulates soluble guanylate cyclase and inhibits hypoxic pulmonary hypertension. Cardiovasc Res. 2007;74:377–387. doi: 10.1016/j.cardiores.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olschewski H. Inhaled iloprost for the treatment of pulmonary hypertension. Eur Respir Rev. 2009;18:29–34. doi: 10.1183/09059180.00011111. [DOI] [PubMed] [Google Scholar]
- Oudiz RJ, Galie N, Olschewski H, Torres F, Frost A, Ghofrani HA, et al. Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:1971–1981. doi: 10.1016/j.jacc.2009.07.033. [DOI] [PubMed] [Google Scholar]
- Parker CR., Jr Dehydroepiandrosterone and dehydroepiandrosterone sulfate production in the human adrenal during development and aging. Steroids. 1999;64:640–647. doi: 10.1016/s0039-128x(99)00046-x. [DOI] [PubMed] [Google Scholar]
- Paulin R, Meloche J, Jacob MH, Bisserier M, Courboulin A, Bonnet S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol. 2011;301:H1798–H1809. doi: 10.1152/ajpheart.00654.2011. [DOI] [PubMed] [Google Scholar]
- Polderman KH, Stehouwer CD, van Kamp GJ, Dekker GA, Verheugt FW, Gooren LJ. Influence of sex hormones on plasma endothelin levels. Ann Intern Med. 1993;118:429–432. doi: 10.7326/0003-4819-118-6-199303150-00006. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2008;118:2372–2379. doi: 10.1172/JCI33452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch M, Gamble WJ, Miettinen OS, Reid L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am J Physiol. 1981;240:H62–H72. doi: 10.1152/ajpheart.1981.240.1.H62. [DOI] [PubMed] [Google Scholar]
- Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, et al. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H1235–H1248. doi: 10.1152/ajpheart.00254.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resta TC, Kanagy NL, Walker BR. Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression. Am J Physiol Lung Cell Mol Physiol. 2001;280:L88–L97. doi: 10.1152/ajplung.2001.280.1.L88. [DOI] [PubMed] [Google Scholar]
- Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–223. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- Roberts KE, Fallon MB, Krowka MJ, Brown RS, Trotter JF, Peter I, et al. Genetic risk factors for portopulmonary hypertension in patients with advanced liver disease. Am J Respir Crit Care Med. 2009;179:835–842. doi: 10.1164/rccm.200809-1472OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowell KO, Hall J, Pugh PJ, Jones TH, Channer KS, Jones RD. Testosterone acts as an efficacious vasodilator in isolated human pulmonary arteries and veins: evidence for a biphasic effect at physiological and supra-physiological concentrations. J Endocrinol Invest. 2009;32:718–723. doi: 10.1007/BF03346526. [DOI] [PubMed] [Google Scholar]
- Rubin LJ. Endothelin receptor antagonists for the treatment of pulmonary artery hypertension. Life Sci. 2012;91:517–521. doi: 10.1016/j.lfs.2012.07.033. [DOI] [PubMed] [Google Scholar]
- Rubtsov AV, Rubtsova K, Kappler JW, Marrack P. Genetic and hormonal factors in female-biased autoimmunity. Autoimmun Rev. 2010;9:494–498. doi: 10.1016/j.autrev.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Irastorza G, Garmendia M, Villar I, Egurbide M-V, Aguirre C. Pulmonary hypertension in systemic lupus erythematosus: prevalence, predictors and diagnostic strategy. Autoimmun Rev. 2012;12:410–415. doi: 10.1016/j.autrev.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Scragg JL, Jones RD, Channer KS, Jones TH, Peers C. Testosterone is a potent inhibitor of L-type Ca(2+) channels. Biochem Biophys Res Commun. 2004;318:503–506. doi: 10.1016/j.bbrc.2004.04.054. [DOI] [PubMed] [Google Scholar]
- Shapiro S, Traiger GL, Turner M, McGoon MD, Wason P, Barst RJ. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest. 2012;141:363–373. doi: 10.1378/chest.10-3114. [DOI] [PubMed] [Google Scholar]
- Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43–S54. doi: 10.1016/j.jacc.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Simpson ER, Misso M, Hewitt KN, Hill RA, Boon WC, Jones ME, et al. Estrogen – the good, the bad, and the unexpected. Endocr Rev. 2005;26:322–330. doi: 10.1210/er.2004-0020. [DOI] [PubMed] [Google Scholar]
- Smith AM, Bennett RT, Jones TH, Cowen ME, Channer KS, Jones RD. Characterization of the vasodilatory action of testosterone in the human pulmonary circulation. Vasc Health Risk Manag. 2008;4:1459–1466. doi: 10.2147/vhrm.s3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney L, Voelkel NF. Estrogen exposure, obesity and thyroid disease in women with severe pulmonary hypertension. Eur J Med Res. 2009;14:433–442. doi: 10.1186/2047-783X-14-10-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapson VF, Torres F, Kermeen F, Keogh AM, Allen RP, Frantz RP, et al. Oral Treprostinil for the Treatment of Pulmonary Arterial Hypertension in Patients on Background Endothelin Receptor Antagonist and/or Phosphodiesterase Type 5 Inhibitor Therapy (The FREEDOM-C Study): a Randomized Controlled Trial. Chest. 2012;142:1383–1390. doi: 10.1378/chest.11-2212. [DOI] [PubMed] [Google Scholar]
- Teoh H, Quan A, Leung SW, Man RY. Differential effects of 17beta-estradiol and testosterone on the contractile responses of porcine coronary arteries. Br J Pharmacol. 2000;129:1301–1308. doi: 10.1038/sj.bjp.0703164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tep-areenan P, Kendall DA, Randall MD. Testosterone-induced vasorelaxation in the rat mesenteric arterial bed is mediated predominantly via potassium channels. Br J Pharmacol. 2002;135:735–740. doi: 10.1038/sj.bjp.0704522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet. 2000;37:741–745. doi: 10.1136/jmg.37.10.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne S, Nelson-Piercy C, MacGregor A, Gibbs S, Crowhurst J, Panay N, et al. Pregnancy and contraception in heart disease and pulmonary arterial hypertension. J Fam Plann Reprod Health Care. 2006;32:75–81. doi: 10.1783/147118906776276486. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Testosterone, cytochrome P450, and cardiac hypertrophy. FASEB J. 2002;16:1537–1549. doi: 10.1096/fj.02-0138com. [DOI] [PubMed] [Google Scholar]
- Tofovic SP, Rafikova O, Champion H, Schneider F. Estrogens exacerbate development of occlusive pulmonary arterial hypertension and formation of plexiform lesions. Am J Respir Crit Care Med. 2012;185:A6803. [Google Scholar]
- Tofovic SP, Zhang X, Zhu H, Jackson EK, Rafikova O, Petrusevska G. 2-Ethoxyestradiol is antimitogenic and attenuates monocrotaline-induced pulmonary hypertension and vascular remodeling. Vascul Pharmacol. 2008;48:174–183. doi: 10.1016/j.vph.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345:325–334. doi: 10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925–1932. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- Umar S, Iorga A, Matori H, Nadadur RD, Li J, Maltese F, et al. Estrogen rescues preexisting severe pulmonary hypertension in rats. Am J Respir Crit Care Med. 2011;184:715–723. doi: 10.1164/rccm.201101-0078OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umar S, Rabinovitch M, Eghbali M. Estrogen paradox in pulmonary hypertension: current controversies and future perspectives. Am J Respir Crit Care Med. 2012;186:125–131. doi: 10.1164/rccm.201201-0058PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander AJ, Moore LG, Brewer G, Menon KM, England BG. Effects of high altitude on plasma concentrations of testosterone and pituitary gonadotropins in man. Aviat Space Environ Med. 1978;49:356–357. [PubMed] [Google Scholar]
- Ventetuolo CE, Ouyang P, Bluemke DA, Tandri H, Barr RG, Bagiella E, et al. Sex hormones are associated with right ventricular structure and function: the MESA-right ventricle study. Am J Respir Crit Care Med. 2011;183:659–667. doi: 10.1164/rccm.201007-1027OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AM, Langleben D, Korelitz JJ, Rich S, Rubin LJ, Strom BL, et al. Temporal trends and drug exposures in pulmonary hypertension: an American experience. Am Heart J. 2006;152:521–526. doi: 10.1016/j.ahj.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Weiss BM, Zemp L, Seifert B, Hess OM. Outcome of pulmonary vascular disease in pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol. 1998;31:1650–1657. doi: 10.1016/s0735-1097(98)00162-4. [DOI] [PubMed] [Google Scholar]
- White K, Dempsie Y, Nilsen M, Wright AF, Loughlin L, MacLean MR. The serotonin transporter, gender, and 17beta oestradiol in the development of pulmonary arterial hypertension. Cardiovasc Res. 2011a;90:373–382. doi: 10.1093/cvr/cvq408. [DOI] [PubMed] [Google Scholar]
- White K, Loughlin L, Maqbool Z, Nilsen M, McClure J, Dempsie Y, et al. Serotonin transporter, sex, and hypoxia: microarray analysis in the pulmonary arteries of mice identifies genes with relevance to human PAH. Physiol Genomics. 2011b;43:417–437. doi: 10.1152/physiolgenomics.00249.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K, Johansen AK, Nilsen M, Ciuclan L, Wallace E, Paton L, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation. 2012;126:1087–1098. doi: 10.1161/CIRCULATIONAHA.111.062927. [DOI] [PubMed] [Google Scholar]
- Williams JR. The effects of dehydroepiandrosterone on carcinogenesis, obesity, the immune system, and aging. Lipids. 2000;35:325–331. doi: 10.1007/s11745-000-0529-7. [DOI] [PubMed] [Google Scholar]
- Williams MR, Ling S, Dawood T, Hashimura K, Dai A, Li H, et al. Dehydroepiandrosterone inhibits human vascular smooth muscle cell proliferation independent of ARs and ERs. J Clin Endocrinol Metab. 2002;87:176–181. doi: 10.1210/jcem.87.1.8161. [DOI] [PubMed] [Google Scholar]
- Williams MR, Dawood T, Ling S, Dai A, Lew R, Myles K, et al. Dehydroepiandrosterone increases endothelial cell proliferation in vitro and improves endothelial function in vivo by mechanisms independent of androgen and estrogen receptors. J Clin Endocrinol Metab. 2004;89:4708–4715. doi: 10.1210/jc.2003-031560. [DOI] [PubMed] [Google Scholar]
- Wort SJ, Woods M, Warner TD, Evans TW, Mitchell JA. Endogenously released endothelin-1 from human pulmonary artery smooth muscle promotes cellular proliferation: relevance to pathogenesis of pulmonary hypertension and vascular remodeling. Am J Respir Cell Mol Biol. 2001;25:104–110. doi: 10.1165/ajrcmb.25.1.4331. [DOI] [PubMed] [Google Scholar]
- Xu DQ, Luo Y, Liu Y, Wang J, Zhang B, Xu M, et al. Beta-estradiol attenuates hypoxic pulmonary hypertension by stabilizing the expression of p27kip1 in rats. Respir Res. 2010;11:182. doi: 10.1186/1465-9921-11-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Wu WH, Gao L, Zheng ZQ, Liu D, Mei HY, et al. Oestradiol ameliorates monocrotaline pulmonary hypertension via NO, PGI2 and ET-1 pathways. Eur Respir J. 2012;41:1116–1125. doi: 10.1183/09031936.00044112. [DOI] [PubMed] [Google Scholar]
- Yue P, Chatterjee K, Beale C, Poole-Wilson PA, Collins P. Testosterone relaxes rabbit coronary arteries and aorta. Circulation. 1995;91:1154–1160. doi: 10.1161/01.cir.91.4.1154. [DOI] [PubMed] [Google Scholar]
- Zhu BT, Han GZ, Shim JY, Wen Y, Jiang XR. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: insights into the structural determinants favoring a differential subtype binding. Endocrinology. 2006;147:4132–4150. doi: 10.1210/en.2006-0113. [DOI] [PubMed] [Google Scholar]
- Zhu D, Ran Y. Role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in hypoxia-induced pulmonary hypertension. J Physiol Sci. 2006;62:163–172. doi: 10.1007/s12576-012-0196-9. [DOI] [PMC free article] [PubMed] [Google Scholar]