

Abstract
Background and Purpose
Osteoclasts play a pivotal role in diseases such as osteoporosis, rheumatoid arthritis and tumour bone metastasis. Thus, searching for natural compounds that may suppress osteoclast formation and/or function is promising for the treatment of osteoclast-related diseases. Here, we examined changes in osteoclastogenesis and LPS-induced osteolysis in response to andrographolide (AP), a diterpenoid lactone isolated from the traditional Chinese and Indian medicinal plant Andrographis paniculata.
Experimental Approach
Effects of AP on osteoclast differentiation and bone resorption were measured in vitro. Western blots and RT-PCR techniques were used to examine the underlying molecular mechanisms. The bone protective activity of AP in vivo was assessed in a mouse model of osteolysis.
Key Results
AP concentration-dependently suppressed RANKL-mediated osteoclast differentiation and bone resorption in vitro and reduced the expression of osteoclast-specific markers, including tartrate-resistant acid phosphatase, calcitonin receptors and cathepsin K. Further molecular analysis revealed that AP impaired RANKL-induced NF-κB signalling by inhibiting the phosphorylation of TGF-β-activated kinase 1, suppressing the phosphorylation and degradation of IκBα, and subsequently preventing the nuclear translocation of the NF-κB p65 subunit. AP also inhibited the ERK/MAPK signalling pathway without affecting p38 or JNK signalling.
Conclusions and Implications
AP suppressed RANKL-induced osteoclastogenesis through attenuating NF-κB and ERK/MAPK signalling pathways in vitro, thus preventing bone loss in vivo. These data indicated that AP is a promising natural compound for the treatment of osteoclast-related bone diseases.
Keywords: andrographolide, osteoclast, osteolysis, NF-κB, ERK
Introduction
Bone is a rigid but dynamic tissue that is continuously shaped and repaired. Its metabolic homeostasis is achieved through a delicate balance between osteoblastic bone formation and osteoclastic bone resorption. Osteoclasts originate from haematopoietic progenitors and their formation is dependent on several key factors, including macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor kappa B ligand (RANKL) (Novack, 2010). While M-CSF is indispensable for osteoclast precursor cell proliferation and survival (Takahashi et al., 2003), RANKL is a key factor for osteoclast differentiation. Upon stimulation with RANKL, its receptor RANK binds to the signalling adaptor molecule, TNF receptor-associated factor 6 (TRAF6) (Suda et al., 1999; Kong et al., 2000; Boyle et al., 2003) and activates the TGF β-activated kinase-1 (TAK1) kinase complex, thereby resulting in the activation of downstream signalling pathways (Takaesu et al., 2001; Keating et al., 2007). NF-κB activation is especially critical for osteoclast development (Khosla, 2001). The initial step of NF-κB activation involves the phosphorylation and degradation of IκBα and this degradation allows the NF-κB proteins (p50, p52 and p65) to enter the nucleus and bind to DNA target sites (Soysa and Alles, 2009), subsequently initiating the expression of certain genes and therefore inducing the formation of functional osteoclasts that are capable of bone resorption (Boyle et al., 2003). Excessive osteoclast formation and bone resorption can cause an imbalance in bone remodelling and thus induce many adult skeletal diseases, including osteoporosis, rheumatoid arthritis, multiple myeloma and metastatic cancers. Therefore, targeting RANKL-induced NF-κB signalling may be a promising strategy for the treatment of osteoclast-related osteolytic diseases (Aoki et al., 2006).
Plant-derived natural products have an important role in the treatment of osteoclast-related diseases. Andrographis paniculata is an important herbal medicine, widely used in Southeast Asia. It has been used clinically for the treatment of fever, inflammation, diarrhoea and other infectious diseases without any obvious side effects (Shen et al., 2002; Reddy et al., 2005). The main components of A. paniculata are diterpene lactones, of which andrographolide (AP) is the major component, comprising 70% of the plant extract (Zhao et al., 2002). Several studies have demonstrated that AP exhibits anti-inflammatory, anticancer and hepatoprotective activities (Chen et al., 2004; Xia et al., 2004; Hidalgo et al., 2005; Wang et al., 2007; Negi et al., 2008; Abu-Ghefreh et al., 2009; Bao et al., 2009). In particular, Xia et al. (2004) showed that AP inhibits NF-κB activation through covalent modification of a cysteine residue on p50 in endothelial cells without affecting IκBα degradation or p50/p65 nuclear translocation.
Given the importance of the NF-κB signalling pathway in osteoclast formation, the effects of AP on NF-κB suppression in other cell types (Xia et al., 2004) and the wide clinical application of AP, we hypothesized that AP may represent a novel treatment for osteoclast-related osteolytic diseases. Therefore, in this study, we aimed to (i) investigate the potential therapeutic benefits of AP on osteoclast-related osteolytic bone diseases, (ii) understand the underlying mechanisms mediating the effects of AP on osteoclast formation and function, and (iii) further elucidate the potential molecular mechanisms of AP in osteoclasts. Our findings suggested that AP may provide a new approach to the prevention of osteolysis caused by enhanced osteoclast formation and activation.
Methods
Animals
All animal care and experimental procedures complied with the guidelines for Ethical Conduct in the Care and Use of Nonhuman Animals in Research by the American Psychological Association and were approved by the Animal Care and Experiment Committee of Shanghai Jiao Tong University School of Medicine. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 28 animals were used in the experiments described here. C57/BL6 mice (females; 23±1.5 g, 8 weeks old) were supplied by Shanghai SLAC Laboratory (Shanghai, China) and maintained in our animal facility under controlled temperature (22-24°C) and humidity (50-60%) conditions and a 12h light/dark cycle with free access to food (standard mouse food supplied by Shanghai SLAC) and water. The mice were for kept for at least 5 days before experimental use.
In vitro osteoclastogenesis assay
In vitro osteoclastogenesis assays were preformed to examine the effects of AP on osteoclast differentiation. Bone marrow macrophages (BMM) cells were prepared as previously described (Qin et al., 2012b; Kogawa et al., 2013; Zou et al., 2013). Briefly, cells extracted from the femur and tibiae of a 6-week-old C57/BL6 mouse were incubated in complete cell culture media and 30 ng·mL−1 M-CSF in a T-75 cm2 flask for proliferation. When changing the medium, the cells were washed in order to deplete residual stromal cells. After reaching 90% confluence, cells were washed with PBS three times and trypsinized for 30 min to harvest BMMs. Cells adhering to the bottom of the dish were classified as BMMs; these BMMs were plated in 96-well plates at a density of 8 × 103 cells per well in triplicate and incubated in a humidified incubator containing 5% CO2 at 37°C for 24 h. The cells were then treated with various concentrations of AP (0, 2.5, 5, or 10 μM) plus M-CSF (30 ng·mL−1) and RANKL (50 ng·mL−1). After 5 days, cells were fixed and stained for tartrate-resistant acid phosphatase (TRAP) activity. TRAP-positive multinucleated cells with more than five nuclei were counted as osteoclasts.
Cytotoxicity assay
Effects of AP on cell proliferation were determined with a CCK-8 following the manufacturer's instructions. BMMs were plated in 96-well plates at a density of 3 × 103 cells per well in triplicate. Twenty-four hours later, the cells were treated with increasing concentrations of AP (0, 2.5, 5, 10 or 20 μM) for 2 days. Next, 10 μL CCK-8 was added to each well, and the plates were then incubated at 37°C for an additional 2 h. The optical density (OD) was then measured with an ELX800 absorbance microplate reader (Bio-Tek Instr., Winooski, VT, USA) at a wavelength of 450 nm (650 nm reference). The cell viability was calculated relative to the control using the following formula: [experimental group OD − zeroing OD]/[control group OD − zeroing OD].
Resorption pit assay
For the bone resorption assay, BMMs were seeded on bone slices in 96-well plates at a density of 8 × 103 cells per well with three replicates and stimulated with M-CSF (30 ng·mL−1) plus RANKL (50 ng·mL−1). Three days later, cells were treated with the indicated concentrations of AP for 48 h post-culture. Cells were then fixed with 2.5% glutaraldehyde. Bone slices were imaged using a scanning electron microscope (FEI Instr., Hillsboro, OR, USA; Quanta 250) with 200× magnification and at 10 kV. Three view fields were randomly selected for each bone slice for further analysis. Pit areas were quantified using Image J software (National Institutes of Health, Bethesda, MD, USA). Similar independent experiments were repeated at least three times.
Western blot analysis
RAW264.7 cells were seeded at 5 × 105 cells per well into 6-well plates and pretreated with or without AP (10 μM) for 4 h before RANKL stimulation (50 ng·mL−1) for the indicated times (0, 5, 10, 30, 40 or 60 min). BMMs were seeded at 5 × 105 cells per well into 6-well plates and treated with or without AP (10 μM) and RANKL (50 ng·mL−1) for the indicated times. Cells were lysed in RIPA lysis buffer containing 50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM sodium fluoride, 1 mM sodium vanadate, 1% deoxycholate and protease inhibitor cocktail. The lysate was centrifuged at 12 000 x g for 10 min, and the protein in the supernatant was collected. Protein concentrations were measured through bicinchoninic acid assay. Thirty micrograms of each protein lysate was resolved by SDS-PAGE using 8–10% gels, and proteins were then transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Non-specific interactions were blocked with 5% skim milk for 1 h, and membranes were then probed with the indicated primary antibodies overnight at 4°C as indicated. Membranes were incubated with the appropriate secondary antibodies conjugated with IRDye 800CW (molecular weight, 1166 Da; LI-COR, Lincoln, NE, USA), and the antibody reactivity was detected by exposure in an Odyssey infrared imaging system (LI-COR).
Confocal microscopy for NF-κB localization
RAW264.7 cells were plated at a density of 1 × 104 cells in 6-well plates containing sterile cover slips and grown at 37°C for 24 h. The medium was then replaced with serum-free medium, and the cells were allowed to grow for another 24 h before treatment. Cells were treated with AP for 4 h, followed by stimulation with RANKL (50 ng·mL−1) for 20 min. After treatment, the cells were washed twice with PBS and fixed onto the cover slips by incubation in 4% paraformaldehyde (PFA) for 30 min. Cells were then washed with PBS three times and permeabilized by 0.1%Triton-X 100 for 30 min at room temperature. Cover slips were blocked in 3% BSA for 1 h at room temperature. Antibodies targeting the NF-κB p65 subunit (1:100) were added to the 1% BSA solution and incubated for 12 h at 4°C. For nuclear staining, DAPI solution (Sigma-Aldrich) was added at a final concentration of 0.1 μg·mL−1 and incubated for 10 min in the dark. Cover slips were then washed three times with PBS. The nuclear translocation of p65 was imaged using a NIKON A1Si spectral detector confocal system (Nikon, Tokyo, Japan).
NF-κB luciferase reporter gene activity assay
The effects of AP on RANKL-induced NF-κB activation were measured using RAW264.7 cells that had been stably transfected with an NF-κB luciferase reporter construct, as previously described (Wang et al., 2003). Briefly, cells were seeded into 48-well plates and maintained in cell culture media for 24 h. Cells were then pretreated with or without the indicated concentrations of AP for 1 h, followed by addition of RANKL (50 ng·mL−1) for 8 h. Luciferase activity was measured using the Promega Luciferase Assay System (Promega, Madison, WI, USA) and normalized to that of the vehicle control.
Quantitative PCR analysis
For real-time PCR, 10 × 104 BMMs were seeded in each well of a 24-well plate and cultured in complete medium containing α-MEM, 10% FBS, 100 U·mL−1 penicillin, M-CSF (30 ng·mL−1) and RANKL (50 ng·mL−1). Cells were then treated with or without AP (10 μM) for the indicated times. Total RNA was prepared using an RNeasy Mini kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions and cDNA was synthesized from 1 μg of total RNA using reverse transcriptase (TaKaRa Biotechnology, Otsu, Japan). Real-time PCR was performed using the SYBR Premix Ex Tag kit (TaKaRa Biotechnology) and an ABI 7500 Sequencing Detection System (Applied Biosystems, Foster City, CA, USA). The detector was programmed with the following PCR conditions: 40 cycles for 5 s denaturation at 95°C and 34 s amplification at 60°C. All reactions were run in triplicate and were normalized to the housekeeping gene β-actin. The following primer sets were used as previously described (Qin et al., 2011; 2012a,): mouse β-actin: forward, 5′-TCTGCTGGAAGGTGGACAGT-3′ and reverse, 5′-CCTCTATGCCAACACAGTGC-3′; mouse NFATc1: forward, 5′-CCGTTGCTTCCAGAAAATAACA-3′ and reverse, 5′-TGTGGGATGTGAACTCGGAA-3′; mouse TRAP: forward, 5′-CTGGAGTGCACGATGCCAGCGACA-3′ and reverse, 5′-TCCGTGCTCGGCGATGGACCAGA-3′; mouse cathepsin K: forward, 5′-CTTCCAATACGTGCAGCAGA-3′ and reverse, 5′-TCTTCAGGGCTTTCTCGTTC-3′; mouse CTR: forward, 5′-TGCAGACAACTCTTGGTTGG-3′ and reverse, 5′-TCGGTTTCTTCTCCTCTGGA-3′; mouse DC-STAMP: forward, 5′-AAAACCCTTGGGCTGTTCTT-3′ and reverse, 5′-AATCATGGACGACTCCTTGG-3′; and mouse v-ATPase d2: forward, 5′-AAGCCTTTGTTTGACGCTGT-3′ and reverse, 5′-TTCGATGCCTCTGTGAGATG-3′.
Effects of AP in vivo on LPS-mediated bone erosion in mouse femurs
C57BL/6 mice (8 weeks old) were divided into four groups of seven mice each. Mice were injected i.p. with AP (5 or 30 mg·kg−1 body weight) or PBS as a control 1 day before injection of LPS (5 μg·g−1 body weight). AP or PBS was injected intraperitoneally every other day for 8 days. LPS was injected intraperitoneally on days one and four. All mice were killed 8 days after the initial LPS injection, and the left femurs of all animals were scanned with a high-resolution micro-CT (Skyscan 1176; Skyscan, Aartselaar, Belgium) at a resolution of 9 μm using the following settings: X-ray voltage, 50 kV; electric current, 500 μA; rotation step, 0.7°. Bone histomorphometric analyses were performed with the microcomputed tomography (micro-CT) data using software described previously (Wedemeyer et al., 2007). The calculation of bone mineral density (BMD), as well as the microstructural indices of trabecular bone density (BV/TV), bone surface/volume ratio (BS/BV), trabecular thickness (Tb.Th), trabecular number (Tb.N) and trabecular space (Tb.Sp) were measured to assess the trabecular bone microstructure of the femurs. In addition, analysis of cortical bone was also carried out by measuring the average cortical thickness for both cortices. Tissues were removed and fixed in 4% PFA (Sigma-Aldrich) for 1 day at 4°C and were then decalcified in 12% EDTA. Decalcified bones were embedded in paraffin and sectioned. For histological examination, sections were stained with haematoxylin and eosin (H&E), and samples that were obtained from the same region within H&E sections were stained for TRAP activity and then counter-stained with methylene blue to identify osteoclasts on the bone surface. The parameters for bone resorption, including the percentage osteoclast surface per bone surface (OcS/BS, %) and number of osteoclasts per field of tissue (No. of OCs/field) in 200× magnification, were quantified using Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA).
Data analysis
Values are presented as the mean ± SD of results obtained from three or more experiments. Statistical analyses were performed using one-way anova followed by the Student-Newman-Keul's test. A P-value of less than 0.05 was considered significant.
Materials
Andrographolide was purchased from Sigma-Aldrich (St. Louis, MO, USA). Alpha-MEM, FBS and penicillin were purchased from Gibco BRL (Gaithersburg, MD, USA). Soluble mouse recombinant M-CSF and RANKL were purchased from R&D Systems (Minneapolis, MN, USA). TRAP staining solution was from Sigma-Aldrich. The Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular Technology (Minato-ku, Tokyo, Japan). Primary antibodies targeting β-actin, phospho-IκBα, IκBα, IKKα, IKKβ and NFATc1 were purchased from Cell Signaling Technology (Danvers, MA, USA), and those targeting phospho-TAK1 and TAK1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Results
AP inhibited RANKL-induced osteoclast formation in vitro
To investigate the effects of AP on osteoclastogenesis, BMMs were treated with various concentrations of AP during the course of osteoclast formation. As shown in Figure 1B, the control group formed numerous TRAP-positive multinucleated osteoclasts. In contrast, the formation of osteoclasts was inhibited after AP treatment, as demonstrated by the concentration-dependent decrease in the number of osteoclasts. Only about 10% the number of osteoclasts found in the control group were formed after treatment with 10 μM AP (Figure 1C). In order to exclude potential cytotoxicity of AP in BMMs, we performed cell viability assays. As shown in Figure 1D, AP did not inhibit BMM proliferation, even at concentrations as high as 10 μM. Furthermore, RAW 264.7 cells, which are derived from murine macrophages, were also used to investigate the effects of AP on osteoclastogenesis. The results showed that AP inhibited osteoclast differentiation of RAW 264.7 cells in a concentration-dependent manner (Supporting Information Figure S1). In addition, AP did not exert any cytotoxic effects on RAW 264.7 cells at concentrations as high as 10 μM (data not shown). Together, these data indicated that AP suppressed osteoclast formation in a concentration-dependent manner without any obvious cytotoxic effects, in both BMMs and RAW 264.7 cells.
Figure 1.
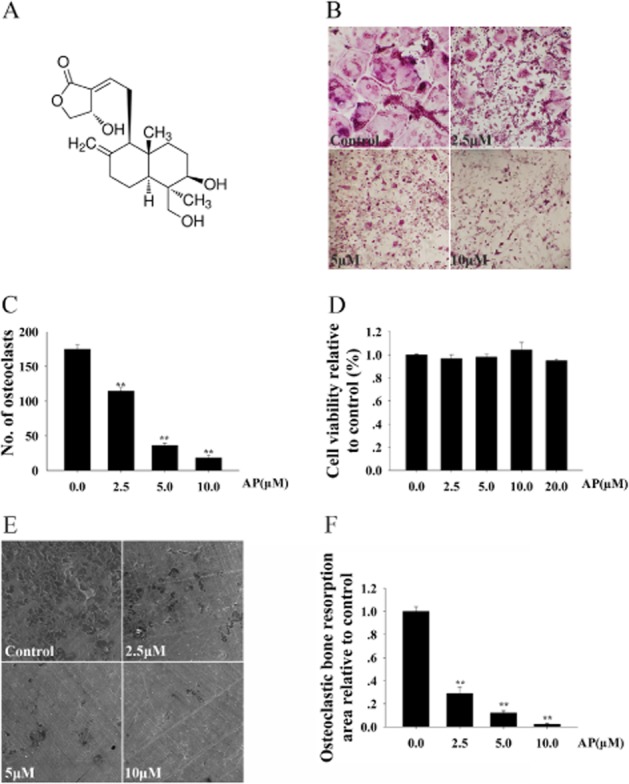
Andrographolide (AP) inhibited RANKL-induced osteoclast formation and bone resorption in a concentration-dependent manner, without cytotoxicity. (A) The structure of AP. (B) Bone marrow-derived monocytes/macrophages (BMMs) were treated with various concentrations of AP followed by M-CSF (30 ng·mL−1) and RANKL (50 ng·mL−1) stimulation for 5 days. Cells were then fixed with 4% PFA and stained for TRAP. (C) TRAP-positive multinuclear cells were counted. (D) BMMs were plated in 96-well plates with 8000 cells per well, stimulated with M-CSF (30 ng·mL−1), RANKL (50 ng·mL−1) and indicated concentrations of AP for 48 h. Cell viability was then measured using CCK-8 assays, as described in the Methods section. (E) BMM-derived pre-osteoclasts were stimulated with M-CSF (30 ng·mL−1) and RANKL (50 ng·mL−1 for three days. Later, cells were cultured in the presence of the indicated concentrations of AP with M-CSF (30 ng·mL−1) and RANKL (50 ng·mL−1) for another 48 h.I Scanning electron micropscopic images of bone resorption pits are shown. (F) Resorption pit areas were measured using Image J and are presented graphically. All experiments were carried out at least three times. ** P < 0.01. significantly different from control; one-way ANOVA, with Student-Newman-Keul's test.
AP impaired osteoclastic bone resorption in vitro
As AP inhibited osteoclast formation, we next investigated whether AP could impair osteoclastic bone resorption in vitro. Thus, osteoclast precursors were treated with the indicated concentrations of AP. As shown in Figure 1E, AP treatment substantially reduced the area of bone resorption. Only approximately 30% of the bone resorption observed in the control group was achieved after treatment with 2.5 μM AP. Osteoclastic bone resorption was almost completely inhibited after treatment with 10 μM AP (Figure 1F). Collectively, these findings suggested that AP impaired osteoclast bone resorption in vitro.
AP suppressed RANKL-induced osteoclast-specific gene expression
Osteoclast differentiation is associated with the up-regulation of specific genes in response to RANKL (Boyle et al., 2003). We further assessed the inhibitory effects of AP on osteoclastogenesis by evaluating the RANKL-induced mRNA expression levels of osteoclast-related genes, such as TRAP, calcitonin receptor (CTR), cathepsin K (Cts k), dendritic cell-specific transmembrane protein (DC-STAMP) and ATP6V0D2. As demonstrated by quantitative real-time PCR, RANKL dramatically induced the expression of all evaluated genes. In contrast, AP substantially suppressed osteoclastic gene expression (Figure 2). Therefore, these data further confirmed that AP suppressed osteoclast formation and gene expression in vitro.
Figure 2.
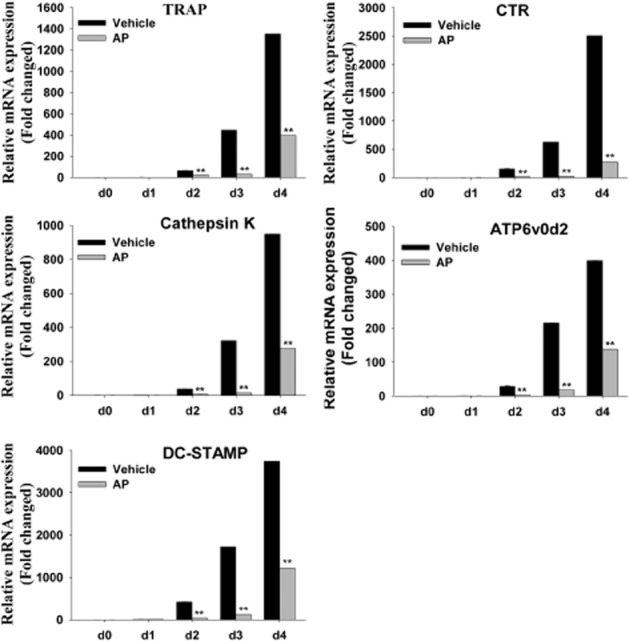
AP suppressed RANKL-induced expression of osteoclast-specific genes. BMMs were cultured with M-CSF (30 ng·mL−1) and RANKL (50 ng·mL−1), with or without 10 μM AP, for 0, 1, 2, 3 or 4 days. Osteoclast-specific gene expression (TRAP, CTR, Cts k, DC-STAMP and ATP6V0D2) was analysed by real-time PCR, and results were normalized to the expression of β-actin. All experiments were performed at least three times. **, P < 0.01, significantly different from initial (d0) values; one-way ANOVA, with Student-Newman-Keul's test.
AP inhibited RANKL-induced NF-κB and ERK/MAPK activation
To further elucidate the mechanisms through which AP mediated osteoclast formation, RANKL-induced signalling pathways were investigated. Upon stimulation with RANKL, its receptor RANK binds the signalling adaptor molecule TRAF6 (Suda et al., 1999; Kong et al., 2000; Boyle et al., 2003), activates the TAK1 kinase complex, and promotes the phosphorylation and degradation of IκBα, leading to the activation of the NF-κB pathway (Takaesu et al., 2001; Keating et al., 2007). In our study, when osteoclast precursor cells were stimulated with RANKL, the phosphorylation of TAK1 was initiated in the control group. However, the addition of AP significantly suppressed TAK1 phosphorylation without affecting IKKα/β expression (Figure 3A). At the same time, RANKL-induced IκBα phosphorylation and degradation was also markedly suppressed by AP (Figure 3B). More specifically, IκBα was phosphorylated and degraded upon RANKL stimulation for 5 min in the control group. In contrast, AP treatment suppressed the phosphorylation and therefore degradation of IκBα (Figure 3C).
Figure 3.

AP inhibited RANKL-induced activation of NF-κB and ERK signalling pathways. (A) and (B) RAW264.7 cells were seeded at 5 × 105 cells per well in 6-well plates and pretreated with or without AP (10 μM) for 4 h prior to RANKL stimulation (50 ng·mL−1) for 5 min. Cells were lysed for Western blotting with specific antibodies against phospho-TAK1, TAK1, IKKα, IKKβ, phospho-IκBα, IκBα and actin. (C) RAW264.7 cells were seeded at 5 × 105 cells per well in 6-well plates and pretreated with or without AP (10 μM) for 4 h prior to RANKL stimulation (50 ng·mL−1) for the indicated times. Cells were lysed for Western blotting with specific antibodies against phospho-IκBα, IκBα and actin. (D) RAW264.7 cells were plated at a density of 1 × 104 cells in 6-well plates and treated with AP for 4 h, followed by stimulation with RANKL (50 ng·mL−1) for 20 min. The localization of p65 was visualized by immunofluorescence analysis. (E) P3K-RAW cells were seeded in 48-well plates and maintained in the cell culture media for 24 h. The cells were then pretreated with or without the indicated concentrations of AP for 1 h, followed by addition of RANKL (50 ng·mL−1) for 8 h. NF-κB luciferase activity was measured. (F) RAW264.7 cells were seeded at 5 × 105 cells per well in 6-well plates and pretreated with or without AP (10 μM) for 4 h prior to RANKL stimulation (50 ng·mL−1) for 5 min. Cells were lysed for Western blotting with specific antibodies against phospho-ERK, ERK, phospho-JNK, JNK, phospho-p38, and p38. (G) BMMs were cultured with M-CSF (30 ng·mL−1), RANKL (50 ng·mL−1) and 10 μM AP for 1, 3 or 5 days. Cells were then lysed for immunoblot analysis with antibodies against NFATc1 and actin. (H) Fold changes in NFATc1 levels were normalized to the expression of actin. (I) BMMs were cultured with M-CSF (30 ng·mL−1) and RANKL (50 ng·mL−1), with or without 10 μM AP for 0, 1, 2, 3 or 4 days. NFATc1 expression was analysed by real-time PCR, and results were normalized to the expression of actin. All experiments were performed at least three times. *, P < 0.05; **, P < 0.01, one-way ANOVA, with Student-Newman-Keul's test.
In unstimulated cells, NF-κB subunits are retained in the cytoplasm by binding to the inhibitory IκB protein. Phosphorylation and subsequent degradation of IκBα liberates NF-κB proteins (such as p65) to enter the nucleus and bind to DNA target sites (Soysa and Alles, 2009). As is shown in Figure 3D, p65 was translocated into nuclei in the control group. However, AP treatment substantially suppressed p65 nuclear translocation, as demonstrated by the retention in the cytoplasm of the p65 proteins (Figure 3D, lower). The inhibitory effects of AP on NF-κB activation were further supported by luciferase assays. The transcriptional activity of NF-κB increased dramatically following treatment with RANKL. However, NF-κB activity was inhibited by AP in a concentration-dependent manner (Figure 3E).
In addition, previous research has revealed that three major subfamilies of MAPKs (p38, ERK1/2 and JNK) play pivotal roles in the development of osteoclasts downstream of RANK signalling (Stevenson et al., 2011). Thus, we further investigated whether these signalling pathways were involved in the suppression of osteoclastogenesis by AP. Interestingly, AP almost completely attenuated ERK phosphorylation, but had no obvious effect on JNK or p38 signalling pathways (Figure 3F).
NFATc1 is well known as a crucial transcription factor during RANKL-induced osteoclastogenesis. NF-κB interacts with the NFATc1 promoter and regulates its expression (Zhao et al., 2010). As expected, our Western blot analysis demonstrated that RANKL induced NFATc1 expression during osteoclast formation. However, the expression of NFATc1 was significantly suppressed after AP treatment (Figure 3G, middle and Figure 3H). This was further confirmed by real-time PCR, which demonstrated the suppression of NFATc1 mRNA after AP treatment (Figure 3I).
Together, our findings demonstrated that AP inhibited RANKL-induced NF-κB and ERK/MAPK activation during osteoclast formation.
AP prevented LPS-induced osteolysis by inhibiting osteoclast activity
Activation of osteoclasts plays a key role in destructive bone diseases. To further explore the potential protective effects of AP under pathological conditions, we used LPS-induced osteolysis in mice to model inflammatory osteolysis (Kim et al., 2010; Kwak et al., 2010; Lee et al., 2012; Khor et al., 2013; Zhang et al., 2013). Mice were intraperitoneally injected with LPS with or without AP. No fatalities were recorded after LPS and AP administration, and the animals retained normal activity throughout the duration of the experiment. Micro-CT confirmed that LPS (5 μg·g−1) injection induced extensive bone loss in mouse femurs, as shown by the markedly reduced trabecular bone mass (Figure 4A). Quantitative analysis of bone parameters confirmed that LPS injection induced osteolysis, with significant reductions in BV/TV, Tb.Th and Tb.N and increased BS/BV and Tb.Sp (Figure 4B). In contrast, treatment with AP (5 or 30 mg·kg−1) reduced the extent of bone loss induced by LPS (Figure 4A, B). Moreover, AP slightly increased the BMD and cortex thickness compared to LPS treatment, although the data did not meet our criteria for significance. Histological examination confirmed the protective effects of AP on LPS-induced bone loss. As shown in Figure 5A, LPS injection led to inflammatory bone erosion and increased numbers of TRAP-positive osteoclasts. However, inflammatory bone erosion was not observed in AP-treated mouse femur tissue sections, which was consistent with the decreased nimber of TRAP-positive osteoclasts (Figure 5A). Furthermore, histomorphometric analysis of OcS/BS and No. of OCs demonstrated that AP treatment attenuated LPS-induced bone loss and reduced osteoclast numbers (Figure 5B, C). Taken together, our data indicated that AP prevented inflammation-induced bone loss in vivo.
Figure 4.

AP prevented LPS-induced bone loss by inhibiting osteoclast activity. Mice were injected intraperitoneally with AP (5 or 30 mg·kg−1 body weight) or PBS as a control 1 day before injection with LPS (5 μg·g−1 body weight). AP or PBS was injected intraperitoneally every other day for 8 days. LPS was injected intraperitoneally on days one and four. All mice were killed 8 days after the initial LPS injection. (A) The left femurs of all animals were scanned with a high-resolution micro-CT. (B) The calculation of the microstructural indices was performed with the micro-CT data as described in the Methods section, including bone volume per tissue volume (BV/TV), bone surface/volume ratio (BS/BV), bone mineral density (BMD), average cortical thickness for both cortices (Cor.Th), trabecular separation (Tb.Sp.), trabecular thickness (Tb.Th.), and trabecular number (Tb.N). *, P < 0.05; **, P < 0.01, significant differences as indicated; one-way ANOVA, with Student-Newman-Keul's test.
Figure 5.
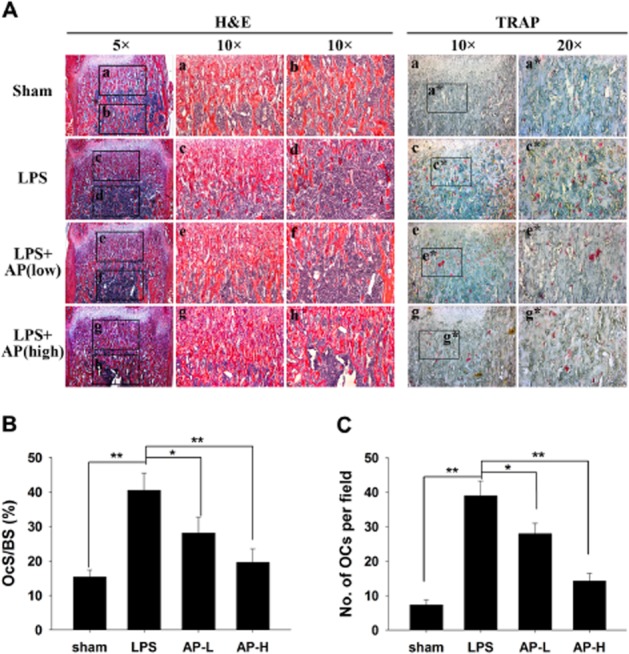
AP prevented LPS-induced bone loss by inhibiting osteoclast activity. (A) Femurs were fixed, decalcified, dehydrated, embedded and sectioned. Sections were stained with H&E (left side, 5× and 10×) and TRAP (right side, 10× and 20×). (B) Percentage osteoclast surface per bone surface (OcS/BS, %) and (C) number of osteoclasts per field of tissue (No. of OCs per field) in 200× magnification were analysed. * P < 0.05; ** P < 0.01, significant differences as indicated; one-way ANOVA, with Student-Newman-Keul's test.
Discussion
Pathological bone destruction occurs when osteoclasts are present in elevated numbers and/or exhibit enhanced activities, leading to excessive bone resorption. Such diseases include osteoporosis, rheumatoid arthritis, multiple myeloma and metastatic cancers (Rodan and Martin, 2000; Kousteni et al., 2001; Rizzoli, 2005; Weitzmann and Pacifici, 2006; Nakamura et al., 2007). During the last two decades, many advances have been made in the treatment of osteolytic diseases, but treatment options are still far from ideal. Although oestrogen-replacement therapy can prevent postmenopausal osteoporosis (Cauley et al., 1995; Çetinkaya et al., 2002), it increases the risk for breast cancer, stroke, heart attack and blood clots, and it is no longer used for long-term treatment (Colditz, 1997; Rossouw et al., 2002). Drugs that either block osteoclast resorption, such as bisphosphonates (BPs), or increase bone formation, such as parathyroid hormone (PTH), have been proposed as alternatives. BPs are the preferred class of antiresorptive agents. Despite their clinical efficacy, an increasing number of clinical reports concerning osteonecrosis of the jaw (ONJ) and atypical fractures are correlated with prolonged use of BPs (Verron and Bouler, 2013). Moreover, PTH, a therapeutic peptide, cannot be given orally, and concerns about osteosarcoma have led to a recommendation of a 2-year maximum treatment course (Cappuzzo and Delafuente, 2004). Recently, the advent of denosumab, a humanized anti-RANKL neutralizing monoclonal antibody, has revolutionized the management of postmenopausal osteoporosis owing to its efficacy, safety and potential to improve adherence rates. Denosumab is now considered an appropriate first-line pharmacological option for the treatment of this disease (Josse et al., 2013). Additionally, denosumab is considered a cost-effective treatment compared with alternative first-line and second-line options (including generic alendronate) in the treatment of women with a high risk of fractures (Hiligsmann et al., 2013). However, while denosumab is a highly efficacious, safe option, further attempts to develop better treatment options should always be pursued.
In this study, we demonstrated, for the first time, that AP was capable of inhibiting osteoclast formation and bone resorption via suppression of RANKL-induced NF-κB and ERK signalling pathways. In addition, we also showed that AP inhibited LPS-induced osteolysis in vivo. Thus, our findings provide additional mechanistic support for the potential application of AP in the treatment of osteoclast-related diseases.
Following stimulation with RANKL, RANK binds to the signalling adaptor molecule TRAF6 (Suda et al., 1999; Kong et al., 2000; Boyle et al., 2003) and activates the TAK1 complex, resulting in activation of the NF-κB pathway (Takaesu et al., 2001; Keating et al., 2007). RANKL-induced NF-κB activation is the dominant mediator of osteoclast differentiation, resorption function and survival (Khosla, 2001). In our study, we demonstrated that AP inhibited TAK1 phosphorylation without affecting IKKα/β expression and attenuated IκBα phosphorylation and degradation, thereby suppressing the nuclear translocation and activation of the p65 subunit of NF-κB. This suggested AP could impair RANKL-induced NF-κB signalling in a concentration-dependent manner, in turn inhibiting efficient transcription of genes involved in osteoclastogenesis.
Interestingly, our molecular analyses were not completely consistent with the results of Xia et al., which revealed that AP attenuated inflammation by inhibition of TNFα-induced NF-κB activation through covalent modification of reduced Cys62 of p50, without affecting IκBα degradation or p50/p65 nuclear translocation (Xia et al., 2004). With respect to this discrepancy, the following points should be taken into consideration: (i) AP may have multiple targets in addition to p50. Indeed, our data suggested that AP inhibited the phosphorylation of TAK1. In addition, AP also inhibited ERK phosphorylation, but not p38 or JNK activation. Together, these data suggested that AP may have multiple targets, and further biological investigations are required to unveil its direct binding sites in osteoclast precursors. (ii) The activation of NF-κB signalling differed between different stimuli. For example, after stimulation with RANKL, RANK binds TRAF6 and induces the formation of a protein complex containing TRAF6, TAK1 and an adaptor protein TAB2, which will then activate the IKK complex, resulting in IκBα phosphorylation and degradation and leading to the activation of the NF-κB pathway (Ninomiya-Tsuji et al., 1999; Mizukami et al., 2002; Ye et al., 2002; Feng, 2005). In contrast to RANKL, TNFα activates NF-κB by relying on TRAF2/5 and cIAP1/2 for activation of IKKβ. There is a strong consensus that TNFα and RANKL can act synergistically to induce osteoclastogenesis (Lam et al., 2000; Komine et al., 2001; Zou et al., 2001) but whether TNFα alone is sufficient for osteoclast formation is still controversial (Kobayashi et al., 2000; Lam et al., 2000). Thus, the discrepancies in IκBα degradation in osteoclast precursors and other cells adopted in the work of Xia et al., 2004 may lie in the use of different stimulation conditions.
NFATc1 is well known as a crucial transcription factor during RANKL-induced osteoclastogenesis. NF-κB interacts with the NFATc1 promoter and regulates its expression (Zhao et al., 2010). Moreover, NFATc1 is a key target gene of NF-κB during osteoclastogenesis. Immediately after NF-κB translocation, NF-κB proteins are recruited to the NFATc1 promoter (Asagiri et al., 2005), leading to induction of NFATc1 auto-amplification during osteoclast formation. This idea is supported by the fact that NFATc1 induction is impaired in p50/p52-deficient cells (Yamashita et al., 2007). Similar to these studies, our findings further demonstrated that inhibition of NF-κB activation and ERK phosphorylation impaired NFATc1 induction after AP treatment, resulting in impaired osteoclast formation.
Binding of NF-κB and NFATc1 to their promoter regions modulates the expression of osteoclast-related genes, including TRAP, CTR, Cts k, DC-STAMP and ATP6V0D2 (Takayanagi et al., 2002), which participate in the osteoclastic bone resorption process (Teitelbaum and Ross, 2003). Here, we found that AP treatment inhibited the expression of these genes and therefore blocked the bone resorption activity of osteoclasts.
LPS is a primary cell wall component of Gram-negative bacteria and elicits inflammatory reactions by stimulating monocytes and macrophages. LPS stimulation leads to phosphorylation of MAPKs as well as NF-κB activation in macrophages and monocytes, thereby promoting the secretion of various inflammatory mediators. The production of inflammatory factors induces pre-osteoclast fusion, supports the survival of mature osteoclasts, and stimulates osteoclastic bone resorption (Huang and Pope, 2009). In the present study, i.p. injection of AP strongly suppressed the bone destruction induced by LPS in our mouse model. As TRAP-positive osteoclast formation was significantly reduced in the group treated with LPS plus AP, the inhibition of bone destruction by AP was thought to be mainly due to the suppression of both LPS-stimulated inflammatory cytokine production and osteoclastogenesis. Further investigations will be needed to understand the signalling mechanisms mediating the effects of AP on osteoblasts.
Taken together, our results demonstrated the inhibitory effects of AP on osteoclastogenesis and osteoclast function in vitro and in vivo. We also clarified that AP functioned through the suppression of NF-κB and ERK signalling pathways. Our data also strongly suggested that AP could be developed as a treatment for osteoclast-related diseases.
Acknowledgments
This work was supported by the Program for Innovative Research Team of Shanghai Municipal Education Commission (Phase I), a grant awarded for Innovative Research from Shanghai Municipal Education Commission (13YZ031), a grant awarded by the Key Disciplines of Shanghai Municipal Education Commission of China (J50206), a grant for scientific research from the National Natural Science Foundation for the Youth of China (No. 81201364) and a grant awarded by the Scientific Research Foundation for Returned Overseas Chinese Scholars from the State Human Resource Ministry.
Glossary
- AP
andrographolide
- BMMs
bone marrow-derived macrophages
- BV/TV
bone volume: tissue volume ratio
- CCK-8
Cell Counting Kit-8
- CTR
calcitonin receptor
- Cts K
cathepsin K
- DC-STAMP
dendritic cell-specific transmembrane protein
- H&E
haematoxylin and eosin
- M-CSF
macrophage colony-stimulating factor
- micro-CT
microcomputed tomography
- PTH
parathyroid hormone
- RANKL
receptor activator of NF-κB ligand
- TAK1
transforming growth factor-β (TGF-β)-activated kinase 1
- Tb.N
trabecular number
- Tb.Sp
trabecular space
- Tb.Th
trabecular thickness
- TRAF6
TNF receptor-associated factor 6
- TRAP
tartrate-resistant acid phosphatase
Conflicts of interest
No authors have any conflicts of interest to declare.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12463
Figure S1 AP inhibited RANKL-induced osteoclastogenesis of RAW 264.7 cells. (A) RAW 264.7 cells were treated with various concentrations of AP, followed by stimulation with RANKL (50 ng·mL−1) for six days. Cells were then fixed with 4% PFA and stained for TRAP. (B) TRAP-positive multinuclear cells were counted (*, P < 0.05; **, P < 0.01).
References
- Abu-Ghefreh AA, Canatan H, Ezeamuzie CI. In vitro and in vivo anti-inflammatory effects of andrographolide. Int Immunopharmacol. 2009;9:313–318. doi: 10.1016/j.intimp.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Aoki K, Saito H, Itzstein C, Ishiguro M, Shibata T, Blanque R, et al. A TNF receptor loop peptide mimic blocks RANK ligand-induced signaling, bone resorption, and bone loss. J Clin Invest. 2006;116:1525–1534. doi: 10.1172/JCI22513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Guan S, Cheng C, Wu S, Wong SH, Kemeny DM, et al. A novel antiinflammatory role for andrographolide in asthma via inhibition of the nuclear factor-κB pathway. Am J Respir Crit Care Med. 2009;179:657–665. doi: 10.1164/rccm.200809-1516OC. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Cappuzzo KA, Delafuente JC. Teriparatide for severe osteoporosis. Ann Pharmacother. 2004;38:294–302. doi: 10.1345/aph.1D353. [DOI] [PubMed] [Google Scholar]
- Cauley JA, Seeley DG, Ensrud K, Ettinger B, Black D, Cummings SR. Estrogen replacement therapy and fractures in older women. Ann Intern Med. 1995;122:9–16. doi: 10.7326/0003-4819-122-1-199501010-00002. [DOI] [PubMed] [Google Scholar]
- Çetinkaya MB, Kökçü A, Yanik FF, Başoğlu T, Malatyalioglu E, Alper T. Comparison of the effects of transdermal estrogen, oral estrogen, and oral estrogen-progestogen therapy on bone mineral density in postmenopausal women. J Bone Miner Metab. 2002;20:44–48. doi: 10.1007/s774-002-8446-8. [DOI] [PubMed] [Google Scholar]
- Chen JH, Hsiao G, Lee AR, Wu CC, Yen MH. Andrographolide suppresses endothelial cell apoptosis via activation of phosphatidyl inositol-3-kinase/Akt pathway. Biochem Pharmacol. 2004;67:1337–1345. doi: 10.1016/j.bcp.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Colditz GA. Hormone replacement therapy increases the risk of breast cancer. Ann N Y Acad Sci. 1997;833:129–136. doi: 10.1111/j.1749-6632.1997.tb48598.x. [DOI] [PubMed] [Google Scholar]
- Feng X. RANKing intracellular signaling in osteoclasts. IUBMB Life. 2005;57:389–395. doi: 10.1080/15216540500137669. [DOI] [PubMed] [Google Scholar]
- Hidalgo MA, Romero A, Figueroa J, Cortés P, Concha II, Hancke JL, et al. Andrographolide interferes with binding of nuclear factor-κB to DNA in HL-60-derived neutrophilic cells. Br J Pharmacol. 2005;144:680–686. doi: 10.1038/sj.bjp.0706105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiligsmann M, Boonen A, Dirksen CD, Sedrine WB, Reginster J-Y. Cost-effectiveness of denosumab in the treatment of postmenopausal osteoporotic women. Expert Rev Pharmacoecon Outcomes Res. 2013;13:19–28. doi: 10.1586/erp.12.76. [DOI] [PubMed] [Google Scholar]
- Huang Q-Q, Pope RM. The role of toll-like receptors in rheumatoid arthritis. Curr Rheumatol Rep. 2009;11:357–364. doi: 10.1007/s11926-009-0051-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josse R, Khan A, Ngui D, Shapiro M. Denosumab, a new pharmacotherapy option for postmenopausal osteoporosis. Curr Med Res Opin. 2013;29:205–216. doi: 10.1185/03007995.2013.763779. [DOI] [PubMed] [Google Scholar]
- Keating SE, Maloney GM, Moran EM, Bowie AG. IRAK-2 participates in multiple toll-like receptor signaling pathways to NFkappaB via activation of TRAF6 ubiquitination. J Biol Chem. 2007;282:33435–33443. doi: 10.1074/jbc.M705266200. [DOI] [PubMed] [Google Scholar]
- Khor EC, Abel T, Tickner J, Chim SM, Wang C, Cheng T, et al. Loss of protein kinase C-δ protects against LPS-induced osteolysis owing to an intrinsic defect in osteoclastic bone resorption. PLoS ONE. 2013;8:e70815. doi: 10.1371/journal.pone.0070815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla S. Minireview: the opg/rankl/rank system. Endocrinology. 2001;142:5050–5055. doi: 10.1210/endo.142.12.8536. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BG, Kwak HB, Choi E-Y, Kim HS, Kim MH, Kim SH, et al. Amorphigenin inhibits osteoclast differentiation by suppressing c-Fos and nuclear factor of activated T cells. Anat Cell Biol. 2010;43:310–316. doi: 10.5115/acb.2010.43.4.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL–RANK interaction. J Exp Med. 2000;191:275–286. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogawa M, Hisatake K, Atkins GJ, Findlay DM, Enoki Y, Sato T, et al. The paired-box domain transcription factor Pax6 binds to the upstream region of the TRAP gene promoter and suppresses RANKL-induced osteoclast differentiation. J Biol Chem. 2013;288:31299–31312. doi: 10.1074/jbc.M113.461848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komine M, Kukita A, Kukita T, Ogata Y, Hotokebuchi T, Kohashi O. Tumor necrosis factor-α cooperates with receptor activator of nuclear factor κB ligand in generation of osteoclasts in stromal cell-depleted rat bone marrow cell culture. Bone. 2001;28:474–483. doi: 10.1016/s8756-3282(01)00420-3. [DOI] [PubMed] [Google Scholar]
- Kong YY, Boyle WJ, Penninger JM. Osteoprotegerin ligand: a regulator of immune responses and bone physiology. Immunol Today. 2000;21:495–502. doi: 10.1016/s0167-5699(00)01718-7. [DOI] [PubMed] [Google Scholar]
- Kousteni S, Bellido T, Plotkin L, O'Brien C, Bodenner D, Han L, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- Kwak HB, Lee BK, Oh J, Yeon JT, Choi SW, Cho HJ, et al. Inhibition of osteoclast differentiation and bone resorption by rotenone, through down-regulation of RANKL-induced c-Fos and NFATc1 expression. Bone. 2010;46:724–731. doi: 10.1016/j.bone.2009.10.042. [DOI] [PubMed] [Google Scholar]
- Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–1488. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-M, Park H, Noh ALSM, Kang J-H, Chen L, Zheng T, et al. 5-Lipoxygenase mediates RANKL-induced osteoclast formation via the cysteinyl leukotriene receptor 1. J Immunol. 2012;189:5284–5292. doi: 10.4049/jimmunol.1003738. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukami J, Takaesu G, Akatsuka H, Sakurai H, Ninomiya-Tsuji J, Matsumoto K, et al. Receptor activator of NF-kappaB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol Cell Biol. 2002;22:992–1000. doi: 10.1128/MCB.22.4.992-1000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, et al. Estrogen prevents bone loss via estrogen receptor [alpha] and induction of fas ligand in osteoclasts. Cell. 2007;130:811–823. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- Negi AS, Kumar J, Luqman S, Shanker K, Gupta M, Khanuja S. Recent advances in plant hepatoprotectives: a chemical and biological profile of some important leads. Med Res Rev. 2008;28:746–772. doi: 10.1002/med.20115. [DOI] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J-I, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-IκB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- Novack DV. Role of NF-κB in the skeleton. Cell Res. 2010;21:169–182. doi: 10.1038/cr.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin A, Cheng TS, Lin Z, Pavlos NJ, Jiang Q, Xu J, et al. Versatile roles of V-ATPases accessory subunit Ac45 in osteoclast formation and function. PLoS ONE. 2011;6:e27155. doi: 10.1371/journal.pone.0027155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin A, Cheng T, Pavlos N, Lin Z, Dai K, Zheng M. V-ATPases in osteoclasts: structure, function and potential inhibitors of bone resorption. Int J Biochem Cell Biol. 2012a;7:1–11. doi: 10.1016/j.biocel.2012.05.014. Issue 4, e34132. [DOI] [PubMed] [Google Scholar]
- Qin A, Cheng TS, Lin Z, Cao L, Chim SM, Pavlos NJ, et al. Prevention of wear particle-induced osteolysis by a novel V-ATPase inhibitor saliphenylhalamide through inhibition of osteoclast bone resorption. PLoS ONE. 2012b;7:e34132. doi: 10.1371/journal.pone.0034132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy VLN, Reddy SM, Ravikanth V, Krishnaiah P, Goud TV, Rao T, et al. A new bis-andrographolide ether from Andrographis paniculata nees and evaluation of anti-HIV activity. Nat Prod Res. 2005;19:223–230. doi: 10.1080/14786410410001709197. [DOI] [PubMed] [Google Scholar]
- Rizzoli R. A new treatment for post-menopausal osteoporosis: strontium ranelate. J Endocrinol Invest. 2005;28(8 Suppl):50–57. [PubMed] [Google Scholar]
- Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289:1508–1514. doi: 10.1126/science.289.5484.1508. [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick M, et al. Writing Group for the Women's Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women's Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- Shen YC, Chen CF, Chiou WF. Andrographolide prevents oxygen radical production by human neutrophils: possible mechanism(s) involved in its anti-inflammatory effect. Br J Pharmacol. 2002;135:399–406. doi: 10.1038/sj.bjp.0704493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soysa N, Alles N. NF-κB functions in osteoclasts. Biochem Biophys Res Commun. 2009;378:1–5. doi: 10.1016/j.bbrc.2008.10.146. [DOI] [PubMed] [Google Scholar]
- Stevenson DA, Schwarz E, Carey JC, Viskochil DH, Hanson H, Bauer S, et al. Bone resorption in syndromes of the Ras/MAPK pathway. Clin Genet. 2011;80:566–573. doi: 10.1111/j.1399-0004.2010.01619.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345–357. doi: 10.1210/edrv.20.3.0367. [DOI] [PubMed] [Google Scholar]
- Takaesu G, Ninomiya-Tsuji J, Kishida S, Li X, Stark GR, Matsumoto K. Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol Cell Biol. 2001;21:2475–2484. doi: 10.1128/MCB.21.7.2475-2484.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Udagawa N, Tanaka S, Suda T. Generating murine osteoclasts from bone marrow. Methods Mol Med. 2003;80:129–144. doi: 10.1385/1-59259-366-6:129. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- Verron E, Bouler J. Is bisphosphonate therapy compromised by the emergence of adverse bone disorders? Drug Discov Today. 2013;116:115–120. doi: 10.1016/j.drudis.2013.08.010. [DOI] [PubMed] [Google Scholar]
- Wang C, Steer JH, Joyce DA, Yip KHM, Zheng MH, Xu J. 12-O-tetradecanoylphorbol-13-acetate (TPA) inhibits osteoclastogenesis by suppressing RANKL-induced NF-κB activation. J Bone Miner Res. 2003;18:2159–2168. doi: 10.1359/jbmr.2003.18.12.2159. [DOI] [PubMed] [Google Scholar]
- Wang YJ, Wang JT, Fan QX, Geng JG. Andrographolide inhibits NF-κB activation and attenuates neointimal hyperplasia in arterial restenosis. Cell Res. 2007;17:933–941. doi: 10.1038/cr.2007.89. [DOI] [PubMed] [Google Scholar]
- Wedemeyer C, Xu J, Neuerburg C, Landgraeber S, Malyar NM, von Knoch F, et al. Particle-induced osteolysis in three-dimensional micro-computed tomography. Calcif Tissue Int. 2007;81:394–402. doi: 10.1007/s00223-007-9077-2. [DOI] [PubMed] [Google Scholar]
- Weitzmann MN, Pacifici R. Estrogen regulation of immune cell bone interactions. Ann N Y Acad Sci. 2006;1068:256–274. doi: 10.1196/annals.1346.030. [DOI] [PubMed] [Google Scholar]
- Xia YF, Ye BQ, Li YD, Wang JG, He XJ, Lin X, et al. Andrographolide attenuates inflammation by inhibition of NF-kappa B activation through covalent modification of reduced cysteine 62 of p50. J Immunol. 2004;173:4207–4217. doi: 10.4049/jimmunol.173.6.4207. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM, et al. NF-kB p50 and p52 regulate RANKL and TNF-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem. 2007;282:18245–18253. doi: 10.1074/jbc.M610701200. [DOI] [PubMed] [Google Scholar]
- Ye H, Arron JR, Lamothe B, Cirilli M, Kobayashi T, Shevde NK, et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature. 2002;418:443–447. doi: 10.1038/nature00888. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wu C, Matesic LE, Li X, Wang Z, Boyce BF, et al. Ubiquitin E3 ligase itch negatively regulates osteoclast formation by promoting deubiquitination of tumor necrosis factor (TNF) receptor-associated factor 6. J Biol Chem. 2013;288:22359–22368. doi: 10.1074/jbc.M112.442459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Yang G, Liu H, Wang D, Song X, Chen Y. Determination of andrographolide, deoxyandrographolide and neoandrographolide in the Chinese herb Andrographis paniculata by micellar electrokinetic capillary chromatography. Phytochem Anal. 2002;13:222–227. doi: 10.1002/pca.644. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Wang X, Liu Y, He A, Jia R. NFATc1: functions in osteoclasts. Int J Biochem Cell Biol. 2010;42:576–579. doi: 10.1016/j.biocel.2009.12.018. [DOI] [PubMed] [Google Scholar]
- Zou W, Hakim I, Tschoep K, Endres S, Bar-Shavit Z. Tumor necrosis factor-alpha mediates RANK ligand stimulation of osteoclast differentiation by an autocrine mechanism. J Cell Biochem. 2001;83:70–83. doi: 10.1002/jcb.1202. [DOI] [PubMed] [Google Scholar]
- Zou W, Izawa T, Zhu T, Chappel J, Otero K, Monkley SJ, et al. Talin1 and Rap1 are critical for osteoclast function. Mol Cell Biol. 2013;33:830–844. doi: 10.1128/MCB.00790-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 AP inhibited RANKL-induced osteoclastogenesis of RAW 264.7 cells. (A) RAW 264.7 cells were treated with various concentrations of AP, followed by stimulation with RANKL (50 ng·mL−1) for six days. Cells were then fixed with 4% PFA and stained for TRAP. (B) TRAP-positive multinuclear cells were counted (*, P < 0.05; **, P < 0.01).