

Abstract
Background and Purpose
Dysregulation of the thromboxane A2 (TP) receptor, resulting in agonist hypersensitivity and hyper-responsiveness, contributes to exaggerated vasoconstriction in the hypoxic pulmonary artery in neonatal persistent pulmonary hypertension. We previously reported that hypoxia inhibits TP receptor phosphorylation, causing desensitization. Hence, we examined the role of PKA-accessible serine residues in determining TP receptor affinity, using site-directed mutational analysis.
Experimental Approach
Vasoconstriction to a thromboxane mimetic and phosphorylation of TP receptor serine was examined in pulmonary arteries from neonatal swine with persistent pulmonary hypertension and controls. Effects of hypoxia were determined in porcine and human TP receptors. Human TPα serines at positions 324, 329 and 331 (C-terminal tail) were mutated to alanine and transiently expressed in HEK293T cells. Saturation binding and displacement kinetics of a TP antagonist and agonist were determined in porcine TP, wild-type human TPα and all TP mutants. Agonist-elicited calcium mobilization was determined for each TP mutant, in the presence of a PKA activator or inhibitor, and in hypoxic and normoxic conditions.
Key Results
The Ser324A mutant was insensitive to PKA activation and hypoxia, had a high affinity for agonist and increased agonist-induced calcium mobilization. Ser329A was no different from wild-type TP receptors. Ser331A was insensitive to hypoxia and PKA with a decreased agonist-mediated response.
Conclusions and Implications
In hypoxic pulmonary hypertension, loss of site-specific phosphorylation of the TP receptor causes agonist hyper-responsiveness. Ser324 is the primary residue phosphorylated by PKA, which regulates TP receptor-agonist interactions. Ser331 mutation confers loss of TP receptor-agonist interaction, regardless of PKA activity.
Keywords: thromboxane, hypoxia, pulmonary hypertension, PKA
Introduction
Thromboxane A2 (TxA2) is an inflammatory prostanoid that contracts vascular myocytes via thromboxane A2 (TP) receptors (Hirata et al., 1991), leading to increased intracellular [Ca2+], force generation and sensitization of the contractile apparatus to Ca2+ (Cogolludo et al., 2003). The balance between vasodilator and vasoconstrictor prostanoid signalling is critical in cardiovascular homeostasis (Gleim et al., 2009; Feletou et al., 2010).
TxA2 is an important participant in neonatal circulatory transition (Reyes, 1993). The disease persistent pulmonary hypertension of the newborn (PPHN) is characterized by hypoxaemia and vasoconstrictor hyper-responsiveness. Hypoxic PPHN increases the serum TxA2 to prostacylin (PGI2) ratio (Fike et al., 2003). Hypoxia also sensitizes the dose–response relationship of pulmonary arteries to TP receptor agonists (Snow et al., 2008; Delannoy et al., 2010).
In humans, TxA2 signals through receptor isoforms TPα and β, splice variants that differ in their C-terminal tails (Hirata et al., 1991; Raychowdhury et al., 1994; 1995,). TPα expression predominates in most tissues, including vascular smooth muscle (Miggin and Kinsella, 1998). Both TP isoforms are rapidly phosphorylated in response to TxA2 agonists (Habib et al., 1999). Functional differences between TP isoforms reflect differing G-protein coupling, desensitization and receptor internalization following agonist stimulation (Hata and Breyer, 2004). The C-terminal sequence variation between TP isoforms results in different patterns of phosphorylation. TPβ, but not TPα, undergoes agonist-induced phosphorylation by G-protein receptor kinase (GRK), leading to receptor internalization (Parent et al., 1999), whereas the C-terminus of TPα is not capable of being phosphorylated by GRKs (Zhou et al., 2001). Desensitization of TP receptors commonly involves a decreased affinity of the receptor for its agonist and is associated with the establishment of an intermediate affinity state (Okwu et al., 1992), requiring TP receptor phosphorylation (Okwu et al., 1994). TPα undergoes desensitization due to phosphorylation of the C-terminus serine residues by PKA and PKC (Murray et al., 1990; Walsh et al., 2000a; Reid and Kinsella, 2003). The association of TPα desensitization with activation of adenylate cyclase-coupled receptors and increased cAMP production suggests that PKA is likely to be involved in this effect (Rosenblum et al., 1987).
We studied TP receptor hyper-reactivity in a porcine model of hypoxic PPHN. Only one porcine TP isoform has been identified; therefore, any variations in porcine TP signalling can be attributed to changes mediated by the hypoxic TP receptor not to receptor isoform differences. Amino acid sequence alignment indicates that the porcine TP receptor is homologous with the human TPα isoform, although the number of phosphorylation-amenable serine residues is greater in porcine TP (five serines) than in human TPα (four serines) (Supporting Information Figure S1). Gα/q co-immunoprecipitates with the porcine TP receptor (Fediuk et al., 2012), suggesting that porcine TP signalling is also similar to that of human TPα. Previously, we showed that serine phosphorylation is decreased in TP receptors from hypoxic myocytes (Hinton et al., 2006); this difference correlates with increased Gα/q coupling and TP agonist hyper-responsiveness in hypoxia, and is prevented by incubation of myocytes with PKA (Hinton et al., 2007). We previously demonstrated that hypoxia attenuates PKA activity in pulmonary arterial myocytes due to hypoxic impairment of prostacyclin receptor activity, thereby inducing dephosphorylation and sensitization of the TP receptor agonist response (Santhosh et al., 2011). However, we did not identify the serine residues responsible for the functional regulation of TP receptors. In this study, using in vivo and in vitro models, we examined agonist responses in hypoxic human and porcine TP receptors, and using site-directed mutagenesis, identified the phosphorylated serine residues that render the TP receptor desensitized in the presence of PKA activation.
Methods
Induction of hypoxic PPHN
Newborn piglets (<24 h age) were obtained from a pathogen-free farm supplier. In the hypoxic animal group (n = 5), pulmonary hypertension was induced by exposure to FiO2 0.10 (by nitrogen washout) for 72 h, in a temperature- and humidity-controlled sealed Plexiglass isolette. Gas concentrations were maintained ±1%. Age-matched normoxic control pigs (n = 5) were identically raised in 21% oxygen. Formula (swine milk replacer) was provided ad libitum. Weights were monitored daily. Any animal unable to maintain adequate oral intake was gavage fed. This protocol was approved by the University of Manitoba and accords with the Canadian Council on Animal Care guidelines. Newborn (<24 h; n = 5) pigs were used as a baseline control. Animals were killed with 480 mg·kg−1 pentobarbital i.p. Heart and lungs were removed en bloc into cold Krebs–Henseleit buffer. Induction of PPHN was confirmed in hypoxic animals by the presence of right ventricular hypertrophy (weight of right ventricle/left ventricle + septum) (Dakshinamurti et al., 2005; Saini-Chohan et al., 2011). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Pulmonary artery isometric myography
Small conduit pulmonary arteries (1 mm in diameter) from newborn, PPHN and control pigs were mounted on a multi-chamber wire myograph (Danish Myo Systems, Aarhus, Denmark) in Krebs–Henseleit buffer at 37°C and pH 7.4, aerated with 95% O2, 5% CO2. Vessels were equilibrated at optimal length (determined from a length/tension curve deriving maximum active tension to KCl stimulation) for measurement of isometric contraction to serial concentrations of the thromboxane mimetic U46619. Force measurements were normalized to maximal contraction to 100 mM KCl.
Tissue TP phosphorylation assay
Second to sixth generation pulmonary arteries microdissected from PPHN or control lungs were recovered in cold HEPES-buffered saline (HBS) supplemented with antibiotic/antimycotic, then washed in HBS with 20 μM CaCl2, finely minced and digested in 1750 U·mL−1 type I collagenase, 1 mM DTT, 2 mg·mL−1 BSA and 9.5 U·mL−1 papain for 15 min at 37°C. Dispersed pulmonary artery myocytes were collected by brief centrifugation and resuspended in Ham's F-12 medium with 10% FBS, 1% penicillin and 1% streptomycin, and then plated at a density of 4.4 × 104 cells·cm–2 (Shimoda et al., 2000). Confluent cells were lysed into lysis buffer (in mM; 50 Tris, 150 NaCl, 1 EDTA, 1 PMSF and 1% Triton-X-100; pH 7.4). Five hundred micrograms of lysate was pre-cleared by incubation with 35 μL of 50% slurry of Protein G Sepharose beads (GE Healthcare, Mississauga, ON, Canada). Beads were removed by centrifugation at 16000× g for 5 min. Pre-cleared lysates were then incubated at 4°C overnight with 2 μg rabbit anti-TP antibody (Cayman Chemicals, Ann Arbor, MI, USA). The immunoprecipitate was pulled down with 30 μL of 50% bead slurry, and proteins were removed from beads by boiling in Laemmli buffer. Eluted proteins were separated by SDS-PAGE, and blots were probed with mouse anti-phospho-serine antisera (Sigma, Oakville, ON, Canada).
TP receptor GTPase assay
Newborn pulmonary artery myocytes in primary culture were grown in hypoxic (10% O2, 5% CO2) or normoxic (21% O2, 5% CO2) conditions for 72 h in vitro, and then challenged with buffer or 10−6 M TP receptor agonist U46619, with or without 10−5 M SQ29548 (TP antagonist), for 2 min before flash freezing. Membrane fractions obtained by centrifugation were added to a reaction mixture containing substrate GTP. The reaction was arrested by the addition of dye (Gold mix), and read at 590 nm excitation and 650 nm emission γ by plate reader. TP receptor-specific live cell GTPase activity was measured as GTP degradation to GDP and inorganic phosphate (Pi), via colorimetric assay of the dye-Pi complex (Innova Biosciences, Babraham, Cambridge, UK). Background enzyme activity was determined in unstimulated membranes. GTPase activity was calculated from a standard curve, expressed as inorganic phosphate liberated·min−1·mg−1 protein.
PKA assay
HEK293T cells were incubated for 72 h in hypoxic or normoxic conditions, as described above. After 3 min of incubation with 10−6 M forskolin, 10−6 M H8, 10−8 M milrinone or diluent, cells were lysed on ice. PKA activity was measured by a 96-well ProFluor assay kit (Promega, Madison, WI, USA). PKA substrate peptide, Bisamide Rhodamine 110 (PKA R110), was pre-incubated with 25 μL ATP, then the PKA assay reaction mixture for 30 min, and the reaction was terminated by 25 μL protease reagent in termination buffer with 25 μL stabilizing reagent. Fluorescence of non-phosphorylated substrate, which inversely correlates with PKA activity, was measured at 485 nm excitation and 530 nm emission λ, against reagent buffer blanks, using a FLUOstar Optima microplate reader. PKA activity was expressed as fold-change compared with (untreated) normoxic lysates.
PKC assay
HEK293T cells grown for 72 h in hypoxic or normoxic conditions were incubated for 3 min with 2°× 10−5 M PMA, 10−5 M Go6983 or vehicle. Cells were lysed, and PKC activity was measured using solid-phase elisa plates coated with PKC substrate and substrate-specific detector antibody (Enzo Life Sciences, Brockville, ON, Canada). Experiments were repeated in the presence of 10 μL H89 (PKA inhibitor) to exclude non-specific kinase activity. PKC activity was expressed as fold-change from normoxic controls.
Identification of potential TP phosphorylation sites
Potential phosphorylation sites on porcine and human TP receptor isoforms were identified using the online NetPhos 2.0 server, a neural network-based method for predicting potential phosphorylation sites at serine, threonine or tyrosine residues in protein sequences with sensitivity reported from 69 to 96% (Blom et al., 1999).
Stable or transient expression of TP receptors and substitution mutants
To establish stable TP expression, wild-type porcine TP or human TPα genes containing a rho-1D4 tag [C-terminal epitope T-E-T-S-Q-V-A-P-A-(COOH) of rhodopsin] were inserted into pcDNA 3.1 and transfected into HEK293T cells (10 mg plasmid per 10 cm plate) using lipofectamine (Invitrogen, Burlington, ON, Canada) as described (Chelikani et al., 2006). Cells were grown in DMEM with 10% FBS, 50 U·mL−1 penicillin and 50 mg·mL−1 streptomycin. After 48 h, stably expressing HTP and PTP clones were selected using 1 mg·mL−1 geneticin (Invitrogen) for 3 days. TP expression was detected by antibody to rho-1D4. One clone was selected for Ca2+ mobilization assay (Goto et al., 2012). Amino acid substitutions with alanines into positions Ser239, Ser324, Ser329 or Ser331 were introduced into a synthetic human TPα gene with rho-1D4 tag, carried by the pMT4 expression vector (Chakraborty et al., 2012). DNA sequences of all TP constructs were verified by automated sequencing. HEK293T cells in 6-well tissue-culture-treated BD-falcon plates were transiently transfected with 10 μg per well plasmid DNA containing wild-type TPα or mutants Ser239A, Ser324A, Ser329A and Ser331A, using lipofectamine as per the manufacturer's instructions. To minimize variation in transfection efficiency, 1 μg cDNA was transfected per 7 × 105 cells. After 12 h of transfection, media was replaced with DMEM-F12 with 5% FBS. Surface TP expression was verified by immunostaining. Membranes were prepared as previously described for binding assays (Chelikani et al., 2007; Chakraborty et al., 2012).
Radioligand binding assays
Saturation binding kinetics were assayed in 2–20 μg of separated membrane protein. Samples were incubated with the TP receptor antagonist [3H]-SQ29548 (0.1–50 nM) in binding buffer, with or without an excess of unlabelled agonist U46619 (10 μM) to determine non-specific binding, in 100 μL total volume for 1 h. Reactions were terminated by vacuum filtration (Millipore, Billerica, MA, USA), and membranes were washed with ice-cold binding buffer. Filters were agitated in distilled water to release adsorbed radioisotope and then equilibrated in 5 mL CytoScint (ICN) overnight. Unbound radioisotope was also collected. Counts min-1 were quantified for 3 min per sample by a liquid scintillation counter. The presence of single receptor populations, with non-cooperative binding, was confirmed by linear Scatchard transformation; equilibrium dissociation constants (Kd) and maximal binding sites (Bmax) were determined from saturation isotherms by non-linear curve fit. Competitive binding kinetics were determined by incubation of membrane fractions with 20 nM [3H]-SQ29548, followed by displacement with serial concentrations of cold U46619. Unbound antagonist was removed by filtration. Data were analysed using a one-site fit method to calculate Ki (Prism 4.03; GraphPad, La Jolla, CA, USA).
Calcium mobilization assay
After transfections, HEK293T cells were incubated for 72 h (stable cell line) or 48 h (transient transfection) in hypoxia or normoxia, prior to calcium mobilization studies. HEK293T cells stably expressing TP receptors were incubated with 10−6 M forskolin, H8, PMA, Go6983 or buffer for 1 h. Cells were loaded with calcium-sensitive dye Fluo-4NW (Invitrogen). Receptor-mediated Ca2+ mobilization was reported as change in fluorescence intensity after being treated with 10−4–10−10 M U46619 (TP agonist), in a Flexstation-3 fluorescence plate reader (Molecular Devices, Sunnyvale, CA, USA) at 525 nm emission following excitation at 494 nm, to generate dose–response curves after subtracting the responses of mock-transfected cells (negative controls, pMT4 vector only) to eliminate baseline fluorescence. EC50 values were calculated by non-linear regression analysis (Prism 4.03; GraphPad).
TP phosphorylation assay
HEK293T cells transiently expressing wild-type or mutant TP receptors were incubated with 10−6 M forskolin or diluent for 1 h, then lysed and TP-immunoprecipitated as described earlier, using an antibody to rho-1d4 tag (Abcam, Toronto, ON, Canada). Eluted fractions separated by SDS-PAGE were immunoblotted for phospho-serine (Sigma). Phosphorylation of wild-type or mutant TP receptors caused specifically by PKA was determined as phospho-serine band density of (TP with forskolin – TP with diluent)/TP with diluent.
Statistical analyses
Normally distributed quantitative data were analysed by Student's t-test or anova with Bonferroni's post test; dose–response curves and binding kinetics by non-linear curve fit. All data are expressed as mean ± SEM; P < 0.05 was considered significant.
Results
Effect of in vivo hypoxia on pulmonary artery TP responses
Pulmonary arteries from PPHN animals were sensitized to TP stimulation, resulting in a hyper-reactive force response to thromboxane challenge, compared with newborn or age-matched controls (Figure 1A). Myocytes from hypoxic PPHN pulmonary arteries exhibited diminished TP serine phosphorylation, even following primary culture under normoxic conditions (Figure 1B).
Figure 1.
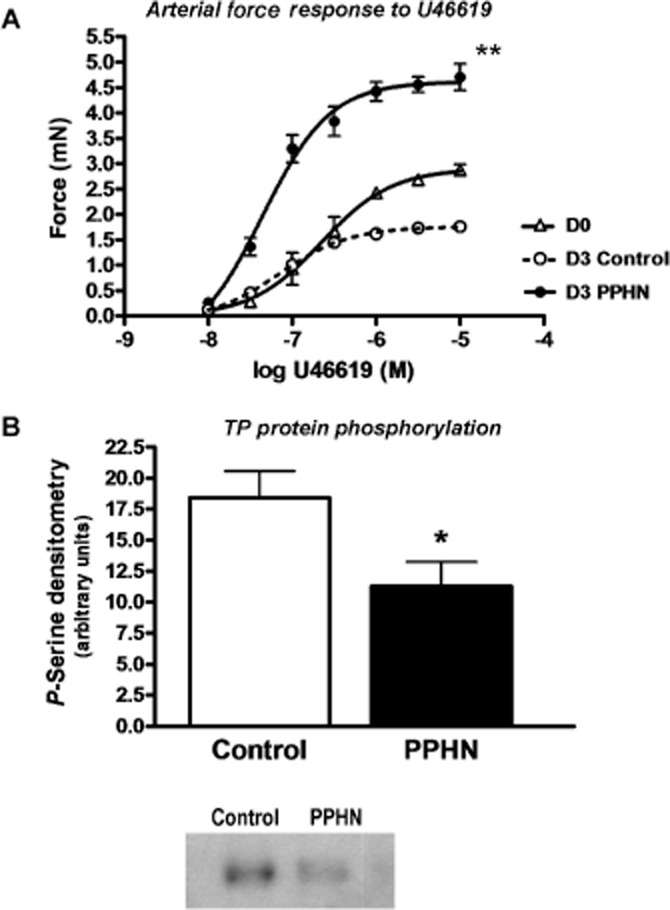
TP receptor phosphorylation is decreased in pulmonary artery myocytes from PPHN animals. (A) Pulmonary arterial rings from newborn (D0), 3-day-old normoxic (D3 Control) and hypoxic pulmonary hypertensive animals (D3 PPHN) were subjected to isometric myography (set at optimal length by length–tension curve; force measurements normalized to maximal KCl-induced force). n = 4 animals per group. Contraction force to TP agonist U46619 declines over the first 3 days of life in normal animals. Hypoxic PPHN increases TP receptor-mediated contraction, **P < 0.001. (B) Representative blot and histogram showing phosphorylation of TP receptors in contractile pulmonary arterial myocytes primary cultured from 3 day hypoxic PPHN swine and age-matched normoxic controls. Cell lysates were subjected to immunoprecipitation using polyclonal antibody (rabbit) to thromboxane receptor, and TP serine phosphorylation quantified in immunoprecipitates by Western blot (mouse monoclonal antibody to phospho-serine; *P < 0.05, n = 8). Total TP receptor protein content was unchanged (not shown).
Effect of in vitro hypoxia on TP receptor-specific GTPase activity
TP receptor-specific membrane GTPase activity was measured as inorganic phosphate release over time (after subtracting basal Pi release), indicative of cumulative GTP hydrolysis during the period of stimulation rather than the rate of hydrolysis. Increased GTP hydrolysis in hypoxic pulmonary artery myocytes infers greater cumulative G protein activation by the TP receptor (Figure 2).
Figure 2.
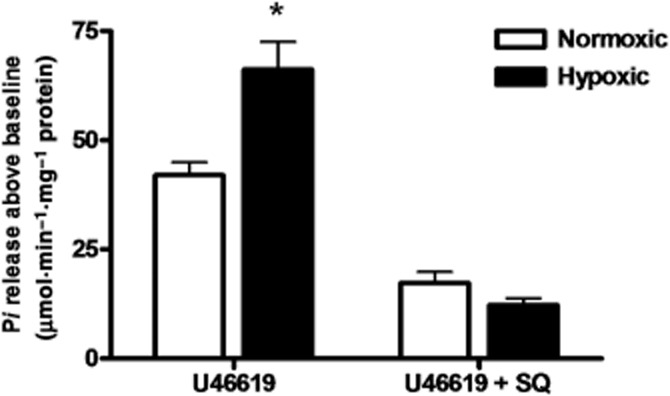
TP receptor-specific GTPase activity is increased in hypoxic pulmonary artery myocytes. Neonatal pulmonary artery myocytes grown in hypoxia or normoxia, challenged for 2 min with TP receptor agonist 10−6 M U46619, with or without TP receptor blockade by 10−5 M SQ29546, then flash-frozen. GTPase activity quantified in membrane fractions. Organic Pi liberated by membrane GTPase from substrate GTP in response to U46619 was calculated from a standard curve as [Pi] min−1·mg−1 protein, after subtraction of baseline (unstimulated) Pi release. Measured GTPase activity represents Pi release specifically due to TP-G-protein complex GTPase activity. This activity is sensitive to TP receptor blockade (+SQ) (data from three separate experiments in triplicate). *P < 0.05.
Effect of in vitro hypoxia on protein kinase and TP receptor activities in HEK293T cells
Wild-type porcine and human TPα receptor were stably expressed in HEK293T cells; comparable protein expression levels were confirmed by Western blot for the rho-1D4 tag, and plasma membrane localization of TP was verified by immunostaining of non-permeabilized cells (data not shown). After 72 h of in vitro hypoxia, porcine and human TP isoforms demonstrated comparable increases in agonist-induced Ca2+ mobilization (Figure 3). Hypoxia reduced PKA activity in HEK293T cells. PKA was activated by an AC activator, forskolin, and a PDE-3 inhibitor, milrinone, in hypoxic and normoxic PKA myocytes. Inhibition with H8 decreased the normoxic PKA activity to the hypoxic level (Figure 4A). Correspondingly, inhibition of PKA increased TP receptor-mediated Ca2+ mobilization in HEK293T cells stably transfected with wild-type TP; PKA activation reduced TP Ca2+ mobilization (Figure 4B). Hypoxia did not significantly reduce PKC activity. PKC was effectively activated by PMA and inhibited by Go6983 (Figure 4C). Both the PKC activator and inhibitor reduced the potency of TP receptor-mediated Ca2+ mobilization (Figure 4D).
Figure 3.
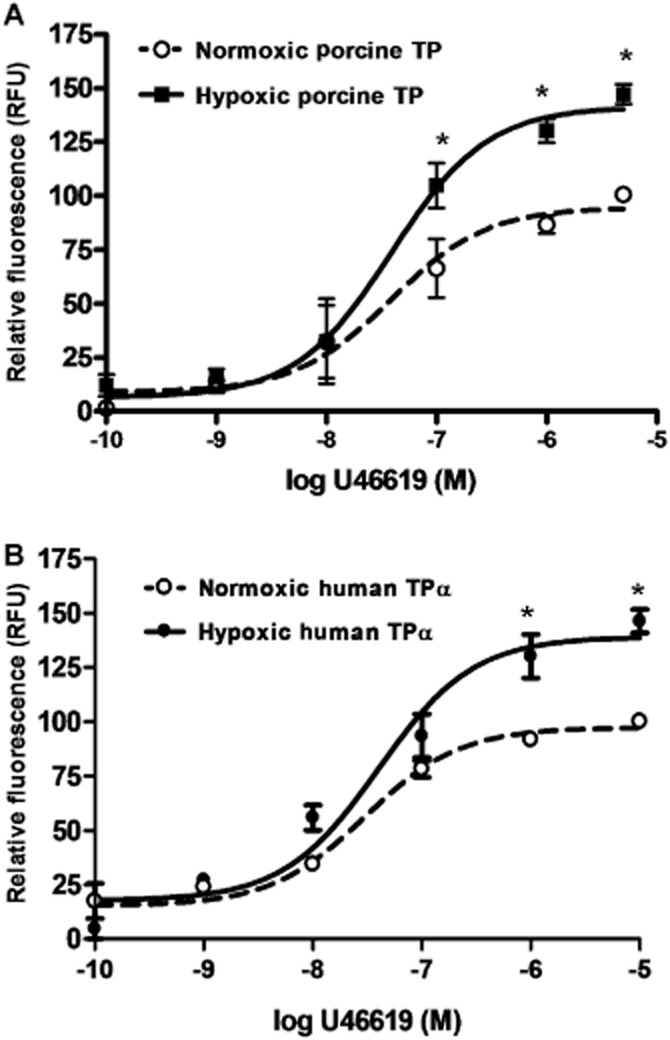
Human and porcine TP receptors expressed in HEK293T cells are hyper-responsive after 72 h of hypoxia. HEK293T clones stably expressing wild-type porcine TP (A) or human TPα (B) were exposed to hypoxic or normoxic culture conditions for 72 h, loaded with calciometric dye fluo-4 and challenged with serial concentrations of the thromboxane mimetic U46619. Ca2+ mobilization response to U46619 challenge under normoxic or hypoxic conditions quantified as relative fluorescence units following baseline subtraction; *P < 0.001, n = 4.
Figure 4.
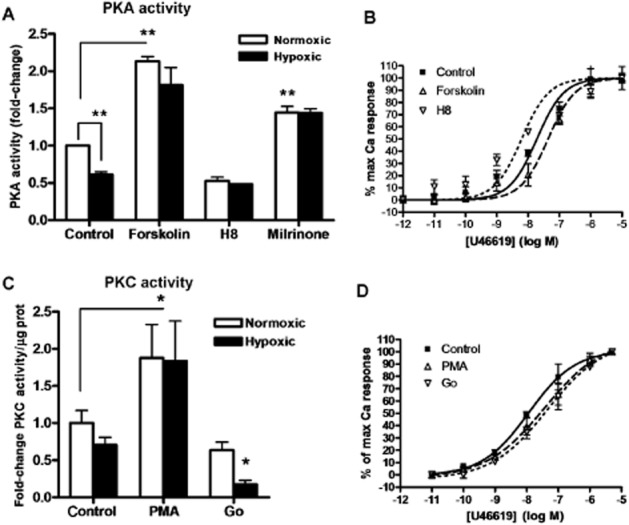
PKA, but not PKC, activity in HEK 293T cells is reduced by hypoxia. After 72 h of hypoxic or normoxic conditions,HEK293T cells were incubated with 10−6 M forskolin (activates PKA), 10−6 M H8 (inhibits PKA), 10−8 M milrinone (PDE-3 inhibitor), PMA (activates PKC), 10−6 M Go6983 (inhibits PKC) or diluent for 30 min. PKA (A) or PKC (C) activity measured in cell lysates, expressed as fold-change from normoxic control. TP-mediated Ca2+ mobilization in the presence of PKA activation or inhibition (B) or PKC activation or inhibition (D) determined using fluo-4, quantified as relative fluorescence units after baseline subtraction, normalized to maximal Ca2+ response. n = 4, **P < 0.001 compared with normoxic control.
Expression and ligand binding properties of TP receptor substitution mutants
Based on the NetPhos sequence analysis, residues Ser239 (located on intracellular loop 3) and Ser324, Ser329 and Ser331 (on the C-terminal tail; Supporting Information Figure S2) of the porcine TP and TPα receptor were predicted for phosphorylation with high probability scores (0.994, 0.985, 0.991 and 0.927 respectively). Immunoprecipitates containing TP mutants Ser324A and Ser331A tended to have decreased PKA-mediated serine phosphorylation compared with wild-type TP receptors, although this did not achieve statistical significance (Supporting Information Figure S3).
TP receptor saturation binding kinetics, maximal receptor binding (Bmax) and dissociation constant (Kd, in nM) were determined using cell membrane fractions expressing wild-type human TPα, porcine TP or TP receptor substitution mutants, using the TP antagonist [3H]-SQ29548. Human TPα and porcine TP receptors had similar Kd (3.03; 3.01 respectively) and Bmax values (783 ± 119; 741 ± 97; data not shown). Among the serine substitution mutants (Table 1), Ser239A and Ser329A had Kd values comparable to the wild-type TP receptor. The Kd of Ser324A was lower (P < 0.05) than that of the wild-type TP receptor, indicating increased radioligand affinity. The Ser331A Kd was increased (P < 0.05) compared with the wild-type receptor. There were no measurable differences in Bmax for any mutant capable of binding SQ29548. Scatchard analysis indicated non-cooperative agonist binding kinetics (not shown).
Table 1.
Radioligand binding and displacement kinetics of human TPα and human TP amino acid substitution mutants
| Receptor | Kda (nM) | 95% Confidence interval | Bmaxb (pM·mg−1) | Normoxic log Ki%c | Hypoxic log Ki%c |
|---|---|---|---|---|---|
| TPα wt | 3.79 | 2.80–4.78 | 783 ± 119 | −8.759 | −10.51 |
| Ser239A | 3.90 | 3.47–4.33 | 969 ± 48 | – | – |
| Ser324A | 2.23 | 1.81–2.65 | 1005 ± 61 | −11.43 | −11.69 |
| Ser329A | 3.80 | 2.21–5.08 | 968 ± 45 | −8.387 | −9.662 |
| Ser331A | 5.80 | 4.81–6.79 | 857 ± 41 | −8.389 | −9.082 |
Radioligand saturation binding kinetics assayed for human wild-type TPα and human TPα serine-to-alanine substitution mutants Ser239A, Ser324A, Ser329A and Ser331A, using labelled TP receptor antagonist [3H]-SQ29548; values from three experiments in duplicate.
Dissociation constant (Kd), expressing inverse of affinity of antagonist SQ29548 for the receptor. Boldface denotes receptor antagonist binding affinity differs from wild type (P < 0.05).
Maximum binding (Bmax) of the antagonist SQ29548 for TP receptor, usually expressed as pmol of the receptor mg-1 total membrane protein; mean ± SEM.
Competitive displacement kinetics under conditions of hypoxic and normoxic cell growth; saturating concentration of labelled antagonist [3H]-SQ29548, displaced with serial concentrations of cold agonist U46619. Ki is the equilibrium dissociation constants, calculated from concentration of agonist inhibiting 50% of antagonist binding. Boldface values differ from normoxic wild type Ki (P < 0.05).
Effects of PKA and in vitro hypoxia on TP responses
Agonist dose–response curves for transiently expressed wild-type TPα, Ser239A and Ser329A were similar. Treatment with 10−6 M forskolin did not alter agonist dose–response relationships of Ser239A or Ser329A, indicating that these receptors remained maximally phosphorylated, whereas 10−6 M H8 treatment sensitized both dose–response curves, indicating that these constructs were amenable to dephosphorylation (Figure 5A,C). Ser324A exhibited a left-shifted agonist response compared with wild type TP receptors. Neither forskolin nor H8 was capable of rectifying the Ser324A Ca2+ response to U46619; it remained elevated compared with wild-type TP (Figure 5B). The Ser331A TP receptor mutant was relatively desensitized to agonist stimulation compared with wild-type TP receptors. Neither forskolin nor H8 treatment altered the response of Ser331A to U46619; it remained desensitized in comparison to wild-type TP receptors (Figure 5D). A comparison of log EC50 values is given in Table 2012. A heterologous competitive displacement assay demonstrated that for wild-type TP receptors, U46619 displaced labelled antagonist more easily in normoxia than in hypoxia (P < 0.01). Ser324A showed high affinity binding to U46619 in both normoxia and hypoxia (Figure 6A). Ser329A bound U46619 with an affinity comparable with that of the native receptor under hypoxic and normoxic conditions (Figure 6B). Both hypoxic and normoxic Ser331A had affinities for U46619 comparable to that of normoxic wild-type TP receptors (Figure 6C). A comparison of log Ki values is included in Table 1. Finally, we examined the effects of C-terminal serine substitution mutants on the hypoxia-induced sensitization of the TP receptor-agonist interaction. Due to loss of transiently transfected cells after 72 h hypoxia exposures, these dose–response curves were obtained after 48 h of hypoxia. Hypoxia still caused relative TP receptor hyper-responsiveness at this time point, although less marked than after 72 h of exposure. Ser324A mutants had a sensitized TP agonist dose–response relationship compared to normoxic cells, which resembled that of hypoxic cells (Figure 7A), although without a discernible effect on maximal reactivity. Ser329A had a normal dose-response relationship with no increased sensitivity or changes in maximal calcium mobilization in either hypoxic or normoxic cells (Figure 7B). Ser331A had a right-shifted (desensitized) dose–response curve and depressed maximal reactivity in both hypoxic and normoxic cells (Figure 7C). A comparison of log EC50 values is presented in Table 3.
Figure 5.
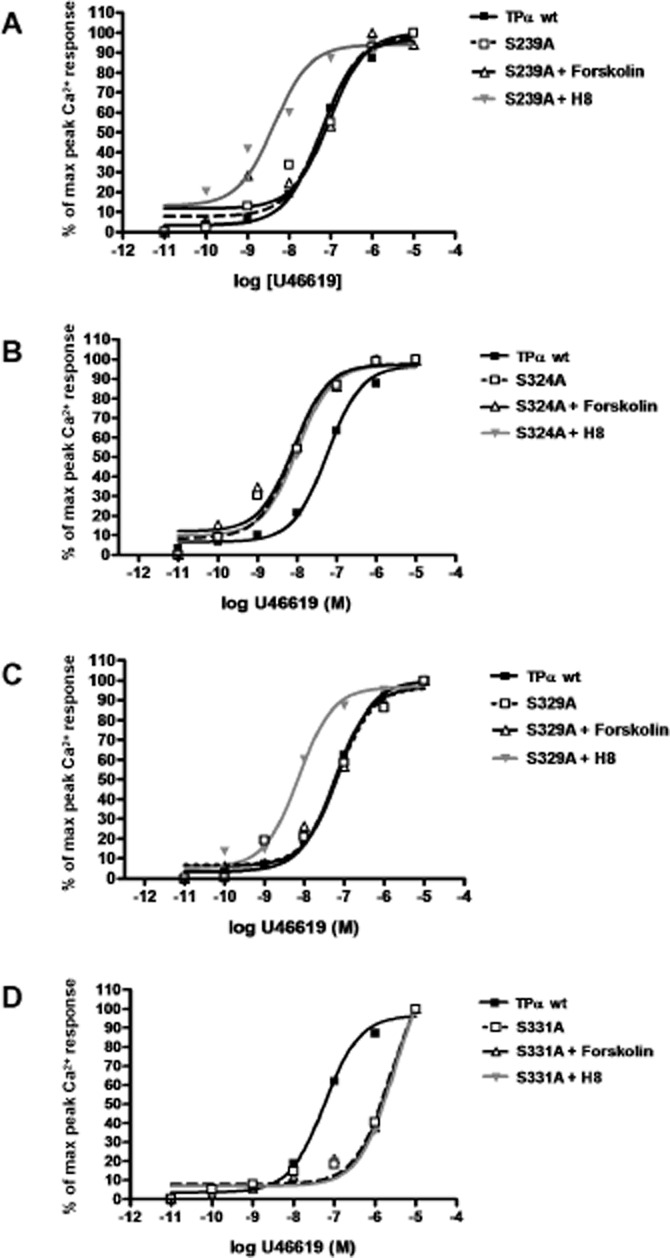
PKA sensitivity of wild-type TPα and amino acid substitution mutants. HEK293T cells transfected with human wild-type TPα, or serine-to-alanine substitution mutants Ser239A, Ser324A, Ser329A or Ser331A were incubated with 10−6 M forskolin (activates PKA), 10−6 M H8 (inhibits PKA) or diluent, then challenged with serial concentrations of TP agonist U46619. (A) Alanine substitution of Ser239 did not alter PKA sensitivity of the TP agonist concentration-response curve relative to the wild-type TP receptor. (B) Substitution of Ser324 sensitized the receptor to the agonist and abolished PKA responsiveness. (C) The Ser329A mutant had PKA sensitivity comparable to wild-type TP receptors. (D) Ser331 substitution reduced the sensitivity of the receptor to the TP receptor agonist, regardless of PKA activation state. Corresponding EC50 values are given in Table 2.
Table 2.
Dose–response relationships of TP substitution mutants in presence of PKA activation or inhibition
| Receptor | log EC50a untreated | log EC50a when PKA activated | log EC50a when PKA inhibited |
|---|---|---|---|
| TPα wt | −7.24 ± 0.08 | – | – |
| Ser239A | −7.18 ± 0.22 | −7.03 ± 0.27 | −8.36 ± 0.30 |
| Ser324A | −8.10 ± 0.18 | −8.09 ± 0.23 | −8.00 ± 0.19 |
| Ser329A | −7.16 ± 0.19 | −7.14 ± 0.13 | −8.15 ± 0.11 |
| Ser331A | −5.63 ± 0.17 | −5.60 ± 0.20 | −5.54 ± 0.20 |
log EC50 = log molar concentration of agonist U46619 that produces 50% of maximal peak calcium mobilization response for TP receptors and TP substitution mutant receptors. Boldface values differ from reference value, untreated wild type (P < 0.05).
Figure 6.
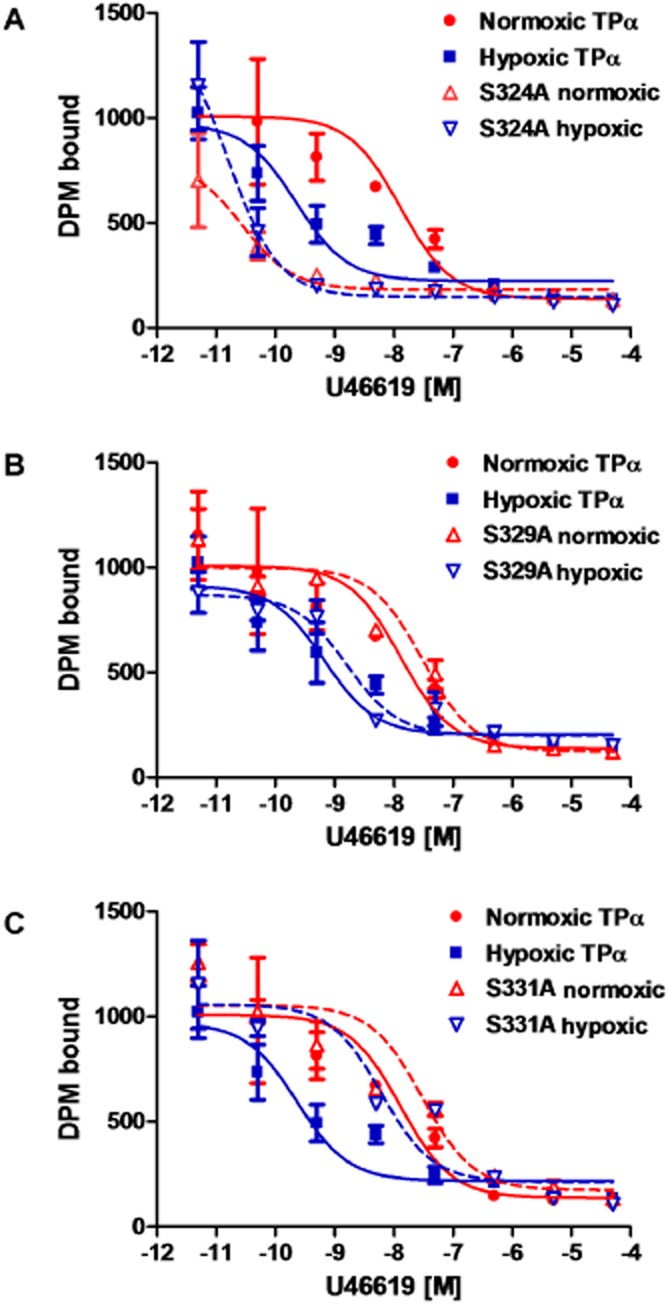
Hypoxia increases competitive binding of agonist to TPα; effect of C-terminal amino acid substitution mutation. HEK293T cells transfected with (A) human wild-type TPα, or serine-to-alanine mutants (B) Ser324A, (C) Ser329A or (D) Ser331A, following 48 h of hypoxic or normoxic exposure, were incubated with a saturating concentration of [3H]-SQ29548, before antagonist displacement with serial concentrations of cold agonist U46619. Data are from three experiments.
Figure 7.
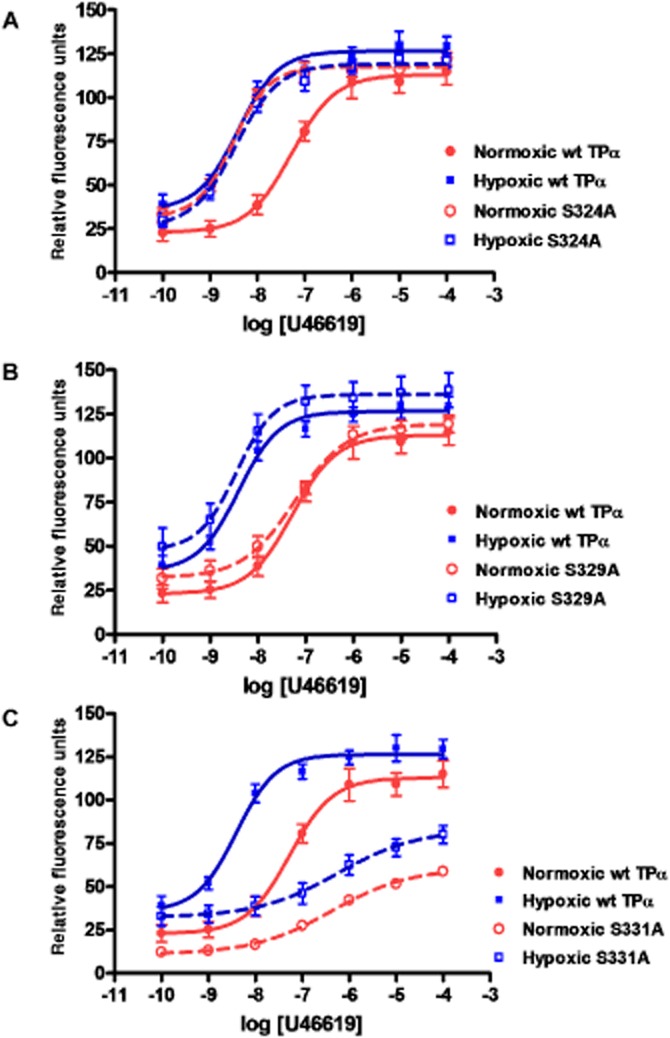
Hypoxia increases agonist-induced Ca2+ mobilization by TPα; effect of C-terminal amino acid substitution mutation. HEK293T cells transfected with human wild-type TPα, or serine-to-alanine substitution mutants Ser324A, Ser329A or Ser331A were incubated for 48 h in hypoxic or normoxic conditions, followed by challenge with serial concentrations of agonist U46619. Absolute Ca2+ mobilization curves showing (A) differing effects of Ser324 substitution on agonist sensitivity in normoxic versus hypoxic conditions, relative to wild-type TP receptors; (B) effect of Ser329 substitution on hypoxia sensitivity of TP receptor-mediated Ca2+ response; and (C) effect of Ser331 substitution on hypoxic and normoxic TP agonist responses.
Table 3.
Dose-response relationships of TP receptors and TP substitution mutants in normoxia or hypoxia
| Receptor | log EC50a in normoxia | log EC50a in hypoxia |
|---|---|---|
| TPα wt | −7.24 ± 0.10 | −8.31 ± 0.09 |
| Ser324A | −8.42 ± 0.08 | −8.35 ± 0.08 |
| Ser329A | −7.21 ± 0.09 | −8.39 ± 0.15 |
| Ser331A | −6.44 ± 0.09 | −6.34 ± 0.17 |
log EC50 = log molar concentration of agonist U46619 that produces 50% of maximal peak calcium mobilization response for TP receptors and TP substitution mutant receptors. Boldface values differ from normoxic wild type (P < 0.05).
Discussion
In this study, the phosphorylation potential of TP serine residues was elucidated using ex vivo experiments on pulmonary arterial rings, and in vitro site-directed mutagenesis of phosphorylation-amenable serine residues.
Hypoxia induces the PPHN phenotype (Dakshinamurti et al., 2005; Saini-Chohan et al., 2011) and renders the pulmonary circuit hyper-responsive to thromboxane. Whereas TP receptor isoform switching has been postulated as a mechanism for sensitization, as only one TP receptor isoform is expressed in swine, isoform switching cannot be cited as a rationale for altered signalling in this PPHN model. We have previously shown that in vitro hypoxia impairs TP receptor phosphorylation in pulmonary artery myocytes. In this study, we demonstrated for the first time that diminished TP receptor phosphorylation exists in the porcine pulmonary artery after in vivo hypoxia, in association with increased force generation to challenge with a thromboxane mimetic. This disease-specific finding provides physiological validation for studying TP receptor phosphorylation in vitro.
The hypoxia and PKA sensitivity of wild-type TP receptors and serine substitution mutants were then assessed to identify specific serine residues responsible for PKA-mediated regulation of TP receptor signalling, which may be important in hypoxic pulmonary hypertension. Multiple regulatory kinases and phosphorylation sites have been studied in relation to TP receptors. The TP receptor is phosphorylated through actions of pulmonary circuit relaxants, particularly PGI2, resulting in TP receptor desensitization (Reid and Kinsella, 2003; O'Meara and Kinsella, 2004). IP-induced TP phosphorylation has been variously ascribed to actions of PKC (Kelley-Hickie and Kinsella, 2004), G-protein-related kinases (Zhou et al., 2001) or PKA (Habib et al., 1997; Foley et al., 2001; Reid and Kinsella, 2003). The major effector of IP receptor-mediated TPα regulation is PKA (Gleim et al., 2009). PKA plays a role in agonist-induced desensitization of TPα (Habib et al., 1997), whereas GRK phosphorylation of Ser239 is implicated in agonist-induced desensitization and internalization of only TPβ (Kelley-Hickie and Kinsella, 2006). The role of PKC in TP receptor desensitization is unclear. PKC causes an initial reduction in TP receptor-ligand binding followed by a time-dependent increase (Kinsella et al., 1994); inhibition of PKC very modestly increases TP ligand binding (Walsh et al., 2000a). Pharmacological PKC inhibitors cross-react with TP-specific GRKs, necessitating cautious evaluation of TP regulation by PKC (Zhou et al., 2001). We previously reported decreased serine phosphorylation of TP in pulmonary arterial myocytes after in vitro hypoxia and established that loss of PKA- (and not PKC-) mediated TP phosphorylation was a mechanism for thromboxane hyper-reactivity in hypoxia. PKC activation did not decrease TP agonist response (Hinton et al., 2007). TP agonist sensitivity is tightly regulated by PKA, giving rise to just two characteristic agonist–response relationships corresponding to phosphorylated (desensitized) or dephosphorylated (sensitized) TP receptors (Santhosh et al., 2011). Our current findings confirm that PKA activation and inhibition reciprocally modulate TP receptor-mediated Ca2+ release. PKC activation and inhibition do not have this precise effect on the TP dose–response curve. We therefore conclude that PKA-mediated phosphorylation determines TP receptor affinity.
We previously reported that hypoxia impairs basal PKA activity in pulmonary arterial myocytes (Santhosh et al., 2011); in the present study, we confirm this phenomenon in hypoxic HEK293T cells, signalling-intact mesenchymal cells that lack native TP and IP receptors. Stably transfected porcine and human TPα are hyper-responsive to U46619 challenge under hypoxic conditions. Transiently transfected HEK293T cells were less robust under hypoxic conditions, so studies of the transiently transfected TP mutants were carried out after 48 h of hypoxic exposure. Both potency and efficacy of U46619 response are increased after 72 h of hypoxia; in studies carried out after 48 h of hypoxia, there was increased agonist potency but little change in efficacy. All TP mutants studied were expressed with similar Bmax and were able to bind labelled antagonist. We focused our attention on C-terminal serines, commonly implicated in receptor regulation. Whereas Ser239A and Ser329A had ligand binding parameters similar to wild-type TP receptors, the Ser324A mutation had increased interaction with both agonist and antagonist. Agonist-mobilized Ca2+ responses of both the Ser239A and Ser329A mutants exhibited sensitivity to PKA activity, as did wild-type TP receptors, indicating that these serines are not associated with regulatory phosphorylation of TP receptors by PKA.
Ser324 has been implicated in PKA-mediated regulation of TP receptors (Walsh et al., 2000b; Foley et al., 2001), although due to differing residue enumeration between research groups, this specific residue is identified in some studies as Ser329. In vitro studies have shown that Ser324 is only marginally phosphorylated by PKC (Yan et al., 2002). In our hands, Ser324A bound to U46619 with high affinity compared with wild-type TPα. The dose–response relationship for Ca2+ mobilization showed that this receptor was hypersensitive to agonist stimulation compared to control TP receptors. Ser324A Ca2+ signalling was insensitive to PKA activation or inhibition, and this construct remained sensitized to agonist stimulation under normoxic conditions, indicating its resistance to regulatory phosphorylation. Hence, we concluded that phosphorylation of Ser324 may be responsible for the PKA-mediated down-regulation in the responsiveness of TP receptors to agonists in the normoxic pulmonary artery.
In contrast, the Ser331A TP mutant showed mildly decreased affinity for agonist and antagonist, compared with wild-type TPα. Substitution of this residue renders it insensitive to PKA inhibition and activation, but with a consistently desensitized agonist dose–response curve and a depressed agonist response in both hypoxia and normoxia. These data suggest that Ser331 is important for regulating TP receptor function, but that loss of phosphorylation at this locus decreases receptor signalling. Other groups have identified Ser331 as being amenable to the phosphorylation and desensitization induced by both PKG (Yamamoto et al., 2001) and PKA (Yamamoto et al., 2002). Our data do not support this finding, but paradoxically suggest that dephosphorylation of Ser331 may serve to decrease TP receptor activity, without significantly altering membrane localization. We speculate that substitution of Ser331 reduces the functionality of the receptor protein. Site-specific receptor phosphorylation can promote positive or negative feedback loops for receptor signalling (Tobin, 2008). PKA-mediated phosphorylation of the β2-adrenoceptor may determine selective G-protein coupling, distinct from mechanisms of desensitization (Daaka et al., 1997). It is therefore possible that loss of Ser331 phosphorylation due to alanine substitution induces the TP receptor to dissociate from its cognate G-protein, or inhibits regulatory phosphorylation occurring at other sites, resulting in the observed loss of activity.
We propose that Ser324 and Ser331 are important residues regulating TP receptor function by phosphorylation. The Ser324A and Ser331A mutants shift the U46619 concentration–response curve in opposite directions, implying that phosphorylation at these two sites has opposite effects. Ser324 phosphorylation by PKA desensitizes the TP receptor. Ser331 phosphorylation has a relatively moderate sensitizing effect, whereas phosphorylation-resistant substitution of this residue markedly reduces TP receptor signalling activity. Cumulatively, the net effect of PKA activation on native TP (human and porcine) receptors is one of diminished receptor-agonist interaction and decreased agonist-mediated Ca2+ mobilization.
Hypoxia induces a loss of PKA-mediated TP receptor phosphorylation in the neonatal pulmonary circuit; the loss of TP receptor phosphorylation and increase in TP receptor-mediated contractility is demonstrable both ex vivo and in vitro. Ser324 appears to be the critical residue for this dysregulation. We previously demonstrated that hypoxic-induced TP receptor hyper-responsiveness may be alleviated by treatment with the PDE inhibitor, milrinone; we now confirm that Ser324 phosphorylation by PKA is the probable mechanism for this pharmacological effect.
Acknowledgments
This work was supported by Canadian Institutes of Health Research (S. D.) and Heart and Stroke Foundation of Canada (S. D.). A. S. holds a graduate studentship from Manitoba Health Research Council.
Glossary
- PPHN
persistent pulmonary hypertension of the newborn
- TP
thromboxane prostanoid receptor
- TxA2
thromboxane A2
Conflict of interest
The authors declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12487
Protein sequence alignment of porcine TP and human TP receptor isoforms. 2D structure of human TPα, indicating position of the four serines targeted for alanine substitution. Comparative sequence alignment of human TP isoforms, TPα and TPβ, with porcine TP; S324, S329 and S331 are the only phosphorylatable C-terminal residues common to human TPα and porcine TP (but not TPβ).
Phosphorylation of TP mutants by PKA. HEK293T cells transfected with human wild-type TPα, or serine-to-alanine substitution mutants S324A, S329A or S331A incubated with 10−6 M forskolin (activates PKA) or diluent; TP immunoprecipitated using antibody to the rho-1D4 tag. Serine phosphorylation quantified in immunoprecipitates by Western blot (mouse monoclonal antibody to phospho-serine; n > 4). Ability of wild-type or alaninesubsitution mutant TP isoforms to be phosphorylated by PKA was determined as density of phospho-serine bands for (TP with forskolin – TP with diluent)/TP with diluent.
Effects of PKA and PKC on maximal TP agonistmediated calcium mobilization. TP-mediated Ca2+ mobilization in the presence of PKA activation or inhibition (A) or PKC activation or inhibition (B), determined using fluo-4, quantified after baseline subtraction. n > 4.
References
- Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Pydi SP, Gleim S, Dakshinamurti S, Hwa J, Chelikani P. Site-directed mutations and the polymorphic variant Ala160Thr in the human thromboxane receptor uncover a structural role for transmembrane helix 4. PLoS ONE. 2012;7:e29996. doi: 10.1371/journal.pone.0029996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelikani P, Reeves PJ, Rajbhandary UL, Khorana HG. The synthesis and high-level expression of a beta2-adrenergic receptor gene in a tetracycline-inducible stable mammalian cell line. Protein Sci. 2006;15:1433–1440. doi: 10.1110/ps.062080006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelikani P, Hornak V, Eilers M, Reeves PJ, Smith SO, RajBhandary UL, et al. Role of group-conserved residues in the helical core of beta2-adrenergic receptor. Proc Natl Acad Sci U S A. 2007;104:7027–7032. doi: 10.1073/pnas.0702024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circ Res. 2003;93:656–663. doi: 10.1161/01.RES.0000095245.97945.FE. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- Dakshinamurti S, Mellow L, Stephens NL. Regulation of pulmonary arterial myosin phosphatase activity in neonatal circulatory transition and in hypoxic pulmonary hypertension: a role for CPI-17. Pediatr Pulmonol. 2005;40:398–407. doi: 10.1002/ppul.20290. [DOI] [PubMed] [Google Scholar]
- Delannoy E, Courtois A, Freund-Michel V, Leblais V, Marthan R, Muller B. Hypoxia-induced hyperreactivity of pulmonary arteries: role of cyclooxygenase-2, isoprostanes, and thromboxane receptors. Cardiovasc Res. 2010;85:582–592. doi: 10.1093/cvr/cvp292. [DOI] [PubMed] [Google Scholar]
- Fediuk J, Gutsol A, Nolette N, Dakshinamurti S. Thromboxane induced actin polymerization in hypoxic pulmonary artery is independent of Rho. Am J Physiol Lung Cell Mol Physiol. 2012;302:L13–26. doi: 10.1152/ajplung.00016.2011. [DOI] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM, Verbeuren TJ. The thromboxane/endoperoxide receptor (TP): the common villain. J Cardiovasc Pharmacol. 2010;55:317–332. doi: 10.1097/fjc.0b013e3181d8bc8a. [DOI] [PubMed] [Google Scholar]
- Fike CD, Kaplowitz MR, Pfister SL. Arachidonic acid metabolites and an early stage of pulmonary hypertension in chronically hypoxic newborn pigs. Am J Physiol Lung Cell Mol Physiol. 2003;284:L316–L323. doi: 10.1152/ajplung.00228.2002. [DOI] [PubMed] [Google Scholar]
- Foley JF, Kelley LP, Kinsella BT. Prostaglandin D(2) receptor-mediated desensitization of the alpha isoform of the human thromboxane A(2) receptor. Biochem Pharmacol. 2001;62:229–239. doi: 10.1016/s0006-2952(01)00661-x. [DOI] [PubMed] [Google Scholar]
- Gleim S, Kasza Z, Martin K, Hwa J. Prostacyclin receptor/thromboxane receptor interactions and cellular responses in human atherothrombotic disease. Curr Atheroscler Rep. 2009;11:227–235. doi: 10.1007/s11883-009-0035-5. [DOI] [PubMed] [Google Scholar]
- Goto S, Saito M, Obara Y, Moriya T, Nakahata N. Involvement of lipid rafts in multiple signal transductions mediated by two isoforms of thromboxane A(2) receptor: dependency on receptor isoforms and downstream signaling types. Eur J Pharmacol. 2012;693:15–24. doi: 10.1016/j.ejphar.2012.07.046. [DOI] [PubMed] [Google Scholar]
- Habib A, Vezza R, Creminon C, Maclouf J, FitzGerald GA. Rapid, agonist-dependent phosphorylation in vivo of human thromboxane receptor isoforms. Minimal involvement of protein kinase C. J Biol Chem. 1997;272:7191–7200. doi: 10.1074/jbc.272.11.7191. [DOI] [PubMed] [Google Scholar]
- Habib A, FitzGerald GA, Maclouf J. Phosphorylation of the thromboxane receptor alpha, the predominant isoform expressed in human platelets. J Biol Chem. 1999;274:2645–2651. doi: 10.1074/jbc.274.5.2645. [DOI] [PubMed] [Google Scholar]
- Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Hinton M, Mellow L, Halayko AJ, Gutsol A, Dakshinamurti S. Hypoxia induces hypersensitivity and hyperreactivity to thromboxane receptor agonist in neonatal pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2006;290:L375–L384. doi: 10.1152/ajplung.00307.2005. [DOI] [PubMed] [Google Scholar]
- Hinton M, Gutsol A, Dakshinamurti S. Thromboxane hypersensitivity in hypoxic pulmonary artery myocytes: altered TP receptor localization and kinetics. Am J Physiol Lung Cell Mol Physiol. 2007;292:L654–L663. doi: 10.1152/ajplung.00229.2006. [DOI] [PubMed] [Google Scholar]
- Hirata M, Hayashi Y, Ushikubi F, Yokota Y, Kageyama R, Nakanishi S, et al. Cloning and expression of cDNA for a human thromboxane A2 receptor. Nature. 1991;349:617–620. doi: 10.1038/349617a0. [DOI] [PubMed] [Google Scholar]
- Kelley-Hickie LP, Kinsella BT. EP1- and FP-mediated cross-desensitization of the alpha (alpha) and beta (beta) isoforms of the human thromboxane A2 receptor. Br J Pharmacol. 2004;142:203–221. doi: 10.1038/sj.bjp.0705695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley-Hickie LP, Kinsella BT. Homologous desensitization of signalling by the beta (beta) isoform of the human thromboxane A2 receptor. Biochim Biophys Acta. 2006;1761:1114–1131. doi: 10.1016/j.bbalip.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella BT, O'Mahony DJ, FitzGerald GA. Phosphorylation and regulated expression of the human thromboxane A2 receptor. J Biol Chem. 1994;269:29914–29919. [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miggin SM, Kinsella BT. Expression and tissue distribution of the mRNAs encoding the human thromboxane A2 receptor (TP) alpha and beta isoforms. Biochim Biophys Acta. 1998;1425:543–559. doi: 10.1016/s0304-4165(98)00109-3. [DOI] [PubMed] [Google Scholar]
- Murray R, Shipp E, FitzGerald GA. Prostaglandin endoperoxide/thromboxane A2 receptor desensitization. Cross-talk with adenylate cyclase in human platelets. J Biol Chem. 1990;265:21670–21675. [PubMed] [Google Scholar]
- O'Meara SJ, Kinsella BT. Investigation of the effect of the farnesyl protein transferase inhibitor R115777 on isoprenylation and intracellular signalling by the prostacyclin receptor. Br J Pharmacol. 2004;143:318–330. doi: 10.1038/sj.bjp.0705956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okwu AK, Ullian ME, Halushka PV. Homologous desensitization of human platelet thromboxane A2/prostaglandin H2 receptors. J Pharmacol Exp Ther. 1992;262:238–245. [PubMed] [Google Scholar]
- Okwu AK, Mais DE, Halushka PV. Agonist-induced phosphorylation of human platelet TXA2/PGH2 receptors. Biochim Biophys Acta. 1994;1221:83–88. doi: 10.1016/0167-4889(94)90220-8. [DOI] [PubMed] [Google Scholar]
- Parent JL, Labrecque P, Orsini MJ, Benovic JL. Internalization of the TXA2 receptor alpha and beta isoforms. Role of the differentially spliced cooh terminus in agonist-promoted receptor internalization. J Biol Chem. 1999;274:8941–8948. doi: 10.1074/jbc.274.13.8941. [DOI] [PubMed] [Google Scholar]
- Raychowdhury MK, Yukawa M, Collins LJ, McGrail SH, Kent KC, Ware JA. Alternative splicing produces a divergent cytoplasmic tail in the human endothelial thromboxane A2 receptor. J Biol Chem. 1994;269:19256–19261. [PubMed] [Google Scholar]
- Raychowdhury MK, Yukawa M, Collins LJ, McGrail SH, Kent KC, Ware JA. Alternative splicing produces a divergent cytoplasmic tail in the human endothelial thromboxane A2 receptor. J Biol Chem. 1995;270:7011. doi: 10.1074/jbc.270.12.7011. [DOI] [PubMed] [Google Scholar]
- Reid HM, Kinsella BT. The alpha, but not the beta, isoform of the human thromboxane A2 receptor is a target for nitric oxide-mediated desensitization. Independent modulation of Tp alpha signaling by nitric oxide and prostacyclin. J Biol Chem. 2003;278:51190–51202. doi: 10.1074/jbc.M309314200. [DOI] [PubMed] [Google Scholar]
- Reyes JL. Arachidonic acid metabolites and haemodynamics of the neonate. Pediatr Nephrol. 1993;7:841–844. doi: 10.1007/BF01213371. [DOI] [PubMed] [Google Scholar]
- Rosenblum WI, el-Sabban F, Nelson GH, Allison TB. Effects in mice of testosterone and dihydrotestosterone on platelet aggregation in injured arterioles and ex vivo. Thromb Res. 1987;45:719–728. doi: 10.1016/0049-3848(87)90082-x. [DOI] [PubMed] [Google Scholar]
- Saini-Chohan HK, Dakshinamurti S, Taylor WA, Shen GX, Murphy R, Sparagna GC, et al. Persistent pulmonary hypertension results in reduced tetralinoleoyl-cardiolipin and mitochondrial complex II + III during the development of right ventricular hypertrophy in the neonatal pig heart. Am J Physiol Heart Circ Physiol. 2011;301:H1415–H1424. doi: 10.1152/ajpheart.00247.2011. [DOI] [PubMed] [Google Scholar]
- Santhosh K, Elkhateeb O, Nolette N, Outbih O, Halayko A, Dakshinamurti S. Milrinone attenuates thromboxane receptor-mediated hyperresponsiveness in hypoxic pulmonary arterial myocytes. Br J Pharmacol. 2011;163:1223–1236. doi: 10.1111/j.1476-5381.2011.01306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda LA, Sham JS, Shimoda TH, Sylvester JT. L-type Ca(2+) channels, resting [Ca(2+)](i), and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol. 2000;279:L884–L894. doi: 10.1152/ajplung.2000.279.5.L884. [DOI] [PubMed] [Google Scholar]
- Snow JB, Kitzis V, Norton CE, Torres SN, Johnson KD, Kanagy NL, et al. Differential effects of chronic hypoxia and intermittent hypocapnic and eucapnic hypoxia on pulmonary vasoreactivity. J Appl Physiol. 2008;104:110–118. doi: 10.1152/japplphysiol.00698.2005. [DOI] [PubMed] [Google Scholar]
- Tobin AB. G-protein-coupled receptor phosphorylation: where, when and by whom. Br J Pharmacol. 2008;153(Suppl 1):S167–S176. doi: 10.1038/sj.bjp.0707662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh M, Foley JF, Kinsella BT. Investigation of the role of the carboxyl-terminal tails of the alpha and beta isoforms of the human thromboxane A(2) receptor (TP) in mediating receptor:effector coupling. Biochim Biophys Acta. 2000a;1496:164–182. doi: 10.1016/s0167-4889(00)00031-8. [DOI] [PubMed] [Google Scholar]
- Walsh MT, Foley JF, Kinsella BT. The alpha, but not the beta, isoform of the human thromboxane A2 receptor is a target for prostacyclin-mediated desensitization. J Biol Chem. 2000b;275:20412–20423. doi: 10.1074/jbc.M907881199. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Yan F, Zhou H, Tai HH. Serine 331 is the major site of receptor phosphorylation induced by agents that activate protein kinase G in HEK 293 cells overexpressing thromboxane receptor alpha. Arch Biochem Biophys. 2001;393:97–105. doi: 10.1006/abbi.2001.2505. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Yan F, Zhou H, Tai HH. Agents that elevate cyclic AMP induce receptor phosphorylation primarily at serine 331 in HEK 293 cells overexpressing human thromboxane receptor alpha. Biochem Pharmacol. 2002;64:375–383. doi: 10.1016/s0006-2952(02)01053-5. [DOI] [PubMed] [Google Scholar]
- Yan FX, Yamamoto S, Zhou HP, Tai HH, Liao DF. Serine 331 is major site of phosphorylation and desensitization induced by protein kinase C in thromboxane receptor alpha. Acta Pharmacol Sin. 2002;23:952–960. [PubMed] [Google Scholar]
- Zhou H, Yan F, Tai HH. Phosphorylation and desensitization of the human thromboxane receptor-alpha by G protein-coupled receptor kinases. J Pharmacol Exp Ther. 2001;298:1243–1251. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Protein sequence alignment of porcine TP and human TP receptor isoforms. 2D structure of human TPα, indicating position of the four serines targeted for alanine substitution. Comparative sequence alignment of human TP isoforms, TPα and TPβ, with porcine TP; S324, S329 and S331 are the only phosphorylatable C-terminal residues common to human TPα and porcine TP (but not TPβ).
Phosphorylation of TP mutants by PKA. HEK293T cells transfected with human wild-type TPα, or serine-to-alanine substitution mutants S324A, S329A or S331A incubated with 10−6 M forskolin (activates PKA) or diluent; TP immunoprecipitated using antibody to the rho-1D4 tag. Serine phosphorylation quantified in immunoprecipitates by Western blot (mouse monoclonal antibody to phospho-serine; n > 4). Ability of wild-type or alaninesubsitution mutant TP isoforms to be phosphorylated by PKA was determined as density of phospho-serine bands for (TP with forskolin – TP with diluent)/TP with diluent.
Effects of PKA and PKC on maximal TP agonistmediated calcium mobilization. TP-mediated Ca2+ mobilization in the presence of PKA activation or inhibition (A) or PKC activation or inhibition (B), determined using fluo-4, quantified after baseline subtraction. n > 4.