

Abstract
Background and Purpose
The aetiology of inflammation in the liver and vessel wall, leading to non-alcoholic steatohepatitis (NASH) and atherosclerosis, respectively, shares common mechanisms including macrophage infiltration. To treat both disorders simultaneously, it is highly important to tackle the inflammatory status. Exendin-4, a glucagon-like peptide-1 (GLP-1) receptor agonist, reduces hepatic steatosis and has been suggested to reduce atherosclerosis; however, its effects on liver inflammation are underexplored. Here, we tested the hypothesis that exendin-4 reduces inflammation in both the liver and vessel wall, and investigated the common underlying mechanism.
Experimental Approach
Female APOE*3-Leiden.CETP mice, a model with human-like lipoprotein metabolism, were fed a cholesterol-containing Western-type diet for 5 weeks to induce atherosclerosis and subsequently treated for 4 weeks with exendin-4.
Key Results
Exendin-4 modestly improved dyslipidaemia, but markedly decreased atherosclerotic lesion severity and area (−33%), accompanied by a reduction in monocyte adhesion to the vessel wall (−42%) and macrophage content in the plaque (−44%). Furthermore, exendin-4 reduced hepatic lipid content and inflammation as well as hepatic CD68+ (−18%) and F4/80+ (−25%) macrophage content. This was accompanied by less monocyte recruitment from the circulation as the Mac-1+ macrophage content was decreased (−36%). Finally, exendin-4 reduced hepatic chemokine expression in vivo and suppressed oxidized low-density lipoprotein accumulation in peritoneal macrophages in vitro, effects dependent on the GLP-1 receptor.
Conclusions and Implications
Exendin-4 reduces inflammation in both the liver and vessel wall by reducing macrophage recruitment and activation. These data suggest that exendin-4 could be a valuable strategy to treat NASH and atherosclerosis simultaneously.
Keywords: cholesterol, exendin-4, inflammation, macrophage content, monocyte recruitment, oxidized LDL
Introduction
Cardiovascular disease (CVD) due to atherosclerosis is the leading cause of morbidity and mortality in the Western world. There is a strong association between atherosclerosis and non-alcoholic fatty liver disease (NAFLD), which raised the interest of the role of the liver in the development of atherosclerosis. NAFLD embraces a pathological spectrum of liver diseases, from steatosis with virtually no evidence of hepatocellular injury or liver inflammation to non-alcoholic steatohepatitis (NASH) and cirrhosis (Brunt, 2010). NASH is characterized by accumulation of fat in the liver in combination with hepatic inflammation (Targher and Arcaro, 2007). This inflammatory response was assumed to be the consequence rather than the cause of the disease. However, compelling data point to a central initiating role of monocyte recruitment and macrophage activation in the progression of hepatic inflammation in a similar way as in the development of atherosclerosis as we recently reviewed (Bieghs et al., 2012a). The potential of a shared aetiology between inflammation in the liver and vessel wall leads to putative intervention therapies to tackle both disorders at the same time by targeting macrophage infiltration. This is particularly interesting as the current standard therapies to reduce inflammation in NASH have only limited effectiveness. Lifestyle intervention, including exercise, is recommended in patients with NASH. However, it appears to be difficult to achieve improvements in NASH in the long run, and pharmacological intervention is ultimately required (Schattenberg and Schuppan, 2011).
Glucagon-like peptide-1 (GLP-1) receptor agonists, such as exendin-4, are currently being validated for the treatment of type 2 diabetes mellitus and have been shown to increase glucose-dependent insulin secretion, regulate gastric emptying, and reduce food intake and body weight (Gallwitz, 2011). In addition to improving glycaemic control, GLP-1 receptor activation improves lipid metabolism. We (Parlevliet et al., 2012) and others (Ding et al., 2006; Svegliati-Baroni et al., 2011; Lee et al., 2012; Mells et al., 2012) have shown that GLP-1 receptor activation reduces hepatic steatosis, explained by increased hepatic lipid oxidation and decreased lipogenesis, and attenuates hepatic very-low-density lipoprotein (VLDL) production. Collectively, these data indicate the potential of GLP-1 receptor activation for the treatment of NAFLD, but its effect on liver inflammation and atherosclerosis is still uncertain. Moreover, contrasting results have been reported regarding the effects of GLP-1 receptor activation on atherosclerosis in apolipoprotein E (ApoE)−/− mice. Whereas Gaspari et al. (2013) showed clear inhibitory effects on the progression of atherosclerosis, Panjwani et al. (2013) did not observe any effects on atherosclerosis development. It should be noted that mice used in these studies generally do not respond to lipid-lowering interventions and lack apoE, a crucial factor for cholesterol efflux from macrophages. Given these controversial effects on atherosclerosis, combined with the reduction in hepatic steatosis, it is essential to further explore the therapeutic potential of GLP-1 receptor activation with regard to its effects on inflammation in both the liver and vessel wall.
The aim of the current study was to evaluate the effect of exendin-4 treatment on hepatic inflammation as well as the development of atherosclerosis, and to elucidate the underlying mechanisms, in APOE*3-Leiden.CETP (E3L.CETP) mice fed a Western-type diet. We showed that exendin-4 reduces the influx of macrophages into both the liver and vessel wall, and thereby limits the progression of hepatic inflammation and atherosclerosis simultaneously.
Methods
The drug/molecular target nomenclature used below conforms to the British Journal of Pharmacology's Concise Guide to PHARMACOLOGY (Alexander et al., 2013).
Animals
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Twelve-week-old female E3L.CETP transgenic mice expressing human cholesteryl ester transfer protein (CETP) under the control of its natural flanking regions were used (Westerterp et al., 2006) and housed in a temperature-controlled room on a 12 h light–dark cycle in conventional cages with free access to food and water, unless indicated otherwise. The animals were fed a semi-synthetic Western-type diet, containing 0.4% (w w−1) cholesterol, 1% (w w−1) corn oil and 15% (w w−1) cacao butter (Hope Farms, Woerden, the Netherlands) for 5 weeks. After randomization according to body weight, plasma total cholesterol (TC) and triglyceride (TG) levels, an osmotic minipump (model 1004; Alzet DURECT Corp., Cupertino, CA, USA) was implanted s.c. into the left back region under light isoflurane anaesthesia for the continuous delivery of exendin-4 (50 μg·kg−1·day−1; Bachem AG, Bubendorf, Switzerland; dissolved in PBS) (n = 17) or PBS as a control (n = 17) for 4 weeks while the Western-type diet was continued. Experiments were performed after 4 h of fasting at 1200 h with food withdrawn at 0800 h. For anaesthesia, mice were placed individually in an induction chamber, and anaesthesia was induced with 4% isoflurane in 100% oxygen with a delivery rate of 5 l min−1. Then, anaesthesia was maintained with 1.5% isoflurane inhalation in 100% oxygen at 1.5 l min−1. The depth of anaesthesia was determined by loss of righting reflex. The Institutional Ethics Committee for Animal Care and Experiments from the Leiden University Medical Center, Leiden, the Netherlands, approved all experiments.
Blood sampling, plasma metabolites and lipoprotein profiles
Blood was obtained via tail vein bleeding into heparin-coated capillary tubes. The tubes were placed on ice and centrifuged, and the plasma obtained was snap-frozen in liquid nitrogen and stored at −20°C until further measurements. Plasma was assayed for glucose (INstruchemie, Delfzijl, the Netherlands) as well as TC, and TG using the commercially available enzymatic kits 236691, 11488872 (Roche Molecular Biochemicals, Indianapolis, IN, USA) respectively. Plasma insulin was measured by elisa (Mercodia AB, Uppsala, Sweden). The distribution of lipids over plasma lipoproteins was determined using fast protein liquid chromatography (FPLC). Plasma was pooled per group, and 50 μL of each pool was injected onto a Superose 6 PC 3.2/30 column (Äkta System, Amersham Pharmacia Biotech, Piscataway, NJ, USA) and eluted at a constant flow rate of 50 μL·min−1 in PBS, 1 mM EDTA, pH 7.4. Fractions of 50 μL were collected and assayed for TC as described earlier.
Atherosclerosis quantification and monocyte adhesion to the endothelium wall
After 4 weeks of treatment, mice were killed and perfused with ice-cold PBS via the heart. Hearts were isolated and fixed in phosphate-buffered 4% formaldehyde, dehydrated, embedded in paraffin and cross-sectioned (5 μm) through the aortic root area. Cross sections were stained with haematoxylin-phloxine-saffron to determine lesion area and lesion severity as described previously (Gijbels et al., 1999; de Haan et al., 2008). Briefly, various types of lesions were discerned: no lesions, mild lesions with fatty streak-like lesions containing foam cells and severe lesions referred to as advanced lesions containing foam cells in the media, presence of fibrosis, cholesterol clefts, mineralization and/or necrosis. Additionally, cross sections were stained with AIA 31420 antiserum (1:3000; Accurate Chemical and Scientific, Westbury, NY, USA) to determine macrophage area and monocyte adhesion to the endothelium wall as described previously (Bieghs et al., 2010). Lesion area and macrophage area were determined using Leica QWin software (Leica Microsystems, Wetzlar, Germany).
Hepatic lipid content
After 4 weeks of treatment, mice were killed and perfused with ice-cold PBS via the heart, and livers were isolated. Lipids were extracted according to a modified protocol from Bligh and Dyer (1959). Briefly, small liver pieces were homogenized in ice-cold methanol. After centrifugation, lipids were extracted by addition of 1800 μL CH3OH : CHCl3 (1:3 v v−1) to 45 μL homogenate, followed by vigorous vortexing and phase separation by centrifugation (5 min at 370x g). The CHCl3 phase was dried and dissolved in 2% Triton X-100 (Sigma-Aldrich, Seelze, Germany). TG and TC concentrations were measured as described previously. Phospholipid (PL) concentration was measured using a commercial kit (phospholipids B; Wako Chemicals, Neuss, Germany). Liver lipids were expressed as nmol mg−1 protein, which was determined using the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA).
Hepatic gene expression analysis
Total RNA was extracted from liver pieces using the NucleoSpin RNAII kit (Macherey-Nagel, Duren, Germany) according to the manufacturer's instructions. RNA quality was examined by the lab-on-a-chip method using Experion StdSens analysis kit (Bio-Rad, Hercules, CA, USA), and RNA concentration was determined by NanoDrop technology (Thermo Scientific, Wilmington, DE, USA). Total RNA was reverse transcribed with iScript cDNA synthesis kit (1708891; Bio-Rad), and the obtained cDNA was purified with NucleoSpin Extract II kit (636973; Macherey-Nagel, Bioké). Real-time PCR was performed on a CFX96 machine (Bio-Rad), the reaction mixture consisting of SYBR-Green SensiMix (QT615; GC Biotech, Alphen aan den Rijn, the Netherlands), cDNA, primers (Biolegio, Nijmegen, the Netherlands; see Supporting Information Table S1 for primer sequences) and nuclease-free water in a total reaction volume of 10 μL. mRNA values of each gene were normalized to mRNA levels of cyclophilin and hypoxanthine ribosyltransferase. Data were calculated as fold difference as compared with the PBS control group.
Liver histology
Paraffin-embedded liver sections were stained for F4/80+ macrophages (1/600; Serotec, Oxford, UK) as described previously (Lanthier et al., 2010). Frozen liver sections (7 μm) were stained for CD68+ resident macrophages (CD68 marker, FA11) and infiltrated macrophages (macrophage marker, Mac-1) as described previously (Bieghs et al., 2010).
Low-density lipoprotein (LDL) isolation, radiolabelling and oxidation
LDL was isolated from human serum by density gradient ultracentrifugation as described previously (Redgrave et al., 1975). LDL was labelled with [3H]-cholesteryl oleoyl ether (COEth) by incubation with donor [3H]-COEth-containing liposomes in the presence of human lipoprotein-deficient serum. In short, liposomes were created by sonication of 1 mg of egg yolk phosphatidylcholine and 200 μCi of [3H]-COEth using a Soniprep 150 (MSE Scientific Instruments, Crawley, UK). Subsequently, LDL was incubated with the liposomes (protein : liposomal PL = 1:8, w w−1) for 24 h at 37°C under argon. [3H]-COEth-labelled LDL was purified by density gradient ultracentrifugation and dialysed overnight at 4°C against PBS, pH 7.4. Both LDL and [3H]-COEth-LDL were oxidized with 5 μM CuSO4 at 37°C for 20 h. Oxidation was terminated by adding 200 μM EDTA. EDTA and CuSO4 were removed by overnight dialysis at 4°C against PBS, pH 7.4. Proper oxidation of LDL was confirmed by a 2.5-fold increased electrophoretic mobility of oxidized LDL (oxLDL) and [3H]-COEth-oxLDL compared with LDL on agarose gel. Protein concentration was determined by the BCA protein assay kit.
Plasma anti-oxLDL antibodies
An enzyme immunoassay/radioimmunoassay high-binding 96-well Costar plate (Corning Inc., Corning, NY, USA) was coated with oxLDL (7.5 μg·mL−1) in PBS. IgM, IgG1 and IgG2a antibodies against oxLDL in serum were measured using an elisa Ig detection kit (Zymed Laboratories, San Francisco, CA, USA) according to the manufacturer's protocol.
oxLDL uptake by peritoneal macrophages and Oil red O staining for foam cells
Three days after i.p. injection of 1 mL of 4% thioglycollate, peritoneal macrophages from E3L.CETP mice were harvested into 10 mL PBS. Subsequently, cells were resuspended in DMEM supplemented with 10% FBS, 1% L-glutamine, 100 U·mL−1 penicillin and 100 mg·mL−1 streptomycin, and incubated at 37°C in a humidified 5% CO2 incubator. Three hours post-plating, cells were washed twice with warm PBS to remove non-adherent cells. Cells were counted and seeded into 24-well plates at a density of 5 × 105 cells per well for 3 days before the experiment. On the experimental day, peritoneal macrophages were washed three times with PBS and incubated in DMEM supplemented with 1% BSA, 100 U·mL−1 penicillin and 100 mg·mL−1 streptomycin for 1 h, followed by DMEM supplemented with 1% BSA, 100 U·mL−1 penicillin, 100 mg·mL−1 streptomycin, 10 μg·mL−1 [3H]-COEth-oxLDL, and exendin-4 (0.05 or 0.5 nM) for 48 h at 37°C in a humidified 5% CO2 incubator. When indicated, exendin-(9–39) (50 nM; Bachem AG) was added 1 h before addition of exendin-4. Exendin-(9–39) is a potent GLP-1 receptor antagonist and acts as a competitive inhibitor of exendin-4. After incubation, macrophages were washed twice with 500 μL PBS, and cell lysates were obtained by adding 500 μL of 0.1 M NaOH. Two hundred fifty microlitres of cell lysates was used for quantification of 3H-radioactivity. Disintegrations per minute (dpm) values were normalized to the total amount of protein (in mg) present in 250 μL of cell lysates. Protein concentration in cell lysates was quantified with BCA protein assay kit.
For Oil red O staining, after incubation, cells were washed twice with PBS and fixed for 30 min in 4% formaldehyde in PBS. Cells were incubated in 60% 2-propanol for 2 min, immediately placed in 60% Oil red O for 30 min, followed by washing with dH2O. Cell nuclei were counterstained with haematoxylin for 1 min.
Statistical analysis
All data are presented as means ± SEM. Statistical differences between groups were assessed with the Mann–Whitney U-test for two independent groups. A P-value of less than 0.05 was considered statistically significant.
Results
Exendin-4 reduces body weight and plasma glucose and insulin levels
E3L.CETP mice were fed a Western-type diet containing 0.4% cholesterol for 5 weeks and thereafter were treated with exendin-4 or PBS via s.c. osmotic minipumps for 4 weeks while continuing the diet (Supporting Information Figure S1A). Exendin-4 decreased body weight after 1 week of treatment, and this difference was sustained throughout the treatment period (Supporting Information Figure S1B). After 2 and 4 weeks of treatment, exendin-4 significantly decreased plasma glucose levels (−21%, P < 0.001 and −24%, P < 0.001, respectively; Supporting Information Figure S1C) and insulin levels (−30%, P < 0.05 and −34%, P < 0.01, respectively; Supporting Information Figure S1D), which is in line with our previous observations in high-fat diet fed E3L mice (Parlevliet et al., 2012).
Exendin-4 decreases plasma VLDL and slightly increases high-density lipoprotein (HDL)
Despite clear beneficial effects on glucose metabolism, exendin-4 only tended to decrease plasma cholesterol levels (−14%, P = 0.05; Figure 1A) and TG levels (−22%, P = 0.07; Figure 1B) after 4 weeks of treatment. Lipoprotein profiling revealed that exendin-4 decreased VLDL cholesterol and slightly increased HDL cholesterol levels (Figure 1C). The latter effect was accompanied by a 26% increased hepatic expression of apo A1 (Apoa1), which encodes the major lipoprotein of HDL (Figure 1D).
Figure 1.
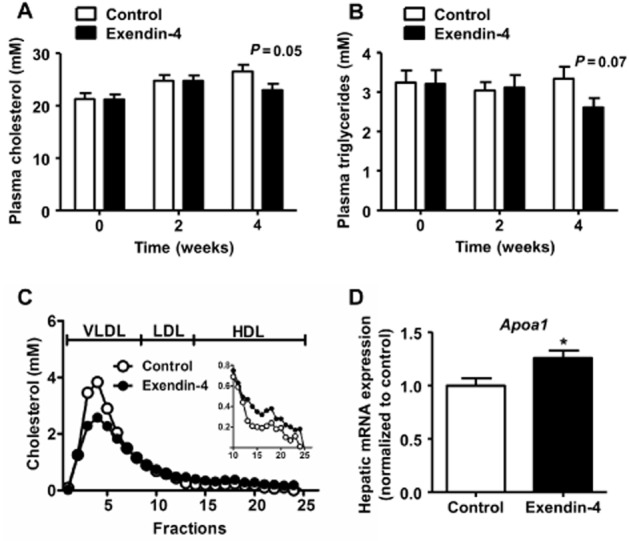
Exendin-4 decreases (V)LDL and slightly increases HDL. After 5 weeks of feeding a Western-type diet containing 0.4% cholesterol, mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (PBS) s.c. for 4 weeks. Blood was collected by tail bleeding after 4 h of fasting before treatment (T = 0), and after 2 (T = 2) and 4 (T = 4) weeks of treatment. Plasma cholesterol (A) and TG (B) levels were determined. After 4 weeks of treatment, group-wise pooled plasma was fractionated using fast protein liquid chromatography on a Superose 6 column, and the individual fractions were assayed for cholesterol (C). Livers were isolated, mRNA was extracted and Apoa1 mRNA was determined as normalized to cyclophilin and hypoxanthine ribosyltransferase mRNA levels. Data were calculated as fold difference compared with vehicle (D). Values are means ± SEM (n = 17 mice per group). *P < 0.05 compared with vehicle.
Exendin-4 largely suppresses atherosclerosis development and monocyte recruitment in the aortic root
Despite modestly attenuating dyslipidaemia, exendin-4 treatment markedly reduced total atherosclerotic lesion area as compared with controls (−33%, P < 0.05; Figure 2A). Additionally, mice treated with exendin-4 showed more lesion-free sections (+63%, P = 0.07) and less severe lesions (−42%, P < 0.05) as compared with PBS controls (Figure 2B). Moreover, exendin-4 significantly reduced the number of monocytes adhering to the endothelium wall in the aortic root (−42%, P < 0.001; Figure 2C) as well as the macrophage area in the plaque (−44%, P < 0.05; Figure 2D).
Figure 2.
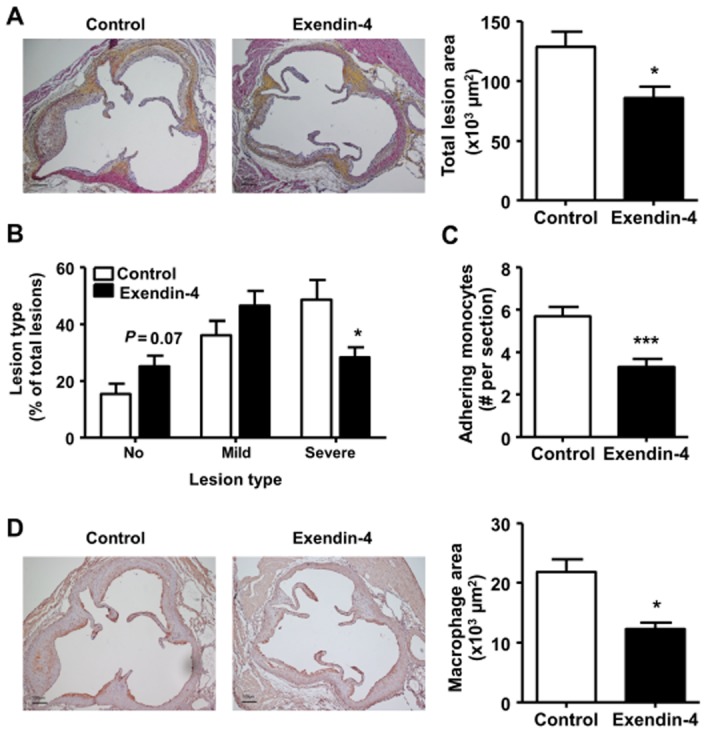
Exendin-4 reduces aortic atherosclerosis development and monocyte recruitment to the endothelium wall. After 5 weeks of feeding a Western-type diet containing 0.4% cholesterol, mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (PBS) s.c. for 4 weeks. Subsequently, hearts were isolated, fixed, dehydrated and embedded in paraffin. Cross sections of the aortic root were stained with haematoxylin-phloxine-saffron (A, B) or anti-AIA serum (C, D). Total lesion area (A), lesion severity (B), the number of adhering monocytes to the endothelium wall (C) and macrophage area (D) were quantified. Values are means ± SEM (n = 17 mice per group). *P < 0.05; ***P < 0.001 compared with vehicle.
Exendin-4 reduces hepatic lipids, inflammation and macrophage content
Four weeks of exendin-4 treatment decreased hepatic TG (−11%, P = 0.057) and TC content (−19%, P < 0.05), without affecting hepatic PL content. In addition, exendin-4 largely reduced the hepatic expression of the inflammatory markers TNF-α, IL-1β and IL-6 (−45%, P < 0.01; −47%, P < 0.001; −40%, P < 0.05, respectively; Figure 3A). Moreover, exendin-4 reduced hepatic mRNA expression of the macrophage markers CD68 (−30%, P < 0.01; Figure 3B) and F4/80 (−28%, P < 0.05; Figure 3C). In line with these data, exendin-4 decreased hepatic CD68+ macrophages (−18%, P < 0.05; Figure 3D) and F4/80+ macrophages (−25%, P < 0.001; Figure 3E).
Figure 3.
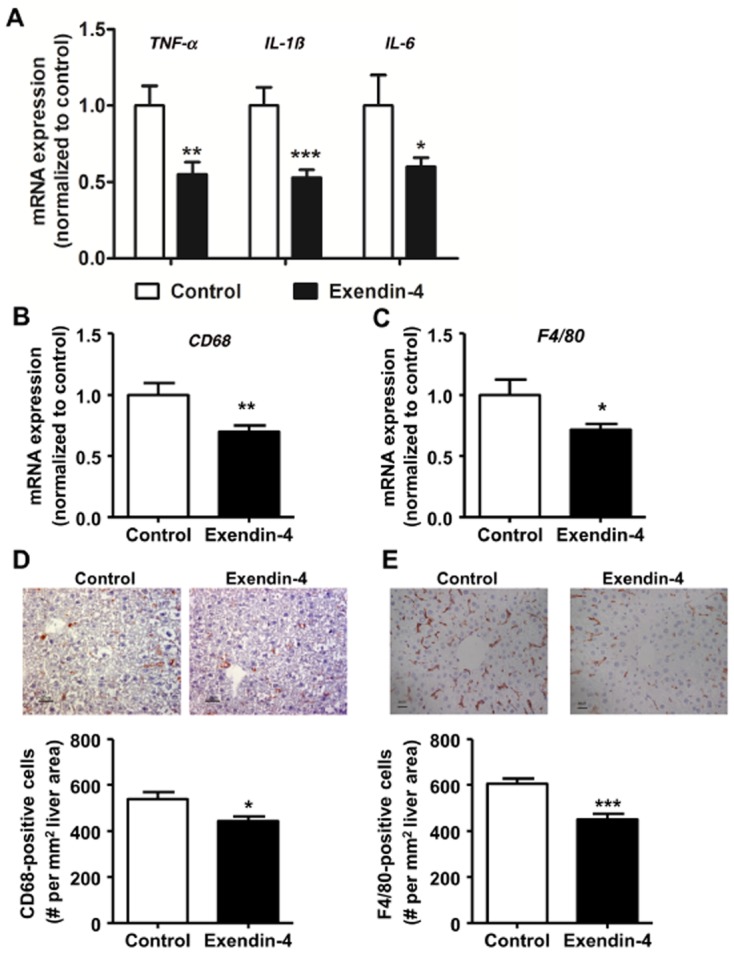
Exendin-4 reduces liver inflammation and macrophage content. After 5 weeks of feeding a Western-type diet containing 0.4% cholesterol, mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (PBS) s.c. for 4 weeks. mRNA was extracted from liver pieces, and mRNA expression of inflammatory markers TNF-α, IL-1β and IL-6 (A); CD68 (B); and F4/80 (C) was determined as normalized to cyclophilin and hypoxanthine ribosyltransferase mRNA levels. Data were calculated as fold difference as compared with vehicle (A–C). Liver sections were immunostained for CD68 and F4/80, and CD68+ (D) F4/80+ (E) macrophages were quantified. Values are means ± SEM (n = 17 mice per group). *P < 0.05; **P < 0.01; ***P < 0.001 compared with vehicle.
Exendin-4 reduces macrophage infiltration into the liver
To elucidate the mechanism underlying the reduction of liver macrophage content by exendin-4, we first determined the hepatic gene expression of monocyte chemotactic protein-1 (Mcp-1), which mediates monocyte recruitment from the circulation to sites of infection and inflammation. As shown in Figure 4A, exendin-4 reduced Mcp-1 expression (−34%, P < 0.05). This was accompanied by a reduction of macrophages positive for Mac-1, an infiltrating macrophage marker (−36%, P < 0.01; Figure 4B).
Figure 4.
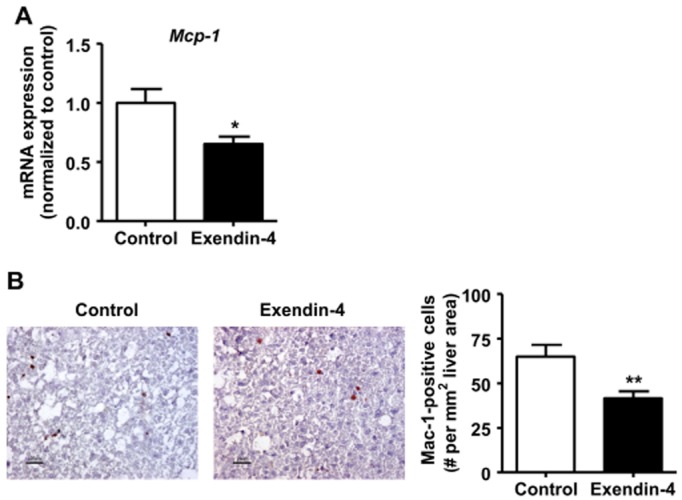
Exendin-4 reduces macrophage infiltration into the liver. After 5 weeks of feeding a Western-type diet containing 0.4% cholesterol, mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (PBS) s.c. for 4 weeks. In the liver, mRNA expression of Mcp-1 (A) was determined as normalized to cyclophilin and hypoxanthine ribosyltransferase mRNA levels. Data were calculated as fold difference as compared with vehicle (A). Liver sections were immunostained for Mac-1, and Mac-1+ macrophages were quantified (B). Values are means ± SEM (n = 17 mice per group). *P < 0.05; **P < 0.01 compared with vehicle.
Exendin-4 does not affect circulating antibodies against oxLDL but reduces oxLDL uptake by macrophages
Because the uptake of oxLDL by macrophages can drive both atherosclerosis and NASH, we first measured specific antibodies against oxLDL in the circulation. Exendin-4 did not affect oxLDL-specific IgG1, IgG2a and IgM levels in plasma (Figure 5A). Next, to determine if exendin-4 can affect oxLDL uptake by macrophages, we incubated peritoneal macrophages with [3H]-COEth-oxLDL (10 μg·mL−1) for 48 h with or without exendin-4 (0.05 and 0.5 nM). Exendin-4 decreased the uptake of [3H]-COEth-oxLDL (up to −33%, P < 0.01) compared with controls (Figure 5B). This effect was completely abolished by pretreatment of the cells with exendin-(9–39) (Figure 5B), which demonstrates that exendin-4 reduces oxLDL uptake by activating the GLP-1 receptor. Oil red O staining confirmed that exendin-4 reduced foam cell formation, which was blocked by exendin-(9–39) (Figure 5C).
Figure 5.
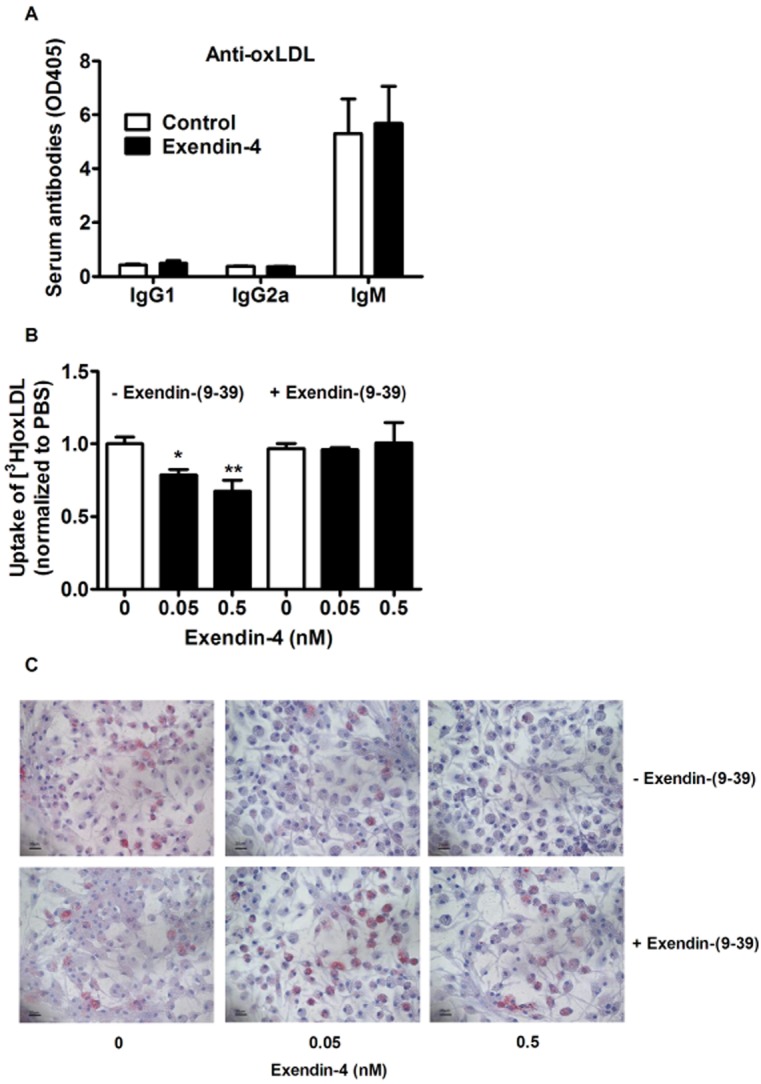
Exendin-4 has no effect on plasma antibodies against oxLDL, but reduces oxLDL-induced foam cell formation in peritoneal macrophages. After 5 weeks of feeding a Western-type diet containing 0.4% cholesterol, mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (PBS) s.c. for 4 weeks. Plasma anti-oxLDL antibodies were determined (A). Peritoneal macrophages were incubated with exendin-4 and/or exendin-(9–39) for 48 h. The uptake of [3H]-COEth-labelled oxLDL was quantified (B). After fixation, lipid accumulation into the cells was visualized by staining with Oil red O (C). Values are means ± SEM (n = 17 mice per group). *P < 0.05; **P < 0.01 compared with vehicle.
Discussion
In this study, we investigated the effects of exendin-4 treatment on liver inflammation in addition to the development of atherosclerosis in E3L.CETP mice fed a Western-type diet. Our data showed that 4 weeks of exendin-4 infusion markedly decreases total atherosclerotic lesion area, accompanied by a reduction in plaque macrophages. In parallel, exendin-4 caused a marked reduction in hepatic lipids and macrophages as well as hepatic inflammation, hallmarks of NASH.
It is interesting to note that short-term treatment with exendin-4, as compared with vehicle, caused a substantial reduction in atherosclerosis even though exendin-4 only modestly affected cholesterol levels and the lipoprotein profile. Therefore, the anti-atherogenic effect of exendin-4 is likely largely independent of modulation of plasma lipid levels. This observation contrasts with the effects of classic lipid-lowering compounds including atorvastatin, which reduce atherosclerosis in E3L.CETP mice mainly by reducing apoB-containing lipoproteins (de Haan et al., 2008). We found that exendin-4 inhibited the adherence of monocytes to the vessel wall and decreased the macrophage area of the plaque, suggesting that exendin-4 decreases the recruitment of monocytes into the vessel wall. Our results add to previous findings demonstrating that 4 weeks of GLP-1 receptor activation decreases atherosclerotic development without reducing plasma lipids in ApoE−/− mice (Arakawa et al., 2010; Nagashima et al., 2011; Gaspari et al., 2013). Although a recent study did not find a reduction in atherosclerosis in ApoE−/− mice (Panjwani et al., 2013), this might be related to potential tissue heterogeneity in GLP-1 receptor expression in the ApoE−/− mice as well as variations in the experimental set-up (e.g. animal age, treatment regimen).
Secondly, we observed that exendin-4 reduced the lipid content of the liver, which was accompanied by a reduction in inflammatory markers and macrophage content of the liver as judged from hepatic gene and protein expression. It is known that feeding hyperlipidaemic mice a diet containing cholesterol increases the hepatic macrophage content even within a few days (Wouters et al., 2008). Because in our study mice were fed the Western-type diet for 5 weeks before starting treatment, exendin-4 may thus have reduced the hepatic macrophage content either by reducing the infiltration of activated macrophages from the circulation or by inducing the elimination of macrophages from the liver. The current study design does not allow us to differentiate between these possibilities. However, we did observe a decrease in hepatic Mcp-1 expression along with a reduction in Mac-1+ infiltrating macrophages, suggesting that reduced recruitment of monocytes/macrophages from the circulation to the liver contributes to the reduction in total macrophages. Collectively, we showed that exendin-4 treatment affects important features of NASH, including reduction of liver macrophages as well as lipid content.
Historically, NASH was thought to be a causal risk factor for CVD as patients with NASH have a higher mortality risk than the general population, mainly due to CVD (Targher and Arcaro, 2007). However, the biological mechanisms linking NASH and accelerated atherosclerosis are still poorly understood. Recently, Bieghs et al. (2012a) put forward the hypothesis that there is a central role for inflammation in the development of NASH and atherosclerosis with common aetiologies involving monocyte recruitment and macrophage foam cell formation. From this perspective, we hypothesized that exendin-4 reduces atherosclerosis as well as hepatic inflammation by acting directly on monocyte/macrophage recruitment into both the vessel wall and the liver.
So what mechanisms may then be involved? In addition to reducing hepatic MCP-1 as observed in our study, exendin-4 reduces the expression of E-selectin, intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in the aorta of ApoE−/− mice (Arakawa et al., 2010). These are all processes that are likely to lead to inhibition of monocyte/macrophage adhesion to the vessel wall and the liver. The importance of blood monocytes in the development of atherosclerosis has been firmly established (Moore and Tabas, 2011), and accumulating evidence demonstrates the role of hepatic infiltration of blood-borne monocytes in liver inflammation (Karlmark et al., 2008; Berres et al., 2010). In fact, the migration of immune cells into the liver appears to be one of the first critical steps in both acute and chronic liver inflammation, mediating innate and adaptive inflammatory responses, which result in ongoing immune cell recruitment, tissue damage and fibrosis (Karlmark et al., 2008).
As a second mechanism explaining the reduction of macrophage recruitment, we showed that exendin-4, via the GLP-1 receptor, reduces the uptake of oxLDL by peritoneal macrophages and, as a consequence, reduced foam cell formation in vitro. These data are in full accordance with previous observations showing that native GLP-1 also reduces oxLDL uptake by peritoneal macrophages, thereby reducing atherosclerosis development from ApoE−/− mice (Nagashima et al., 2011). We did not observe any changes in plasma anti-oxLDL antibodies after exendin-4 treatment, indicating that exendin-4 probably does not affect the oxLDL levels in plasma. Because native GLP-1 decreased the macrophage protein levels of CD36 (Nagashima et al., 2011), involved in the uptake of modified LDL, it is likely that in our study exendin-4 exerts its protective effects on macrophage foam cell formation by a similar mechanism. The relevance of oxLDL uptake via scavenger receptors by macrophages in atherosclerosis development has been confirmed by many studies (Moore and Freeman, 2006). Recent studies have shown that oxLDL uptake via scavenger receptors is also an important risk factor for the progression to hepatic inflammation (Bieghs et al., 2010). In fact, inhibition of oxLDL uptake and thus foam cell formation by reducing the number of scavenger receptors CD36 and scavenger receptor class A in haematopoietic cells reduces hepatic macrophage infiltration (Bieghs et al., 2012b), confirming that oxLDL uptake by macrophages not only plays a vital role in atherosclerosis development but also regulates hepatic inflammation.
Collectively, we hypothesize that the protective effect of exendin-4 against inflammation in both the vessel wall and the liver can be explained by (i) reduced monocyte/macrophage recruitment from the circulation and by (ii) inhibition of the uptake of oxLDL by macrophages, and the subsequent activation and formation of foam cells at these locations (see Figure 6).
Figure 6.
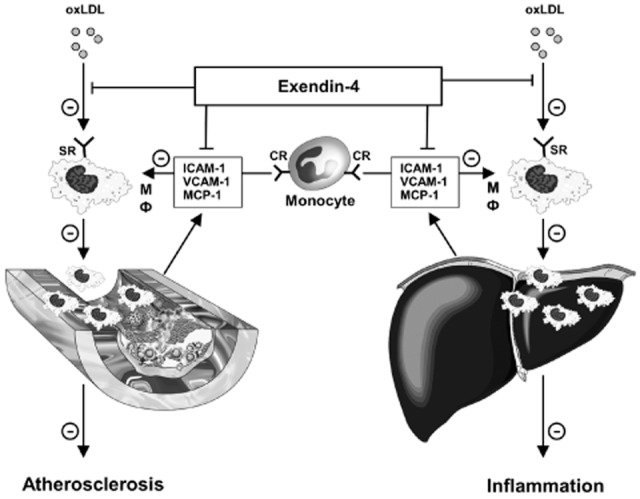
Proposed mechanism underlying the beneficial effects of exendin-4 on atherosclerosis development and NASH. Exendin-4 attenuates the development of atherosclerosis and NASH by (i) reducing the expression of chemokines and adhesion molecules, which leads to less recruitment of circulating monocytes/macrophages, and (ii) by inhibiting the uptake of oxLDL by macrophages and the subsequent formation of foam cells in the vessel wall and liver respectively. For further explanations, see text. CR, chemokine receptor(s); ICAM-1, intercellular adhesion molecule-1; mΦ, macrophage; MCP-1, monocyte chemotactic protein-1; oxLDL, oxidized LDL; SR, scavenger receptor; VCAM, vascular cell adhesion molecule-1.
Interestingly, we also observed that exendin-4 decreased hepatic CETP gene expression (−34%, P < 0.05) and plasma CETP concentration (−15%, P < 0.01; Supporting Information Figure S2). Recently, we demonstrated similar effects for niacin, which were explained by a reduction in macrophages that largely contribute to hepatic CETP expression (Li et al., 2012). In our present study, hepatic CETP expression positively correlated with the hepatic expression of the macrophage markers CD68 (R2 = 0.332, P < 0.001) and F4/80 (R2 = 0.607, P < 0.001; Supporting Information Figure S3), indicating that exendin-4 also reduces CETP expression by reducing the hepatic macrophage content. As CETP is involved in the transfer of cholesteryl esters from HDL to (V)LDL, the reduction in CETP may have contributed to the slight decrease in (V)LDL/HDL ratio, and thereby to reduced atherosclerosis development. Taken together, these data suggest that reducing NASH in general, by reducing the macrophage content, may reduce CETP expression, thereby improving the lipoprotein profile and therefore decrease the risk of developing atherosclerosis.
Thus far, there is no established pharmacological compound to treat NASH. Lifestyle modifications, such as weight loss, exercise and restriction of nutrition intake, are still the mainstays for the treatment of NASH (McCarthy and Rinella, 2012). Although lipid-lowering agents (e.g. statins and fibrates) and antioxidants (e.g. vitamins C, E) have beneficial effects on atherosclerosis development, none of them have shown adequate and convincing benefits in the treatment of NASH (Musso et al., 2010; Bhatia et al., 2012). Thiazolidinediones, especially pioglitazone, have properties that affect both atherosclerosis and NASH; however, its clinical use is currently disputed due to potential long-term side effects (Yau et al., 2013). Exendin-4 has been approved for the treatment of type 2 diabetes mellitus (Gallwitz, 2011) and has also been shown to possess cardioprotective actions that include, in addition to reducing atherogenesis, suppression of arrhythmias, heart failure, myocardial infarction and death (Mundil et al., 2012). Based on our collective findings that exendin-4 reduces high-fat diet-induced hepatic steatosis by decreasing lipogenesis (Parlevliet et al., 2012) and suppresses macrophage content in both vessel wall and liver (in the current study), we propose that exendin-4 is a suitable candidate to concomitantly treat atherosclerosis and NASH. Evidently, future clinical trials should be undertaken to prove the potential of exendin-4 to treat patients with atherosclerosis and NASH. Moreover, they should identify whether the effects of exendin-4 on atherosclerosis and NASH are merely related to improvements in metabolic parameters, such as weight loss, or are independent of these changes.
In conclusion, our findings show that exendin-4 treatment reduces inflammation in both the liver and the vessel wall in E3L.CETP mice fed a Western-type diet, by reducing monocyte/macrophage recruitment and inhibiting the uptake of oxLDL by macrophages. We anticipate that exendin-4 can be used as a valuable strategy to treat NASH and atherosclerosis in addition to type 2 diabetes mellitus, especially in patients who display a combination of these diseases.
Acknowledgments
This research was supported by the Netherlands Heart Foundation (NHS grant 2007B81 to P. C. N. R.), the Dutch Diabetes Foundation (DFN grant 2007.00.010 to P. C. N. R.), the Center for Translational Molecular Medicine (CTMM; http://www.ctmm.nl), project PREDICCt (grant 01C-104 to K. W. v. D.), the Center of Medical Systems Biology (CMSB), the Netherlands Consortium for Systems Biology (NCSB) established by the Netherlands Genomics Initiative/Netherlands Organisation for Scientific Research (NGI/NWO) and the Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centres, the Netherlands Organisation for Health Research and Development and the Royal Netherlands Academy of Sciences for the GENIUS project ‘Generating the best evidence-based pharmaceutical targets for atherosclerosis’ (CVON2011-19). P. C. N. R. is an Established Investigator of the Netherlands Heart Foundation (Grant No. 2009T038).
Glossary
- apoE
apolipoprotein E
- CETP
cholesteryl ester transfer protein
- CVD
cardiovascular disease
- E3L.CETP mice
APOE*3-Leiden.CETP transgenic mice
- GLP-1
glucagon-like peptide-1
- HDL
high-density lipoprotein
- LDL
low-density lipoprotein
- MCP-1
monocyte chemotactic protein-1
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- oxLDL
oxidized LDL
- PL
phospholipids
- TC
total cholesterol
- TG
triglycerides
- VLDL
very low-density lipoprotein
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12490
Exendin-4 decreases body weight and plasma glucose and insulin levels. After 5 weeks of feeding aWesterntype diet containing 0.4% cholesterol, mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (PBS) s.c. for 4 weeks. Body weight was determined weekly (B). Blood was collected by tail bleeding after 4 h of fasting before treatment (T = 0), and after 2 (T = 2) and 4 (T = 4) weeks of treatment (A). Plasma glucose (C) and insulin (D) levels were determined. Values are means ± SEM (n = 17 mice per group). * P < 0.05, ** P < 0.01, *** P < 0.001 compared with vehicle.
Exendin-4 decreases hepatic expression and plasma level of CETP. After 5 weeks of feeding a Western-type diet containing 0.4% cholesterol, mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (PBS) s.c. for 4 weeks. mRNA was extracted from liver pieces, and mRNA expression of CETP (A) was determined as normalized to Cyclo and Hprt mRNA levels. Data were calculated as fold difference as compared with vehicle. Plasma CETP levels were determined (B). Values are means ± SEM ( n = 17 mice per group). * P < 0.05, ** P < 0.01 compared with vehicle.
Hepatic CETP expression is positively correlated with hepatic CD68 and F4/80 expression. After 5 weeks of feeding a Western-type diet containing 0.4% cholesterol, E3L.CETP mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (Control) s.c. for 4 weeks. Subsequently, livers were isolated and mRNA was extracted from liver pieces. mRNA expression of CD68, F4/80 and CETP was determined as normalized to Cyclo and Hprt mRNA levels. Data were calculated as fold difference as compared with vehicle and the correlation between hepatic CETP expression, and hepatic CD68 (A) or F4/80 (B) expression was linearly plotted.
Primer sequences used for RT-qPCR.
References
- Alexander SPH, et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. doi: 10.1111/bph.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, et al. Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes. 2010;59:1030–1037. doi: 10.2337/db09-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berres ML, Nellen A, Wasmuth HE. Chemokines as immune mediators of liver diseases related to the metabolic syndrome. Dig Dis. 2010;28:192–196. doi: 10.1159/000282085. [DOI] [PubMed] [Google Scholar]
- Bhatia LS, Curzen NP, Calder PC, Byrne CD. Non-alcoholic fatty liver disease: a new and important cardiovascular risk factor? Eur Heart J. 2012;33:1190–1200. doi: 10.1093/eurheartj/ehr453. [DOI] [PubMed] [Google Scholar]
- Bieghs V, Wouters K, van Gorp PJ, Gijbels MJ, de Winther MP, Binder CJ, et al. Role of scavenger receptor A and CD36 in diet-induced nonalcoholic steatohepatitis in hyperlipidemic mice. Gastroenterology. 2010;138:2477–2486. doi: 10.1053/j.gastro.2010.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieghs V, Rensen PC, Hofker MH, Shiri-Sverdlov R. NASH and atherosclerosis are two aspects of a shared disease: central role for macrophages. Atherosclerosis. 2012a;220:287–293. doi: 10.1016/j.atherosclerosis.2011.08.041. [DOI] [PubMed] [Google Scholar]
- Bieghs V, Verheyen F, van Gorp PJ, Hendrikx T, Wouters K, Lutjohann D, et al. Internalization of modified lipids by CD36 and SR-A leads to hepatic inflammation and lysosomal cholesterol storage in Kupffer cells. PLoS ONE. 2012b;7:e34378. doi: 10.1371/journal.pone.0034378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Brunt EM. Pathology of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7:195–203. doi: 10.1038/nrgastro.2010.21. [DOI] [PubMed] [Google Scholar]
- Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43:173–181. doi: 10.1002/hep.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallwitz B. Glucagon-like peptide-1 analogues for type 2 diabetes mellitus: current and emerging agents. Drugs. 2011;71:1675–1688. doi: 10.2165/11592810-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Gaspari T, Welungoda I, Widdop RE, Simpson RW, Dear AE. The GLP-1 receptor agonist liraglutide inhibits progression of vascular disease via effects on atherogenesis, plaque stability and endothelial function in an ApoE–/– mouse model. Diab Vasc Dis Res. 2013;10:353–360. doi: 10.1177/1479164113481817. [DOI] [PubMed] [Google Scholar]
- Gijbels MJ, van der Cammen M, van der Laan LJ, Emeis JJ, Havekes LM, Hofker MH, et al. Progression and regression of atherosclerosis in APOE*3-Leiden transgenic mice: an immunohistochemical study. Atherosclerosis. 1999;143:15–25. doi: 10.1016/s0021-9150(98)00263-9. [DOI] [PubMed] [Google Scholar]
- de Haan W, de Vries-van der Weij J, van der Hoorn JW, Gautier T, van der Hoogt CC, Westerterp M, et al. Torcetrapib does not reduce atherosclerosis beyond atorvastatin and induces more proinflammatory lesions than atorvastatin. Circulation. 2008;117:2515–2522. doi: 10.1161/CIRCULATIONAHA.107.761965. [DOI] [PubMed] [Google Scholar]
- Karlmark KR, Wasmuth HE, Trautwein C, Tacke F. Chemokine-directed immune cell infiltration in acute and chronic liver disease. Expert Rev Gastroenterol Hepatol. 2008;2:233–242. doi: 10.1586/17474124.2.2.233. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanthier N, Molendi-Coste O, Horsmans Y, van Rooijen N, Cani PD, Leclercq IA. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2010;298:G107–G116. doi: 10.1152/ajpgi.00391.2009. [DOI] [PubMed] [Google Scholar]
- Lee J, Hong SW, Chae SW, Kim DH, Choi JH, Bae JC, et al. Exendin-4 improves steatohepatitis by increasing Sirt1 expression in high-fat diet-induced obese C57BL/6J mice. PLoS ONE. 2012;7:e31394. doi: 10.1371/journal.pone.0031394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wang Y, van der Sluis RJ, van der Hoorn JW, Princen HM, Van Eck M, et al. Niacin reduces plasma CETP levels by diminishing liver macrophage content in CETP transgenic mice. Biochem Pharmacol. 2012;84:821–829. doi: 10.1016/j.bcp.2012.06.020. [DOI] [PubMed] [Google Scholar]
- McCarthy EM, Rinella ME. The role of diet and nutrient composition in nonalcoholic fatty liver disease. J Acad Nutr Diet. 2012;112:401–409. doi: 10.1016/j.jada.2011.10.007. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mells JE, Fu PP, Sharma S, Olson D, Cheng L, Handy JA, et al. Glp-1 analog, liraglutide, ameliorates hepatic steatosis and cardiac hypertrophy in C57BL/6J mice fed a Western diet. Am J Physiol Gastrointest Liver Physiol. 2012;302:G225–G235. doi: 10.1152/ajpgi.00274.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711. doi: 10.1161/01.ATV.0000229218.97976.43. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundil D, Cameron-Vendrig A, Husain M. GLP-1 receptor agonists: a clinical perspective on cardiovascular effects. Diab Vasc Dis Res. 2012;9:95–108. doi: 10.1177/1479164112441526. [DOI] [PubMed] [Google Scholar]
- Musso G, Gambino R, Cassader M, Pagano G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology. 2010;52:79–104. doi: 10.1002/hep.23623. [DOI] [PubMed] [Google Scholar]
- Nagashima M, Watanabe T, Terasaki M, Tomoyasu M, Nohtomi K, Kim-Kaneyama J, et al. Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia. 2011;54:2649–2659. doi: 10.1007/s00125-011-2241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, Brown TJ, et al. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE(–/–) mice. Endocrinology. 2013;154:127–139. doi: 10.1210/en.2012-1937. [DOI] [PubMed] [Google Scholar]
- Parlevliet ET, Wang Y, Geerling JJ, Schroder-Van der Elst JP, Picha K, O'Neil K, et al. GLP-1 receptor activation inhibits VLDL production and reverses hepatic steatosis by decreasing hepatic lipogenesis in high-fat-fed APOE*3-Leiden mice. PLoS ONE. 2012;7:e49152. doi: 10.1371/journal.pone.0049152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redgrave TG, Roberts DC, West CE. Separation of plasma lipoproteins by density-gradient ultracentrifugation. Anal Biochem. 1975;65:42–49. doi: 10.1016/0003-2697(75)90488-1. [DOI] [PubMed] [Google Scholar]
- Schattenberg JM, Schuppan D. Nonalcoholic steatohepatitis: the therapeutic challenge of a global epidemic. Curr Opin Lipidol. 2011;22:479–488. doi: 10.1097/MOL.0b013e32834c7cfc. [DOI] [PubMed] [Google Scholar]
- Svegliati-Baroni G, Saccomanno S, Rychlicki C, Agostinelli L, De MS, Candelaresi C, et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31:1285–1297. doi: 10.1111/j.1478-3231.2011.02462.x. [DOI] [PubMed] [Google Scholar]
- Targher G, Arcaro G. Non-alcoholic fatty liver disease and increased risk of cardiovascular disease. Atherosclerosis. 2007;191:235–240. doi: 10.1016/j.atherosclerosis.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Westerterp M, van der Hoogt CC, de Haan W, Offerman EH, Dallinga-Thie GM, Jukema JW, et al. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden mice. Arterioscler Thromb Vasc Biol. 2006;26:2552–2559. doi: 10.1161/01.ATV.0000243925.65265.3c. [DOI] [PubMed] [Google Scholar]
- Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–486. doi: 10.1002/hep.22363. [DOI] [PubMed] [Google Scholar]
- Yau H, Rivera K, Lomonaco R, Cusi K. The future of thiazolidinedione therapy in the management of type 2 diabetes mellitus. Curr Diab Rep. 2013;13:329–341. doi: 10.1007/s11892-013-0378-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Exendin-4 decreases body weight and plasma glucose and insulin levels. After 5 weeks of feeding aWesterntype diet containing 0.4% cholesterol, mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (PBS) s.c. for 4 weeks. Body weight was determined weekly (B). Blood was collected by tail bleeding after 4 h of fasting before treatment (T = 0), and after 2 (T = 2) and 4 (T = 4) weeks of treatment (A). Plasma glucose (C) and insulin (D) levels were determined. Values are means ± SEM (n = 17 mice per group). * P < 0.05, ** P < 0.01, *** P < 0.001 compared with vehicle.
Exendin-4 decreases hepatic expression and plasma level of CETP. After 5 weeks of feeding a Western-type diet containing 0.4% cholesterol, mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (PBS) s.c. for 4 weeks. mRNA was extracted from liver pieces, and mRNA expression of CETP (A) was determined as normalized to Cyclo and Hprt mRNA levels. Data were calculated as fold difference as compared with vehicle. Plasma CETP levels were determined (B). Values are means ± SEM ( n = 17 mice per group). * P < 0.05, ** P < 0.01 compared with vehicle.
Hepatic CETP expression is positively correlated with hepatic CD68 and F4/80 expression. After 5 weeks of feeding a Western-type diet containing 0.4% cholesterol, E3L.CETP mice were treated with exendin-4 (50 μg·kg−1·day−1) or vehicle (Control) s.c. for 4 weeks. Subsequently, livers were isolated and mRNA was extracted from liver pieces. mRNA expression of CD68, F4/80 and CETP was determined as normalized to Cyclo and Hprt mRNA levels. Data were calculated as fold difference as compared with vehicle and the correlation between hepatic CETP expression, and hepatic CD68 (A) or F4/80 (B) expression was linearly plotted.
Primer sequences used for RT-qPCR.