

Abstract
Background and Purpose
The COX-2/PGE2 pathway in hypoxic cancer cells has important implications for stimulation of inflammation and tumourigenesis. However, the mechanism by which glucocorticoid receptors (GRs) inhibit COX-2 during hypoxia has not been elucidated. Hence, we explored the mechanisms underlying glucocorticoid-mediated inhibition of hypoxia-induced COX-2 in human distal lung epithelial A549 cells.
Experimental Approach
The expressions of COX-2 and glucocorticoid-induced leucine zipper (GILZ) in A549 cells were determined by Western blot and/or quantitative real time-PCR respectively. The anti-invasive effect of GILZ on A549 cells was evaluated using the matrigel invasion assay.
Key Results
The hypoxia-induced increase in COX-2 protein and mRNA levels and promoter activity were suppressed by dexamethasone, and this effect of dexamethasone was antagonized by the GR antagonist RU486. Overexpression of GILZ in A549 cells also inhibited hypoxia-induced COX-2 expression levels and knockdown of GILZ reduced the glucocorticoid-mediated inhibition of hypoxia-induced COX-2 expression, indicating that the inhibitory effects of dexamethasone on hypoxia-induced COX-2 are mediated by GILZ. GILZ suppressed the expression of hypoxia inducible factor (HIF)-1α at the protein level and affected its signalling pathway. Hypoxia-induced cell invasion was also dramatically reduced by GILZ expression.
Conclusion and Implications
Dexamethasone-induced upregulation of GILZ not only inhibits the hypoxic-evoked induction of COX-2 expression and cell invasion but further blocks the HIF-1 pathway by destabilizing HIF-1α expression. Taken together, these findings suggest that the suppression of hypoxia-induced COX-2 by glucocorticoids is mediated by GILZ. Hence, GILZ is a potential key therapeutic target for suppression of inflammation under hypoxia.
Keywords: cyclooxygenase-2, hypoxia, hypoxia-inducible factor-1, glucocorticoid receptor, glucocorticoid-induced leucine zipper
Introduction
Hypoxia is a state of reduced overall tissue oxygen availability and causes many pathological states, including ischaemic disease, chronic inflammatory disease and tumour progression (Semenza, 2012). The expression of COX-2 is induced by the hypoxic microenvironment in various systems, such as colorectal tumours, pulmonary artery smooth muscle and epithelial cells (Kaidi et al., 2006; Fredenburgh et al., 2009; Lee et al., 2010; Zhao et al., 2012). This up-regulation of COX-2 is transcriptional and is mediated by the master regulator of transcription during hypoxia, hypoxia inducible factor (HIF)-1α (Kaidi et al., 2006; Lee et al., 2010). COX-2 is overexpressed in various cancer tissues (Kargman et al., 1995; Ristimaki et al., 1997; Wolff et al., 1998) and this has been linked to inflammation, inflammatory disorders and tumourigenesis (Gilroy et al., 2001; Mantovani et al., 2008). COX-2 and/or PGs have pleiotropic effects in tumours, promoting proliferation, survival, angiogenesis, migration and invasion (Greenhough et al., 2009).
Several studies have shown that hypoxia is associated with glucocorticoid-mediated cellular responses. Furthermore, glucocorticoids have been used to treat many pathological states where the perpetuation of hypoxia plays a major role, such as inflammatory bowel disease and rheumatoid arthritis (Baschant et al., 2012; Reuter et al., 2012; Sidoroff and Kolho, 2012). Hypoxia and glucocorticoid signalling converge to regulate the expression of the pro-inflammatory regulator, macrophage migration inhibitory factor (Elsby et al., 2009). Huang et al. (2009) reported that hypoxia reduces the glucocorticoid receptor (GR) and attenuates the anti-inflammatory effects of glucocorticoids, whereas others have reported that dexamethasone impairs the function of HIF-1 (Wagner et al., 2008; Elsby et al., 2009).
Recently, glucocorticoid-induced leucine zipper (GILZ) was identified (D'Adamio et al., 1997). GILZ is a member of the leucine zipper protein family and belongs to the transforming growth factor β-stimulated clone-22 family of transcription factors (Shibanuma et al., 1992; Ayroldi et al., 2002). GILZ has been shown to interact with and inhibit the activities of the key inflammatory signalling mediators NF-κB and activator protein-1 (AP-1) (Ayroldi et al., 2001; Mittelstadt and Ashwell, 2001). Glucocorticoids strongly suppress the expression of COX-2 induced by inflammatory stimuli. GILZ mediates this effect of glucocorticoids, inhibiting inflammatory cytokine-induced expression of COX-2 (Yang et al., 2008). One of the mechanisms by which glucocorticoids exert their anti-inflammatory effects is by impeding the MAPK signalling pathways (Ayroldi et al., 2012). However, the mechanism by which glucocorticoids inhibit the expression of COX-2 during hypoxia has not been elucidated. Hence, in this study, we investigated the mechanisms by which glucocorticoids inhibit hypoxia-induced COX-2. We found that glucocorticoid-induced GILZ plays a major role in the suppression of hypoxia-induced COX-2; GILZ inhibited the expression of HIF-1, which induces COX-2 under hypoxia. Furthermore, the expression of GILZ was found to be effective at inhibiting hypoxia-induced cell invasion. Our results suggest that GILZ is an important molecular therapeutic target for the inflammatory response and tumourigenesis in lung cancer.
Methods
Materials
Aldosterone, spironolactone, dexamethasone, RU486 and MG132 were purchased from Sigma (St. Louis, MO, USA). FBS, Trizol Reagent and penicillin/streptomycin were purchased from Gibco Invitrogen (Grand Island, NY, USA). Anti-HIF-1α was obtained from BD Biosciences (San Jose, CA, USA). Anti-β-actin was purchased from Sigma. Anti-COX-2 and anti-GILZ were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture and hypoxic conditions
Human pulmonary epithelial A549 cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were maintained in DMEM containing 10% FBS and penicillin/streptomycin. Cells were grown at 37°C in a humidified atmosphere of 95% air/5% CO2 and fed every 2–3 days. Before treatment, the cells were washed with PBS and cultured in DMEM/5% charcoal–dextran stripped FBS (CD-FBS) for 2 days. All treatments were done with DMEM/5% CD-FBS. For the hypoxic condition, cells were incubated at a CO2 level of 5% with 1% O2 balanced with N2 using a hypoxic chamber (Thermo Fisher Scientific, Waltham, MA, USA).
Plasmids
COX-2-Luc, a firefly luciferase reporter construct containing the mouse COX-2 gene promoter fragment −327/+59, a firefly luciferase reporter deletion construct of the human COX-2 promoter, were kindly provided by Dr Hiroyasu Inoue (Nara Women's University, Nara, Japan). The hypoxia-response element (HRE)-Luc reporter plasmid contains four copies of the erythropoietin HRE, the SV40 promoter and the luciferase gene. GFP-HIF-1α vector was kindly provided by Dr Kyu-Won Kim (Seoul National University, Seoul, Korea). A 3x (NF-κB)tk-Luc, a firefly luciferase reporter construct containing three repeated NF-κB-responsive elements, were kindly provided by Dr Sam Okret (Karolinska University Hospital Huddinge, Huddinge, Sweden). pcDNA3-Myc-GILZ and pSUPER-GILZ were kindly provided by Dr Marc Pallardy (Institut National de la Santé et de la Recherche Médicale, Paris, France).
Transfection and luciferase assays
A549 cells were transiently transfected with plasmids by using polyethylenimine (Polysciences, Warrington, PA, USA). Luciferase activity was determined 24 or 48 h after treatment with an AutoLumat LB9507 luminometer (EG & G Berthold, Bad Widbad, Germany) using the luciferase assay system (Promega Corp., Madison, WI, USA) and expressed as relative light units. For GILZ knockdown, pSUPER-GILZ was linearized before transfection together with a pTK-Hygromycine resistance plasmid. Hygromycine B–resistant cells were subcloned and selected for their levels of GILZ expression.
Reverse transcription (RT)- PCR
Total RNA was extracted using Trizol Reagent according to the manufacturer's instruction. RNA pellets were dissolved in diethylpyrocarbonate-treated water. The yield of RNA was quantified by spectroscopy at 260 nm. Samples were aliquoted and stored at −80°C until further processing. To synthesize first strand cDNA, 3 μg total RNA was incubated at 70°C for 5 min with 0.5 μg of random hexamer and deionized water (up to 11 μL). The RT reaction was performed using 40 U of M-MLV reverse transcriptase (Promega) in 5 × reaction buffer (250 mmol l−1 Tris-HCl; pH 8.3, 375 mM KCl, 15 mM MgCl2, 50 mM DTT), RNase inhibitor at 1 U μL−1, and 1 mM dNTP mixtures at 37°C for 60 min. Quantitative real-time PCR (qPCR) was performed using iQ™ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). The primers used were: β-actin sense primer, 5′- CAAATGCTTCTAGGCGGACTATG-3′; β-actin anti-sense primer, 5′- TGCGCAAGTTAGGTTTTGTCA-3′; VEGF sense primer, 5′- CTGCTGTCTTGGGTGCATTGG-3′; VEGF anti-sense primer, 5′- GTTTGATCCGCATAATCTGCAT-3′; COX-2β sense primer, 5′- TGAAGAACTTACAGGAGAAAA-3′; COX-2 anti-sense primer, 5′- TACCAGAAGGGCAGGATACA-3′. A final volume was 25 μL, and an iCycler iQ Real-time PCR Detection System (Bio-Rad) was used for qPCR. The amplification data were analysed by iQ™5 optical system software version 2.1 and calculated using the ΔΔCT method. The ΔΔCT method was used to calculate relative mRNA expression.
Western blot analysis
Protein was isolated in lysis buffer (150 mM NaCl, 50 mM Tris-HCl, 5 mM EDTA, 1% Nonidet P-40, 0.5% deoxycholate, 1% SDS) with protease inhibitor cocktail (Sigma) on ice for 1 h and then centrifuged for 20 min at 13 000 × g. Supernatant was collected and protein concentrations were measured using the Bradford method (Bio-Rad). Proteins were dissolved in sample buffer and boiled for 5 min before being loaded onto an acrylamide gel. After SDS-PAGE, proteins were transferred to a PVDF membrane, blocked with 5% non-fat dry milk in TBST for 60 min at room temperature. The membranes were incubated for 2 h at room temperature with antibody. Equal lane loading was assessed using β-actin monoclonal antibody (Sigma). After being washed with TBST, the blots were incubated with 1:5000 dilution of the HRP conjugated-secondary antibody (Gibco Invitrogen), and washed again three times with TBST. The transferred proteins were visualized with an enhanced chemiluminescence detection kit (Amersham Pharmacia Biotech, Buckinghamshire, UK).
Immunoprecipitation
Five hundred micrograms of the cell lysates were mixed with 1 μg of antibody and incubated overnight at 4°C with constant rotation. To recover immunoprecipitated complexes, 50 μL of protein A-sepharose, diluted 1:1 in PBS, were then added to the samples and incubated on ice. The beads were pelleted by centrifugation and the bound proteins were eluted by incubation in 2X SDS loading buffer by boiling. The eluted proteins were analysed by immunoblot analysis.
Cell migration and invasion assays
The migration assay was performed with Transwell inserts that have 6.5-mm polycarbonate membranes with 8.0-μm pores. Matrigel invasion assay was performed using membranes coated with Matrigel matrix (BD Science, Sparks, MD, USA). A549 cells were seeded into the upper chamber in serum-free media. The lower chambers consisted of DMEM media containing 10% FBS. After incubation under normoxia or hypoxia for 48 h, non-invasive cells present on the upper surface of the membrane were scraped with cotton swabs and the invasive cells present on the lower side of the membrane were fixed with ice-cold methanol, stained with 0.1% crystal violet. The cells that migrated and invaded to the lower side of the filter were observed using a light microscope and counted.
Statistical analysis
Data are expressed as means ± SD, and statistical analysis for single comparison was performed using the Student's t-test. The criterion for statistical significance was P < 0.05.
Results
Hypoxia induces COX-2 expression and GR inhibits hypoxia induction of COX-2 expression in A549 cells
COX-2 is transcriptionally induced by hypoxia and has been implicated in tumour progression and angiogenesis in colorectal tumour cells (Kaidi et al., 2006). However, no report has shown COX-2 behaviour in response to glucocorticoids under hypoxia in lung cancer cells, where COX-2 is also strongly implicated in tumourigenesis. Therefore, we first determined the effects of hypoxia on COX-2 expression in A549 cells. A549 cells were exposed to hypoxia for 0, 2, 6, 12 or 24 h. VEGF mRNA was examined in parallel as a control. COX-2 mRNA was increased at 2 h and continued to accumulate until 24 h, the latest time examined (Figure 1A). To assess the role of the GR in the hypoxia induction of COX-2 in A549 cells, we pre-incubated cells with dexamethasone and/or the GR antagonist RU486 for 1 h and co-treated with hypoxia. Ten nanomolar dexamethasone blocked the hypoxia-induced increase in COX-2 mRNA (Figure 1B) and protein (Figure 1C) expression and COX-2 promoter activity (Figure 1D). This suppressive effect of dexamethasone was antagonized by the GR antagonist RU486 (Figure 1C,D), suggesting that the response is mediated by GRs. To further determine the specificity of the inhibitory effect of dexamethasone on hypoxia-induced COX-2 regulation, we determined whether aldosterone exhibits a similar suppressive effects. Cells were pre-incubated with aldosterone and/or the mineralocorticoid receptor antagonist spironolactone for 1 h and were co-treated with hypoxia for 24 h. As shown in Figure 1E, aldosterone did not suppress the hypoxia-mediated induction of COX-2 protein expression and spironolactone failed to antagonize the dexamethasone-induced suppression of COX-2 in A549 cells. These results indicate that dexamethasone specifically inhibits hypoxia-induced COX-2 regulation through the GR in A549 cells.
Figure 1.

Hypoxia induces COX-2 expression and the GR inhibits hypoxia induction of COX-2 expression in A549 cells. (A,B) A549 cells were treated with hypoxia as indicated for 24 h and analysed by qPCR. (C) A549 cells were pretreated with dexamethasone (0.1 μM) and/or RU486 (1 μM) for 1 h before treatment with hypoxia for 24 h and analysed by Western blot. (D) A549 cells were transfected with COX-2-Luc and treated as indicated. After treatment, luciferase expression was determined. (E) A549 cells were pretreated with aldosterone (0.1 μM) and/or spironolactone (1 μM) for 1 h before treatment with hypoxia for 24 h. Immunoblots were probed with a COX-2 antibody and reprobed with a β-actin antibody. Dex, dexamethasone; Aldo, aldosterone. Spiro, spironolactone. Values represent the mean ± SD (n = 3). *P < 0.05. All experiments were repeated at least three times.
GILZ inhibits hypoxia-induced COX-2 expression
To test the hypothesis that glucocorticoid inhibition of hypoxia-induced COX-2 is mediated by GILZ, we first determined whether glucocorticoid induces GILZ expression in A549 cells. As shown in Figure 2A, dexamethasone significantly increased GILZ mRNA expression in A549 cells. Next, to determine whether hypoxia-induced COX-2 expression is inhibited by GILZ, A549 cells were transfected with a GILZ expression vector and exposed to hypoxia. As shown in Figure 2B, transfection of GILZ significantly reduced the hypoxia-induced increase in COX-2 mRNA expression. The hypoxia-induced increase in COX-2 protein expression was also completely eliminated by overexpression of GILZ (Figure 2C). Then, to determine whether the regulation of COX-2 is mediated by HIF-1α and GILZ, we examined the effect of HIF-1α and GILZ on COX-2 promoter activity. The expression of GILZ effectively inhibited COX-2 promoter activity both during hypoxia and when HIF-1α was overexpressed under normoxia (Figure 2D). These results demonstrate that the mere expression of GILZ blocked hypoxia-induced COX-2 expression and had an effect equivalent to that of dexamethasone treatment. The transcription of COX-2 depends on NF-κB binding to the COX-2 promoter (Ayroldi et al., 2012). We next investigated the effects of GILZ expression on NF-κB mediated gene transcription using a NF-κB responsive reporter gene. GILZ significantly decreased the hypoxia-induced transcriptional activity of NF-κB in a dose-dependent manner (Figure 2E).
Figure 2.

GILZ inhibits hypoxia-induced COX-2 expression. (A) A549 cells were pretreated with dexamethasone (0.1 μM) and/or RU486 (1 μM) for 1 h before treatment with hypoxia for 24 h and analysed by qPCR. (B) A549 cells were transfected with pcDNA-GILZ expression plasmids as indicated. At 24 h post-transfection, cells were incubated for 24 h under normoxic or hypoxic conditions and analysed by qPCR. (C) A549 cells were transfected with 2 μg of pcDNA-GILZ expression plasmids, treated as indicated and analysed by Western blot. (D) A549 cells were cotransfected with COX-2-Luc along with pcDNA-GILZ expression plasmids and/or HIF-1α plasmids. At 24 h post-transfection, cells were incubated for 24 h under normoxic or hypoxic conditions and luciferase activity was analysed. (E) A549 cells were transfected with pNF-κB-Luc reporter with or without pcDNA-GILZ expression plasmids. At 24 h post-transfection, cells were incubated for 24 h under normoxic or hypoxic conditions and analysed. Values represent the mean ± SD (n = 3). *P < 0.05. All experiments were repeated at least three times.
GILZ knockdown reduces glucocorticoid inhibition of hypoxia-induced COX-2 expression
To confirm that glucocorticoid inhibition of hypoxia-induced COX-2 expression is mediated by GILZ, we examined whether the expression of COX-2 was affected by knockdown of GILZ. As shown in Figure 3A, shGILZ significantly reduced dexamethasone-induced GILZ mRNA expression. The inhibitory effect of dexamethasone on COX-2 protein expression was impaired in GILZ-depleted cells (Figure 3A). Also, GILZ depletion completely blocked glucocorticoid inhibition of hypoxia-induced COX-2 promoter activity (Figure 3B), which confirms that GILZ mediates the inhibitory effects of glucocorticoids on the hypoxia-induced increase in COX-2.
Figure 3.
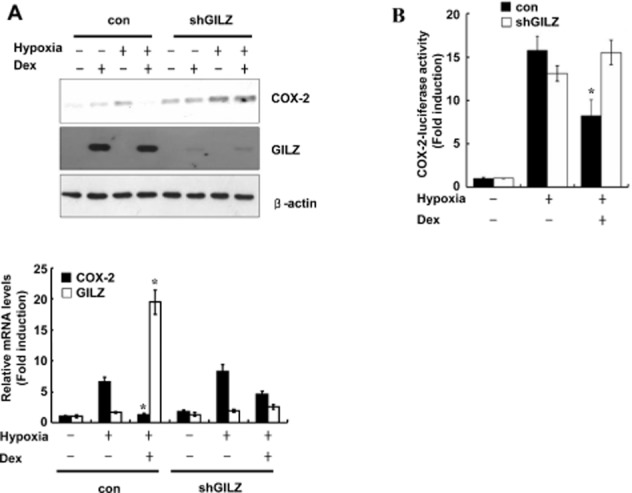
GILZ knockdown reduces glucocorticoid inhibition of hypoxia-induced COX-2 expression. (A) A549 cells were transfected with plasmid expressing shRNA targeting GILZ and pretreated with dexamethasone (0.1 μM) for 1 h before treatment with hypoxia for 24 h. Immunoblots were probed with a COX-2 antibody and reprobed with actin antibody (top). Total RNA from A549 cells were analysed for GILZ or COX-2 mRNA expression by qPCR (bottom). (B) A549 cells were transfected with COX-2-Luc reporter plasmid and/or plasmid shGILZ and treated as indicated. After treatment, luciferase expression was determined. Values represent the mean ± SD (n = 3). *P < 0.05. All experiments were repeated at least three times.
GILZ physically interacts with HIF-1α and serves as a negative regulator of HIF-1α
A few studies have demonstrated the existence of cross-talk between GRs and hypoxia. Dexamethasone impairs HIF-1 function in liver cells (Wagner et al., 2008). In contrast, HIF-1 was induced by dexamethasone in rat cardiac tissues (Roy et al., 2009). Conversely, hypoxia has been also shown to attenuate the effects of dexamethasone (Huang et al., 2009). However, the influence of GILZ on the HIF-1 pathway has not yet been elucidated. Therefore, we assessed the effects of glucocorticoids and GILZ on HIF-1 activity. Firstly, we determined whether GILZ expression influences HIF-1α expression. HIF-1α protein levels were decreased by the overexpression of GILZ as well as by dexamethasone treatment (Figure 4A). To investigate the involvement of the ubiquitin-proteasome pathway in GILZ-induced degradation of HIF-1α, A549 cells were transfected with GILZ and treated with or without the proteasome inhibitor MG132 for 24 h. As shown in Figure 4B, MG132 significantly reduced HIF-1α degradation by GILZ, suggesting that GILZ degrades HIF-1α via the proteasomal pathway. To assess the ubiquitination of HIF-1α by GILZ, we performed a ubiquitination assay in A549 cells transfected with Ubi and GILZ. As shown in Figure 4C, ubiquitination of the HIF-1α protein was enhanced by GILZ expression, indicating that this process is mediated through the ubiquitin-proteasome pathway. The GILZ-induced degradation of HIF-1α prompted us to investigate the possibility that GILZ may interact with HIF-1α. To test this hypothesis, co-immunoprecipitation assays were performed in A549 cells. GILZ was specifically precipitated by anti-HIF-1α antibody, but not by control IgG (Figure 4D). To further characterize GILZ inhibition of hypoxia-induced transcription activation, we investigated the effect of GILZ expression on HIF-1α-mediated gene transcription using an HRE-driven reporter gene. As shown in Figure 4E, GILZ significantly inhibited this hypoxic activation of HRE-luciferase reporter gene in a dose-dependent manner, indicating that the expression of GILZ significantly decreased HIF-1α transcriptional activity. Then, we investigated the effect of GILZ overexpression on an endogenous HIF-1α target, VEGF gene transcription. As shown in Figure 4F, the expression of GILZ significantly decreased VEGF steady-state mRNA levels under hypoxic conditions. As a positive control, COX-2 levels were examined simultaneously. This shows that GILZ physically interacts with HIF-1α and is a negative regulator of HIF-1.
Figure 4.

GILZ physically interacts with HIF-1α and serves as a negative regulator of HIF-1. (A,B) A549 cells were transfected with pcDNA-GILZ expression plasmids. At 24 h post-transfection, cells were pretreated with dexamethasone (0.1 μM) for 1 h before treatment with hypoxia for 24 h and analysed by Western blot (WB). (C) A549 cells were transfected with Ubi or pcDNA-GILZ plasmid as indicated. At 36 h post-transfection, cells were treated with 10 μM MG132 for 12 h. Ubi-conjugated HIF-1 was detected using anti-ubiquitin antibody. Arrows indicate the ubiquitinated HIF-1 protein bands. (D) After transfection with pcDNA-GILZ and HIF-1α, the whole cell lysates were immunoprecipitated (IP) with GILZ or HIF-1α antibody, and WB was performed with GILZ or HIF-1α antibody after immunoprecipitation. The expression of proteins was analysed by WB using GILZ or HIF-1α antibody as input. (E) A549 cells were transfected with HRE-Luc reporter with or without pcDNA-GILZ expression plasmids, treated as indicated, and luciferase activity was assayed. (F) A549 cells were transfected with pcDNA-GILZ expression plasmids and treated as indicated. COX-2 and VEGF mRNA expression was quantified by qPCR. Values represent the mean ± SD (n = 3). *P < 0.05. All experiments were repeated at least three times.
The GR suppresses hypoxia-mediated induction of COX-2 via effects on CREB binding protein (CBP)
Glucocorticoid-mediated transrepression has been reported to be regulated by histone acetylation (Kagoshima et al., 2001). Deacetylation of the GR induces its association with NF-κB and the attenuation of pro-inflammatory gene transcription (Ito et al., 2006). Increased levels of CBP, which possesses an intrinsic histone acetylase function, prevent the inhibition of glucocorticoid-mediated supression of NF-κB activity (Sheppard et al., 1998). We therefore investigated whether acetylation affected glucocorticoid-mediated inhibition of hypoxia-induced COX-2. As shown in Figure 5A, the ability of dexamethasone to inhibit the hypoxic induction of COX-2 was attenuated by the histone deacetylase inhibitor trichostatin A (TSA). In addition, TSA in the absence of hypoxia significantly increased the COX-2 protein level and luciferase activity of the full-length COX-2 promoter in A549 cells (Figure 5B). Furthermore, the ability of dexamethasone to inhibit the hypoxic induction of COX-2 was markedly attenuated in CBP-overexpressing cells (Figure 5C). These results suggest that deacetylation plays an important role in GR-mediated inhibition of hypoxia-induced COX-2 and controls the GR-GILZ-mediated signalling pathway.
Figure 5.
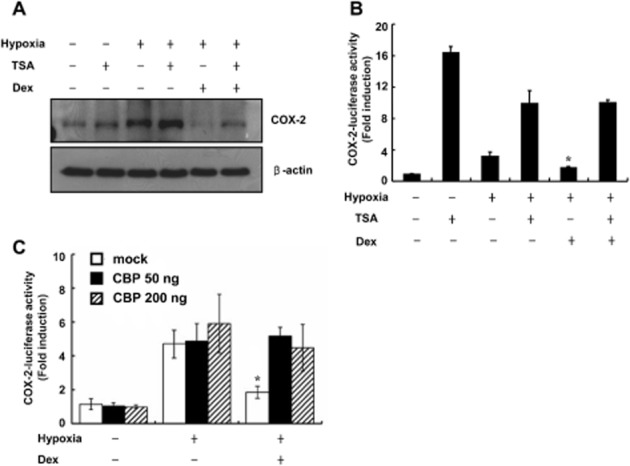
GR suppresses hypoxia-mediated induction of COX-2 via effects of on CBP. (A) A549 cells were pretreated with dexamethasone (0.1 μM) and/or TSA (1 μM) for 1 h before treatment with hypoxia for 24 h and analysed by Western. (B) A549 cells were transfected with 0.5 μg of COX-2-Luc reporter plasmid and treated as indicated, and luciferase activity was assayed. (C) A549 cells were transfected with 0.5 μg of COX-2-Luc reporter plasmid with or without CBP expression plasmids, treated as indicated, and luciferase expression was determined. Values represent the means ± SD (n = 3). *P < 0.05. Dex, dexamethasone. All experiments were repeated at least three times.
GILZ inhibits cellular migration and invasion of A549 cells under hypoxia
The hypoxic microenvironment within solid tumours has been increasingly recognized as an important driver of local invasion and metastasis (Arsenault et al., 2013). To evaluate the biological relevance of the reduced COX-2 expression induced by GILZ in A549 cells, we examined the effect of GILZ overexpression on A549 cell migration and invasion capacity during hypoxia. As shown in Figure 6, the migration and invasion capacities of A549 cells were increased under hypoxic conditions as compared with those under normoxic conditions (Figure 6A,B). The expression of GILZ significantly decreased the migratory potential of A549 cells under hypoxic conditions by 40% (Figure 6A). Similarly, GILZ also inhibited the invasion of A549 cells under hypoxic conditions by 25% (Figure 6B). Although, GILZ overexpression did show a dramatic induction of GILZ mRNA (Figure 6C), it was not as effective as dexamethasone at reducing the migratory potential and invasion of these cells, suggesting that other dexamethasone-induced factors may influence cell migration and invasion. These findings indicate that GILZ has anti-invasive and antimigratory properties under a hypoxic microenvironment.
Figure 6.

GILZ inhibits cellular migration and invasion of A549 cells under hypoxia. A549 cells were transfected with pcDNA-GILZ and treated as indicated. Transwell migration assays (A) and matrigel invasion assays (B) were done under normoxia or hypoxia. The cells that migrated and invaded were counted and are shown in the graph below. (C) A549 cells were transfected with 2 μg of pcDNA-GILZ expression plasmids, treated as indicated. GILZ mRNA expression was quantified by qPCR. Values represent the means ± SD (n = 3). *P < 0.05. All experiments were repeated at least three times.
Discussion
In this study, we sought to understand the mechanism by which glucocorticoids suppress hypoxia-induced COX-2. Our data showed that dexamethasone-induced the expression of GILZ, which supressed both hypoxia-induced COX-2 and HIF-1α expression. Also the overexpression of GILZ was sufficient to mimic the inhibitory effects of glucocorticoids on COX-2 and on hypoxia-induced invasion. Furthermore, GILZ degraded the hypoxia-stabilized HIF-1α via an effect on the proteasome pathway, which interrupts the initial stimuli inducing COX-2 under hypoxia. Glucocorticoids play a key role in the suppression of inflammation by inhibiting the transcription of the cytokines through binding to the GR and the activated GR interacts with transcription factors, such as AP-1, NF-κB and CCAAT/enhancer-binding protein-β (De Bosscher et al., 2003). Induction of GILZ under hypoxia has been shown to play an important role in hypoxic adaptation and inhibition of IL-1β and IL-6 secretions (Wang et al., 2012). Yang et al. (2008) reported that GILZ inhibits cytokine-induced COX-2 expression in bone marrow mesenchymal stem cells by blocking NF-κB nuclear translocation. However, the interaction between hypoxia and GR- and GILZ-mediated anti-inflammatory actions is not fully understood. Our data clearly demonstrated that the induction of GILZ is a key event in the inhibitory effect of glucocorticoids on hypoxia-induced COX-2, which is an indirect result of GR activation, through synthesis of new proteins. These data suggest that the regulation of GILZ has a significant effect on the hypoxia and glucocorticoid signalling pathways.
Studies have shown the existence of cross-talk between hypoxia and steroid hormone receptor pathways, including the GR. Hypoxia causes a down-regulation of GRs, which attenuates the anti-inflammatory actions in A549 cells and contributes to the glucocorticoid insensitivity observed in some respiratory diseases associated with hypoxia (Huang et al., 2009). In contrast, Leonard et al. (2005) reported a potentiation of glucocorticoid effects due to hypoxia through the induction of GR, increasing glucocorticoid sensitivity. Dexamethasone has also been shown to attenuate HIF activity in a GR-dependent manner (Wagner et al., 2008), but the function of GILZ was not investigated in this study. Others have shown that HIF-1α interacts with the GR, by using an artificial approach with GAL4-fusion reporter assays (Kodama et al., 2003), and that GR activation is associated with enhanced HIF-1 activity, which is partly in contrast to our findings (Kodama et al., 2003). Our data showed that dexamethasone treatment led to a marked dose-dependent and rapid inhibition of HIF-1α expression and affected HIF-1α-mediated target gene expression, suggesting that targeting HIF-1α may promote antitumor immune responses. Furthermore, we demonstrated that GILZ is responsible for the GR-mediated downregulation of HIF-1α establishing an inhibitory loop for COX-2 inhibition. Our results suggest that the cross-talk between the GR and HIF-1α involves an interaction between GILZ and HIF-1α. In addition, it is possible that cross-talk between HIF-1 and steroid hormone receptors, such as the oestrogen and androgen receptor, is mediated by a common regulator rather than direct interaction of HIF-1α with a nuclear hormone receptor. The interplay between pro-inflammatory transcription factors and GR may reflect differing effects on histone acetylation. Hypoxia alters global histone H3 lysine 9 acetylation and methylation in A549 cells, and dexamethasone suppresses IL-1β-stimulated histone acetylation by direct inhibition of CBP-associated histone acetyltransferase activity (Ito et al., 2000; Chen et al., 2006). The dynamic pattern of histone H4 acetylation has been demonstrated to be associated with the induction of COX-2 transcription by bradykinin and IL-1β (Nie et al., 2003). Indeed, we found that TSA and CBP overexpression significantly reduced the glucocorticoid-mediated inhibition of hypoxia-induced COX-2 protein expression and COX-2 promoter activity. Thus, histone modification by hypoxia may result in conformational changes in chromatin and selective association of the transcription factor with the COX-2 promoter, leading to enhanced expression of the COX-2 gene. Moreover, the GR may modulate hypoxia-induced COX-2 expression through histone acetylation. Our data suggest that acetylation overcomes the necessity for GILZ induction by glucocorticoid. Additional studies are required to elucidate the exact mechanisms underlying the hypoxia-induced alterations in glucocorticoid-GR interactions at multiple levels.
Hypoxia is a hallmark of solid tumours that leads to cell invasion and metastasis (Arsenault et al., 2013). HIF-1 transcriptional activity was proposed to be in part responsible for the enhanced invasive properties of cancer cell (Krishnamachary et al., 2003; McMahon et al., 2005). Most solid tumours exhibit inflammatory properties characterized by increased levels of prostaglandins (Stasinopoulos et al., 2013). Hypoxia has been shown to induce the expression of COX-2 (Bradbury et al., 2002). COX-2 increases the metastatic potential of cancer cells and silencing of COX-2 inhibits metastasis and delays tumour onset of poorly differentiated metastatic cancer cells (Tsujii et al., 1997; Stasinopoulos et al., 2013). These observations indicate the importance of inhibiting COX-2 to prevent hypoxia-induced cell invasion. Only a few previous studies have demonstrated the effect of glucocorticoids on cell invasion but no direct observations have been made under hypoxia (Zheng et al., 2012). Hence, the results of the present study are the first to demonstrate that GILZ inhibits cell invasion most likely through the destabilization of HIF-1α and the induction of GILZ could be crucial for inhibition of HIF-1α-mediated metastasis and cancer progression.
Until now, not much attention has been paid to the role of GILZ in hypoxia-induced inflammatory responses and cell metastasis. The results of the present study clearly show that GILZ is a major player in glucocorticoid suppression of hypoxia-induced COX-2 in A549 cells and provide mechanistic evidence for the anti-inflammatory effect of GILZ under hypoxia. We suggest that the induction of GILZ has potential therapeutic value and could be a key therapeutic target for suppression of inflammation and cancer therapy under hypoxic conditions.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2012R1A2A2A06044458; 2012R1A1B5001291) to Y. J. L.
Glossary
- GILZ
glucocorticoid-induced leucine zipper
- GR
glucocorticoid receptor
- HIF
hypoxia-inducible factor
Conflict of interest
None.
References
- Arsenault D, Brochu-Gaudreau K, Charbonneau M, Dubois CM. HDAC6 deacetylase activity is required for hypoxia-induced invadopodia formation and cell invasion. PLoS One. 2013;8:e55529. doi: 10.1371/journal.pone.0055529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, et al. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood. 2001;98:743–753. doi: 10.1182/blood.v98.3.743. [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C. Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol. 2002;22:7929–7941. doi: 10.1128/MCB.22.22.7929-7941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroldi E, Cannarile L, Migliorati G, Nocentini G, Delfino DV, Riccardi C. Mechanisms of the anti-inflammatory effects of glucocorticoids: genomic and nongenomic interference with MAPK signaling pathways. FASEB J. 2012;26:4805–4820. doi: 10.1096/fj.12-216382. [DOI] [PubMed] [Google Scholar]
- Baschant U, Lane NE, Tuckermann J. The multiple facets of glucocorticoid action in rheumatoid arthritis. Nat Rev Rheumatol. 2012;8:645–655. doi: 10.1038/nrrheum.2012.166. [DOI] [PubMed] [Google Scholar]
- Bradbury DA, Newton R, Zhu YM, Stocks J, Corbett L, Holland ED, et al. Effect of bradykinin, TGF-beta1, IL-1beta, and hypoxia on COX-2 expression in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L717–L725. doi: 10.1152/ajplung.00070.2002. [DOI] [PubMed] [Google Scholar]
- Chen H, Yan Y, Davidson TL, Shinkai Y, Costa M. Hypoxic stress induces dimethylated histone H3 lysine 9 through histone methyltransferase G9a in mammalian cells. Cancer Res. 2006;66:9009–9016. doi: 10.1158/0008-5472.CAN-06-0101. [DOI] [PubMed] [Google Scholar]
- D'Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, et al. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. 1997;7:803–812. doi: 10.1016/s1074-7613(00)80398-2. [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- Elsby LM, Donn R, Alourfi Z, Green LM, Beaulieu E, Ray DW. Hypoxia and glucocorticoid signaling converge to regulate macrophage migration inhibitory factor gene expression. Arthritis Rheum. 2009;60:2220–2231. doi: 10.1002/art.24659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredenburgh LE, Ma J, Perrella MA. Cyclooxygenase-2 inhibition and hypoxia-induced pulmonary hypertension: effects on pulmonary vascular remodeling and contractility. Trends Cardiovasc Med. 2009;19:31–37. doi: 10.1016/j.tcm.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilroy DW, Saunders MA, Sansores-Garcia L, Matijevic-Aleksic N, Wu KK. Cell cycle-dependent expression of cyclooxygenase-2 in human fibroblasts. FASEB J. 2001;15:288–290. doi: 10.1096/fj.00-0573fje. [DOI] [PubMed] [Google Scholar]
- Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–386. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- Huang Y, Zhao JJ, Lv YY, Ding PS, Liu RY. Hypoxia down-regulates glucocorticoid receptor alpha and attenuates the anti-inflammatory actions of dexamethasone in human alveolar epithelial A549 cells. Life Sci. 2009;85:107–112. doi: 10.1016/j.lfs.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, et al. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagoshima M, Wilcke T, Ito K, Tsaprouni L, Barnes PJ, Punchard N, et al. Glucocorticoid-mediated transrepression is regulated by histone acetylation and DNA methylation. Eur J Pharmacol. 2001;429:327–334. doi: 10.1016/s0014-2999(01)01332-2. [DOI] [PubMed] [Google Scholar]
- Kaidi A, Qualtrough D, Williams AC, Paraskeva C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 2006;66:6683–6691. doi: 10.1158/0008-5472.CAN-06-0425. [DOI] [PubMed] [Google Scholar]
- Kargman SL, O'Neill GP, Vickers PJ, Evans JF, Mancini JA, Jothy S. Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res. 1995;55:2556–2559. [PubMed] [Google Scholar]
- Kodama T, Shimizu N, Yoshikawa N, Makino Y, Ouchida R, Okamoto K, et al. Role of the glucocorticoid receptor for regulation of hypoxia-dependent gene expression. J Biol Chem. 2003;278:33384–33391. doi: 10.1074/jbc.M302581200. [DOI] [PubMed] [Google Scholar]
- Krishnamachary B, Berg-Dixon S, Kelly B, Agani F, Feldser D, Ferreira G, et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003;63:1138–1143. [PubMed] [Google Scholar]
- Lee JJ, Natsuizaka M, Ohashi S, Wong GS, Takaoka M, Michaylira CZ, et al. Hypoxia activates the cyclooxygenase-2-prostaglandin E synthase axis. Carcinogenesis. 2010;31:427–434. doi: 10.1093/carcin/bgp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard MO, Godson C, Brady HR, Taylor CT. Potentiation of glucocorticoid activity in hypoxia through induction of the glucocorticoid receptor. J Immunol. 2005;174:2250–2257. doi: 10.4049/jimmunol.174.4.2250. [DOI] [PubMed] [Google Scholar]
- McMahon S, Grondin F, McDonald PP, Richard DE, Dubois CM. Hypoxia-enhanced expression of the proprotein convertase furin is mediated by hypoxia-inducible factor-1: impact on the bioactivation of proproteins. J Biol Chem. 2005;280:6561–6569. doi: 10.1074/jbc.M413248200. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem. 2001;276:29603–29610. doi: 10.1074/jbc.M101522200. [DOI] [PubMed] [Google Scholar]
- Nie M, Pang L, Inoue H, Knox AJ. Transcriptional regulation of cyclooxygenase 2 by bradykinin and interleukin-1beta in human airway smooth muscle cells: involvement of different promoter elements, transcription factors, and histone h4 acetylation. Mol Cell Biol. 2003;23:9233–9244. doi: 10.1128/MCB.23.24.9233-9244.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter KC, Grunwitz CR, Kaminski BM, Steinhilber D, Radeke HH, Stein J. Selective glucocorticoid receptor agonists for the treatment of inflammatory bowel disease: studies in mice with acute trinitrobenzene sulfonic acid colitis. J Pharmacol Exp Ther. 2012;341:68–80. doi: 10.1124/jpet.111.183947. [DOI] [PubMed] [Google Scholar]
- Ristimaki A, Honkanen N, Jankala H, Sipponen P, Harkonen M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. 1997;57:1276–1280. [PubMed] [Google Scholar]
- Roy SG, De P, Mukherjee D, Chander V, Konar A, Bandyopadhyay D, et al. Excess of glucocorticoid induces cardiac dysfunction via activating angiotensin II pathway. Cell Physiol Biochem. 2009;24:1–10. doi: 10.1159/000227803. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2012;365:537–547. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- Sheppard KA, Phelps KM, Williams AJ, Thanos D, Glass CK, Rosenfeld MG, et al. Nuclear integration of glucocorticoid receptor and nuclear factor-kappaB signaling by CREB-binding protein and steroid receptor coactivator-1. J Biol Chem. 1998;273:29291–29294. doi: 10.1074/jbc.273.45.29291. [DOI] [PubMed] [Google Scholar]
- Shibanuma M, Kuroki T, Nose K. Isolation of a gene encoding a putative leucine zipper structure that is induced by transforming growth factor beta 1 and other growth factors. J Biol Chem. 1992;267:10219–10224. [PubMed] [Google Scholar]
- Sidoroff M, Kolho KL. Glucocorticoid sensitivity in inflammatory bowel disease. Ann Med. 2012;44:578–587. doi: 10.3109/07853890.2011.590521. [DOI] [PubMed] [Google Scholar]
- Stasinopoulos I, Shah T, Penet MF, Krishnamachary B, Bhujwalla ZM. COX-2 in cancer: Gordian knot or Achilles heel? Front Pharmacol. 2013;4:1–7. doi: 10.3389/fphar.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujii M, Kawano S, DuBois RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A. 1997;94:3336–3340. doi: 10.1073/pnas.94.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AE, Huck G, Stiehl DP, Jelkmann W, Hellwig-Bürgel T. Dexamethasone impairs hypoxia-inducible factor-1 function. Biochem Biophys Res Commun. 2008;372:336–340. doi: 10.1016/j.bbrc.2008.05.061. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ma YY, Song XL, Cai HY, Chen JC, Song LN, et al. Upregulations of glucocorticoid-induced leucine zipper by hypoxia and glucocorticoid inhibit proinflammatory cytokines under hypoxic conditions in macrophages. J Immunol. 2012;188:222–229. doi: 10.4049/jimmunol.1002958. [DOI] [PubMed] [Google Scholar]
- Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res. 1998;58:4997–5001. [PubMed] [Google Scholar]
- Yang N, Zhang W, Shi XM. Glucocorticoid-induced leucine zipper (GILZ) mediates glucocorticoid action and inhibits inflammatory cytokine-induced COX-2 expression. J Cell Biochem. 2008;103:1760–1771. doi: 10.1002/jcb.21562. [DOI] [PubMed] [Google Scholar]
- Zhao L, Wu Y, Xu Z, Wang H, Zhao Z, Li Y, et al. Involvement of COX-2/PGE2 signalling in hypoxia-induced angiogenic response in endothelial cells. J Cell Mol Med. 2012;16:1840–1855. doi: 10.1111/j.1582-4934.2011.01479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Izumi K, Li Y, Ishiguro H, Miyamoto H. Contrary regulation of bladder cancer cell proliferation and invasion by dexamethasone-mediated glucocorticoid receptor signals. Mol Cancer Ther. 2012;11:2621–2632. doi: 10.1158/1535-7163.MCT-12-0621. [DOI] [PubMed] [Google Scholar]