

Abstract
Background and Purpose
Receptor activity-modifying proteins (RAMPs) define the pharmacology of the calcitonin receptor-like receptor (CLR). The interactions of the different RAMPs with this class B GPCR yield high-affinity calcitonin gene-related peptide (CGRP) or adrenomedullin (AM) receptors. However, the mechanism for this is unclear.
Experimental Approach
Guided by receptor models, we mutated residues in the N-terminal helix of CLR, RAMP2 and RAMP3 hypothesized to be involved in peptide interactions. These were assayed for cAMP production with AM, AM2 and CGRP together with their cell surface expression. Binding studies were also conducted for selected mutants.
Key Results
An important domain for peptide interactions on CLR from I32 to I52 was defined. Although I41 was universally important for binding and receptor function, the role of other residues depended on both ligand and RAMP. Peptide binding to CLR/RAMP3 involved a more restricted range of residues than that to CLR/RAMP1 or CLR/RAMP2. E101 of RAMP2 had a major role in AM interactions, and F111/W84 of RAMP2/3 was important with each peptide.
Conclusions and Implications
RAMP-dependent effects of CLR mutations suggest that the different RAMPs control accessibility of peptides to binding residues situated on the CLR N-terminus. RAMP3 appears to alter the role of specific residues at the CLR-RAMP interface compared with RAMP1 and RAMP2.
Keywords: adrenomedullin, CGRP, GPCR, receptor activity-modifying protein, RAMP
Introduction
Adrenomedullin (AM) and AM2 (intermedin) are 52 and 47 amino acid peptides that are members of the calcitonin peptide family (Poyner et al., 2002; Hong et al., 2012). This family also includes calcitonin gene-related peptide (CGRP) and amylin. AM is involved in the development of the lymphatic and blood vasculature (Hinson et al., 2000; Fritz-Six et al., 2008; Ichikawa-Shindo et al., 2008). AM2 is a more recently discovered member of the AM family (Roh et al., 2004; Takei et al., 2004a,b2004b) and, along with AM, has extensive effects on the CVS including vasodilatation, cardioprotection, modulation of vascular tone and stimulation of angiogenesis (Hinson et al., 2000; Fujisawa et al., 2004; Takei et al., 2004a,b2004b).
The AM1 and AM2 receptors are class B (secretin family) GPCR (Poyner et al., 2002). However, they differ from many of the other members of this class, as they are formed from the obligate co-expression of the calcitonin receptor-like receptor (CLR) and receptor activity-modifying proteins (RAMP) 2 or 3 respectively. The CGRP receptor is formed of a complex between CLR and RAMP1 (McLatchie et al., 1998). AM and AM2 both activate AM1 and AM2 receptors, although AM2 has a 10-fold lower potency than AM at the AM1 receptor (Hong et al., 2012). CGRP also activates AM1 and AM2 receptors although with reduced potency compared to AM (McLatchie et al., 1998).
Class B GPCRs consist of a seven transmembrane helical region connected by extra- and intracellular loops; they are characterized by a large extracellular N-terminal domain (ECD). Although to date no full length, peptide-bound class B GPCR structures have been solved, the recent structures of the transmembrane domains of the glucagon- and corticotropin-releasing hormone receptor have given us great insight into the configuration of the transmembrane domain (Hollenstein et al., 2013; Siu et al., 2013). However, neither of these structures captures the native orientation of the ECD with respect to the transmembrane bundle and extracellular loops, although the modelling of the position of the ECD using the glucagon receptor structure may give some indication of its relative orientation (Siu et al., 2013). Crystal structures of isolated ECDs including the glucagon-like peptide-1 receptor (Runge et al., 2008; Underwood et al., 2010), parathyroid hormone (PTH) receptor (Pioszak and Xu, 2008), glucose-dependent insulinotropic polypeptide receptor (Parthier et al., 2007) and the corticotropin-releasing hormone receptors (Pioszak et al., 2008) are available. These structures support the two-domain model of peptide ligand binding (Hoare, 2005; Parthier et al., 2009). This model proposes that the C-terminal of the peptide is captured by the receptor ECD, which induces the formation of an α-helix in the peptide, stabilized by hydrophobic interactions with the receptor ECD. These receptor ECDs share the common ‘secretin family recognition fold’ consisting of two antiparallel β-sheets and an N-terminal α-helix stabilized by three disulphide bonds. The C-termini of the peptides bind in a common binding groove in the ECD composed of the N-terminal helix, ECD loops 2, 4 and the C-terminal region.
It has long been a challenge to understand how RAMPs allow different peptide preferences. It has been suggested that RAMPs do this by directly interacting with peptides, by altering the conformation of CLR or a combination of both. However, in attempts to identify the RAMP residues involved very few candidates have been identified (Qi and Hay, 2010). Thus far, in RAMP1 only W84 has a substantial effect on CGRP interactions (Moore et al., 2013). Its equivalent in RAMP2 (F111) has been implicated in AM binding, as has E101 (Kusano et al., 2012). Mutagenesis studies of RAMP1 and RAMP3 indicate that W74 and E74 (the analogous residue in each protein to E101 of RAMP2) are involved in AM selectivity (Hay et al., 2006a; Qi et al., 2008).
The recently determined crystal structures of the CGRP and AM1 receptor ECDs show that RAMPs interact with CLR by making extensive contacts with its N-terminal α-helix. Although no peptide is bound in any of these structures (ter Haar et al., 2010; Kusano et al., 2012), it has been proposed that due to the high structural similarity of the CLR ECD to the ECD of the parathyroid hormone (PTH)1 receptor, the peptide-binding groove may be in a similar position in CGRP and AM1 receptors (Archbold et al., 2011). This is broadly supported by alanine substitution experiments in the CGRP receptor, indicating that CLR residues I32, G35, T37 and I41 in the N-terminal α-helix form a cluster involved in the modification of CGRP interactions (Barwell et al., 2010). Furthermore, in the AM1 receptor, W72, F92 and W121 of CLR are thought to be involved in AM binding by virtue of the structural similarity of the CLR ECD to other class B ECD structures, which contain bound peptide (Kusano et al., 2012). If this is the case, then most RAMP residues would not be in a position to make major contacts with AM and CGRP, consistent with the lack of residues identified by RAMP mutagenesis. Interestingly, the structures reveal that RAMP2 is slightly rotated with respect to CLR, compared with RAMP1 (Kusano et al., 2012). This RAMP–CLR interaction could affect the exact position of the peptide-binding groove of CLR. An AM2 (CLR/RAMP3) receptor ECD structure is not available for comparison.
Therefore, there is much evidence pointing towards the notion that RAMP influences the orientation of key residues along the N-terminal α-helix of CLR, although the current ECD structures do not have sufficient resolution to provide details of this. In turn, this could affect the affinity of different peptide ligands for the CGRP, AM1 and AM2 receptors. In addition, the conserved aromatic residue at position 84/111 of the three RAMPs may also play a major role in influencing the peptide-binding groove, given its position in the crystal structures of the CGRP and AM1 receptor ECD.
In order to clarify the role of the RAMPs in ligand binding we compared the effect of mutating residues along the N-terminal α-helix of CLR on different peptides in the presence of different RAMPs. We also mutated the RAMP2 and RAMP3 residues implicated in influencing peptide interactions. We observed striking RAMP-dependent effects on the function of CLR mutants and confirm that the conserved aromatic residue at position 84/111 of RAMP is also a key determinant of peptide interactions. However, we conclude that the RAMPs are particularly important in the control of accessibility to a peptide-binding groove located at the interface between CLR and the RAMP.
Methods
Materials
Human AM (AM 1–52), rat AM (rAM) and human αCGRP were from American Peptide (Sunnyvale, CA, USA). Human AM2 (AM2-47) was from Bachem, Bubendorf, Switzerland. Forskolin was from Tocris Bioscience (Wiltshire, UK). AlphaScreen cAMP assay kits and all reagents and plates were from PerkinElmer (Boston, MA, USA).
Expression constructs and mutagenesis
Human CLR with an N-terminal haemagglutinin (HA) epitope tag, human RAMP2 with an N-terminal FLAG epitope tag (Qi et al., 2013) and untagged human RAMP3 were mutated using a method based on the Quik Change II site-directed mutagenesis kit (Stratagene, Cambridge, UK) as described previously (Conner et al., 2005; Bailey and Hay, 2007). The HA-CLR mutants have previously been described (Barwell et al., 2010).
Cell culture and transfection
Culture of Cos7 cells was performed as previously described (Bailey and Hay, 2006). Cells were cultured in DMEM supplemented with 8% heat-inactivated FBS and 5% (v/v) penicillin/streptomycin and kept in a 37°C humidified 95% air, 5% CO2 incubator. For cAMP assays, cells were seeded into 96-well plates at a density of 15 000 cells per well (determined using a Countess Counter™, Invitrogen, Carlsbad, CA, USA) 1 day before transfection. Cells were transiently transfected using polyethylenimine (PEI) as described previously (Bailey and Hay, 2006) using a 1:1 ratio of CLR to RAMP. For radioligand binding assays, cells were seeded into 24-well poly-d-lysine-coated plates (BD Biosciences, San Jose, CA, USA) at a density of 75 000 cells per well 1 day before transfection and transfected using PEI; total amount used, 1 μg of DNA per well.
cAMP assay
cAMP assays were performed as previously described (Gingell et al., 2010). Briefly, on the day of the assay, cells were serum-deprived in DMEM containing 1 mM IBMX and 0.1% BSA for 30 min. Peptides, reconstituted to 1 mM in ultra-pure water, were diluted in the same medium to give a final concentration range of 1 pM to 1 μM. Peptides were added to cells and incubated at 37°C for 15 min. The contents of the wells were then aspirated, and 50 μL of ice-cold absolute ethanol was added and allowed to evaporate. cAMP was extracted by adding 40 μL of lysis buffer, and the plates were gently shaken at room temperature for 15 min. Ten microlitres of each cell lysate was transferred to a 384 well plate, followed by 5 μL of acceptor beads, and the plate was sealed and incubated in the dark for 30 min at room temperature. Five microlitres of the donor bead mix was added to all wells; the plate was resealed and incubated in the dark for 16 h. The plates were read using an Envision plate reader (PerkinElmer). The quantity of cAMP produced was determined from the raw data using the cAMP standard curve.
Radioiodination and radioligand binding
rAM was iodinated using iodobeads (Pierce, Rockford, IL, USA). One iodobead was incubated with 100 μg (20 nmol) rAM and 1 mCi Na125I (PerkinElmer) for 5 min at room temperature. Iodinated rAM was purified by reverse phase HPLC. [125I]-rAM eluted as a single peak at 26% acetonitrile. Homologous competition radioligand binding assays were conducted on Cos7 cells in 24-well plates. Cells were washed once with warm-binding buffer (DMEM, 0.5% BSA). Wash media were replaced with ice-cold binding buffer followed by binding buffer containing [125I]-rAM (approximately 3 pmol, 30 000 cpm per well) and depending on experimental design increasing concentrations of unlabelled peptide. These ranged from 3 μM (non-specific binding) to 10 pM for human AM (hAM) and 10 μM to 10 nM for hAM2 and hαCGRP; each concentration was in duplicate or triplicate. Total binding was assessed in the absence of unlabelled peptide. Cells were then incubated at 4°C for 1 h followed by one wash with ice-cold PBS, pH 7.4 and solubilized in 0.2 M NaOH. Radioactivity was determined using a Wizard2 gamma counter (PerkinElmer).
Analysis of cell surface expression of mutants by elisa
Cell surface expression of all RAMP/HA-CLR receptor complexes was assessed by measuring HA-CLR expression in an elisa as previously described (Bailey and Hay, 2007). For selected mutants, FLAG-RAMP2 expression was also measured. Values were normalized to wild-type (WT) RAMP/CLR as 100% and empty vector transfected cells as 0%. In most assays, HA-CLR expression was used for normalization purposes, except in assays specifically measuring FLAG-RAMP2. Statistical significance between WT and mutants was determined by the Kruskal–Wallis test followed by a post hoc Dunnʼs test.
Generation of an AM2 receptor ECD model
Previously reviewed RAMP1 and RAMP3 full-length sequences were downloaded from the UniProt server, and a multiple sequence alignment was generated using the T-Coffee server (Poirot et al., 2003). Modeller9v8 (Sali and Blundell, 1993; Fiser and Sali, 2003) was used to generate 500 models of CLR/RAMP3 ECD using chains A and D from the protein database (PDB) accession 3N7S as templates. RAMP3 had a standard N-terminus cap whereas CLR contained an acetylated N-terminus. Both RAMP3 and CLR contained N-methylamide C-termini. This modification prevents unnecessary large electrostatic attractive forces between the ends of the protein fragment during energy minimization. The models were ranked by the Modeller9v8 energy objective function. The top 10 structures were retained, and the stereochemical quality was assessed by PROCHECK v3.5.4 (Laskowski et al., 1993; Laskowski, 2001). Based on overall and residue-by-residue geometry a structure was selected. The ProPka programme (Li et al., 2005) via the PDBQPR server (see Dolinsky et al., 2007) was used to assign the protonation states of the titratable groups in CLR ECD, using the Chemistry at HARvard Molecular Mechanics (CHARMM) parameters set at pH 7.0. The CHARMM (c35b3) module Screened Coulomb Potentials Implicit Solvent Model was used to minimize the model. One hundred steps of steepest descent were conducted followed by adopted basis Newton–Raphson minimization until convergence was met.
Data analysis
Data analysis was performed in GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA, USA). Data were fitted to obtain concentration–response curves using a four parameter logistic equation, and an F-test was conducted to compare whether the Hill slope of the curves was significantly different from 1. In most analyses, the Hill slope was not different from 1 and was therefore constrained to 1. From these curves, pEC50 values were obtained. Data were normalized in each experiment to the fitted minimum and maximum of the WT curve on each assay plate. Emax values from these curves are reported. The means of replicates from these individual experiments were combined to generate the curves that are shown. pEC50 and Emax values are presented as the mean ± SEM of values from individual data sets and were tested for statistical significance versus WT using a paired t-test. For radioligand binding assays specific binding was calculated by subtraction of non-specific binding (the highest unlabelled hAM concentration). Binding curves were analysed as for cAMP assays and normalized to their fitted minimum and maximum. pIC50 values from these curves are reported and tested for statistical significance using an unpaired t-test. Curves are presented as the combined means of data from each unlabelled hAM concentration for each individual experimental repeat. For the determination of relative binding for each of the mutant receptors compared with WT, specific binding data were normalized to the mean of specific binding for the WT receptor and an overall mean value calculated and analysed for statistical significance using the Kruskal–Wallis test followed by a post hoc Dunnʼs test. For all assays, significance was accepted at P < 0.05.
Results
Selection of residues in CLR, RAMP2 and RAMP3
We used the CGRP and AM1 receptor structures along with the residues involved in CGRP binding at the CGRP receptor to guide this study of residues potentially involved in peptide–receptor interactions (Barwell et al., 2010; ter Haar et al., 2010; Kusano et al., 2012). Residues along the exposed face of the CLR ECD helix ranging from V36 to I41 were selected for alanine substitution. We also focused on a region of importance in the CGRP receptor N-terminal to this helix and beyond the extent of the crystal structures between residues I32 and G35. Proceeding further along the same face of the ECD helix as I41, we did not test C48 as this forms a disulphide bond with C74, which is integral to the structural integrity of the ECD. Y49 is predicted to form part of the RAMP2/CLR interface (ter Haar et al., 2010; Kusano et al., 2012). We therefore tested this for cell surface expression. To further refine the limits of the binding pocket, the next residue whose effect we tested along the ECD helix was I52. We also tested two RAMP2 residues that are in the vicinity of I41 (E101, F111) and the corresponding RAMP3 residue (W84). Mutant receptors were assayed for cAMP production at the AM1, AM2 and CGRP receptors when stimulated with hAM or hAM2. The AM2 receptor was also stimulated with hαCGRP. hαCGRP was not used to stimulate the AM1 receptor due to its very low binding affinity and potency at this receptor (McLatchie et al., 1998; Aiyar et al., 2001; Poyner et al., 2002).
Residues in the CLR ECD in the AM1 receptor – stimulation of cAMP production
At the AM1 receptor the point mutants I32A, Q33A, G35A and V36A had no effect on AM potency (Table 1). L34A, T37A, I41A, M42A, Q45A and I52A all showed a decrease in AM potency, although the effect of T37A was not statistically significant (Table 1; Figure 1). I41A had the largest effect. AM2 potency at the AM1 receptor was affected in a similar manner to AM; however, in this case very large reductions in potency were seen for T37A, I41A and Q45A. No change was seen in AM2 potency for I52A, and a small reduction in potency was observed for G35A. Emax was substantially decreased for I41A and Q45A when the AM1 receptor was stimulated with AM and AM2. The Emax for Q33A with both ligands was substantially increased. Emax data are also presented in Supporting Information Figure S1 for easy comparison between receptors and peptides.
Table 1.
Summary of pEC50 and Emax values for mutants of CLR in the AM1 receptor when stimulated with hAM or hAM2
| hAM | hAM2 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | Mutant | Emax %WT | n | WT | Mutant | Emax %WT | n | |||||||
| CLR I32A | 9.07 ± 0.21 | 9.21 ± 0.27 | 133.9 ± 19.4 | 3 | 8.23 ± 0.03 | 8.24 ± 0.09 | 110.7 ± 15.5 | 3 | ||||||
| CLR Q33A | 9.06 ± 0.06 | 9.15 ± 0.05 | 269.4 ± 44.2* | 4 | 8.12 ± 0.09 | 7.89 ± 0.12 | 281.0 ± 92.5 | 4 | ||||||
| CLR L34A | 8.89 ± 0.03 | 8.50 ± 0.09* | 104.7 ± 42.9 | 3 | 8.25 ± 0.16 | 7.64 ± 0.20%** | 117.9 ± 17.9 | 4 | ||||||
| CLR G35A | 9.07 ± 0.22 | 8.91 ± 0.23 | 139.9 ± 26.8 | 3 | 8.22 ± 0.02 | 7.92 ± 0.03* | 92.9 ± 3.07 | 3 | ||||||
| CLR V36A | 9.07 ± 0.22 | 8.92 ± 0.25 | 138.2 ± 19.8 | 3 | 8.22 ± 0.02 | 7.89 ± 0.18 | 153.9 ± 50.8 | 3 | ||||||
| CLR T37A | 8.94 ± 0.13 | 8.25 ± 0.19 | 89.0 ± 9.96 | 3 | 8.12 ± 0.26 | 6.99 ± 0.21 | 70.6 ± 12.1 | 2a | ||||||
| CLR I41A | 9.14 ± 0.18 | 7.91 ± 0.34* | 33.7 ± 2.37%*** | 4 | 8.21 ± 0.34 | No curve | 3 | |||||||
| CLR M42A | 9.11 ± 0.05 | 8.48 ± 0.08* | 139.6 ± 36.9 | 3 | 8.21 ± 0.10 | 7.86 ± 0.13 | 89.9 ± 26.6 | 4 | ||||||
| CLR Q45A | 9.03 ± 0.19 | 8.45 ± 0.22%*** | 26.1 ± 8.61%** | 5 | 8.48 ± 0.24 | No curve | 3 | |||||||
| CLR I52A | 9.05 ± 0.16 | 8.66 ± 0.20* | 136.1 ± 33.6 | 6 | 8.03 ± 0.17 | 8.11 ± 0.24 | 94.6 ± 9.00 | 3 | ||||||
Data are the mean ± SEM of n individual data sets.
P < 0.05;
P < 0.01;
P < 0.001 versus WT receptor by paired t-test.
In two other experiments, the mutant did not produce any measurable response. ‘No curve’ indicates that the mutation abolished measurable receptor function.
Figure 1.
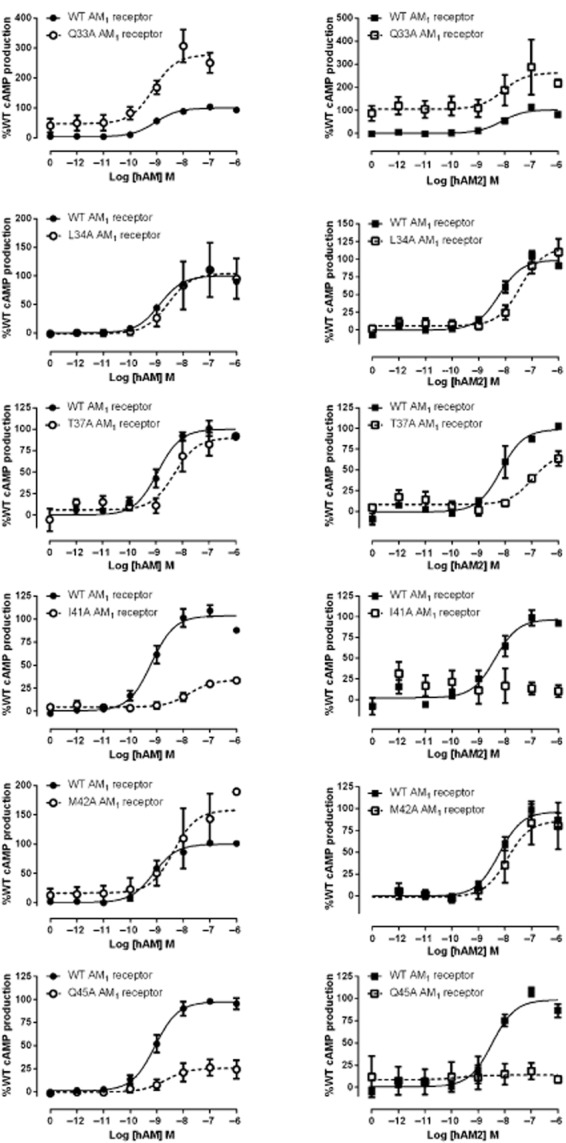
hAM and hAM2 stimulated cAMP responses of CLR mutant AM1 receptors. Normalized data were combined and data points shown are mean ± SEM of at least three individual data sets. Normalization is relative to the paired fitted maximal and basal cAMP production of the WT receptor.
Residues in the CLR ECD in the AM2 receptor – stimulation of cAMP production
At the AM2 receptor I32A, Q33A, V36A, M42A, Q45A and I52A had no effect on the potency of AM, AM2 or hαCGRP (Table 2). L34A and G35A decreased AM potency. T37A and I41A had a substantial effect on AM and AM2 potency. Only T37A and I41A had major effects on hαCGRP, with large reductions in Emax (Table 2, Figure 2). No significant effects on Emax were observed with any ligand for Q33A and Q45A.
Table 2.
Summary of pEC50 and Emax values for mutants of CLR in the AM2 receptor when stimulated with hAM, hAM2 or hαCGRP
| hAM | hAM2 | hαCGRP | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | Mutant | Emax %WT | n | WT | Mutant | Emax %WT | n | WT | Mutant | Emax %WT | n | |||||||||||
| CLR I32A | 9.54 ± 0.11 | 9.36 ± 0.13 | 135.2 ± 13.8 | 4 | 9.83 ± 0.13 | 9.47 ± 0.22 | 129.3 ± 30.7 | 3 | 7.20 ± 0.27 | 7.33 ± 0.15 | 72.7 ± 20.3 | 3 | ||||||||||
| CLR Q33A | 9.46 ± 0.11 | 9.61 ± 0.09 | 127.2 ± 19.2 | 3 | 8.59 ± 0.22 | 8.56 ± 0.14 | 155.2 ± 23.2 | 3 | 7.68 ± 0.26 | 7.42 ± 0.26 | 196.6 ± 40.7 | 4 | ||||||||||
| CLR L34A | 9.59 ± 0.06 | 8.88 ± 0.15** | 139.5 ± 19.3 | 4 | 9.42 ± 0.11 | 9.13 ± 0.16 | 104.5 ± 22.0 | 3 | 7.13 ± 0.11 | 7.24 ± 0.08 | 99.8 ± 7.96 | 4 | ||||||||||
| CLR G35A | 9.29 ± 0.24 | 8.77 ± 0.18* | 71.0 ± 11.8 | 3 | 9.09 ± 0.22 | 8.78 ± 0.12 | 97.9 ± 21.0 | 3 | 7.15 ± 0.10 | 6.50 ± 0.42 | 53.3 ± 16.3 | 3 | ||||||||||
| CLR V36A | 9.29 ± 0.24 | 9.06 ± 0.25 | 89.9 ± 5.71 | 3 | 9.09 ± 0.22 | 8.62 ± 0.19 | 121.0 ± 17.1 | 3 | 7.16 ± 0.12 | 6.91 ± 0.07 | 70.5 ± 9.21 | 3 | ||||||||||
| CLR T37A | 9.61 ± 0.20 | 8.63 ± 0.03* | 90.1 ± 17.1 | 3 | 9.13 ± 0.25 | 7.70 ± 0.14** | 68.5 ± 10.6 | 3 | 7.21 ± 0.13 | 7.17 ± 0.28 | 59.1 ± 10.2* | 4 | ||||||||||
| CLR I41A | 9.80 ± 0.13 | 8.51 ± 0.15** | 77.6 ± 9.69 | 4 | 9.32 ± 0.28 | 8.47 ± 0.34** | 87.4 ± 11.8 | 3 | 7.30 ± 0.37 | No curve | 3 | |||||||||||
| CLR M42A | 9.66 ± 0.30 | 9.52 ± 0.17 | 150.1 ± 32.0 | 4 | 8.82 ± 0.17 | 8.61 ± 0.11 | 262.8 ± 95.2 | 3 | 7.36 ± 0.16 | 7.33 ± 0.25 | 67.4 ± 19.0 | 4 | ||||||||||
| CLR Q45A | 9.61 ± 0.23 | 9.40 ± 0.16 | 138.3 ± 30.0 | 3 | 8.95 ± 0.10 | 8.94 ± 0.09 | 104.6 ± 16.9 | 4 | 7.50 ± 0.15 | 7.21 ± 0.26 | 112.2 ± 35.4 | 3 | ||||||||||
| CLR I52A | 9.55 ± 0.17 | 9.36 ± 0.22 | 97.5 ± 5.32 | 4 | 9.29 ± 0.14 | 9.12 ± 0.16 | 71.8 ± 22.4 | 3 | 7.42 ± 0.20 | 6.91 ± 0.34 | 108.8 ± 36.2 | 3 | ||||||||||
Data are the mean ± SEM of n individual data sets.
P < 0.05;
P < 0.01;
P < 0.001 versus WT receptor by paired t-test. ‘No curve’ indicates that the mutation abolished measurable receptor function.
Figure 2.

hAM, hAM2 and hαCGRP stimulated cAMP responses of CLR mutant AM2 receptors. Normalized data were combined and data points shown are mean ± SEM of at least three individual data sets. Normalization is relative to the paired fitted maximal and basal cAMP production of the WT receptor.
Residues in the CLR ECD in the CGRP receptor – stimulation of cAMP production
A similar study has been carried out to assess the effects of these mutations on CGRP potency at the CGRP receptor (Barwell et al., 2010). To provide an accurate comparison with all three RAMPs, we additionally studied the effects of selected mutations on AM and AM2 in the CGRP receptor. For AM, only I41A affected potency and only Q45A affected Emax (Table 3; Figure 3). More effects were seen with AM2; G35A, T37A, I41A and Q45A all showed a decrease in potency. Emax values were also decreased for I41A and Q45A. The Emax was increased for AM2 at the I32A CGRP receptor.
Table 3.
Summary of pEC50 and Emax values for mutants of CLR in the CGRP receptor when stimulated with hAM or hAM2
| hAM | hAM2 | |||||||
|---|---|---|---|---|---|---|---|---|
| WT | Mutant | Emax %WT | n | WT | Mutant | Emax %WT | n | |
| CLR I32A | 8.35 ± 0.14 | 8.33 ± 0.14 | 82.8 ± 16.5 | 4 | 8.42 ± 0.22 | 8.52 ± 0.33 | 141.9 ± 6.01* | 3 |
| CLR L34A | 8.49 ± 0.25 | 8.64 ± 0.09 | 87.8 ± 6.35 | 3 | 8.51 ± 0.20 | 8.44 ± 0.10 | 358.9 ± 118.4 | 3 |
| CLR G35A | 8.41 ± 0.17 | 8.21 ± 0.14 | 112.1 ± 11.1 | 3 | 8.40 ± 0.20 | 8.04 ± 0.20* | 149.8 ± 41.1 | 4 |
| CLR V36A | 8.22 ± 0.06 | 8.25 ± 0.08 | 124.3 ± 26.3 | 3 | 8.42 ± 0.22 | 8.21 ± 0.25 | 127.3 ± 19.8 | 3 |
| CLR T37A | 8.36 ± 0.27 | 8.09 ± 0.21 | 125.4 ± 20.6 | 3 | 8.31 ± 0.19 | 7.47 ± 0.26* | 83.0 ± 15.2 | 3 |
| CLR I41A | 8.54 ± 0.16 | 7.47 ± 0.13* | 68.4 ± 11.4 | 4 | 8.31 ± 0.19 | 7.23 ± 0.34* | 57.4 ± 8.66* | 3 |
| CLR M42A | 8.36 ± 0.27 | 8.25 ± 0.21 | 106.9 ± 26.8 | 3 | 8.33 ± 0.20 | 8.27 ± 0.20 | 92.5 ± 9.76 | 3 |
| CLR Q45A | 8.23 ± 0.25 | 7.87 ± 0.13 | 49.9 ± 4.27** | 3 | 8.65 ± 0.07 | 7.97 ± 0.08* | 34.7 ± 9.56* | 3 |
Data are the mean ± SEM of n individual data sets.
P < 0.05;
P < 0.01 versus WT receptor by paired t-test. To confirm cross-compatibility of assays, an experiment with CGRP under the same assay conditions as for AM and AM2 gave values of 10.1 for the WT receptor and 9.09 for the I41A CGRP receptor, consistent with the findings of Barwell et al. (2010).
Figure 3.
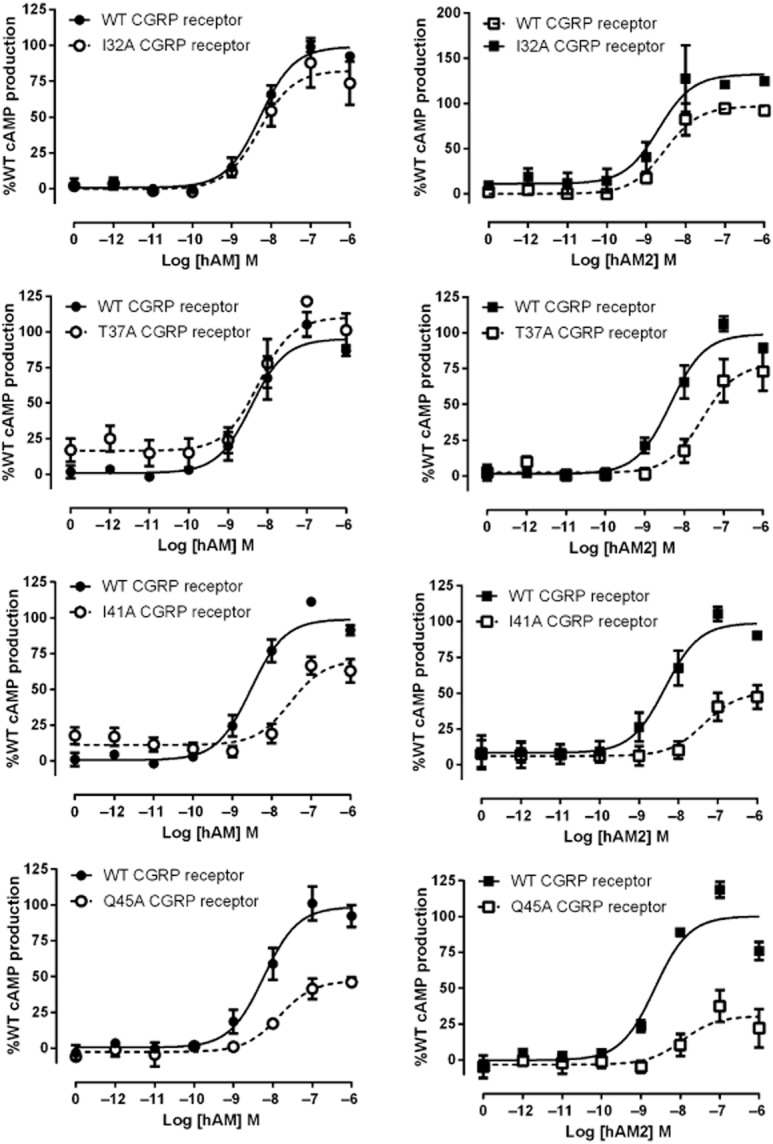
hAM and hAM2 stimulated cAMP responses of CLR mutant CGRP receptors. Normalized data were combined and data points shown are mean ± SEM of at least three individual data sets. Normalization is relative to the paired fitted maximal and basal cAMP production of the WT receptor.
Residues in the RAMP2 ECD in the AM1 receptor – stimulation of cAMP production
E101A and F111A mutations in RAMP2 both substantially affected receptor function (Table 4; Figure 4). For E101A this was evident as a 20-fold reduction in AM potency. With F111A, potency was apparently preserved but there was a large reduction in Emax. Due to the low Emax, the potency estimate should be treated with caution. Both residues also affected AM2-mediated receptor activation in a similar fashion.
Table 4.
Summary of pEC50 and Emax values for RAMP2 and RAMP3 mutants in the AM1 and AM2 receptors, respectively, when stimulated with hAM, hAM2 or hαCGRP
| hAM | hAM2 | hαCGRP | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WT | Mutant | Emax %WT | n | WT | Mutant | Emax %WT | n | WT | Mutant | Emax %WT | n | |||||||||||
| RAMP2 E101A | 9.06 ± 0.15 | 7.75 ± 0.30* | 71.1 ± 14.7 | 4 | 8.10 ± 0.05 | 7.39 ± 0.13 | 74.1 ± 14.8 | 3 | – | – | – | – | ||||||||||
| RAMP2 F111A | 9.06 ± 0.15 | 8.94 ± 0.29 | 39.2 ± 10.6* | 4 | 8.10 ± 0.05 | No curve | 3 | – | – | – | – | |||||||||||
| RAMP3 W84A | 9.64 ± 0.13 | 8.78 ± 0.23* | 59.0 ± 5.99** | 4 | 9.62 ± 0.08 | 9.02 ± 0.06** | 42.3 ± 3.34*** | 4 | 6.91 ± 0.15 | No curve | 4 | |||||||||||
| RAMP3 W84F | 9.58 ± 0.04 | 9.08 ± 0.07** | 72.6 ± 8.90 | 4 | 9.63 ± 0.08 | 9.38 ± 0.07 | 61.8 ± 7.21* | 4 | 7.06 ± 0.07 | 6.12 ± 0.09*** | 54.1 ± 5.34 | 4 | ||||||||||
Data are the mean ± SEM of n individual data sets.
P < 0.05;
P < 0.01;
P < 0.001 versus WT receptor by paired t-test. ‘No curve’ indicates that the mutation abolished measurable receptor function. – indicates experiment not performed due to low-ligand potency.
Figure 4.
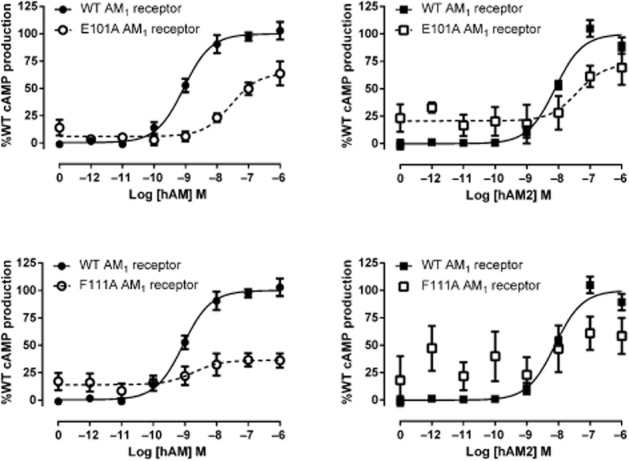
hAM and hAM2 stimulated cAMP responses of RAMP2 mutant AM1 receptors. Normalized data were combined and data points shown are mean ± SEM of at least three individual data sets. Normalization is relative to the paired fitted maximal and basal cAMP production of the WT receptor.
Residues in the RAMP3 ECD in the AM2 receptor – stimulation of cAMP production
We have previously extensively characterized mutations at E74 of RAMP3, the equivalent to E101 in RAMP2 (Hay et al., 2006b; Qi et al., 2008). The RAMP3 equivalent to F111 is W84. W84A resulted in a large decrease in AM potency and Emax, a smaller decrease in AM2 potency but a large decrease in Emax and complete abolition of hαCGRP responses (Table 4; Figure 5). The conservative mutation of W84F partially restored receptor function, with less perturbation of potency and Emax. AM potency was reduced only threefold with this mutation, and AM2 potency was not significantly reduced. hαCGRP responses were measurable with this mutation. A summary of all cAMP data is presented in Figure 6.
Figure 5.
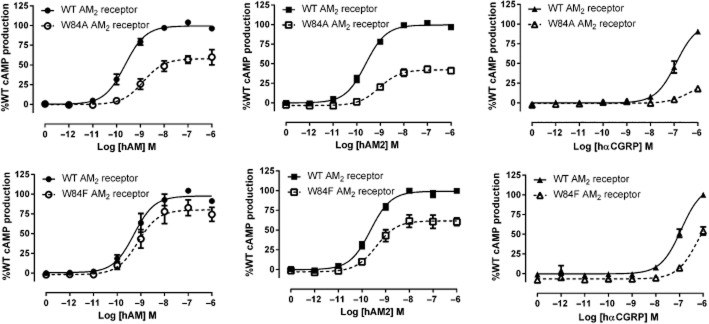
hAM, hAM2 and hαCGRP stimulated cAMP responses of RAMP3 mutant AM2 receptors. Normalized data were combined and data points shown are mean ± SEM of at least three individual data sets. Normalization is relative to the paired fitted maximal and basal cAMP production of the WT receptor.
Figure 6.

Effect of mutations in the N-terminal CLR residues on pEC50 values. Data are ordered to show the effect induced by RAMP (left) or peptide (right). The ΔlogEC50 values were calculated by subtracting the mean pEC50 for a given ligand at a given mutation from the mean of its paired WT control. For comparisons with the CGRP receptor, these values were calculated from the data presented in Barwell et al. (2010). Values above the line are an increase in potency and below the line are a decrease. Where the mutation abolished measurable receptor function, an arbitrary value of two has been assigned.
Residues in the CLR ECD in the AM1 and AM2 receptors – ligand binding
We used a whole-cell 125IrAM binding assay to determine the relative affinities of AM, AM2 and hαCGRP at the AM1 receptor. hAM had a mean pIC50 of 8.29 ± 0.25 (n = 4) and hαCGRP showed very weak binding (Figure 7), consistent with literature reports (Aiyar et al., 2001). hAM2 showed a 10-fold lower pIC50 than hAM (7.25 ± 0.05, n = 4, P < 0.01 by unpaired t-test), consistent with their rank order of potency (Hong et al., 2012).
Figure 7.
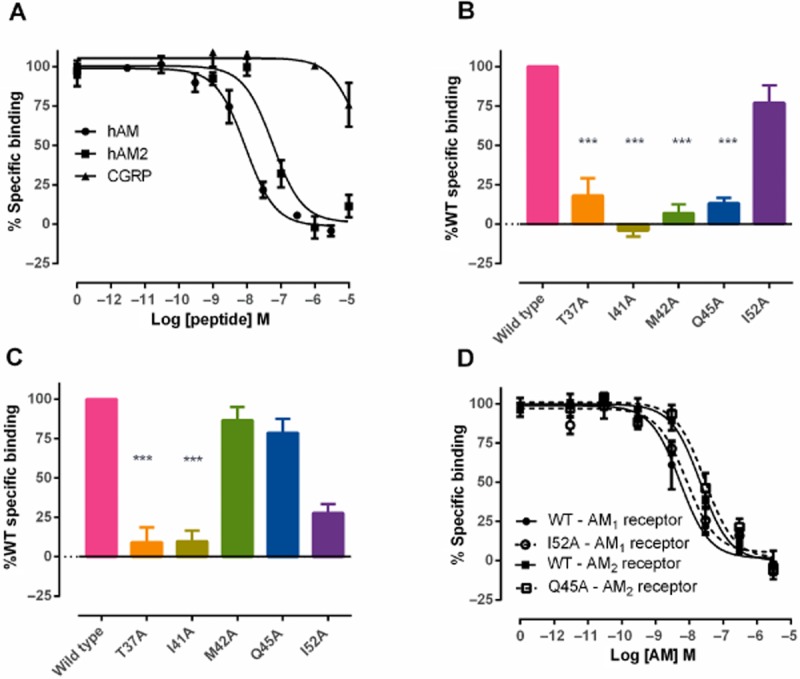
Effect of mutations in N-terminal CLR residues on ligand binding. (A) Competitive binding curves showing the displacement of [125I]-rAM by hAM, hAM2 and hαCGRP at the AM1 receptor. (B) Specific binding of [125I]-rAM to selected CLR mutants at the AM1 receptor expressed as a percentage of WT binding (100%). (C) Specific binding of [125I[-rAM to selected CLR mutants at the AM2 receptor expressed as a percentage of WT binding (100%). (D) Competitive binding curves showing the displacement of [125I]-rAM by hAM for I52A in the AM1 receptor and Q45A in the AM2 receptor, compared with WT controls. Data were combined and data points shown are mean ± SEM of at least three individual data sets.
Our cAMP assays indicated some differences in the role of CLR ECD residues between the AM1 and AM2 receptors. To further investigate this, we performed radioligand binding assays on selected mutants. Specific binding was abolished at both receptors for T37A and I41A (Figure 7). Binding was also abolished at the AM1 receptor with M42A and Q45A. Interestingly, specific binding was retained for both of these mutants at the AM2 receptor, in line with the cAMP data. Given that Q45A also showed a large differential effect in cAMP assays, we determined the pIC50 for AM at the AM2 receptor. This was 7.51 ± 0.21 (n = 3). The WT pIC50 was 7.69 ± 0.11 (n = 3); there was no significant difference between these values. Only 152A retained binding at the AM1 receptor and full-curve analysis showed no difference in pIC50 for AM compared with WT [8.02 ± 0.03 (n = 3) for I52A vs. 8.29 ± 0.25 (n = 4) for WT].
Cell surface expression
The CLR and RAMP2 or RAMP3 components are individually expressed inefficiently at the cell surface (McLatchie et al., 1998); however, when either RAMP2 or RAMP3 is expressed together with CLR, they are translocated to the cell surface to form functional AM1 or AM2 receptors respectively. The presence of HA-CLR at the cell surface can therefore be used as an estimate of AM receptor complex expression. To determine whether a change in binding or peptide responsiveness with the mutations was due to perturbed cell surface expression, we performed whole-cell elisas. For almost all mutants, the mean expression was within 50% of WT and any reduction was not statistically significant (Table 5). Previously, we have shown that reductions of less than 50% in cell surface expression in our Cos7 cells do not substantially perturb cAMP responsiveness (Bailey and Hay, 2007). Only Y49A at the AM1 receptor and W84A RAMP3 in the AM2 receptor (Table 5) caused a significant reduction in HA-CLR expression. As Q45A and Y49A are found at the interface between CLR and RAMP2, we additionally tested FLAG-RAMP2 expression for these two mutants to determine whether any reduction in CLR expression was likely to be caused by a disruption in CLR/RAMP2 complex formation. The results were consistent with the HA-CLR measurements with FLAG-RAMP2 expression only significantly reduced with the Y49A mutation (13.1 ± 12.5%, n = 4, P < 0.01 by Kruskal–Wallis test followed by Dunnʼs test) and reduced to a lesser degree with the Q45A mutation (73.5 ± 10.7%, n = 3).
Table 5.
Summary of cell surface expression data for HA-CLR in mutant AM1 and AM2 receptors
| AM1 receptor | AM2 receptor | |
|---|---|---|
| HA-CLR | HA-CLR | |
| Mutant | %WT | %WT |
| CLR I32A | 83.1 ± 6.71 (3) | 113.7 ± 25.8 (3) |
| CLR Q33A | 115.8 ± 33.9 (3) | 116.2 ± 14.2 (5) |
| CLR L34A | 95.5 ± 7.54 (4) | 107.2 ± 7.15 (3) |
| CLR G35A | 90.8 ± 10.4 (3) | 94.7 ± 8.95 (3) |
| CLR V36A | 78.5 ± 8.74 (3) | 96.9 ± 2.28 (3) |
| CLR T37A | 80.5 ± 10.3 (3) | 126.5 ± 12.5 (5) |
| CLR I41A | 65.7 ± 9.52 (5) | 86.4 ± 9.69 (3) |
| CLR M42A | 123.7 ± 20.9 (3) | 112.8 ± 19.4 (5) |
| CLR Q45A | 61.5 ± 5.91 (3) | 104.9 ± 12.8 (3) |
| CLR Y49A | 25.9 ± 7.91 (4)** | 111.6 ± 8.25 (4) |
| CLR I52A | 98.7 ± 7.11 (3) | 116.2 ± 19.5 (3) |
| RAMP2 E101A | 98.78 ± 10.4 (4) | – |
| RAMP2 F111A | 95.79 ± 13.1 (4) | – |
| RAMP3 W84A | – | 47.7 ± 4.67 (6)* |
| RAMP3 W84F | – | 63.6 ± 3.79 (4) |
Data are the mean ± SEM of n individual data sets (in parentheses).
P < 0.05;
P < 0.01 versus WT receptor by the Kruskal–Wallis test followed by Dunnʼs multiple comparison test. –, not applicable.
Discussion
The recently solved crystal structures of the AM1 and CGRP receptor ECDs have greatly expanded our understanding of these receptors and the interaction of their common CLR subunit with these two different RAMPs. Of these, the CGRP receptor ECD is the most complete, especially at the N-terminus of CLR. However, the lack of bound peptides in these structures gives us limited insight into the manner of peptide binding to these receptors. Mutagenesis studies pinpointed the extreme N-terminus of CLR as a key determinant of CGRP binding to the CGRP receptor ECD. We have now defined an essential region for peptide binding in the three CLR-based receptors (Figure 6; Supporting Information Figure S1). This centres around I41 with contributions from T37 and Q45, and lesser contributions from I32, Q33, L34, G35, M42 and I52, depending on the RAMP co-expressed. These residues mainly contribute to agonist binding and thus potency, although, especially for I32 and Q33, there are effects on Emax. We observed potentially important differences in the function of CLR residues in the presence of RAMP3, compared with RAMP1 and RAMP2.
The N-terminus of CLR is predominantly helical, although the region N-terminal to T37 in the CGRP receptor structure is somewhat disordered. In one of the three structures (3N7P) of the CGRP receptor ECD (3N7P, 3N7R and 3N7S), it is not helical and in all structures the atoms have high b-factors, indicating significant mobility (ter Haar et al., 2010; Barwell et al., 2012). These extreme N-terminal residues lie beyond the extent of the AM1 receptor structure. Interestingly, this region has very low-sequence identity across species compared with the rest of the N-terminal region (Supporting Information Figure S2), and the periodicity of these residues implies that this region may not be helical. The ability of I32 and Q33 to increase Emax may mean that these residues are able to influence ligand binding to the transmembrane core or the conformation of CLR to promote coupling to Gs, possibly via an interaction with one of the extracellular loops of the receptor. However, without a full-length (ligand-bound) receptor structure their steric relationship to the transmembrane bundle and extracellular loops is speculative.
T37, I41 and Q45 are highly conserved across species (Supporting Information Figure S2), and each has interesting effects upon mutation in the three CLR-based receptors. These residues all lie on the same face of the N-terminal CLR helix, and in the CGRP and AM1 receptor ECD crystal structures have their side chains projecting upwards into a groove between CLR and the RAMP, in a good position to interact with a ligand that might bind in this area.
I41 is universally important for cAMP accumulation and binding; it has also been shown to be important for the binding of CGRP to the CGRP receptor (Barwell et al., 2010). The equivalent residue is important for glucagon binding to the glucagon receptor (Siu et al., 2013). The hydrophobic side chain of I41 is in an exposed position, at the entrance to the interface between CLR and all three RAMPs (Figure 8). We suggest that it acts as a pivot point for peptide binding and is probably a direct contact for all three agonists.
Figure 8.
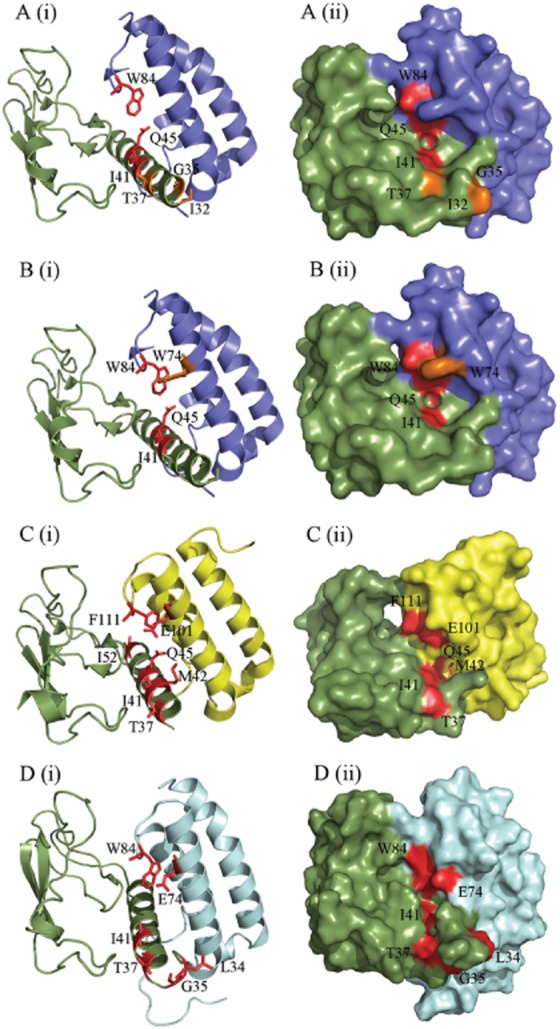
Figure depicting the amino acid residues likely to be involved in peptide binding, generated using PyMol. (A) CGRP-binding residues in the CGRP receptor ECD (3N7S; a representative structure for the CGRP receptor), as previously determined by alanine scanning mutagenesis (Barwell et al., 2010). (B) AM-binding residues in the CGRP receptor. (C) AM-binding residues in the AM1 receptor ECD (3AQF). The AM1 receptor structure is N-terminally truncated to V36; thus, many residues involved in AM binding could not be depicted. This may indicate increased mobility of this region of the receptor ECD. (D) AM-binding residues in our AM2 receptor ECD model. RAMP1 is shown in blue, RAMP2 in yellow, RAMP3 in pale blue and CLR in green. Residues where substitution resulted in a decrease in peptide potency are shown in red; those that cause an increase in potency are shown in orange. In each part of the figure, (i) shows secondary structural elements with residues involved represented as sticks and (ii) shows the surface map.
Beyond I41, the accessibility of the residues on the CLR helix sharply decreases (Figure 8), although the details of this are RAMP-dependent (ter Haar et al., 2010; Kusano et al., 2012). The side chain of M42 is perpendicular to those of T37, I41 and Q45 and points towards the RAMP. Although it is sufficiently accessible to be important for the binding of non-peptide antagonists to the CGRP receptor (ter Haar et al., 2010; Miller et al., 2010), this change of angle is likely to make it particularly inaccessible to any peptide orientated parallel to the main axis of the CLR N-terminal helix. Its effect on hAM binding and small effect on hAM potency in the AM1 receptor may not therefore be due to a direct interaction with the peptide but to a localized disruption of the structure of the AM-binding pocket.
The side chains of Q45 and Y49, although favourably orientated for peptide binding, are also deep enough within the CLR/RAMP interface to interact with conserved tyrosine residues in RAMP1 and RAMP2. The effects of Y49 are particularly notable; mutation of this residue abolishes binding and substantially reduces expression in the CGRP receptor (Barwell et al., 2010). Similar effects on cell surface expression are seen in the AM1 receptor. These data suggest that the main effect of Y49 is to stabilize the CLR/RAMP interface in the CGRP and AM1 receptors, rather than having a direct effect on ligand binding. In contrast, the major effect of Q45A on the AM1 receptor is most likely to be due to an effect on the peptide-binding pocket, perhaps as a contact for AM. Like Y49A, this residue shows intriguing differential behaviour in the presence of RAMP3. This will be explored further below.
I52 is situated on the opposite face of the ECD to the proposed peptide-binding cleft and is not readily accessible to ligand, consistent with its very minor role in binding and peptide responsiveness at each of the three receptors (Barwell et al., 2010). The crystal structures of ligand-free or antagonist-bound receptors in the absence of the transmembrane domains of either CLR or RAMP may of course not be an accurate reflection of the agonist-bound state of the intact receptors; in particular, there may be rearrangements to increase accessibility of the binding groove in the CLR-RAMP interface. Nonetheless, it is reasonable to assume that the residues C-terminal to I41A will remain partly buried.
The proposed essential binding residues of the three receptors are presented in Figure 8. The RAMP residue W84/F111 is similarly involved in all of the binding pockets and is conserved within each RAMP across a wide range of species (Hay et al., 2006a). Interestingly, the W74/E101/E74 position is only hydrophobic in RAMP1 where its mutation causes an increase in AM and AM2 binding but has no effect on CGRP binding (Figure 8) (Qi et al., 2008; Moore et al., 2013). The conserved polar residue E101 (W74 in RAMP1) is not particularly prominent, being situated further back in the AM1 receptor binding pocket. However, it may still form the top of our putative peptide-binding pocket as it does have a substantial role to play in AM potency at the AM1 and AM2 receptors. These effects correspond with the decrease in binding affinity of AM to these mutants at the AM1 receptor ECD, as determined using surface plasmon resonance (Kusano et al., 2012). Mutation of this position has fewer effects on AM2 potency at both receptors (Qi et al., 2011). Taken together with the position of all of these residues in the AM1 receptor ECD crystal structure, the data collectively suggest that the CLR residues from I41 and the RAMP2 residue F111 form a hydrophobic area at the back of the peptide-binding groove. E101 forms a polar sub-region of this area that may contribute to ligand selectivity between the AM1, AM2 and CGRP receptors. A similar hydrophobic region is formed by I41 and the corresponding RAMP1 residues W74 and W84 in the CGRP receptor crystal structure (ter Haar et al., 2010). Our data and model of the AM2 receptor ECD suggest that the top of the pocket may be formed in an analogous manner. There are a number of hydrophobic regions in the C-terminal region of CGRP, AM and AM2 that could dock in such an environment. For example, the C-terminal amide residue of the three peptides is a conserved aromatic residue (Tyr or Phe), which is essential for the binding of AM22–52 and CGRP derivatives (Watkins et al., 2012; 2012; Moad and Pioszak, 2013). The aromatic side chain of this residue could form hydrophobic interactions or pi–pi stacking in this area (Archbold et al., 2011). Peptide binding to class B GPCRs ECDs involves the formation of hydrophobic interactions that result in the formation of an α-helix in the peptide ligand (Parthier et al., 2009). The mutation of hydrophobic residues along the CLR ECD helix and at the top of our proposed binding pocket could therefore affect ligand structure as well as peptide binding.
It is possible to comment on some general features of RAMP action and their implications for peptide binding. A greater number of residues in CLR investigated in this study are involved in peptide interactions with RAMP1 and RAMP2, compared with RAMP3 (Figure 6). This is particularly striking for M42 and Q45, which do not influence peptide binding to the AM2 receptor. We propose that RAMP3 either orientates all three peptides at an angle so as to miss the main part of the groove between it and CLR, or it changes the shape of the groove. The latter proposition is supported by the differential effect of Y49A on CLR cell surface expression with RAMP1, RAMP2 and RAMP3. The reduction with RAMP1 and RAMP2 confirm that this is an important interface residue between these RAMPs and CLR. However, the data strongly suggest that Y49 may not assume the same role in CLR/RAMP3 complexes. However, our CLR/RAMP3 ECD model cannot inform us as to any possible differences in the interface between CLR and RAMP3 compared with the other RAMPs, because it is a homology model based on the CGRP receptor ECD structure. A crystal structure of the CLR/RAMP3 ECD is needed to determine the specific position of residues at the interface between these proteins. Nonetheless, the idea that the interface is different between each CLR/RAMP complex is also supported by mutation of the RAMP residue (W84/F111), which has modest effects on cell surface expression with RAMP1 and RAMP3 but not RAMP2 (Moore et al., 2013). This position may have a dual role in ligand binding and stabilization of the RAMP/CLR interface, depending on the RAMP.
In contrast to RAMP3, RAMP1 and RAMP2 support the involvement of more N-terminal helix residues, suggesting that the peptides lie parallel to these. CGRP and AM2 may sit slightly further forward (to the N-terminus of the CLR helix) than AM, depending on the RAMP. This is unlike the situation found for the majority of other class B GPCR ECDs that do not require a RAMP. In these cases, the bound peptide is at approximately 45° to the ECD helix, with the C-terminal amide (where present in the structure) binding to a residue in loop 4 of the receptor ECD, although the precise orientation of the individual peptides differs (Pioszak and Xu, 2008; Pioszak et al., 2009; Pal et al., 2010; Archbold et al., 2011). Regardless of the precise peptide orientation in each class B GPCR, binding to the receptor ECD helix occurs at the approximate position of I41 of CLR, as we have proposed for cognate peptide binding to the CGRP, AM1 and AM2 receptors.
We suggest that the binding mode of CGRP, AM and AM2 to their receptors is broadly similar to other class B GPCRs, but that the RAMP alters the conformation of the CLR to influence ligand affinity and thus potency. In particular, it controls accessibility of the peptide-binding epitope on the N-terminus of the CLR, particularly where this is itself part of the CLR-RAMP interface. In addition, there are direct interactions from two key residues positioned at 74/101 and 84/111 in the RAMPs (Figure 8). This results in a slightly different manner of binding for the AM, AM2 and CGRP peptides across the three receptors. Subtle differences in CLR ECD conformation with each RAMP could affect the function of the receptor as a whole. This could be reflected further downstream in terms of differential receptor signalling. Structure-based drug design at these receptors will need to take these factors into account.
Acknowledgments
We thank the New Zealand Lotteries Commission (Health), Maurice and Phyllis Paykel Trust, National Heart Foundation of New Zealand and Health Research Council of New Zealand for support. D. R. P. and J. B. acknowledge support of the Wellcome Trust (grant 091496).
Glossary
- AM
adrenomedullin
- CGRP
calcitonin gene-related peptide
- CLR
calcitonin receptor-like receptor
- ECD
extracellular N-terminal domain
- RAMP
receptor activity-modifying protein
Conflict of interest
None for any author.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisherʼs web-site: http://dx.doi.org/10.1111/bph.12508
Effect of mutations in N-terminal CLR residues on E max values. Data are ordered to show the effect by RAMP (left) or peptide (right). For comparisons with the CGRP receptor, these values were calculated from the data presented in Barwell et al. (2010). *P < 0.05; **P < 0.01; ***P < 0.001 versus relevant WT receptor by paired t-test.
Amino acid sequence alignment of CLR sequences from selected species: human (homo sapiens), chicken (Gallus gallus), mouse (Mus musculus), rat (Rattus norvegicus), pufferfish CLR1 (Takifugu obscurus), salmon (Salmo salar) and pig (Sus scrofa) showing only the ECD helix region. The alignment was generated using Clustal Omega, and ESPript total conservation of residues is shown in red, partial conservation in yellow. The sequence was aligned using the % equivalence algorithm on the basis of physicochemical parameters.
References
- Aiyar N, Disa J, Pullen M, Nambi P. Receptor activity modifying proteins interaction with human and porcine calcitonin receptor-like receptor (CRLR) in HEK-293 cells. Mol Cell Biochem. 2001;224:123–133. doi: 10.1023/a:1011907328682. [DOI] [PubMed] [Google Scholar]
- Archbold JK, Flanagan JU, Watkins HA, Gingell JJ, Hay DL. Structural insights into RAMP modification of secretin family G protein-coupled receptors: implications for drug development. Trends Pharmacol Sci. 2011;32:591–600. doi: 10.1016/j.tips.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Bailey RJ, Hay DL. Pharmacology of the human CGRP1 receptor in Cos 7 cells. Peptides. 2006;27:1367–1375. doi: 10.1016/j.peptides.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Bailey RJ, Hay DL. Agonist-dependent consequences of proline to alanine substitution in the transmembrane helices of the calcitonin receptor. Br J Pharmacol. 2007;151:678–687. doi: 10.1038/sj.bjp.0707246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwell J, Miller PS, Donnelly D, Poyner DR. Mapping interaction sites within the N-terminus of the calcitonin gene-related peptide receptor; the role of residues 23–60 of the calcitonin receptor-like receptor. Peptides. 2010;31:170–176. doi: 10.1016/j.peptides.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwell J, Gingell JJ, Watkins HA, Archbold JK, Poyner DR, Hay DL. Calcitonin and calcitonin receptor-like receptors: common themes with family B GPCRs? Br J Pharmacol. 2012;166:51–65. doi: 10.1111/j.1476-5381.2011.01525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner AC, Hay DL, Simms J, Howitt SG, Schindler M, Smith DM, et al. A key role for transmembrane prolines in calcitonin receptor-like receptor agonist binding and signalling: implications for family B G-protein-coupled receptors. Mol Pharmacol. 2005;67:20–31. [PubMed] [Google Scholar]
- Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, et al. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- Fritz-Six KL, Dunworth WP, Li M, Caron KM. Adrenomedullin signaling is necessary for murine lymphatic vascular development. J Clin Invest. 2008;118:40–50. doi: 10.1172/JCI33302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa Y, Nagai Y, Miyatake A, Takei Y, Miura K, Shoukouji T, et al. Renal effects of a new member of adrenomedullin family, adrenomedullin2, in rats. Eur J Pharmacol. 2004;497:75–80. doi: 10.1016/j.ejphar.2004.06.039. [DOI] [PubMed] [Google Scholar]
- Gingell JJ, Qi T, Bailey RJ, Hay DL. A key role for tryptophan 84 in receptor activity-modifying protein 1 in the amylin 1 receptor. Peptides. 2010;31:1400–1404. doi: 10.1016/j.peptides.2010.03.027. [DOI] [PubMed] [Google Scholar]
- ter Haar E, Koth CM, Abdul-Manan N, Swenson L, Coll JT, Lippke JA, et al. Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure. 2010;18:1083–1093. doi: 10.1016/j.str.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Hay DL, Christopoulos G, Christopoulos A, Sexton PM. Determinants of 1-piperidinecarboxamide, N-[2-[[5-amino-l-[[4-(4-pyridinyl)-l-piperazinyl]carbonyl]pentyl]amino]-1-[(3,5-d ibromo-4-hydroxyphenyl)methyl]-2-oxoethyl]-4-(1,4-dihydro-2-oxo-3(2H)-quinazoliny l) (BIBN4096BS) affinity for calcitonin gene-related peptide and amylin receptors – the role of receptor activity modifying protein 1. Mol Pharmacol. 2006a;70:1984–1991. doi: 10.1124/mol.106.027953. [DOI] [PubMed] [Google Scholar]
- Hay DL, Poyner DR, Sexton PM. GPCR modulation by RAMPs. Pharmacol Ther. 2006b;109:173–197. doi: 10.1016/j.pharmthera.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Hinson JP, Kapas S, Smith DM. Adrenomedullin, a multifunctional regulatory peptide. Endocr Rev. 2000;21:138–167. doi: 10.1210/edrv.21.2.0396. [DOI] [PubMed] [Google Scholar]
- Hoare SR. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov Today. 2005;10:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, Jazayeri A, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- Hong Y, Hay DL, Quirion R, Poyner DR. The pharmacology of adrenomedullin 2/intermedin. Brit J Pharmacol. 2012;166:110–120. doi: 10.1111/j.1476-5381.2011.01530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa-Shindo Y, Sakurai T, Kamiyoshi A, Kawate H, Iinuma N, Yoshizawa T, et al. The GPCR modulator protein RAMP2 is essential for angiogenesis and vascular integrity. J Clin Invest. 2008;118:29–39. doi: 10.1172/JCI33022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano S, Kukimoto-Niino M, Hino N, Ohsawa N, Okuda K, Sakamoto K, et al. Structural basis for extracellular interactions between calcitonin receptor-like receptor and receptor activity-modifying protein 2 for adrenomedullin-specific binding. Protein Sci. 2012;21:199–210. doi: 10.1002/pro.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA. PDBsum: summaries and analyses of PDB structures. Nucleic Acids Res. 2001;29:221–222. doi: 10.1093/nar/29.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Macarthur MW, Moss DS, Thornton JM. (PROCHECK): a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Li H, Robertson AD, Jensen JH. Very fast empirical prediction and interpretation of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- Miller PS, Barwell J, Poyner DR, Wigglesworth MJ, Garland SL, Donnelly D. Non-peptidic antagonists of the CGRP receptor, BIBN4096BS and MK-0974, interact with the calcitonin receptor-like receptor via methionine-42 and RAMP1 via tryptophan-74. Biochem Biophys Res Commun. 2010;391:437–442. doi: 10.1016/j.bbrc.2009.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moad HE, Pioszak AA. Selective CGRP and adrenomedullin peptide binding by tethered RAMP-calcitonin receptor-like receptor extracellular domain fusion proteins. Protein Sci. 22:1775–1785. doi: 10.1002/pro.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore EL, Gingell JJ, Kane SA, Hay DL, Salvatore CA. Mapping the CGRP receptor ligand binding domain: tryptophan-84 of RAMP1 is critical for agonist and antagonist binding. Biochem Biophys Res Commun. 2013-2010;394:141–145. doi: 10.1016/j.bbrc.2010.02.131. [DOI] [PubMed] [Google Scholar]
- Pal K, Swaminathan K, Xu HE, Pioszak AA. Structural basis for hormone recognition by the Human CRFR2(alpha) G protein-coupled receptor. J Biol Chem. 2010;285:40351–40361. doi: 10.1074/jbc.M110.186072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthier C, Kleinschmidt M, Neumann P, Rudolph R, Manhart S, Schlenzig D, et al. Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc Natl Acad Sci U S A. 2007;104:13942–13947. doi: 10.1073/pnas.0706404104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthier C, Reedtz-Runge S, Rudolph R, Stubbs MT. Passing the baton in class B GPCRs: peptide hormone activation via helix induction? Trends Biochem Sci. 2009;34:303–310. doi: 10.1016/j.tibs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Pioszak AA, Xu HE. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc Natl Acad Sci U S A. 2008;105:5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak AA, Parker NR, Suino-Powell K, Xu HE. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J Biol Chem. 2008;283:32900–32912. doi: 10.1074/jbc.M805749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak AA, Parker NR, Gardella TJ, Xu HE. Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. J Biol Chem. 2009;284:28382–28391. doi: 10.1074/jbc.M109.022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirot O, OʼToole E, Notredame C. Tcoffee@igs: a web server for computing, evaluating and combining multiple sequence alignments. Nucleic Acids Res. 2003;31:3503–3506. doi: 10.1093/nar/gkg522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, et al. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54:233–246. doi: 10.1124/pr.54.2.233. [DOI] [PubMed] [Google Scholar]
- Qi T, Hay DL. Structure-function relationships of the N-terminus of receptor activity-modifying proteins. Br J Pharmacol. 2010;159:1059–1068. doi: 10.1111/j.1476-5381.2009.00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi T, Christopoulos G, Bailey RJ, Christopoulos A, Sexton PM, Hay DL. Identification of N-terminal receptor activity-modifying protein residues important for calcitonin gene-related peptide, adrenomedullin, and amylin receptor function. Mol Pharmacol. 2008;74:1059–1071. doi: 10.1124/mol.108.047142. [DOI] [PubMed] [Google Scholar]
- Qi T, Ly K, Poyner DR, Christopoulos G, Sexton PM, Hay DL. Structure–function analysis of amino acid 74 of human RAMP1 and RAMP3 and its role in peptide interactions with adrenomedullin and calcitonin gene-related peptide receptors. Peptides. 2011;32:1060–1067. doi: 10.1016/j.peptides.2011.03.004. [DOI] [PubMed] [Google Scholar]
- Qi T, Dong M, Watkins HA, Wootten D, Miller LJ, Hay DL. Receptor activity-modifying protein-dependent impairment of calcitonin receptor splice variant Δ(1-47)hCT((a)) function. Br J Pharmacol. 2013;168:644–657. doi: 10.1111/j.1476-5381.2012.02197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh J, Chang CL, Bhalla A, Klein C, Hsu SY. Intermedin is a calcitonin/calcitonin gene-related peptide family peptide acting through the calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes. J Biol Chem. 2004;279:7264–7274. doi: 10.1074/jbc.M305332200. [DOI] [PubMed] [Google Scholar]
- Runge S, Thogersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Siu FY, He M, de Graaf C, Han GW, Yang D, Zhang Z, et al. Structure of the human glucagon class B G-protein-coupled receptor. Nature. 2013;499:444–449. doi: 10.1038/nature12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Hyodo S, Katafuchi T, Minamino N. Novel fish-derived adrenomedullin in mammals: structure and possible function. Peptides. 2004a;25:1643–1656. doi: 10.1016/j.peptides.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Takei Y, Inoue K, Ogoshi M, Kawahara T, Bannai H, Miyano S. Identification of novel adrenomedullin in mammals: a potent cardiovascular and renal regulator. FEBS Lett. 2004b;556:53–58. doi: 10.1016/s0014-5793(03)01368-1. [DOI] [PubMed] [Google Scholar]
- Underwood CR, Garibay P, Knudsen LB, Hastrup S, Peters GH, Rudolph R, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem. 2010;285:723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins HA, Rathbone DL, Barwell J, Hay DL, Poyner DR. Structure–activity relationships for alpha calcitonin gene-related peptide. Br J Pharmacol. 170:1308–1322. doi: 10.1111/bph.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins HA, Au M, Bobby R, Archbold JK, Abdul-Manan N, Moore JM, et al. Identification of key residues involved in adrenomedullin binding to the AM1 receptor. Br J Pharmacol. 2012-2013;169:143–155. doi: 10.1111/bph.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of mutations in N-terminal CLR residues on E max values. Data are ordered to show the effect by RAMP (left) or peptide (right). For comparisons with the CGRP receptor, these values were calculated from the data presented in Barwell et al. (2010). *P < 0.05; **P < 0.01; ***P < 0.001 versus relevant WT receptor by paired t-test.
Amino acid sequence alignment of CLR sequences from selected species: human (homo sapiens), chicken (Gallus gallus), mouse (Mus musculus), rat (Rattus norvegicus), pufferfish CLR1 (Takifugu obscurus), salmon (Salmo salar) and pig (Sus scrofa) showing only the ECD helix region. The alignment was generated using Clustal Omega, and ESPript total conservation of residues is shown in red, partial conservation in yellow. The sequence was aligned using the % equivalence algorithm on the basis of physicochemical parameters.