

Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease where motor neurons in cortex, brain stem, and spinal cord die progressively, resulting in muscle wasting, paralysis, and death. Currently, effective therapies for ALS are lacking; however, identification of pathological TAR DNA-binding protein 43 (TDP-43) as the hallmark lesion in sporadic ALS suggests new therapeutic targets for pharmacological intervention. Pathological TDP-43 phosphorylation appears to drive the onset and progression of ALS and may result from upregulation of the protein kinase CK-1 in affected neurons, resulting in postranslational TDP-43 modification. Consequently, brain penetrant specific CK-1 inhibitors may provide a new therapeutic strategy for treating ALS and other TDP-43 proteinopathies. Using a chemical genetic approach, we report the discovery and further optimization of a number of potent CK-1δ inhibitors. Moreover, these small heterocyclic molecules are able to prevent TDP-43 phosphorylation in cell cultures, to increase Drosophila lifespan by reduction of TDP-43 neurotoxicity, and are predicted to cross the blood–brain barrier. Thus, N-(benzothiazolyl)-2-phenyl-acetamides are valuable drug candidates for further studies and may be a new therapeutic approach for ALS and others pathologies in which TDP-43 is involved.
Introduction
Described in 1874 by the French neurologist Charcot, amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease where no effective treatment exists today. Riluzole is the only palliative drug approved by the U.S. Food and Drug Administration (FDA), which moderates disease progression by extending survival 2–3 months without benefits to motor function.1
ALS affects lower and upper neurons in the brain stem, spinal cord and in the motor cortex, respectively.2 This neuron loss causes atrophy and paralysis of skeletal muscles because of the lack of communication between the nervous system and voluntary muscles of the body. In general, patients die between 3 and 5 years after symptom onset.3
ALS can be classified as familial (fALS) or sporadic (sALS), although the majority of the cases are sporadic (90%). The comparative lack of understanding of ALS etiology has hindered effective therapy development.4
Among the fALS, different mutations in many different genes have been discovered including the genes encoding super oxide dismutase (SOD1),5 the Tar DNA binding protein 43 (TDP-43),6 and the recently discovered C9ORF72.7,8
TDP-43 was identified in 2006 as the major component of protein aggregates of ALS and frontotemporal lobar degeneration (FTLD).9 In 2008, the role of TDP-43 in both sporadic and familial ALS was confirmed by the identification of mutations in the exon 6 of the TDP-43 encoding gene.10 The pathogenesis of TDP-43 mutation in ALS has been validated in a variety of animal and cell models. Overexpression of mutant TDP-43 causes neuronal death in worms (Caenorhabditis elegans),11 flies (Drosophila melanogaster),12 zebrafish,13 mice,14 rats,15 monkeys,16 and cultured human motor neurons differentiated from reprogrammed stem cells.17 Insoluble intracellular aggregates of phosphorylated TDP-43 are observed in fALS and sALS patients.18 Pathological TDP-43 phosphorylation seems to be a relatively early event in the onset and progression of ALS and FTLD-TDP. There is some evidence that TDP-43 phosphorylation may result in differential degradation and/or toxicity of the protein, but the precise role of TDP-43 phosphorylation in mechanisms of disease remains unclear.19 Regardless, TDP-43 phosphorylation at S409/410 is the most robust and consistent marker of pathological TDP-43 in human patients20 and drives neurodegeneration in animal models of TDP-43 proteinopathy.21−23
The protein kinase CK-1 was the first kinase reported to phosphorylate TDP-43 directly,24 and up-regulation of its activity is reported on ALS spinal cord tissue.25 CK-1 is a Ser/Thr kinase that is ubiquitously expressed in eukaryotic organisms.26 At least seven isoforms (α, β, γ1–3, δ, and ε) and their various splice variants have been characterized in different organisms.27 During recent years, several studies have highlighted the importance of CK-1 in neurodegenerative diseases28 and CK-1δ has been determined to phosphorylate many different sites on TDP-43 in vitro.29 Consequently, brain penetrant specific CK-1δ inhibitors may provide a new therapeutic strategy for treating ALS and other TDP-43 proteinopathies. We have developed a number of potent CK-1δ inhibitors and demonstrate that CK-1δ inhibition prevents TDP-43 phosphorylation in vitro and neurotoxicity in vivo.
Results and Discussion
Discovery of New CK-1δ Inhibitors
Biological screening of chemical libraries has been shown to be an effective methodology to discover new and chemically diverse hit compounds for a specific target. Here, we have followed a forward chemical genetic approach30 exploiting the use of small molecules as pharmacological tools to discover and validate new biological targets for further pharmacological intervention. Moreover, this chemogenomic approach has the advantage of revealing simultaneously some new lead compounds for further pharmacological development. First, we performed a blind screening of 25 chemically diverse heterocyclic small molecules from our in-house chemical library (Figure 1). A luminescent-based method to determine CK-1 inhibition using CK-1δ human recombinant enzyme was used.31 For the initial screening, we tested all the compounds at the same concentration (10 μM), and only two compounds, MR-3.55 and MR-2.16, showed significant inhibition in the micromolar range, with IC50 calculated values of 10.12 and 7.29 μM, respectively.
Figure 1.

Chemically diverse heterocyclic compounds selected from in-house chemical library in the first CK-1δ screening.
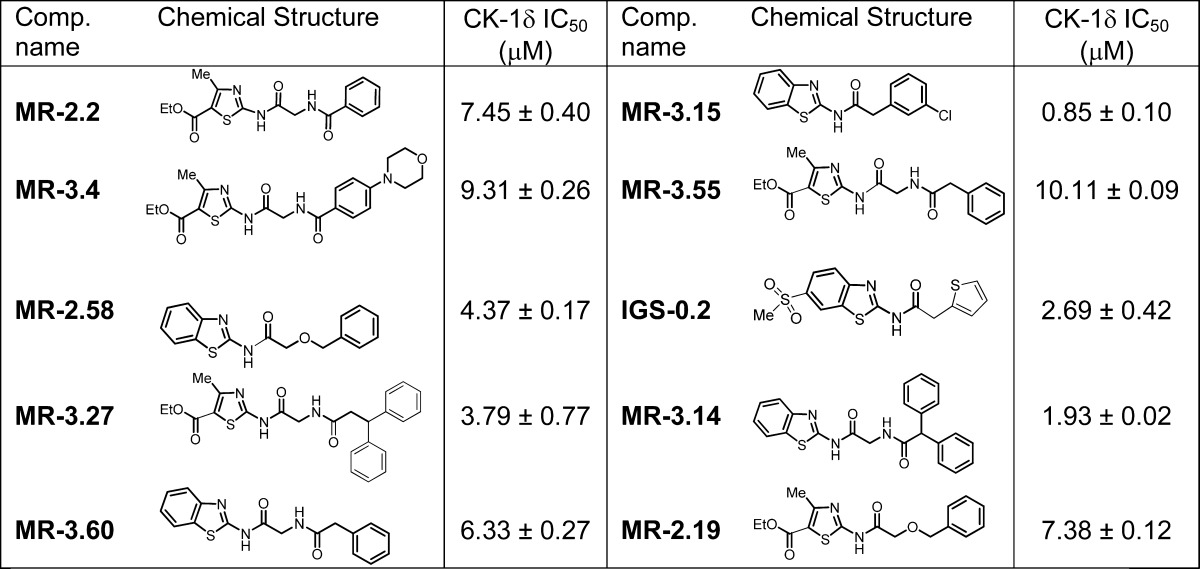
On the basis of the chemical structure of these two hits (MR-3.55 and MR-2.16), we selected a small focused subset from another chemical library. Thus, five more compounds with similar thiazole and benzothiazole scaffolds in their chemical structure bearing different linkers between the heterocycle and the aromatic ring were selected and evaluated against the CK-1δ enzyme (Table 1). In this second screening, almost all the evaluated compounds inhibited CK-1δ at low micromolar level, with MR-3.15 being the most potent hit, with an IC50 value of 0.85 μM.
Table 1. Focalized Structures Subset Chosen from Our In-House Chemical Library and Biological Evaluation on CK-1δ.
Hit-to-Lead Optimization
Given its potency against CK-1δ, N-(benzothiazol-2-yl)-2-(3-chlorophenyl) acetamide (MR-3.15) was selected for further biological activity optimization. Representative chemical features of MR-3.15 are the 2-aminobenzothiazole moiety linked to a substituted phenyl ring through a carbonyl and methylene group spacer. On the basis of that scaffold, we designed different structures depicted in Figure 2 to determine the relationship between chemical structure and biological activity toward CK-1δ enzyme. Several substituents such halides, alkyloxy, or trifluoromethyl were introduced in the heterocycle core or the phenyl ring. Moreover, the influence of the nature of the heterocyclic moiety in CK-1δ inhibition was studied. Thus, the benzothiazole scaffold was changed by benzimidazole, 1,3,4-thiadiazole, pyrimidine, 1,3-thiazole, and benzoxazole. Finally, the length and nature of the linker between the heterocycle and the phenyl ring was also analyzed.
Figure 2.
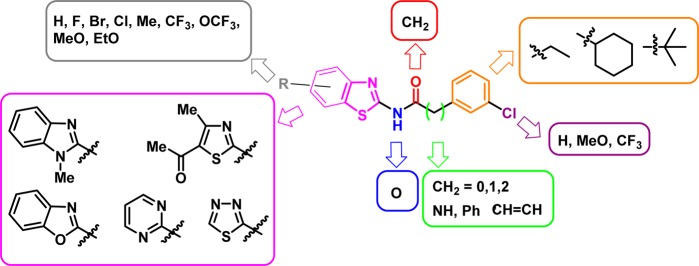
Structural modifications proposed from the hit compounds MR-3.15.
The preparation of all the proposed derivatives was performed by a convergent synthesis (Scheme 1), using as starting material the corresponding amino heterocycle. The coupling reaction with different organic acid derivatives was accomplished under microwave irradiation. In that case, reactions were performed at 110–150 °C for 5–20 min with excellent yields (see Experimental Section). In other cases, reaction of the amino heterocycle with carboxylic acids or N-aryl-isocyanates yielded the desired compound. All the synthesized derivatives were characterized using NMR, HPLC, and elemental analysis techniques.
Scheme 1. General Synthetic Procedure for the New Designed CK-1 Inhibitors.

Reactions conditions: (i) THF or solvent free, MW, 110–150 °C, 5–20 min; (ii) DMF or CH2Cl2, EDC, DMAP, N(Et)3, rt or reflux, 5–30 h; (iii) THF, MW, 110 °C, 0.5–4 h.
All the synthesized compounds were evaluated on CK-1δ human recombinant enzyme at a fixed concentration of 10 μM. In the cases where the inhibitory effect toward the enzyme was higher than 60%, the IC50 was calculated. All the data are collected in Tables 2, 3, and 4.
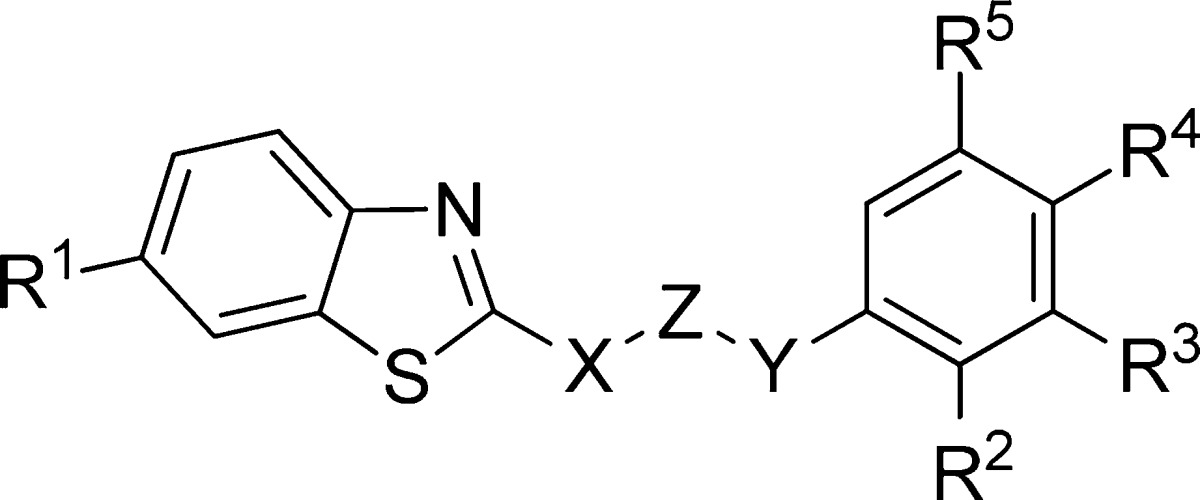
Table 2. N-Heteroaryl-phenyl-acetamides Derivatives Synthesized and Evaluated as CK-1δ Inhibitors.
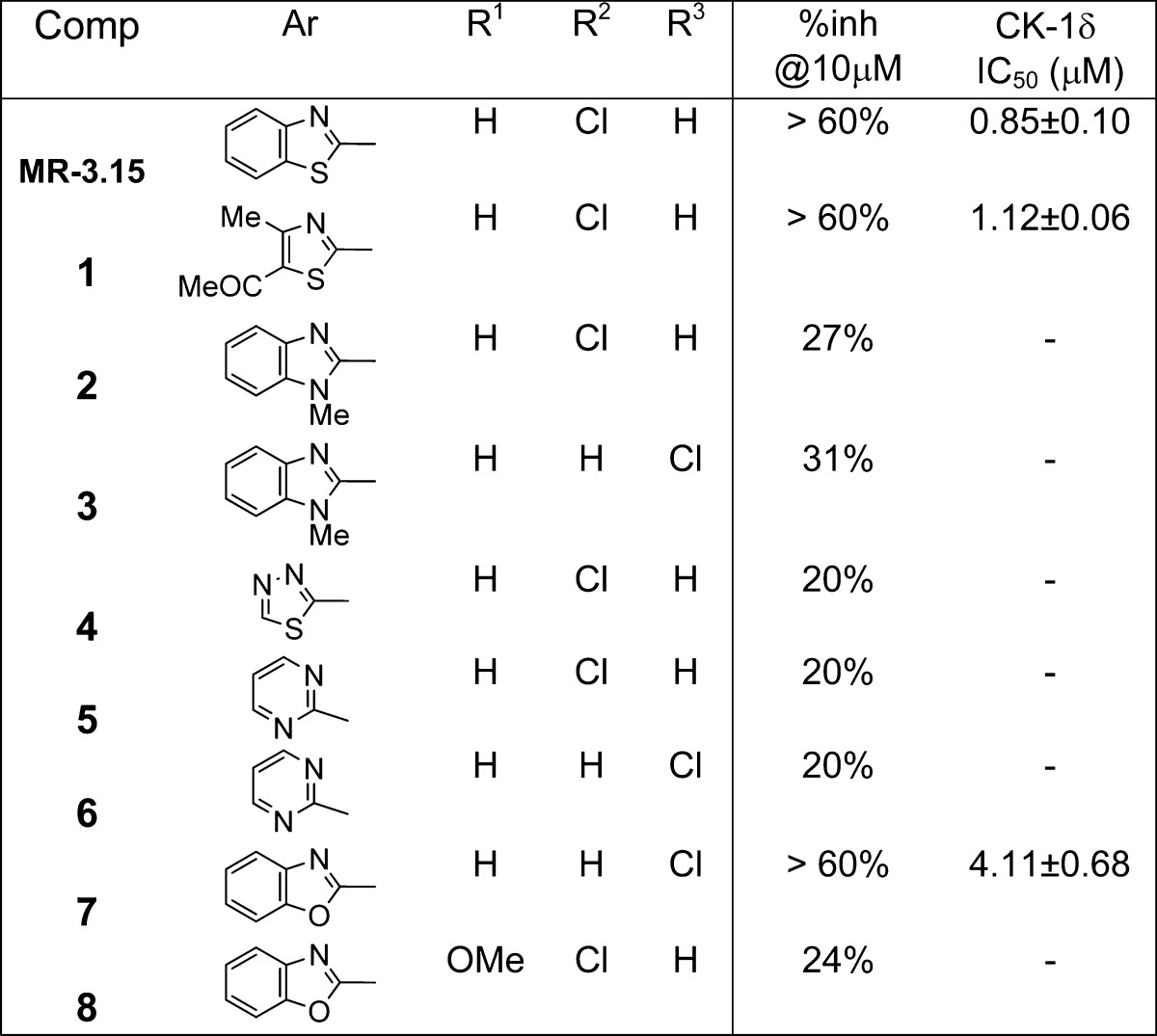
Table 3. Benzothiazoles Derivatives Synthesized and Evaluated as CK-1δ Inhibitors.
| compd | X | Z | Y | R1 | R2 | R3 | R4 | %inh @10 μM (%) | CK-1δ IC50 (μM) |
|---|---|---|---|---|---|---|---|---|---|
| MR-3.15 | NH | CO | CH2 | H | H | Cl | H | >60 | 0.85 ± 0.10 |
| 9 | O | CO | CH2 | H | H | H | Cl | 20 | |
| 10 | NH | CO | CF3 | OMe | H | H | 25 | ||
| 11 | NH | CH2 | CH2 | CF3 | OMe | H | H | 25 | |
| 12 | NH | CO | CHPh | H | H | H | H | >60 | 1.96 ± 0.83 |
| 13 | NH | CO | CHPh | OEt | H | H | H | >60 | 2.82 ± 0.43 |
| 14 | NH | CO | CH2CH2 | H | H | Cl | H | >60 | 3.58 ± 0.21 |
| 15 | NH | CO | CH2CHPh | H | H | H | H | >60 | 2.50 ± 0.33 |
| 16 | NH | CO | NH | CF3 | H | H | OMe | >60 | 5.50 ± 0.11 |
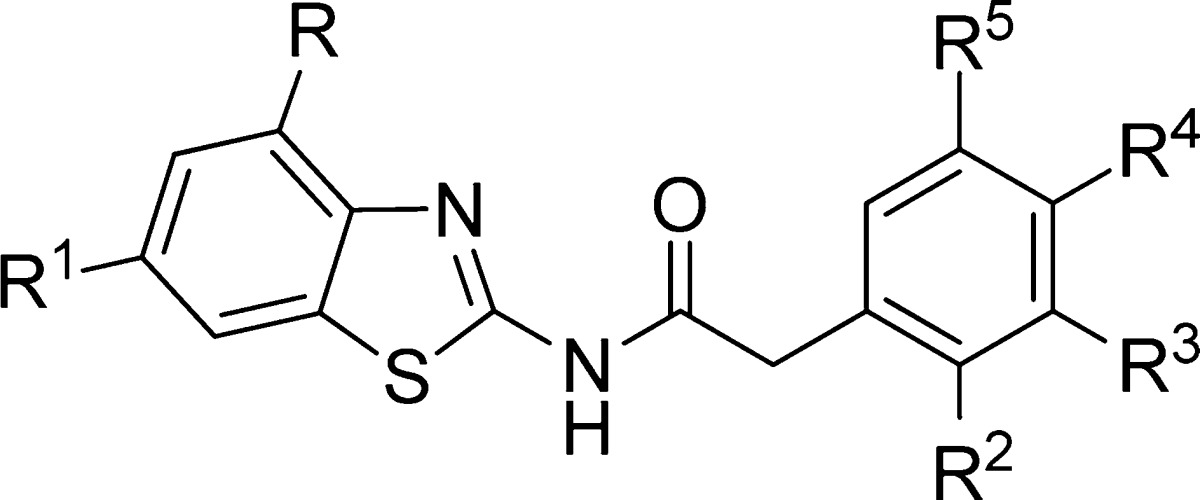
Table 4. N-Benzothiazolyl-2-phenyl-acetamides Derivatives Synthesized and Evaluated as CK-1δ Inhibitors.
| compd | R | R1 | R2 | R3 | R4 | R5 | %inh @10 μM (%) | CK-1δ IC50 (μM) |
|---|---|---|---|---|---|---|---|---|
| MR-3.15 | H | H | H | Cl | H | H | >60 | 0.85 ± 0.10 |
| 17 | H | Me | H | Cl | H | H | >60 | 0.083 ± 0.003 |
| 18 | Me | H | H | Cl | H | H | 20 | |
| 19 | Cl | H | H | Cl | H | H | 20 | |
| 20 | H | CF3 | H | Cl | H | H | >60 | 0.023 ± 0.002 |
| 21 | H | OMe | H | Cl | H | H | >60 | 0.53 ± 0.07 |
| 22 | H | OCF3 | H | Cl | H | H | >60 | 0.54 ± 0.02 |
| 23 | H | OEt | H | Cl | H | H | >60 | 1.21 ± 0.09 |
| 24 | H | CF3 | Cl | H | H | H | >60 | 0.068 ± 0.007 |
| 25 | H | OMe | Cl | H | H | H | >60 | 9.71 ± 0.99 |
| 26 | H | OEt | Cl | H | H | H | >60 | 17.43 ± 1.21 |
| 27 | H | CF3 | H | H | Cl | H | >60 | 0.065 ± 0.003 |
| 28 | H | OMe | H | H | Cl | H | >60 | 0.75 ± 0.09 |
| 29 | H | OEt | H | H | Cl | H | >60 | 1.11 ± 0.29 |
| 30 | H | Br | OMe | H | H | H | >60 | 0.26 ± 0.02 |
| 31 | H | Cl | OMe | H | H | H | >60 | 0.32 ± 0.03 |
| 32 | H | F | OMe | H | H | H | >60 | 1.17 ± 0.51 |
| 33 | H | Me | OMe | H | H | H | >60 | 0.29 ± 0.03 |
| 34 | H | CF3 | OMe | H | H | H | >60 | 0.010 ± 0.001 |
| 35 | H | OMe | OMe | H | H | H | >60 | 2.22 ± 0.30 |
| 36 | H | OCF3 | OMe | H | H | H | >60 | 0.62 ± 0.06 |
| 37 | H | OEt | OMe | H | H | H | >60 | 5.76 ± 0.61 |
| 38 | H | CF3 | H | OMe | H | H | >60 | 0.042 ± 0.050 |
| 39 | H | OMe | H | OMe | H | H | >60 | 0.42 ± 0.06 |
| 40 | H | OEt | H | OMe | H | H | >60 | 0.99 ± 0.07 |
| 41 | H | CF3 | H | CF3 | H | H | >60 | 0.087 ± 0.033 |
| 42 | H | CF3 | H | H | OMe | H | >60 | 0.033 ± 0.002 |
| 43 | H | OMe | H | H | OMe | H | >60 | 0.57 ± 0.08 |
| 44 | H | OEt | H | H | OMe | H | >60 | 1.09 ± 0.12 |
| 45 | H | H | H | H | H | H | >60 | 0.33 ± 0.03 |
| 46 | H | CF3 | H | H | H | H | >60 | 0.047 ± 0.005 |
| 47 | H | CF3 | H | Cl | Cl | H | >60 | 0.056 ± 0.002 |
| 48 | H | OMe | H | Cl | Cl | H | >60 | 1.24 ± 0.17 |
| 49 | H | OEt | H | Cl | Cl | H | >60 | 3.43 ± 0.69 |
| 50 | H | OCF3 | H | Cl | Cl | H | >60 | 0.59 ± 0.04 |
| 51 | H | CF3 | OMe | H | H | OMe | >60 | 0.19 ± 0.05 |
| 52 | H | OCF3 | H | OMe | OMe | OMe | >60 | 0.079 ± 0.007 |
| 53 | H | OMe | H | OMe | OMe | OMe | >60 | 1.12 ± 0.41 |
| 54 | H | OEt | H | OMe | OMe | OMe | >60 | 1.43 ± 0.35 |
| 55 | H | CF3 | H | OMe | OMe | OMe | >60 | 0.015 ± 0.007 |
Modification of the nature of the heterocyclic moiety present in the hit compound MR-3.15 by introduction of N-methyl-benzimidazole, benzoxazole, pyrimidine, 1,3,4-thiadiazole, or thiazole led to inactive or less potent derivatives (compounds 1–8) (Table 2). Thus, the benzothiazole framework was selected as fixed scaffold for the following compound set synthesis in which the nature and length of the central linker was explored. Data collected in Table 3 show that the inhibitory potency on CK-1δ is completely lost when the nitrogen directly attached to the benzothiazole ring is substituted by an oxygen atom (compound 9), when the carbonyl group of the linker is directly attached to the aromatic moiety (compound 10), or when the carbonyl group is replaced by a methylene group (compound 11). Although other possibilities such as the increase in the linker length and/or steric volume are tolerated by the enzyme (compounds 12–16), a reduction in the inhibitory potency of these compounds, in comparison with the hit MR-3.15, is observed. Thus we maintained the atom sequence NHCOCH2 as a spacer in the third group of synthesized compounds to explore different substitutions both in the benzothiazole and phenyl moiety (Table 4).
Several N-(benzothiazolyl)-2-phenylacetamides were then prepared which incorporate diverse substituents in different positions of both aromatic rings. One striking observation is the influence that the introduction of a substituent at position 6 of benzothiazole has in the CK-1δ inhibition. Thus, while a methyl or trifluoromethyl group increases 1 order of magnitude, the inhibitory potency of the acetamides (compounds 17 and 20), the methoxy or trifluoromethoxy moieties maintain the biological activity (compounds 21 and 22) and the sterically greater ethoxy group (compound 23) slightly decrease the enzymatic inhibition in comparison with the hit compound MR-3.15. This part of the inhibitor chemical structure may fit in a hydrophobic cavity of the enzyme, and when a methyl group or chlorine atom is attached to position 4 of benzothiazole, the enzymatic inhibition is lost (compounds 18 and 19) due to steric hindrance. On the other hand, substituents in the phenyl ring have little influence on biological activity, suggesting that hydrophobic interactions may be more relevant in the interaction with the enzyme than electrostatic interactions (compounds 24–55). Moreover, derivatives 56 and 57 were synthesized (Figure 3) to test this hypothesis. Both compounds are CK-1δ inhibitors in the submicromolar range, suggesting hydrophobic interactions produced by the phenyl ring are crucial for enzymatic inhibition.
Figure 3.
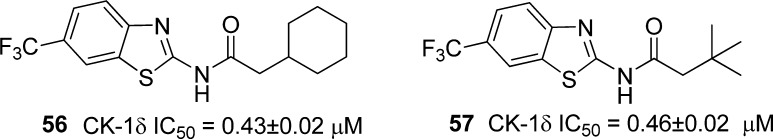
N-Benzothiadizolyl-2-alkyl-acetamides 56 and 57 as CK-1δ inhibitors.
In the light of these results, we conclude that N-(benzothiazolyl)-phenyl acetamides derivatives are potent CK-1δ inhibitors, with IC50 values in the low micromolar to nanomolar range. The preferred benzothiazole ring substituent is the trifluoromethyl group attached to the 6 position, and almost all the compounds bearing this moiety have IC50 values for CK-1δ inhibition in the nanomolar range. These compounds have emerged as promising leads for further studies.
Binding Mode Studies of CK-1δ Inhibitors
Once we established that N-(benzothiazolyl)-2-phenylacetamides are potent CK-1δ inhibitors, we began to explore structural reasons for their inhibitory activity. A two-step docking study was performed to gain insight into the nature of small molecule/enzyme interactions. First, a blind/ensemble docking was done following by a docking refinement (see Experimental Section for details).
There are three human CK-1δ crystallographic structures collected in the Protein Data Bank (PDBs entries: 3UYS, 3UYT, 3UZP). The docking study was performed over the whole CK-1δ (blind docking), with the potent CK-1δ inhibitor 20 (IC50 = 23 nM) and the inactive compound 11 (25% inhibition at 10 μM) employing the crystal structure of CK-1δ (PDB code: 3UYS). Docking analysis of the best solutions for compound 20 showed the most populated cluster and lowest energy poses in the ATP binding site, giving us a clue of the preferred location for N-(benzothiazolyl)-phenylacetamides inhibitors (Figure 4A). The same docking study applied to the CK-1δ inhibitors 24 and 34 confirms that the catalytic enzyme site is the preferred binding site for this new family of compounds (Figure S1 Supporting Information). A detailed inspection of the ATP site shows that Ile 23 and 148, Asp 91, 132, and 149, Leu 135, Lys 130, and Asn 133 are the most important amino acids for the drug interaction with the enzyme. The amide group present in this class of inhibitors is essential for the interaction with the aspartyl residues 132 and 91 (inhibitor 20) and Asp 149, Asn 133, and Lys 130 (inhibitors 24 and 34). This fact is corroborated by the analysis of the best docking found for the inactive compound 11. It is also located in the ATP region (Figure 4B), but in that case, the absence of the carbonyl group in the linker does not allow stable interaction between the compound and enzyme, leading to a very weak drug–enzyme complex. Moreover, the benzothiazole moiety with trifluoromethyl group substituent facilitates the hydrophobic interactions between 20 and the hydrophobic area created by Leu 135, Ile 23, and Ile 148. Inhibitors 24 and 34 also presented affinity to hydrophobic area but differently to compound 20 showed hydrogen bonding interaction with Asn 133 and Lys 130.
Figure 4.

Docking studies with CK-1δ (PDB code: 3UYS) of compound 20 (A) and 11(B).
To confirm the results obtained with the “blind docking” study, experimental enzymatic kinetic studies were performed. Three different CK-1δ inhibitors were chosen, 20, 24, and 34, to analyze the competition with ATP. As standard reference, the commercially available compound IC261, a known ATP competitive CK1δ inhibitor (IC50 = 1.0 μM), was used. Kinetic experiments were performed by varying concentrations of both ATP (from 1 to 50 μM) and compounds. Double reciprocal plotting of the data is depicted in Figure 5. The intercept of the plot in the vertical axis (1/V) does not change when the IC261 concentration increases (from 0.25 to 0.5 μM), which means that this compound acts as an ATP competitive inhibitor as it was described in the literature.32
Figure 5.

Kinetic data determined for IC261 and the N-benzothiazolyl-phenyl-amides derivatives 20, 24 and 34. ATP concentrations in the reaction mixture varied from 1 to 50 μM. Compound concentrations used are depicted in the plot, and the concentration of casein, the substrate used in the phosphorylation reaction, was kept constant at 12.5 μM. Each point is the mean of two different experiments, each one analyzed in duplicate.
The same experiments were done with the three new compounds 20, 24, and 34 here reported (Figure 5). In all the cases, these compounds have a similar behavior than IC261, showing an ATP competitive inhibition of the enzyme and validating the in silico docking studies of these compounds.
Kinases Selectivity Study
The human ‘‘Kinome’’ describes the total protein kinase component encoded by the human genome and includes more than 500 genes.33 The close homology within the ATP binding pocket is a defining characteristic for most protein kinases and constitutes the promise and challenge for kinase drug discovery. Considering that compounds here reported are ATP competitive, it is important to determine their selectivity against an extensive panel of kinases to avoid unexpected off-target effects and undesirable side effects in future drug development.
Compounds 20 and 24 were screened at 10 μM using the KinomeScan approach on a panel of 456 protein kinases (Tables S2 and S3 Supporting Information). Results are depicted in Figure 6. Both compounds were highly selective for CK-1δ inhibition, with a final selectivity score or “S” score (a quantitative measure of compound selectivity which is calculated by dividing the number of kinases that compounds bind to by the total number of distinct kinases tested, excluding mutant variants) of 0.04. Other commercialized kinases inhibitors such as Sunitinib and Lapatinib, present an S score of 0.57 and 0.010, respectively, in a 290 kinases panel.34
Figure 6.
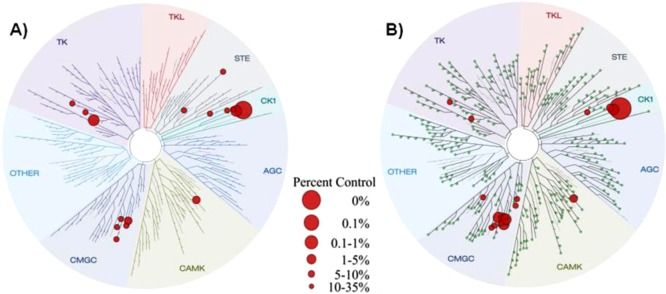
Kinome tree of compounds 20 and 24 (A and B, respectively). Figures were generated with DiscoverRx TREEspot version 4. The original results were shown as percent control to DMSO, and targets exhibiting less than 1% remaining activity were selected in the figures. The sizes of the red circles are proportional to the strength of the binding; the larger circles imply higher affinity.
Both compounds also inhibited CDC like kinase 1 and 4 (CLK1, CLK4), the protein kinase CK-1 family (CK-1α1, CK-1δ, CK-1ε, CK-1γ2), the dual-specificity tyrosine-(Y)-phosphorylation regulated kinase (DYRK1A, DYRK1B), fms-related tyrosine kinase 1 (FLT1), myosin light chain kinase 3 (MLCK), and platelet-derived growth factor receptor (PDGFRB). These results delineated an excellent selectivity kinase profile for the N-(benzothiazolyl)-phenylamides, 20 and 24, in spite of their competition for the ATP.
Cellular Assays of CK-1δ Inhibitors
To determine whether small molecule inhibition of CK-1 kinase activity by novel CK-1δ inhibitors will prevent TDP-43 phosphorylation in vitro, we utilized a mammalian cultured cell model of induced TDP-43 phosphorylation driven by glutathione depletion. HEK293 cells were pretreated with CK-1δ inhibitors for 1.5 h. Phosphorylation of endogenous cellular TDP-43 was then induced with exposure to ethacrynic acid.22,35 Cells were harvested, lysed, and tested for changes in TDP-43 phosphorylation state by immunoblotting. Candidate inhibitors were tested at an extended concentration range between 1.25 and 160 μM, with levels of TDP-43 phosphorylation determined by immunoblotting (Figure 7). Compound 46 exhibited the strongest inhibition of TDP-43 phosphorylation, with nearly complete absence of phosphorylation at 2.5 μM and higher. Amide 38 exhibited significant reduction of TDP-43 phosphorylation by 5 μM inhibitor, while 41 had greater than 2-fold reduction in TDP-43 phosphorylation by 10 μM. Compound 24 had moderate inhibition at 1.25 μM and above, while compounds 20 and 34 demonstrated inhibition at concentrations above 80 μM. Relatively low doses of the selected CK-1δ inhibitors are required to dramatically reduce TDP-43 phosphorylation in mammalian cells, indicating that inhibition of CK-1δ may be viable strategy to reduce neurotoxic TDP-43 phosphorylation in more complex in vivo systems as well.
Figure 7.

CK-1δ inhibitors prevent TDP-43 phosphorylation in HEK293 cells. HEK293 cells are incubated with increasing concentrations of CK-1δ inhibitors for 1.5 h, then treated with 150 μM ethacrynic acid to induce TDP-43 phosphorylation. Increasing concentrations of compounds (A) 20, (B) 24, (C) 34, (D) 38, (E) 41, or (F) 46 prevent ethacrynic acid induced TDP-43 phosphorylation.
Blood–Brain Barrier Penetration
Blood–brain barrier (BBB) penetration is an essential property for any compound developed to target neurodegenerative diseases. Thus the next step in development for CK-1δ inhibitors is to determine CNS activity and BBB penetration, so selection of lead compounds for in vivo studies can proceed. The BBB is a unique barrier, controlling the selective and specific transport of both exogenous and endogenous materials to the brain. Because of its biological structure, lipophilic compounds with low molecular weight (MW< 550 Da) are more likely to cross the BBB and may pass directly through via passive diffusion between the capillary walls. Determination of BBB penetration and other druglike properties at early stages during drug discovery is of utmost importance to select good candidates for in vivo studies and further pharmacological development. Parallel artificial membrane permeability assay (PAMPA) is a high-throughput technique developed to predict passive permeability through biological membranes.36 To explore the ability of N-(benzothiazolyl)-phenylacetamides to penetrate into the brain, we used the PAMPA-BBB method employing a porcine brain lipid membrane. First, an assay validation was made comparing the reported permeabilities (Pe) values of commercial drugs with the experimental data obtained by using this methodology (Figure S2 Supporting Information). A good correlation between experimental-described values was obtained Pe (exp) = 1.1202 (bibl) – 0.7413 (R2 = 0.9689). From this equation and following the pattern established in the literature for BBB permeation prediction,37 we could classify compounds as CNS+ when they present a permeability >3.74 × 10–6 cm·s–1. The in vitro permeabilities (Pe) of commercial drugs through lipid membrane extract together with those belonging to several CK-1δ inhibitors with IC50 values below 90 nM were determined and described in Table 5. Prediction of BBB permeability for compound 24 was not possible due to the low solubility of this compound in the assay vehicle. With exception of derivative 47, all the N-(benzothiazolyl)-phenylacetamides tested were predicted to cross the BBB by PAMPA assay. These compounds are thus the preferred candidates for further pharmaceutical development including in vivo studies.
Table 5. Permeability (Pe 10–6 cm·s–1) in the PAMPA-BBB Assay for 10 Commercial Drugs, Used in the Experiment Validation, and Different N-(Benzothiazolyl)-phenylamides with Their Predictive Penetration in the CNS.
| compd | bibl | Pea (10–6 cm·s–1) | BBB prediction |
|---|---|---|---|
| atenolol | 0.8 | 0.2 ± 0.1 | |
| caffeine | 1.3 | 0.9 ± 0.1 | |
| desipramine | 12 | 14.6 ± 0.6 | |
| enoxacine | 0.9 | 0.2 ± 0.1 | |
| hydrocortisone | 1.9 | 1.1 ± 0.8 | |
| ofloxacine | 0.8 | 0.4 ± 0.3 | |
| piroxicam | 2.5 | 0.5 ± 0.1 | |
| promazine | 8.8 | 12.7 ± 1.3 | |
| testosterone | 17 | 21.8 ± 4.0 | |
| verapamil | 16 | 24.6 ± 1.5 | |
| 17 | 5.6 ± 0.8 | CNS+ | |
| 20 | 11.2 ± 2.0 | CNS+ | |
| 27 | 9.6 ± 0.1 | CNS+ | |
| 34 | 11.2 ± 2.0 | CNS+ | |
| 38 | 11.2 ± 2.0 | CNS+ | |
| 41 | 6.4 ± 3.0 | CNS+ | |
| 42 | 14.6 ± 0.1 | CNS+ | |
| 46 | 4.4 ± 2.9 | CNS+ | |
| 47 | 1.4 ± 1.5 | CNS– | |
| 52 | 2.6 ± 0.8 | CNS+/CNS– | |
| 55 | 7.2 ± 2.5 | CNS+ |
PBS:EtOH (70:30) was used as solvent. Data are the mean ± SD of three independent experiments.
Efficacy on Drosophila Transgenic TDP-43 flies
As the N-(benzothiazolyl)-phenylacetamides here described target CK-1δ and are able to cross the BBB, we decided to evaluate their therapeutic potential in vivo, using a new transgenic Drosophila model of TDP-43 proteinopathies.38 Several Drosophila models of TDP-43 proteinopathies, based on the expression of human TDP-43 (hTDP-43) protein by the Gal4/UAS binary expression system, were recently characterized.39 Collectively, these models showed that in flies, hTDP-43 expression recapitulates several key features of the human TDP-43 proteinopathies, including axon and neuron degeneration, impaired motor behavior, cognitive deficits, and reduced lifespan. Additionally, biochemical data showed that hTDP-43 proteins undergo processing and abnormal phosphorylation at disease-specific sites in flies. In this study, we used the Drosophila lifespan as a phenotypic test to evaluate the neuroprotective role of N-(benzothiazolyl)-phenylacetamides against hTDP-43 potentially produced by decreasing its phosphorylation by inhibition of CK-1δ. Reduction of the lifespan of flies is a phenotype closely related to the neurodegenerative process that reflects an alteration of either neuronal functioning or cell viability. Previously, we showed that specific expression of hTDP-43 in adult differentiated neurons, using the panneuronal inducible GeneSwitch-GAL4 system (elav-Gal4GS),40 drastically reduced Drosophila lifespan.38 To check our hypothesis, we selected four compounds as chemical probes, (20, 24, 35, and 9), with different CK-1δ inhibition potency (IC50 values of 23 nM, 68 nM, and 2.22 μM for compounds 20, 24, and 35, respectively, and the inactive N-benzothiazolyl-phenylacetamide 9). Induction of hTDP-43 expression on adult flies was started nine days after hatching by feeding with RU486 (Mifepristone). From this moment, the gene-switch was “ON” and the UAS-transgene was expressed. As shown in Figure 8, feeding flies with food supplemented with 100 nM of compounds 20, 24, or 35 significantly extended fly lifespan (20, mean lifespan = 37.84 days, p = 0.0 × 10+00, N = 178; 24, mean lifespan = 38.63 days, p = 0.0 × 10+00, N = 163; 35, mean lifespan = 36.17 days, p = 4.2 × 10–6, N = 173), compared with the control group (DMSO, mean lifespan = 33.17 days, N = 151). Interestingly, in direct correlation with their inhibitory potency on CK-1δ in vitro (Table 3), the benzothiazoles 20 and 24 were more efficient in reducing hTDP-43 toxicity than 35. This compound is 100-fold less potent than 20 and 24 as CK-1δ inhibitor. Furthermore, the chemically similar inactive compound 9 did not significantly modify fly longevity (N = 102). From these experiments, we can conclude that CK-1δ inhibitors here reported have a protective effect on in vivo hTDP-43 neurotoxicity, showing their potential for the pharmacological treatment of human TDP-43 proteinopathies such ALS.
Figure 8.
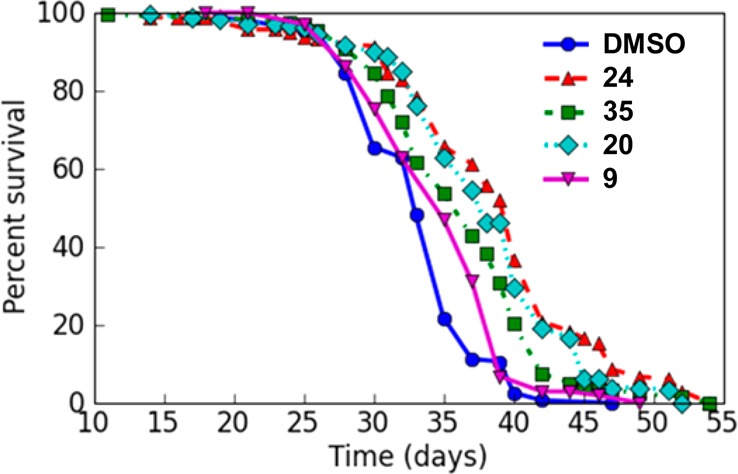
CK-1δ inhibitors decrease TDP-43 toxicity in flies. Lifespan of elav-Gal4GS > UAS-hTDP-43 transgenic flies expressing hTDP-43 proteins specifically in adult differentiated neurons and treated with candidate drugs or vehicle (DMSO, control flies). The survival curves show the proportion of living flies as a function of age (days). The longevity assay was performed on a large cohort (N > 150/experimental condition, see text). Statistical data relative to longevity experiments are described in the text. The lifespan of the flies was significantly increased when they were treated with 100 nM of compounds 20, 24, or 35, as judged by the log rank test.
Conclusions
The search of new therapies for ALS is an urgent need. The identification of pathological TDP-43 as the hallmark lesion in sporadic ALS open new avenues for pharmacological intervention. Our library screening methodology has led to the discovery and further optimization of a new family of potent CK-1δ inhibitors able to reduce TDP-43 phosphorylation in a cellular-based assay. These compounds are heterocyclic small molecules with IC50 on the selected kinase in the nanomolar range and selective on a 456 kinases panel. They are predicted to cross the blood–brain barrier, making them excellent tools for further pharmacological studies, and they have a protective effect on in vivo hTDP-43 neurotoxicity Drosophila model. Collectively, all these data show that N-benzothiazolyl amides represent a promising family of new drugs with potential for the pharmacological treatment of human TDP-43 proteinopathies such as ALS.
Experimental Section
Chemistry
Substrates were purchased from commercial sources and used without further purification. Melting points were determined with a Mettler Toledo MP70 apparatus. Crude residues were purified with the indicated solvent as eluent by flash column chromatography carried out at medium pressure using silica gel (E. Merck, grade 60, particle size 0.040–0.063 mm, 230–240 mesh ASTM) or IsoleraOne flash purification system from Biotage. Compounds were detected with UV light (254 nm). 1H NMR spectra were obtained on the Bruker AVANCE-300 spectrometer working at 300 MHz or on a Varian INOVA 400 spectrometer working at 400 MHz. Typical spectral parameters: spectral width 16 ppm, pulse width 9 μs (57°), data size 32 K. 13C NMR experiments were carried out on the Bruker AVANCE-300 spectrometer operating at 75 MHz or on a Varian INOVA 400 spectrometer working at 100 MHz. The acquisition parameters: spectral width 16 kHz, acquisition time 0.99 s, pulse width 9 μs (57°), data size 32 K. Chemical shifts are reported in values (ppm) relative to internal Me4Si, and J values are reported in Hz. HPLC analyses were performed on Alliance Waters 2690 equipment, with a UV detector photodiode array Waters 2996 with MS detector MicromassZQ (Waters), using an Sunfire column C18, 3.5 μm (50 mm × 4.6 mm) and acetonitrile and Milli-Q water (with 0.1% formic acid) as mobile phase. The standard gradient consisted of a 5 min run from 15% to 95% of acetonitrile at a flow rate of 1 mL/min. Elemental analysis results of all the new compounds were recorded on Heraeus CHN-O-rapid analyzer performed by the analytical department at CENQUIOR (CSIC), and values were within ±0.4% of the theoretical values for all compounds; therefore, these compounds meet the criteria of ≥95%. Additionally, purity of all final compounds was found to be ≥95% by LC/MS analysis. The microwave assisted syntheses were carried out using a Biotage Initiator 8 single-mode cavity instrument from Biotage. Experiments were performed with temperature control mode in sealed microwave process vials. The temperature was measured with an IR sensor on the outside of the reaction vessel. Stirring was provided by an in situ magnetic stirrer.
General Procedure for 1–8, 10, 13, 17–44, and 46–57
A mixture of the corresponding acid (1 equiv) and SOCl2 (1.5 equiv) was heated at 80 °C for 6 h. Afterward, the excess of SOCl2 was removed under reduced pressure and the acyl chloride obtained was used in the next step of the synthesis without further purification.
A mixture of the amine derivative (1 equiv), the corresponding acyl chloride previously synthesized (1 equiv), and THF if necessary, was heated under microwave irradiation (5–20 min, 110–150 °C). After cooling to room temperature, 50 mL of CH2Cl2 were added and the mixture was extracted with HCl 0.1 M (50 mL × 3). Afterward, the organic layer was washed with a saturated solution of NaHCO3 (50 mL × 3) and with a saturated solution of NaCl (50 mL × 3). Finally, the organic phase was dried over sodium sulfate and the solvent evaporated under reduced pressure. The residue was chromatographed as indicated in each case.
N-(5-Acetyl-4-methylthiazol-2-yl)-2-(3-chlorophenyl)acetamide (1)
Reagents: 1-(2-amino-4-methylthiazol-5-yl)ethanone (1.6 mmol), 2-(3-chlorophenyl)acetyl chloride (1.6 mmol), and THF (1 mL). Reaction conditions: 15 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a white solid (352.8 mg, 72%), mp 195–196 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.71 (s, 1H), 7.40 (s, 1H), 7.38–7.29 (m, 2H), 7.27 (dt, J = 7.0, 1.6 Hz, 1H), 3.82 (s, 2H), 2.55 (s, 3H), 2.45 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 190.6, 169.5, 159.3, 154.3, 136.8, 132.9, 130.2, 129.2, 128.1, 126., 125.5, 41.1, 30.0, 18.0. HPLC: purity >99%. m/z (ES) 309 [M + H]. Anal. (C14H13ClN2O2S) C, H, N, S.
2-(3-Chlorophenyl)-N-(1-methyl-1H-benzimidazol-2-yl)acetamide (2)
Reagents: 1-methyl-1H-benzimidazol-2-amine (1.7 mmol), 2-(3-chlorophenyl)acetyl chloride (1.7 mmol), and THF (0.3 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a gray solid (19.9 mg, 4%), mp 154–156 °C. 1H NMR (300 MHz, CDCl3): δ 7.44 (s, 1H), 7.37–7.11 (m, 7H), 3.80 (s, 2H), 3.66 (s, 3H). 13C NMR (75 MHz, CDCl3): δ 182.3, 153.3, 139.2, 133.9, 130.0, 129.6, 129.4, 128.1, 127.7, 126.4, 123.2, 111.2, 109.1, 46.9, 28.3. HPLC: purity >99%. m/z (ES) 300 [M + H]. Anal. (C16H14ClN3O) C, H, N.
2-(4-Chlorophenyl)-N-(1-methyl-1H-benzimidazol-2-yl)acetamide (3)
Reagents: 1-methyl-1H-benzimidazol-2-amine (1.7 mmol), 2-(4-chlorophenyl)acetyl chloride (1.7 mmol), and THF (0.5 mL). Reaction conditions: 20 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:1) to afford a white solid (158.0 mg, 31%), mp 195–197 °C. 1H NMR (300 MHz, CDCl3): δ 7.35–7.11 (m, 8H), 3.79 (s, 2H), 3.66 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 182.8, 153.6, 136.0, 132.3, 131.1, 130.2, 128.5, 128.4, 123.4, 111.5, 109.3, 46.8, 28.5. HPLC: purity >99%. m/z (ES) 301 [M + H]. Anal. (C16H14ClN3O) C, H, N.
2-(3-Chlorophenyl)-N-(1,3,4-thiadiazol-2-yl)acetamide (4)
Reagents: 1,3,4-thiadiazol-2-amine (2.5 mmol), 2-(3-chlorophenyl)acetyl chloride (2.5 mmol), and THF (1 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (75.7 mg, 12%), mp 240–242 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.80 (s, 1H), 9.13 (s, 1H), 7.42–7.36 (m, 1H), 7.36–7.29 (m, 2H), 7.25 (dt, J = 6.9, 1.8 Hz, 1H), 3.83 (s, 2H). 13C NMR (100 MHz, DMSO): δ 169.8, 159.2, 149.5, 137.9, 133.6, 130.9, 130.0, 128.8, 127.6, 41.6. HPLC: purity >99%. m/z (ES) 254 [M + H]. Anal. (C10H8ClN3OS) C, H, N, S.
2-(3-Chlorophenyl)-N-(pyrimidin-2-yl)acetamide (5)
Reagents: pyrimidin-2-amine (2.6 mmol), 2-(3-chlorophenyl)acetyl chloride (2.6 mmol), and THF (1 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (CH2Cl2/MeOH 9:1) to afford a brown solid (217.8 mg, 34%), mp 172–174 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.81 (s, 1H), 8.64 (d, J = 4.8 Hz, 2H), 7.43–7.23 (m, 4H), 7.17 (t, J = 4.8 Hz, 1H), 3.82 (s, 2H). 13C NMR (100 MHz, DMSO-d6): δ 169.4, 159.0, 158.2, 138.7, 133.4, 130.7, 129.9, 128.8, 127.2, 117.5, 43.3. HPLC: purity >99%. m/z (ES) 248 [M + 2H]. Anal. (C12H10ClN3O) C, H, N.
2-(4-Chlorophenyl)-N-(pyrimidin-2-yl)acetamide (6)
Reagents: pyrimidin-2-amine (2.6 mmol), 2-(4-chlorophenyl)acetyl chloride (2.6 mmol), and THF (0.7 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (CH2Cl2/MeOH 9:1) to afford a brown solid (256.4 mg, 40%), mp 179–180 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.79 (s, 1H), 8.64 (dd, J = 4.8, 0.8 Hz, 2H), 7.42–7.27 (m, 4H), 7.16 (td, J = 4.8, 0.8 Hz, 1H), 3.80 (s, 2H). 13C NMR (100 MHz, DMSO-d6): δ 169.6, 159.0, 158.3, 135.3, 131.9, 131.9, 128.8, 117.5, 43.1. HPLC: purity >99%. m/z (ES) 248 [M + 2H]. Anal. (C12H10ClN3O) C, H, N.
N-(Benzoxazol-2-yl)-2-(4-chlorophenyl)acetamide (7)
Reagents: benzoxazol-2-amine (1.9 mmol) and 2-(4-chlorophenyl)acetyl chloride (1.9 mmol). Reaction conditions: 10 min at 150 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:1) to afford a white solid (331.9 mg, 62%), mp 191–193 °C. 1H NMR (300 MHz, DMSO-d6): δ 11.90 (s, 1H), 7.58 (td, J = 7.3, 4.9 Hz, 2H), 7.49–6.79 (m, 6H), 3.85 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 168.5, 155.0, 147.6, 140.6, 133.7, 131.5, 131.3, 128.2, 124.5, 123.6, 118.2, 110.0, 41.7. HPLC: purity >99%. m/z (ES) 287 [M + H]. Anal. (C15H11ClN2O2) C, H, N.
N-(Benzoxazol-2-yl)-2-(2-methoxyphenyl)acetamide (8)
Reagents: benzoxazol-2-amine (1.6 mmol), 2-(2-methoxyphenyl)acetyl chloride (1.6 mmol), and THF (0.5 mL). Reaction conditions: 15 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a brown solid (281.9 mg, 29%), mp 135–137 °C. 1H NMR (300 MHz, DMSO-d6): δ 11.75 (s, 1H), 7.64–7.51 (m, 2H), 7.35–7.17 (m, 4H), 6.98 (d, J = 7.8 Hz, 1H), 6.90 (dd, J = 7.9, 6.9 Hz, 1H), 3.82 (s, 2H), 3.74 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 168.8, 157.2, 155.1, 147.6, 140.7, 131.1, 128.3, 124.4, 123.4, 123.1, 120.1, 118.1, 110.6, 109.9, 55.4, 37.4. HPLC: purity >99%. m/z (ES) 283 [M + H]. Anal. (C16H14N2O3) C, H, N.
N-(6-(Trifluoromethyl)benzothiazol-2-yl)-2-methoxybenzamide (10)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol) and 2-methoxybenzoyl chloride (1.1 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a yellow solid (164.1 mg, 41%), mp 215–217 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.29 (s, 1H), 8.53 (s, 1H), 7.93 (d, J = 8.5 Hz, 1H), 7.75 (dd, J = 7.7, 1.7 Hz, 2H), 7.66–7.53 (m, 1H), 7.24 (d, J = 8.3 Hz, 1H), 7.11 (t, J = 7.5 Hz, 1H), 3.94 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 165.3, 160.9, 157.3, 151.3, 133.8, 132.1, 130.3, 124.6 (q, J = 272.1 Hz), 123.8 (q, J = 31.8 Hz), 123.0 (q, J = 3.6 Hz), 121.3, 121.0, 120.7, 119.9 (q, J = 4.2 Hz), 112.3, 56.1. HPLC: purity >99%. m/z (ES) 353 [M + H]. Anal. (C16H11F3N2O2S) C, H, N, S.
N-(6-Ethoxybenzothiazol-2-yl)-2,2-diphenylacetamide (13)
Reagents: 6-ethoxy-benzothiazol-2-amine (1.3 mmol), 2,2-diphenylacetyl chloride (1.3 mmol) and THF (0.4 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (388.4 mg, 67%), mp 223–224 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.70 (s, 1H), 7.61 (d, J = 8.8 Hz, 1H), 7.54 (d, J = 2.4 Hz, 1H), 7.44–7.31 (m, 10H), 7.00 (dd, J = 8.8, 2.5 Hz, 1H), 5.36 (s, 1H), 4.05 (q, J = 6.9 Hz, 2H), 1.33 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.6, 155.6, 155.4, 142.5, 138.8, 132.7, 128.5, 127.2, 121.2, 115.4, 105.3, 63.6, 56.1, 14.7. HPLC: purity >99%. m/z (ES) 389 [M + H]. Anal. (C23H20N2O2S) C, H, N, S.
2-(3-Chlorophenyl)-N-(6-methylbenzothiazol-2-yl)acetamide (17)
Reagents: 6-methylbenzothiazol-2-amine (1.5 mmol), 2-(3-chlorophenyl)acetyl chloride (1.5 mmol), and THF (0.3 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a white solid (195.6 mg, 41%), mp 206–208 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.53 (s, 1H), 7.74 (s, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.43 (s, 1H), 7.39–7.27 (m, 3H), 7.23 (d, J = 8.3 Hz, 1H), 3.84 (s, 2H), 2.39 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.4, 156.8, 146.4, 136.9, 132.9, 132.8, 131.4, 130.1, 129.2, 128.1, 127.3, 126.8, 121.2, 120.1, 41.1, 20.8. HPLC: purity >99%. m/z (ES) 317 [M + H]. Anal. (C16H13ClN2OS) C, H, N, S.
2-(3-Chlorophenyl)-N-(4-methylbenzothiazol-2-yl)acetamide (18)
Reagents: 4-methylbenzothiazol-2-amine (1.5 mmol), 2-(3-chlorophenyl)acetyl chloride (1.5 mmol), and THF (1 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:7) to afford a white solid (206.2 mg, 43%), mp 165–166 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.68 (s, 1H), 7.75 (dd, J = 7.6, 1.5 Hz, 1H), 7.43 (s, 1H), 7.40–7.12 (m, 5H), 3.84 (s, 2H), 2.56 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.6, 156.9, 147.5, 137.0, 132.9, 131.0, 130.2, 129.8, 129.3, 128.1, 126.9, 126.6, 123.5, 119.1, 41.2, 17.9. HPLC: purity >99%. m/z (ES) 317 [M + H]. Anal. (C16H13ClN2OS) C, H, N, S.
N-(4-Chlorobenzothiazol-2-yl)-2-(3-chlorophenyl)acetamide (19)
Reagents: 4-chlorobenzothiazol-2-amine (1.4 mmol), 2-(3-chlorophenyl)acetyl chloride (1.4 mmol), and THF (1 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:8) to afford a white solid (144.9 mg, 32%), mp 160– 161 °C. 1H NMR (300 MHz, DMSO-d6): δ 13.00 (s, 1H), 7.94 (dd, J = 8.0, 1.1 Hz, 1H), 7.51 (dd, J = 7.9, 1.1 Hz, 1H), 7.44 (d, J = 2.1 Hz, 1H), 7.40–7.20 (m, 4H), 3.87 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 170.0, 158.9, 145.4, 136.8, 133.0, 132.9, 130.2, 129.3, 128.2, 127.0, 126.2, 124.4, 124.4, 120.9, 41.2. HPLC: purity >99%. m/z (ES) 337 [M]. Anal. (C15H10Cl2N2OS) C, H, N, S.
2-(3-Chlorophenyl)-N-(6-(trifluoromethyl)benzothiazol-2-yl)acetamide (20)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (2.3 mmol), 2-(3-chlorophenyl)acetyl chloride (2.3 mmol), and THF (0.7 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a white solid (340 mg, 40%), mp 190–191 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.85 (s, 1H), 8.48 (s, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.44 (s, 1H), 7.35 (t, J = 8.1 Hz, 3H), 3.90 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 170.2, 161.1, 151.3, 136.8, 132.9, 132.0, 130.2, 129.4, 128.3, 127.0, 124.7 (q, J = 277.6 Hz), 123.8 (q, J = 31.8 Hz), 122.9 (q, J = 3.2 Hz), 121.0, 119.9 (q, J = 4.5 Hz), 41.2. HPLC: purity >99%. m/z (ES) 371 [M + H]. Anal. (C16H10ClF3N2OS) C, H, N, S.
2-(3-Chlorophenyl)-N-(6-methoxybenzothiazol-2-yl)acetamide (21)
Reagents: 6-methoxybenzothiazol-2-amine (1.7 mmol), 2-(3-chlorophenyl)acetyl chloride (1.7 mmol), and THF (0.7 mL). Reaction conditions: 15 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a white solid (418 mg, 76%), mp 179–180 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.47 (s, 1H), 7.63 (d, J = 8.9 Hz, 1H), 7.52 (d, J = 2.5 Hz, 1H), 7.41 (m, 1H) 7.38–7.2 (m, 3H), 7.00 (dd, J = 8.9, 2.6 Hz, 1H,), 3.82 (s, 2H), 3.77 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.7, 156.4, 156.0, 142.7, 137.3, 133.1, 132.9, 130.5, 129.5, 128.4, 127.1, 121.4, 115.2, 104.9, 55.8, 41.4. HPLC: purity >99%. m/z (ES) 333 [M + H]. Anal. (C16H13ClN2O2S) C, H, N, S.
2-(3-Chlorophenyl)-N-(6-(trifluoromethoxy)benzothiazol-2-yl)acetamide (22)
Reagents: 6-(trifluoromethoxy)benzothiazol-2-amine (1.1 mmol) and 2-(3-chlorophenyl)acetyl chloride (1.1 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (386.7 mg, 48%), mp 150–151 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.74 (s, 1H), 8.11 (t, J = 1.8 Hz, 1H), 7.82 (dd, J = 8.8, 1.7 Hz, 1H), 7.47–7.25 (m, 5H), 3.88 (s, 2H). 13C NMR (100 MHz, DMSO-d6): δ 170.7, 160.0, 148.2, 144.7, 137.5, 133.6, 133.3, 130.9, 130.1, 128.9, 127.6, 122.2, 120.9 (c, J = 256.1 Hz), 119.6, 115.7, 41.8. HPLC: purity >99%. m/z (ES) 387 [M + H]. Anal. (C16H10ClF3N2O2S) C, H, N, S.
2-(3-Chlorophenyl)-N-(6-ethoxybenzothiazol-2-yl)acetamide (23)
Reagents: 6-ethoxybenzothiazol-2-amine (1.5 mmol), 2-(3-chlorophenyl)acetyl chloride (1.5 mmol), and THF (1 mL). Reaction conditions: 15 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a white solid (373.5 mg, 70%), mp 163–164 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.7 (s, 1H), 7.61 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 2.4 Hz, 1H), 7.42 (s, 1H), 7.40–7.31 (m, 2H), 7.29 (dt, J = 7.1, 1.5 Hz, 1H), 7.00 (dd, J = 8.9, 2.5 Hz, 1H), 4.04 (q, J = 7.0 Hz, 2H), 3.83 (s, 2H), 1.32 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.1, 156.4, 156.1, 143.2, 137.8, 133.6, 133.4, 131.9, 128.9, 128.2, 127.6, 121.8, 116.0, 106.0, 64.3, 41.9, 15.4. HPLC: purity >99%. m/z (ES) 347 [M + H]. Anal. (C17H15ClN2O2S) C, H, N, S.
2-(2-Chlorophenyl)-N-(6-(trifluoromethyl)benzothiazol-2-yl)acetamide (24)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.5 mmol), 2-(2-chlorophenyl)acetyl chloride (1.5 mmol), and THF (1 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a white solid (66.8 mg, 12%), mp 226–228 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.92 (s, 1H), 8.49 (s, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.74 (dd, J = 8.6, 1.9 Hz, 1H), 7.48–7.45 (m, 2H), 7.38–7.22 (m, 2H), 4.06 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 169.7, 161.1, 151.3, 133.7, 132.6, 132.5, 132.0, 129.1, 127.3, 124.6 (q, J = 271.9 Hz), 123.8 (q, J = 31.8 Hz), 123.0 (q, J = 3.5 Hz), 121.0, 119.9 (q, J = 4.0 Hz), 40.4. HPLC: purity >99%. m/z (ES) 371 [M + H]. Anal. (C16H10ClF3N2OS) C, H, N, S.
2-(2-Chlorophenyl)-N-(6-methoxybenzothiazol-2-yl)acetamide (25)
Reagents: 6-methoxybenzothiazol-2-amine (1.4 mmol) and 2-(2-chlorophenyl)acetyl chloride (1.4 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a white solid (216.3 mg, 47%), mp 216–218 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.52 (s, 1H), 7.65 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 2.3 Hz, 1H), 7.52–7.39 (m, 2H), 7.35–7.32 (m, 2H), 7.03 (dd, J = 8.8, 2.5 Hz, 1H), 4.02 (s, 2H), 3.80 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.2, 156.5, 156.2, 143.0, 134.1, 133.3, 133.1, 132.8, 129.4, 129.3, 127.6, 121.5, 115.3, 105.1, 56.0, 40.2. HPLC: purity >99%. m/z (ES) 333 [M + H]. Anal. (C16H13ClN2O2S) C, H, N, S.
2-(2-Chlorophenyl)-N-(6-ethoxybenzothiazol-2-yl)acetamide (26)
Reagents: 6-ethoxybenzothiazol-2-amine (1.3 mmol) and 2-(2-chlorophenyl)acetyl chloride (1.3 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a white solid (234.6 mg, 53%), mp 206–207 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.51 (s, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.54 (s, 1H), 7.50–7.40 (m, 2H), 7.38–7.27 (m, 2H), 7.02 (m, 1H), 4.05 (c, J = 6.9 Hz, 2H), 4.01 (s, 2H), 1.34 (t, J = 6.8 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.2, 156.2, 155.7, 142.9, 134.1, 133.3, 133.1, 132.8, 129.4, 129.3, 127.6, 121.5, 115.7, 105.7, 63.9, 39.6, 15.1. HPLC: purity >99%. m/z (ES) 348 [M + 2H]. Anal. (C17H15ClN2O2S) C, H, N, S.
2-(4-Chlorophenyl)-N-(6-(trifluoromethyl)benzothiazol-2-yl)acetamide (27)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.2 mmol) and 2-(4-chlorophenyl)acetyl chloride (1.2 mmol). Reaction conditions: 5 min at 110 °C. The desired product was obtained directly after the extractions affording a white solid (404.1 mg, 95%), mp 182–183 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.84 (s, 1H), 8.48 (s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.49–7.27 (m, 4H), 3.87 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 170.4, 161.1, 151.2, 134.0, 133.4, 132.0, 131.7, 131.3, 128.1, 124.5 (q, J = 272.0 Hz), 123.7 (q, J = 31.8 Hz), 122.9 (q, J = 3.9 Hz), 120.9, 119.9 (q, J = 4.3 Hz), 41.0. HPLC: purity >99%. m/z (ES) 371 [M + H]. Anal. (C16H10ClF3N2OS) C, H, N, S.
2-(4-Chlorophenyl)-N-(6-methoxybenzothiazol-2-yl)acetamide (28)
Reagents: 6-methoxybenzothiazol-2-amine (1.4 mmol) and 2-(4-chlorophenyl)acetyl chloride (1.4 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a white solid (36.6 mg, 8%), mp 246–247 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.47 (s, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 2.4 Hz, 1H), 7.43–7.35 (m, 4H), 7.03 (dd, J = 8.8, 2.6 Hz, 1H), 3.83 (s, 2H), 3.80 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.9, 156.5, 156.2, 142.9, 134.1, 133.1, 132.0, 131.7, 128.7, 121.5, 115.3, 105.1, 56.0, 41.3. m/z (ES) 333 [M + H]. Anal. (C16H13ClN2O2S) C, H, N, S.
2-(4-Chlorophenyl)-N-(6-ethoxybenzothiazol-2-yl)acetamide (29)
Reagents: 6-ethoxybenzothiazol-2-amine (1.3 mmol) and 2-(4-chlorophenyl)acetyl chloride (1.3 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a white solid (165.4 mg, 37%), mp 192–193 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.46 (s, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.53 (d, J = 2.4 Hz, 1H), 7.46–7.31 (m, 4H), 7.01 (dd, J = 8.8, 2.5 Hz, 1H), 4.05 (q, J = 6.9 Hz, 2H), 3.83 (s, 2H), 1.34 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.9, 156.1, 155.7, 142.9, 134.1, 133.1, 132.0, 131.6, 128.7, 121.5, 115.6, 105.7, 63.9, 41.3, 15.1. HPLC: purity >99%. m/z (ES) 348 [M + 2H]. Anal. (C17H15ClN2O2S) C, H, N, S.
N-(6-Bromobenzothiazol-2-yl)-2-(2-methoxyphenyl)acetamide (30)
Reagents: 6-bromobenzothiazol-2-amine (1.1 mmol) and 2-(2-methoxy-phenyl)acetyl chloride (1.1 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a yellow solid (172.5 mg, 42%), mp 169–170 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.58 (s, 1H), 8.24 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.57 (dd, J = 8.6, 2.0 Hz, 1H), 7.34–7.19 (m, 2H), 6.99 (d, J = 8.0 Hz, 1H), 6.92 (t, J = 7.4 Hz, 1H), 3.82 (s, 2H), 3.75 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.9, 159.2, 157.6, 148.1, 134.0, 131.5, 129.5, 128.9, 124.6, 123.3, 122.4, 120.6, 115.7, 111.2, 55.8, 37.0. HPLC: purity >99%. m/z (ES) 377 [M + 2H]. Anal. (C16H13BrN2O2S) C, H, N, S.
N-(6-Chlorobenzothiazol-2-yl)-2-(2-methoxyphenyl)acetamide (31)
Reagents: 6-chlorobenzothiazol-2-amine (1.5 mmol) and 2-(2-methoxyphenyl)acetyl chloride (1.5 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a yellow solid (265.1 mg, 59%), mp 168–169 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.57 (s, 1H), 8.09 (d, J = 2.1 Hz, 1H), 7.73 (d, J = 8.6 Hz, 1H), 7.44 (dd, J = 8.6, 2.2 Hz, 1H), 7.32–7.19 (m, 2H), 6.98 (d, J = 7.8 Hz, 1H), 6.91 (t, J = 7.4 Hz, 1H), 3.81 (s, 2H), 3.73 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.5, 158.9, 157.3, 147.5, 133.1, 131.2, 128.5, 127.5, 126.4, 122.9, 121.7, 121.4, 120.2, 110.8, 55.5, 36.6. HPLC: purity >99%. m/z (ES) 333 [M + 2H]. Anal. (C16H13ClN2O2S) C, H, N, S.
N-(6-Fluorobenzothiazol-2-yl)-2-(2-methoxyphenyl)acetamide (32)
Reagents: 6-fluorobenzothiazol-2-amine (1.4 mmol) and 2-(2-methoxyphenyl)acetyl chloride (1.4 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford an orange solid (153.2 mg, 33%), mp 154–156 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.51 (s, 1H), 7.88 (dd, J = 8.8, 2.6 Hz, 1H), 7.75 (dd, J = 8.9, 4.8 Hz, 1H), 7.33–7.20 (m, 3H), 6.99 (d, J = 7.8 Hz, 1H), 6.92 (td, J = 7.4, 0.9 Hz, 1H), 3.81 (s, 2H), 3.75 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.8, 159.0 (d, J = 239.6 Hz), 158.4, 157.6, 145.7, 133.0 (d, J = 11.0 Hz), 131.5, 128.8, 123.4, 121.9 (d, J = 9.3 Hz), 120.6, 114.5 (d, J = 24.3 Hz), 111.2, 108.5 (d, J = 26.9 Hz), 55.8, 37.0. HPLC: purity >99%. m/z (ES) 317 [M + 2H]. Anal. (C16H13FN2O2S) C, H, N, S.
2-(2-Methoxyphenyl)-N-(6-methylbenzothiazol-2-yl)acetamide (33)
Reagents: 6-methylbenzothiazol-2-amine (1.5 mmol) and 2-(2-methoxyphenyl)acetyl chloride (1.5 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford an orange solid (93.5 mg, 20%), mp 165–167 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.38 (s, 1H), 7.73 (s, 1H), 7.61 (d, J = 8.2 Hz, 1H), 7.32–7.16 (m, 3H), 6.98 (d, J = 7.7 Hz, 1H), 6.91 (td, J = 7.4, 1.0 Hz, 1H), 3.79 (s, 2H), 3.74 (s, 3H), 2.39 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.1, 157.2, 157.1, 146.5, 132.8, 131.5, 131.1, 128.4, 127.3, 123.0, 121.2, 120.2, 120.1, 110.8, 55.4, 36.6, 20.9. HPLC: purity >99%. m/z (ES) 312 [M]. Anal. (C17H16N2O2S) C, H, N, S.
N-(6-(Trifluoromethyl)benzothiazol-2-yl)-2-(2-methoxyphenyl)acetamide (34)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.2 mmol) and 2-(2-methoxyphenyl)acetyl chloride (1.2 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (226.9 mg, 54%), mp 177–179 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.72 (s, 1H), 8.47 (s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.48–7.09 (m, 2H), 6.99 (d, J = 8.2 Hz, 1H), 6.92 (t, J = 7.4 Hz, 1H), 3.84 (s, 2H), 3.74 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.8, 161.2, 157.3, 151.4, 132.0, 131.2, 128.5, 124.6 (q, J = 271.8 Hz), 123.6 (q, J = 31.8 Hz), 122.9 (q, J = 5.2 Hz), 122.8, 120.8, 120.2, 119.9 (q, J = 4.2 Hz), 110.8, 55.5, 36.7. HPLC: purity 97%. m/z (ES) 367 [M + H]. Anal. (C17H13F3N2O2S) C, H, N, S.
N-(6-Methoxybenzothiazol-2-yl)-2-(2-methoxyphenyl)acetamide (35)
Reagents: 6-methoxybenzothiazol-2-amine (1.4 mmol) and 2-(2-methoxyphenyl)acetyl chloride (1.4 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a white solid (166.8 mg, 37%), mp 159–160 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.34 (s, 1H), 7.64 (d, J = 8.8 Hz, 1H), 7.55 (d, J = 2.4 Hz, 1H), 7.33–7.18 (m, 2H), 7.07–6.85 (m, 3H), 3.80 (s, 5H), 3.75 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.4, 157.7, 156.4, 156.3, 143.0, 133.1, 131.5, 128.8, 123.5, 121.4, 120.6, 115.2, 111.2, 105.1, 56.0, 55.8, 36.9. HPLC: purity >99%. m/z (ES) 330 [M + 2H]. Anal. (C17H16N2O3S) C, H, N, S.
N-(6-(Trifluoromethoxy)benzothiazol-2-yl)-2-(2-methoxyphenyl)acetamide (36)
Reagents: 6-(trifluoromethoxy)benzothiazol-2-amine (1.1 mmol), 2-(2-methoxyphenyl)acetyl chloride (1.1 mmol), and THF (0.6 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a yellow solid (187.0 mg, 46%), mp 143–145 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.60 (s, 1H), 8.09 (d, J = 1.3 Hz, 1H), 7.81 (d, J = 8.8 Hz, 1H), 7.40 (dd, J = 8.8, 1.6 Hz, 1H), 7.31–7.19 (m, 2H), 6.98 (d, J = 7.8 Hz, 1H), 6.91 (m, 1H), 3.82 (s, 2H), 3.73 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.6, 159.5, 157.3, 147.6, 145.0, 132.6, 131.2, 128.5, 122.9, 121.4, 120.2, 120.1 (q, J = 255.6 Hz), 119.8, 115.0, 110.8, 55.4, 36.6. HPLC: purity >99%. m/z (ES) 383 [M + H]. Anal. (C17H13F3N2O3S) C, H, N, S.
N-(6-Ethoxybenzothiazol-2-yl)-2-(2-methoxyphenyl)acetamide (37)
Reagents: 6-ethoxybenzothiazol-2-amine (1.3 mmol) and 2-(2-methoxyphenyl)acetyl chloride (1.3 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a white solid (222.7 mg, 51%), mp 148–150 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.33 (s, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.53 (d, J = 2.4 Hz, 1H), 7.32–7.19 (m, 2H), 7.05–6.96 (m, 2H), 6.92 (t, J = 7.4 Hz, 1H), 4.06 (q, J = 6.9 Hz, 2H), 3.79 (s, 2H), 3.75 (s, 3H), 1.34 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.4, 157.7, 156.3, 155.7, 143.0, 133.1, 131.5, 128.8, 123.5, 121.4, 120.6, 115.6, 111.2, 105.7, 63.9, 55.8, 36.9, 15.1. HPLC: purity >99%. m/z (ES) 343 [M + H]. Anal. (C18H18N2O3S) C, H, N, S.
N-(6-(Trifluoromethyl)benzothiazol-2-yl)-2-(3-methoxyphenyl)acetamide (38)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.5 mmol), 2-(3-methoxyphenyl)acetyl chloride (1.5 mmol), and THF (1 mL). Reaction conditions: 15 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a white solid (108.1 mg, 26%), mp 154–156 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.83 (s, 1H), 8.48 (s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.73 (dd, J = 8.5, 1.5 Hz, 1H), 7.25 (t, J = 7.9 Hz, 1H), 6.92 (m, 2H), 6.84 (dd, J = 7.9, 2.2 Hz, 1H), 3.82 (s, 2H), 3.74 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 171.2, 161.8, 160.0, 152.0, 136.5, 132.7, 130.2, 125.2 (q, J = 271.8 Hz), 124.4 (q, J = 31.9 Hz), 123.6 (q, J = 3.6 Hz), 122.2, 121.6, 120.6 (q, J = 4.1 Hz), 115.9, 113.0, 55.7, 42.6. HPLC: purity 98%. m/z (ES) 367 [M + H]. Anal. (C17H13F3N2O2S) C, H, N, S.
N-(6-Methoxybenzothiazol-2-yl)-2-(3-methoxyphenyl)acetamide (39)
Reagents: 6-methoxybenzothiazol-2-amine (1.4 mmol), 2-(3-methoxy-phenyl)acetyl chloride (1.4 mmol), and THF (1 mL). Reaction conditions: 15 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:3) to afford a yellow solid (284.4 mg, 62%), mp 147–149 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.43 (s, 1H), 7.62 (d, J = 8.8 Hz, 1H), 7.54 (d, J = 2.5 Hz, 1H), 7.24 (t, J = 7.8 Hz, 1H), 7.01 (dd, J = 8.8, 2.6 Hz, 1H), 6.91–6.89 (m, 2H), 6.83 (dd, J = 8.3, 1.5 Hz, 1H), 3.78 (s, 3H), 3.76 (s, 2H), 3.73 (s, 3H). 13C NMR (100 MHz, DMSO-d6): δ 170.4, 159.9, 156.8, 156.5, 143.3, 136.8, 133.4, 130.1, 122.2, 121.8, 115.8, 115.6, 112.9, 105.4, 56.3, 55.7, 42.5. HPLC: purity 97%. m/z (ES) 330 [M + 2H]. Anal. (C17H16N2O3S) C, H, N, S.
N-(6-Ethoxybenzothiazol-2-yl)-2-(3-methoxyphenyl)acetamide (40)
Reagents: 6-ethoxybenzothiazol-2-amine (1.3 mmol), 2-(3-methoxy-phenyl)acetyl chloride (1.3 mmol), and THF (1 mL). Reaction conditions: 15 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a white solid (194.1 mg, 44%), mp 162–164 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.42 (s, 1H), 7.61 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 2.4 Hz, 1H), 7.24 (t, J = 7.8 Hz, 1H), 7.00 (dd, J = 8.8, 2.5 Hz, 1H), 6.96–6.77 (m, 1H), 6.92 (s, 1H), 6.83 (ddd, J = 8.3, 2.5, 1.2 Hz, 1H), 4.04 (q, J = 6.9 Hz, 2H), 3.76 (s, 2H), 3.74 (s, 3H), 1.33 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.7, 159.2, 155.8, 155.3, 142.5, 136.1, 132.7, 129.4, 121.5, 121.1, 115.3, 115.1, 112.2, 105.3, 63.6, 55.0, 41.8, 14.7. HPLC: purity >99%. m/z (ES) 343 [M + H]. Anal. (C18H18N2O3S) C, H, N, S.
N-(6-(Trifluoromethyl)benzothiazol-2-yl)-2-(3-(trifluoromethyl)phenyl)acetamide (41)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol), 2-(3-trifluoromethylphenyl)acetyl chloride (1.1 mmol), and THF (0.5 mL). Reaction conditions: 20 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a white solid (194.4 mg, 42%), mp 139–141 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.87 (s, 1H), 8.48–8.43 (m, 1H), 7.88 (d, J = 8.5 Hz, 1H), 7.74–7.51 (m, 5H), 3.99 (s, 2H). 13C NMR (100 MHz, DMSO-d6): δ 170.9, 161.7, 151.9, 136.4, 134.5, 132.7, 130.1, 129.7 (q, J = 31.5 Hz), 126.9 (q, J = 3.9 Hz), 125.2 (q, J = 271.8 Hz), 124.9 (q, J = 272.1 Hz), 124.4 (q, J = 3.8 Hz), 124.4 (q, J = 31.8 Hz), 123.6 (q, J = 4.0 Hz), 121.7, 120.6 (q, J = 4.4 Hz), 41.9. HPLC: purity 97%. m/z (ES) 405 [M + H]. Anal. (C17H10F6N2OS) C, H, N, S.
N-(6-(Trifluoromethyl)benzothiazol-2-yl)-2-(4-methoxyphenyl)acetamide (42)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol), 2-(4-methoxyphenyl)acetyl chloride (1.1 mmol), and THF (0.5 mL). Reaction conditions: 20 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a white solid (184.8 mg, 42%), mp 134–136 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.79 (s, 1H), 8.47 (s, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.26 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.7 Hz, 2H), 3.77 (s, 2H), 3.72 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 171.0, 161.1, 158.3, 151.3, 132.0, 130.4, 126.3, 124.5 (q, J = 272.2 Hz), 123.7 (q, J = 31.9 Hz), 122.9 (q, J = 3.7 Hz), 120.9, 119.8 (q, J = 4.3 Hz), 113.9, 55.0, 40.9. HPLC: purity >99%. m/z (ES) 367 [M + H]. Anal. (C17H13F3N2O2S) C, H, N, S.
N-(6-Methoxybenzothiazol-2-yl)-2-(4-methoxyphenyl)acetamide (43)
Reagents: 6-methoxybenzothiazol-2-amine (1.4 mmol), 2-(4-methoxy-phenyl)acetyl chloride (1.4 mmol), and THF (1 mL). Reaction conditions: 20 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a beige solid (192.4 mg, 42%), mp 183–184 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.40 (s, 1H), 7.62 (d, J = 8.8 Hz, 1H), 7.54 (d, J = 2.4 Hz, 1H), 7.25 (d, J = 8.5 Hz, 2H), 7.01 (dd, J = 8.8, 2.6 Hz, 1H), 6.89 (d, J = 8.7 Hz, 2H), 3.78 (s, 3H), 3.72 (s, 5H). 13C NMR (75 MHz, DMSO-d6): δ 170.2, 158.2, 156.1, 155.9, 142.5, 132.7, 130.3, 126.6, 121.1, 114.8, 113.8, 104.7, 55.6, 55.0, 40.9. HPLC: purity >99%. m/z (ES) 330 [M + 2H]. Anal. (C17H16N2O3S) C, H, N, S.
N-(6-Ethoxybenzothiazol-2-yl)-2-(4-methoxyphenyl)acetamide (44)
Reagents: 6-ethoxybenzothiazol-2-amine (1.3 mmol), 2-(4-methoxy-phenyl)acetyl chloride (1.3 mmol), and THF (1 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:3) to afford a beige solid (297.0 mg, 67%), mp 177–178 °C. 1H NMR (300 MHz, DMSO): δ 12.39 (s, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.51 (d, J = 2.4 Hz, 1H), 7.25 (d, J = 8.6 Hz, 2H), 6.99 (dd, J = 8.8, 2.5 Hz, 1H), 6.89 (d, J = 8.7 Hz, 2H), 4.04 (q, J = 6.9 Hz, 2H), 3.72 (s, 5H), 1.33 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, DMSO): δ 170.2, 158.2, 155.9, 155.3, 142.5, 132.7, 130.3, 126.6, 121.1, 115.2, 113.9, 105.3, 63.6, 55.0, 40.9, 14.7. HPLC: purity >99%. m/z (ES) 343 [M + H]. Anal. (C18H18N2O3S) C, H, N, S.
N-(6-(Trifluoromethyl)benzothiazol-2-yl)-2-phenylacetamide (46)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol) and 2-phenylacetyl chloride (1.1 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white–yellow solid (234.3 mg, 61%), mp 214–216 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.85 (s, 1H), 8.49 (s, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.74 (dd, J = 7.9, 1.9 Hz, 1H), 7.40–7.22 (m, 6H), 3.87 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 171.1, 161.5, 151.6, 134.8, 132.4, 129.7, 128.8, 127.3, 125.8 (q, J = 36.1 Hz), 124.9 (q, J = 267.0 Hz), 123.3 (q, J = 3.3 Hz), 121.3, 120.3 (q, J = 3.8 Hz), 42.2. HPLC: purity >99%. m/z (ES) 336 [M + H]. Anal. (C16H11F3N2OS) C, H, N, S.
2-(3,4-Dichlorophenyl)-N-(6-(trifluoromethyl)benzothiazol-2-yl)acetamide (47)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol), 2-(3,4-dichlorophenyl)acetyl chloride (1.1 mmol), and THF (0.4 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:1) to afford a white solid (68 mg, 65%), mp 163–165 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.85 (s, 1H), 8.49 (s, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.64 (d, J = 1.8 Hz, 1H), 7.61 (d, J = 8.3 Hz, 1H), 7.34 (dd, J = 8.3, 1.9 Hz, 1H), 3.92 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 170.0, 161.0, 151.3, 135.4, 132.0, 131.7, 130.8, 130.4, 130.1, 129.7, 124.5 (q, J = 272.0 Hz), 123.8 (q, J = 31.8 Hz), 122.9 (q, J = 3.4 Hz), 121.0, 119.9 (q, J = 4.1 Hz), 40.5. HPLC: purity 97%. m/z (ES) 406 [M + H]. Anal. (C16H9F3Cl2N2OS) C, H, N, S.
2-(3,4-Dichlorophenyl)-N-(6-methoxybenzothiazol-2-yl)acetamide (48)
Reagents: 6-methoxybenzothiazol-2-amine (1.4 mmol), 2-(3,4-dichlorophenyl)acetyl chloride (1.4 mmol), and THF (0.4 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a beige solid (100.0 mg, 20%), mp 198–199 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.47 (s, 1H), 7.64–7.58 (m, 3H), 7.55 (d, J = 2.7 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.01 (dd, J = 8.8, 2.7 Hz, 1H), 3.85 (s, 2H), 3.78 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.8, 156.9, 156.4, 143.3, 136.5, 133.4, 132.3, 131.5, 131.1, 130.7, 130.3, 121.9, 115.7, 105.4, 56.3, 41.2. HPLC: purity >99%. m/z (ES) 368 [M + H]. Anal. (C16H12Cl2N2O2S) C, H, N, S.
2-(3,4-Dichlorophenyl)-N-(6-ethoxybenzothiazol-2-yl)acetamide (49)
Reagents: 6-ethoxybenzothiazol-2-amine (1.3 mmol), 2-(3,4-dichloro-phenyl)acetyl chloride (1.3 mmol), and THF (0.4 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a brown solid (109.6 mg, 22%), mp 148–149 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.48 (s, 1H), 7.68–7.57 (m, 3H), 7.52 (d, J = 1.6 Hz, 1H), 7.32 (d, J = 7.6 Hz, 1H), 7.00 (d, J = 8.8 Hz, 1H), 4.04 (q, J = 6.9 Hz, 2H), 3.85 (s, 2H), 1.32 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.1, 155.6, 155.3, 142.4, 135.7, 132.7, 131.5, 130.7, 130.4, 129.9, 129.6, 121.1, 115.2, 105.3, 63.5, 40.4, 14.6. HPLC: purity >99%. m/z (ES) 381 [M]. Anal. (C17H14Cl2N2O2S) C, H, N, S.
2-(3,4-Dichlorophenyl)-N-(6-(trifluoromethoxy)benzothiazol-2-yl)acetamide (50)
Reagents: 6-(trifluoromethoxy)benzothiazol-2-amine (1.1 mmol), 2-(3,4-dichlorophenyl)acetyl chloride (1.1 mmol), and THF (0.3 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (203.1 mg, 45%), mp 170–172 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.85 (s, 1H), 8.49 (s, 1H), 7.92 (d, J = 8.5 Hz, 1H), 7.78–7.70 (m, 1H), 7.40–7.22 (m, 6H), 3.87 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 171.4, 162.0, 148.1, 143.5, 136.7, 132.9, 131.5, 130.6, 130.3, 130.0, 129.3, 121.9, 120.2 (q, J = 255.9 Hz), 118.5, 114.6, 41.7. HPLC: purity >99%. m/z (ES) 421 [M]. Anal. (C16H9Cl2F3N2O2S) C, H, N, S.
N-(6-(Trifluoromethyl)benzothiazol-2-yl)-2-(2,5-dimethoxyphenyl)acetamide (51)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol) and 2-(2,5-dimethoxyphenyl)acetyl chloride (1.1 mmol). Reaction conditions: 7 min at 150 °C. The crude product was purified by IsoleraOne (CH2Cl2/MeOH 15:1) to afford a beige solid (141.7 mg, 31%), mp 146–147 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.26 (d, J = 24.7 Hz, 1H), 7.63 (dd, J = 32.9, 7.4 Hz, 2H), 6.82 (dd, J = 30.0, 9.1 Hz, 3H), 3.68 (s, 8H). 13C NMR (75 MHz, DMSO-d6): δ 174.0, 166.0, 152.9, 152.6, 151.5, 132.5, 128.6 (q, J = 272.1 Hz), 125.9, 121.9 (q, J = 2.25 Hz), 121.6 (q, J = 37.2 Hz), 119.2, 118.8 (q, J = 3.6 Hz), 117.4, 111.7, 56.0, 55.3, 38.4. HPLC: Purity >99%. m/z (ES) 397 [M + H]. Anal. (C18H15F3N2O3S) C, H, N, S.
N-(6-(Trifluoromethoxy)benzothiazol-2-yl)-2-(3,4,5-trimethoxyphenyl)acetamide (52)
Reagents: 6-(trifluoromethoxy)benzothiazol-2-amine (1.1 mmol), 2-(3,4,5-trimethoxyphenyl)acetyl chloride (1.1 mmol), and THF (1.5 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a brown solid (89.3 mg, 19%), mp 224–227 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.66 (s, 1H), 8.10 (d, J = 1.2 Hz, 1H), 7.81 (d, J = 8.8 Hz, 1H), 7.41 (ddd, J = 8.8, 2.4, 0.9 Hz, 1H), 6.67 (s, 2H), 3.76 (s, 8H), 3.62 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 170.4, 159.5, 152.8, 147.5, 144.1, 136.5, 132.6, 129.9, 121.8, 120.2 (q, J = 255.8 Hz), 118.5, 115.0, 106.8, 59.9, 55.8, 42.1. HPLC: purity >99%. m/z (ES) 443 [M + H]. Anal. (C19H17F3N2O5S) C, H, N, S.
N-(6-Methoxybenzothiazol-2-yl)-2-(3,4,5-trimethoxyphenyl)acetamide (53)
Reagents: 6-methoxybenzothiazol-2-amine (1.4 mmol), 2-(3,4,5-trimethoxyphenyl)acetyl chloride (1.4 mmol), and THF (0.4 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by column chromatography (AcOEt/hexane 1:1) to afford a white solid (44.2 mg, 8%), mp 251–252 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.39 (s, 1H), 7.62 (d, J = 8.8 Hz, 1H), 7.54 (d, J = 2.4 Hz, 1H), 7.01 (dd, J = 8.8, 2.6 Hz, 1H), 6.66 (s, 2H), 3.79 (s, 3H), 3.76 (s, 6H), 3.72 (s, 2H), 3.63 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.7, 156.1, 155.8, 152.7, 142.5, 136.4, 132.7, 130.1, 121.0, 114.8, 106.7, 104.6, 59.9, 55.8, 55.5, 42.0. HPLC: purity >99%. m/z (ES) 389 [M + H]. Anal. (C19H20N2O5S) C, H, N, S.
N-(6-Ethoxybenzothiazol-2-yl)-2-(3,4,5-trimethoxyphenyl)acetamide (54)
Reagents: 6-ethoxybenzothiazol-2-amine (1.3 mmol), 2-(3,4,5-trimethoxy-phenyl)acetyl chloride (1.3 mmol), and THF (0.4 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a gray solid (149.0 mg, 29%), mp 215–217 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.38 (s, 1H), 7.61 (d, J = 8.8 Hz, 1H), 7.52 (d, J = 2.4 Hz, 1H), 7.00 (dd, J = 8.8, 2.5 Hz, 1H), 6.66 (s, 2H), 4.04 (q, J = 6.9 Hz, 2H), 3.76 (s, 6H), 3.72 (s, 2H), 3.62 (s, 3H), 1.33 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 169.7, 155.8, 155.3, 152.7, 142.5, 136.5, 132.7, 130.2, 121.1, 115.2, 106.6, 105.3, 63.6, 59.9, 55.8, 42.1, 14.7. HPLC: purity >99%. m/z (ES) 403 [M + H]. Anal. (C20H22N2O5S) C, H, N, S.
N-(6-(Trifluoromethyl)benzothiazol-2-yl)-2-(3,4,5-trimethoxyphenyl)acetamide (55)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol), 2-(3,4,5-trimethoxyphenyl)acetyl chloride (1.1 mmol), and THF (0.4 mL). Reaction conditions: 10 min at 110 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (89.5 mg, 18%), mp 223–224 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.80 (s, 1H), 8.49 (s, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.74 (d, J = 8.9 Hz, 1H), 6.69 (s, 2H), 3.79 (s, 2H), 3.77 (s, 6H), 3.64 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 171.0, 161.5, 153.1, 151.7, 136.9, 132.4, 130.2, 124.9 (q, J = 272.0 Hz), 124.1 (q, J = 31.8 Hz), 123.3 (q, J = 3.5 Hz), 121.3, 120.3 (q, J = 4.2 Hz), 107.2, 60.3, 56.2, 42.5. HPLC: purity 98%. m/z (ES) 427 [M + H]. Anal. (C19H17F3N2O4S) C, H, N, S.
2-Cyclohexyl-N-(6-(trifluoromethyl)benzothiazol-2-yl)acetamide (56)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol) and 2-cyclohexylacetyl chloride (1.1 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (208.3 mg, 53%), mp 187–189 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.57 (s, 1H), 8.47 (s, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.72 (d, J = 9.0 Hz, 1H), 2.39 (d, J = 7.1 Hz, 2H), 1.91–1.52 (m, 6H), 1.39–0.87 (m, 5H). 13C NMR (75 MHz, DMSO-d6): δ 172.7, 161.7, 152.0, 132.6, 125.3 (q, J = 271.8 Hz), 124.3 (q, J = 32.0 Hz), 123.6 (q, J = 3.5 Hz), 121.5, 120.5 (q, J = 4.3 Hz), 43.5, 35.2, 33.0, 26.8, 26.2. HPLC: purity >99%. m/z (ES) 342 [M + H]. Anal. (C16H17F3N2OS) C, H, N, S.
N-(6-(Trifluoromethyl)benzothiazol-2-yl)-3,3-dimethylbutanamide (57)
Reagents: 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol) and 3,3-dimethylbutanoyl chloride (1.1 mmol). Reaction conditions: 5 min at 150 °C. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (146.4 mg, 40%), mp 189–191 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.50 (s, 1H), 8.47 (s, 1H), 7.88 (d, J = 8.5 Hz, 1H), 7.72 (dd, J = 8.6, 1.6 Hz, 1H), 2.40 (s, 2H), 1.02 (s, 9H). 13C NMR (75 MHz, DMSO-d6): δ 171.4, 160.9, 151.2, 131.9, 124.6 (q, J = 271.7 Hz), 123.6 (q, J = 31.9 Hz), 122.8 (q, J = 3.0 Hz), 120.8, 119.8 (q, J = 4.1 Hz), 48.0, 31.0, 29.4. HPLC: purity >99%. m/z (ES) 317 [M + H]. Anal. (C14H15F3N2OS) C, H, N, S.
General Procedure for 12, 14–15, and 45
A mixture of the corresponding acid derivative (1 equiv), the coupling reagent 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) (1.3 equiv), 4-dimethylaminopyridine (DMAP) (0.2 equiv), and triethylamine (TEA) (1 equiv) if necessary, in CH2Cl2 or DMF (20–40 mL) was stirred at room temperature for 1 h. Afterward, the amine derivative (1 equiv) was added and the reaction mixture was heated at the temperature and during the time indicated in each case. After cooling, the solvent was evaporated under reduced pressure. The residue was dissolved in AcOEt (50 mL) and extracted with HCl 0.1 M (50 mL × 3). Then the organic layer was washed with a saturated solution of NaHCO3 (50 mL × 3) and with a saturated solution of NaCl (50 mL × 3). Finally, the organic phase was dried over sodium sulfate and the solvent evaporated under reduced pressure. The residue was chromatographed as indicated in each case.
N-(Benzothiazol-2-yl)-2,2-diphenylacetamide (12)
Reagents: 2,2-diphenyl-acetic acid (1.7 mmol), EDC (2.2 mmol), DMAP (0.3 mmol), CH2Cl2 (40 mL), and benzothiazol-2-amine (1.7 mmol). Reaction conditions: 48 h at room temperature. The desired product was obtained directly after the extractions affording a white solid (417.9 mg, 73%), mp 212–213 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.83 (s, 1H), 7.98 (d, J = 7.4 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.57–7.08 (m, 12H), 5.42 (s, 1H). 13C NMR (75 MHz, DMSO-d6): δ 171.4, 158.2, 148.8, 139.2, 131.8, 128.9, 127.6, 126.6, 124.1, 122.1, 121.0, 56.6. HPLC: purity 97%. m/z (ES) 345 [M + H]. Anal. (C21H16N2OS) C, H, N, S.
N-(Benzothiazol-2-yl)-3-phenylpropanamide (14)
Reagents: 3-(3-chloro-phenyl)propanoic acid (1.7 mmol), EDC (2.2 mmol), DMAP (0.3 mmol), CH2Cl2 (40 mL), and benzothiazol-2-amine (1.7 mmol). Reaction conditions: 17 h at reflux temperature. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (166.2 mg, 32%), mp 158–156 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.37 (s, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.73 (d, J = 8.1 Hz, 1H), 7.48–7.39 (m, 1H), 7.36 (s, 1H), 7.34–7.19 (m, 4H), 3.02–2.90 (m, 2H), 2.88–2.77 (m, 2H). 13C NMR (75 MHz, DMSO-d6): δ 171.6, 158.1, 148.9, 143.7, 133.3, 131.8, 130.6, 128.6, 127.4, 126.5, 126.4, 123.8, 122.0, 120.8, 36.8, 30.1. HPLC: purity >99%. m/z (ES) 317 [M + H]. Anal. (C16H13ClN2OS) C, H, N, S.
N-(Benzothiazol-2-yl)-3,3-diphenylpropanamide (15)
Reagents: 3,3-diphenyl-propanoic acid (1.7 mmol), EDC (2.2 mmol), DMAP (0.3 mmol), CH2Cl2 (40 mL), and benzothiazol-2-amine (1.7 mmol). Reaction conditions: 17 h at reflux temperature. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a white solid (195.7 mg, 33%), mp 197–198 °C. 1H NMR (300 MHz, DMSO-d6): δ 12.43 (s, 1H), 7.92 (d, J = 7.9 Hz, 1H), 7.72 (d, J = 8.2 Hz, 1H), 7.42 (dd, J = 10.7, 4.6 Hz, 2H), 7.37–7.22 (m, 8H), 7.22–7.11 (m, 2H), 4.65 (t, J = 8.0 Hz, 1H), 3.31 (d, J = 8.1 Hz, 2H). 13C NMR (75 MHz, DMSO-d6): δ 170.7, 158.0, 148.8, 144.1, 131.7, 127.9, 126.7, 126.4, 123.9, 122.0, 120.8, 46.7, 41.3. HPLC: purity 97%. m/z (ES) 359 [M + H]. Anal. (C22H18N2OS) C, H, N, S.
N-(Benzothiazol-2-yl)-2-phenylacetamide (45)
Reagents: 2-phenylacetic acid (1.7 mmol), EDC (2.2 mmol), DMAP (0.3 mmol), DMF (20 mL), and benzothiazol-2-amine (1.7 mmol). Reaction conditions: 17 h at reflux temperature. The crude product was purified by IsoleraOne (AcOEt/hexane 1:1) to afford a yellow solid (159.1 mg, 34%), mp 162–164 °C (lit.41 160–161 °C). 1H NMR (300 MHz, DMSO-d6): δ 12.58 (s, 1H), 7.95 (d, J = 7.9 Hz, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 7.50–7.18 (m, 6H), 3.82 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 170.2, 157.9, 148.5, 134.6, 131.4, 129.3, 128.4, 126.9, 126.1, 123.5, 121.7, 120.5, 41.8. HPLC: purity >99%. m/z (ES) 269 [M + H]. Anal. (C15H12N2OS) C, H, N, S.
(Benzothiazol-2-yl)-2-(4-chlorophenyl)acetate (9)
To a solution of benzothiazol-2-ol (1.2 mmol) and pyridine (4.6 mmol) in anhydrous CH2Cl2 (17 mL), 2-(4-chlorophenyl)acetyl chloride (1.2 mmol) was added drop by drop at room temperature. The reaction was heated vigorously under reflux for 6 h. To get reaction to completion, a catalytic amount of DMAP was added and stirred overnight at room temperature. Afterward, the mixture was extracted with a 0.1 M solution of HCl (50 mL × 3). The organic phase was dried over sodium sulfate and the solvent evaporated under reduced pressure. The residue was purified by column chromatography using AcOEt/hexane (1:2) as eluent to afford a white solid (74.1 mg, 21%), mp 137–139 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.15 (dd, J = 7.8, 1.8 Hz, 1H), 7.72 (dd, J = 7.1, 2.1 Hz, 1H), 7.49–7.19 (m, 6H), 4.42 (s, 2H). 13C NMR (75 MHz, DMSO-d6): δ 172.3, 169.9, 136.3, 134.1, 131.3, 128.1, 126.4, 123.2, 122.7, 122.6, 111.4, 39.8. m/z (ES) 304 [M + H]. Anal. (C15H10ClNO2S) C, H, N, S.
N-(2-Methoxyphenethyl)-6-(trifluoromethyl)benzothiazol-2-amine (11)
A solution of 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol) and potassium carbonate (1.1 mmol) in anhydrous DMF (20 mL) was heated vigorously under reflux for 30 min. Afterward, 1-(2-bromoethyl)-2-methoxybenzene (1.1 mmol) was added and the reaction was heated under reflux overnight. After cooling, the solvent was evaporated under reduced pressure. The residue was dissolved in AcOEt (50 mL) and extracted with HCl 0.1 M (50 mL × 3). The organic phase was dried over sodium sulfate and the solvent evaporated under reduced pressure. The residue was purified by column chromatography using AcOEt/hexane (1:2) as eluent to afford a beige solid (169.0 mg, 44%), mp 158–160 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.45 (t, J = 5.3 Hz, 1H), 8.10 (s, 1H), 7.51 (s, 2H), 7.26–7.11 (m, 2H), 6.97 (d, J = 7.9 Hz, 1H), 6.87 (t, J = 7.4 Hz, 1H), 3.79 (s, 3H), 3.55 (c, J = 6.7 Hz, 2H), 2.90 (t, J = 7.2 Hz, 2H). 13C NMR (75 MHz, DMSO-d6): δ 169.0, 157.6, 155.9, 131.2, 130.4, 128.1, 127.0, 125.1 (q, J = 271.3 Hz), 123.0 (q, J = 3.7 Hz), 121.2 (q, J = 31.8 Hz), 120.6, 118.8 (q, J = 4.1 Hz), 118.0, 111.0, 55.6, 44.3, 29.8. HPLC: purity >99%. m/z (ES) 353 [M + H]. Anal. (C17H15F3N2OS) C, H, N, S.
1-(4-Methoxyphenyl)-3-(6-(trifluoromethyl)benzothiazol-2-yl)urea (16)
A solution of 6-(trifluoromethyl)benzothiazol-2-amine (1.1 mmol) and 1-isocyanate-4-methoxybenzene (1.1 mmol) in THF (0.4 mL) was heated under microwave irradiation (1 h, 110 °C). After cooling to room temperature, 50 mL of CH2Cl2 were added and the mixture was extracted with a saturated solution of NaCl (50 mL × 3). The organic phase was dried over sodium sulfate and the solvent evaporated under reduced pressure. The residue was purified by column chromatography using AcOEt/hexane (1:3) as eluent to afford a white solid (30.0 mg, 7%), mp 194–196 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.97 (s, 1H), 8.99 (s, 1H), 8.39 (s, 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.72–7.62 (m, 1H), 7.41 (d, J = 8.9 Hz, 2H), 6.91 (d, J = 9.0 Hz, 2H), 3.72 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 162.6, 160.8, 155.4, 151.8, 132.3, 131.1, 125.3 (q, J = 39.6 Hz), 124.8 (q, J = 242.6 Hz), 122.8 (q, J = 2.7 Hz), 120.9, 119.5 (q, J = 4.2 Hz), 119.5, 114.1, 55.2. HPLC: purity >99%. m/z (ES) 368 [M + H]. Anal. (C16H12F3N3O2S) C, H, N, S.
Molecular Modeling
Ligand Preparation
Molecular modeling of the compounds (12, 20, 24, and 34) was performed using Spartan 10 V.1.1.0 (Wave function Inc. Irvine, CA, 2000) software in order to obtain the most favorable conformations for each ligand. Structures were minimized in vacuum with mechanical molecular force field MMFF.42 Then the lower energy compounds were submitted an equilibrium geometry calculation using a semiempirical PM6 method.43 Optimized structures models were used to the docking studies.
Docking Studies
Initial protein structures were set up using Sybyl 8.0 software44 adding hydrogens, removing ligands, cofactors, waters, and capping N and C-terminal residues and finally removing clashes and amide bumps. To carry out blind docking experiments, the resulting proteins structures were used with Autodock Vina software.45,46 To this end, grids of points covering the whole protein were generated with Box size: 126 × 126 × 126 points with a standard space of 0.375 Å. The center of the box is the protein center. The resulting docking solutions were clustered, and only the lower energy binding pose in each protein structure was considered for further analysis. Visual inspection of solutions for all ligands in all structures led us to select only the solutions obtained for structure 3UYS, which provided a common binding mode for all ligands compatible with the substrate binding site. Finally, the most interesting complexes were minimized with MMFF94 force field implemented in Sybyl 8.0 until a 0.01 kcal/mol gradient was reached in order to carry out a detailed analysis.
Biology
CK-1δ Assay Protocol
The “Kinase-Glo” Kit from Promega was used to screen compounds for activity against CK-1δ. Kinase-Glo assays were performed in assay buffer using black 96-well plates. In a typical assay, 10 μL of test compound (dissolved in dimethyl sulfoxide [DMSO] at 1 mM concentration and diluted in advance in assay buffer to the desired concentration) and 10 μL (16 ng) of enzyme were added to each well followed by 20 μL of assay buffer containing 0.1% casein as substrate and 4 μM ATP. The final DMSO concentration in the reaction mixture did not exceed 1%. After 60 min incubation at 30 °C, the enzymatic reaction was stopped with 40 μL of Kinase-Glo reagent. Glow-type luminescence was recorded after 10 min using a FLUOstar Optima (BMG Labtechnologies GmbH, Offenburg, Germany) multimode reader. The activity is proportional to the difference of the total and consumed ATP. The inhibitory activities were calculated on the basis of maximal activities measured in the absence of inhibitor. The IC50 was defined as the concentration of each compound that reduces a 50% the enzymatic activity with respect to that without inhibitors.
Kinetic Studies on CK-1δ
To investigate the inhibitory mechanism of ATP competitivity of benzothiazoles on CK-1δ, several kinetic experiments were performed. Lineweaver–Burk plots of enzyme kinetics are shown in Figure 5. Kinetic experiments varying both ATP (from 1 to 50 μM), compound used as control IC261 (from 0.25 to 0.5 μM), and benzothiazoles 20, 24, and 34 (from 0.025 to 0.05 μM) concentrations were performed using the ADP-Glo Kinase Assay.47
Cell Cultures Assays
HEK293 cells (ATCC, Manassas, VA) were cultured in 6-well dishes under standard culture conditions in Dulbecco Modified Eagle Medium (DMEM), 10% defined fetal bovine serum (FBS), and penicillin (50 IU/mL)–streptomycin (50 mg/mL). CK-1δ inhibitors were diluted in dimethyl sulfoxide (DMSO), and 8 μL of inhibitor + DMSO or DMSO alone (control) were added to cells at the indicated concentrations. After 1.5 h of exposure to the CK-1δ inhibitor alone, ethacrynic acid was added at a final concentration of 150 μM and cells were incubated for 4 h to induce phosphorylation of endogenous TDP-43. Cells were harvested, washed in phosphate buffered saline, and snap frozen prior to preparation for immunoblot. Cell lysates were loaded and resolved on precast 4–15% gradient sodium dodecyl sulfate polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride membrane as recommended by the manufacturer (Bio-Rad, Hercules, CA). On immunoblots, phosphorylated human TDP-43 (pS409/410) was detected with a monoclonal antibody (TIP-PTD-M01, Cosmo Bio, Carlsbad, CA). Actin (load control) was detected with a monoclonal antibody (A4700, Sigma-Aldrich, St. Louis, MO).
In Vitro Parallel Artificial Membrane Permeability Assay (PAMPA)
Ten commercial drugs, phosphate buffer saline solution at pH 7.4 (PBS), ethanol, and dodecane were purchased from Sigma, Acros Organics, Merck, Aldrich, and Fluka. The porcine polar brain lipid (PBL) (catalogue no. 141101) was from Avanti Polar Lipids. The donor plate was a 96-well filtrate plate (Multiscreen IP Sterile Plate PDVF membrane, pore size 0.45 μM, catalogue no. MAIPS4510) and the acceptor plate was an indented 96-well plate (Multiscreen, catalogue no. MAMCS9610), both from Millipore. Filter PDVF membrane units (diameter 30 mm, pore size 0.45 μm) from Symta were used to filtered the samples. A 96-well plate UV reader (Thermoscientific, Multiskan spectrum) was used for the UV measurements. Test compounds [(3–5 mg of caffeine, enoxacine, hydrocortisone, desipramine, ofloxacine, piroxicam, and testosterone), (12 mg of promazine), and 25 mg of verapamil and atenolol] were dissolved in EtOH (1000 μL). Then 100 μL of this compound stock solution was taken and 1400 μL of EtOH and 3500 μL of PBS pH 7.4 buffer were added to reach 30% of EtOH concentration in the experiment. These solutions were filtered. The acceptor 96-well microplate was filled with 180 μL of PBS/EtOH (70/30). The donor 96-well plate was coated with 4 μL of porcine brain lipid in dodecane (20 mg mL–1), and after 5 min, 180 μL of each compound solution was added. Then 1–2 mg of every compound to be determined their ability to pass the blood–brain barrier were dissolved in 1500 μL of EtOH and 3500 μL of PBS pH = 7.4 buffer, filtered, and then added to the donor 96-well plate. Then the donor plate was carefully put on the acceptor plate to form a “sandwich”, which was left undisturbed for 2 h and 30 min at 25 °C. During this time the compounds diffused from the donor plate through the brain lipid membrane into the acceptor plate. After incubation, the donor plate was removed. UV plate reader determined the concentration of compounds and commercial drugs in the acceptor and the donor wells. Every sample was analyzed at three to five wavelengths, in three wells and in two independent runs. Results are given as the mean [standard deviation (SD)], and the average of the two runs is reported. Ten quality control compounds (previously mentioned) of known BBB permeability were included in each experiment to validate the analysis set.
Drosophila Strains and Evaluation of Lifespan
To generate the UAS-hTDP-43 transgenic line, PCR-amplification of human TDP-43 cDNA was achieved with the primers 5′-CGCAGGGCCGGACGGGCCCAAAATGTCTGAATATATTCGGGTAACCG-3′ and 5′-CGCAGGGCCCCAGTGGCC CTACATTCCCCAGCCAGAAGACTTAGAATCC-3′. The PCR product was sub cloned into the pUASTattBSfiI vector as a Sfi fragment and then sequenced. The pUASTattBSfiI, kindly provided by Dr. Hervé Tricoire, had been generated by subcloning the SfiI cassette (described in Lasbleiz et al., 2005)48 into the pUASTattB vector digested with EcoRI/BglII. Transgenic strain was generated by BestGene Inc. (Chino Hills, CA, USA), according to standard methods, using the y1M ZH-2A w*; M ZH-51C (cytological region 51C on the second chromosome, strain identifier at BestGene: 24482) as recipient strain. Drosophila were maintained on a 12:12 light/dark cycle on standard cornmeal–yeast agar medium at 25 °C. UAS-hTDP43 transgenic males were crossed with elav-GAL4-GeneSwitch (ElavGal4GS) females.40 Then 50:50 males and females (0–2 days old) of the progeny were collected (clusters of 25–30) into food tubes containing instant Drosophila medium (Carolina Biological Supply Company, Burlington, NC). Vials were changed on a 2–3 day cycle. After 7 days, flies were treated with 100 μg/mL RU486 (Mifepristone, Betapharma-Shanghai Co., Ltd., China) in the presence of 100 nM candidate drugs or vehicle (DMSO). When flies were fed with RU486, Gene-Switch was “On” and the UAS-transgene was expressed. Every 2–3 days, the flies were transferred to new tubes with fresh media and dead flies were counted. Survival curves were established using the Kaplan–Meier method, and differences between curves were assessed using a Log Rank Test (OASIS web application).49
Acknowledgments
This work was supported by grants from MINECO (SAF2012-37979-C03-01 to A.M.), Association Francaise contre les Myopathies (AFM-16169 to D.I.P.), the Department of Veterans Affairs (Merit Review grant no. 1147891 to B.K.) and CDA2 no. I01BX007080 to N.L.), Ligue Européenne Contre la Maladie d’Alzheimer (LECMA to M.L.), and National Institutes of Health (R01NS064131 to B.K.). I.G.S. and D.I.P. acknowledge a pre- and postdoctoral fellowship from MICINN (FPI program) and CSIC (JAE program), respectively. M.L.B. acknowledges a travel grant from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). We thank Elaine Loomis for outstanding technical assistance with immunoblotting
Glossary
Abbreviations Used
- ALS
amyotrophic lateral sclerosis
- TDP-43
Tar DNA binding protein 43
- SOD1
super oxide dismutase 1
- CK-1
casein kinase 1
- PDB
Protein Data Bank
- BBB
blood–brain barrier
- PAMPA
parallel artificial membrane permeability assay
- CNS
central nervous system
Supporting Information Available
Elemental analyses of all synthesized compounds, binding mode on CK-1δ for derivatives 24 and 34, kinases panel screening for compounds 20 and 24, and calibration test for BBB permeability determination. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Miller R. G.; Mitchell J. D.; Lyon M.; Moore D. H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2007, CD001447. [DOI] [PubMed] [Google Scholar]
- Robberecht W.; Philips T. The changing scene of amyotrophic lateral sclerosis. Nature Rev. Neurosci. 2013, 14, 248–264. [DOI] [PubMed] [Google Scholar]
- Regal L.; Vanopdenbosch L.; Tilkin P.; Van den Bosch L.; Thijs V.; Sciot R.; Robberecht W. The G93C mutation in superoxide dismutase 1: clinicopathologic phenotype and prognosis. Arch. Neurol. 2006, 63, 262–267. [DOI] [PubMed] [Google Scholar]
- Venkova-Hristova K.; Christov A.; Kamaluddin Z.; Kobalka P.; Hensley K.. Progress in therapy development for amyotrophic lateral sclerosis. Neurol. Res. Int. 2012, 2012, Article ID 187234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen D. R.; Siddique T.; Patterson D.; Figlewicz D. A.; Sapp P.; Hentati A.; Donaldson D.; Goto J.; O’Regan J. P.; Deng H. X.; Rahman Z.; Krizus A.; McKenna-Yasek D.; Cayabyab A.; Gaston S. M.; Berger R.; Tanzi R. E.; Halperin J. J.; Herzfeldt B.; Van Den Bergh R.; Hung W. Y.; Bird T.; Deng D.; Mulder D. W.; Smyth C.; Laing N. G.; Soriano E.; Pericak-Vance M. A.; Haines J.; Rouleau G. A.; Gusella J. S.; Horvitz H. R.; Brown R. H. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [DOI] [PubMed] [Google Scholar]
- Kabashi E.; Valdmanis P. N.; Dion P.; Spiegelman D.; McConkey B. J.; Vande Velde C.; Bouchard J. P.; Lacomblez L.; Pochigaeva K.; Salachas F.; Pradat P. F.; Camu W.; Meininger V.; Dupre N.; Rouleau G. A. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nature Genet. 2008, 40, 572–574. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M.; MacKenzie I. R.; Boeve B. F.; Boxer A. L.; Baker M.; Rutherford N. J.; Nicholson A. M.; Finch N. A.; Flynn H.; Adamson J.; Kouri N.; Wojtas A.; Sengdy P.; Hsiung G. Y.; Karydas A.; Seeley W. W.; Josephs K. A.; Coppola G.; Geschwind D. H.; Wszolek Z. K.; Feldman H.; Knopman D. S.; Petersen R. C.; Miller B. L.; Dickson D. W.; Boylan K. B.; Graff-Radford N. R.; Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton A. E.; Majounie E.; Waite A.; Simon-Sanchez J.; Rollinson S.; Gibbs J. R.; Schymick J. C.; Laaksovirta H.; van Swieten J. C.; Myllykangas L.; Kalimo H.; Paetau A.; Abramzon Y.; Remes A. M.; Kaganovich A.; Scholz S. W.; Duckworth J.; Ding J.; Harmer D. W.; Hernandez D. G.; Johnson J. O.; Mok K.; Ryten M.; Trabzuni D.; Guerreiro R. J.; Orrell R. W.; Neal J.; Murray A.; Pearson J.; Jansen I. E.; Sondervan D.; Seelaar H.; Blake D.; Young K.; Halliwell N.; Callister J. B.; Toulson G.; Richardson A.; Gerhard A.; Snowden J.; Mann D.; Neary D.; Nalls M. A.; Peuralinna T.; Jansson L.; Isoviita V. M.; Kaivorinne A. L.; Holtta-Vuori M.; Ikonen E.; Sulkava R.; Benatar M.; Wuu J.; Chio A.; Restagno G.; Borghero G.; Sabatelli M.; Consortium I.; Heckerman D.; Rogaeva E.; Zinman L.; Rothstein J. D.; Sendtner M.; Drepper C.; Eichler E. E.; Alkan C.; Abdullaev Z.; Pack S. D.; Dutra A.; Pak E.; Hardy J.; Singleton A.; Williams N. M.; Heutink P.; Pickering-Brown S.; Morris H. R.; Tienari P. J.; Traynor B. J. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M.; Sampathu D. M.; Kwong L. K.; Truax A. C.; Micsenyi M. C.; Chou T. T.; Bruce J.; Schuck T.; Grossman M.; Clark C. M.; McCluskey L. F.; Miller B. L.; Masliah E.; MacKenzie I. R.; Feldman H.; Feiden W.; Kretzschmar H. A.; Trojanowski J. Q.; Lee V. M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [DOI] [PubMed] [Google Scholar]
- Sreedharan J.; Blair I. P.; Tripathi V. B.; Hu X.; Vance C.; Rogelj B.; Ackerley S.; Durnall J. C.; Williams K. L.; Buratti E.; Baralle F.; de Belleroche J.; Mitchell J. D.; Leigh P. N.; Al-Chalabi A.; Miller C. C.; Nicholson G.; Shaw C. E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash P. E.; Zhang Y. J.; Roberts C. M.; Saldi T.; Hutter H.; Buratti E.; Petrucelli L.; Link C. D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010, 19, 3206–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Ray P.; Rao E. J.; Shi C.; Guo W.; Chen X.; Woodruff E. A. III; Fushimi K.; Wu J. Y. A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E.; Lin L.; Tradewell M. L.; Dion P. A.; Bercier V.; Bourgouin P.; Rochefort D.; Bel Hadj S.; Durham H. D.; Vande Velde C.; Rouleau G. A.; Drapeau P. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2010, 19, 671–683. [DOI] [PubMed] [Google Scholar]
- Wegorzewska I.; Bell S.; Cairns N. J.; Miller T. M.; Baloh R. H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18809–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Huang C.; Chen H.; Wang D.; Landel C. P.; Xia P. Y.; Bowser R.; Liu Y. J.; Xia X. G. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010, 6, e1000887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida A.; Sasaguri H.; Kimura N.; Tajiri M.; Ohkubo T.; Ono F.; Sakaue F.; Kanai K.; Hirai T.; Sano T.; Shibuya K.; Kobayashi M.; Yamamoto M.; Yokota S.; Kubodera T.; Tomori M.; Sakaki K.; Enomoto M.; Hirai Y.; Kumagai J.; Yasutomi Y.; Mochizuki H.; Kuwabara S.; Uchihara T.; Mizusawa H.; Yokota T. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain 2012, 135, 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilican B.; Serio A.; Barmada S. J.; Nishimura A. L.; Sullivan G. J.; Carrasco M.; Phatnani H. P.; Puddifoot C. A.; Story D.; Fletcher J.; Park I. H.; Friedman B. A.; Daley G. Q.; Wyllie D. J.; Hardingham G. E.; Wilmut I.; Finkbeiner S.; Maniatis T.; Shaw C. E.; Chandran S. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5803–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie I. R.; Rademakers R.; Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [DOI] [PubMed] [Google Scholar]
- Lee E. B.; Lee V. M.; Trojanowski J. Q. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nature Rev. Neurosci. 2012, 13, 38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M.; Kwong L. K.; Lee E. B.; Kremmer E.; Flatley A.; Xu Y.; Forman M. S.; Troost D.; Kretzschmar H. A.; Trojanowski J. Q.; Lee V. M. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009, 117, 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko N. F.; Guthrie C. R.; Kraemer B. C. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 2010, 30, 16208–16219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liachko N. F.; McMillan P. J.; Guthrie C. R.; Bird T. D.; Leverenz J. B.; Kraemer B. C. CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann. Neurol. 2013, 74, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choksi D. K.; Roy B.; Chatterjee S.; Yusuff T.; Bakhoum M. F.; Sengupta U.; Ambegaokar S.; Kayed R.; Jackson G. R. TDP-43 phosphorylation by casein kinase I{varepsilon} promotes oligomerization and enhances toxicity in vivo. Hum. Mol. Genet. 2013, 23, 1025–1035. [DOI] [PubMed] [Google Scholar]
- Hasegawa M.; Arai T.; Nonaka T.; Kametani F.; Yoshida M.; Hashizume Y.; Beach T. G.; Buratti E.; Baralle F.; Morita M.; Nakano I.; Oda T.; Tsuchiya K.; Akiyama H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J. H.; Zhang H.; Wagey R.; Krieger C.; Pelech S. L. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J. Neurochem. 2003, 85, 432–442. [DOI] [PubMed] [Google Scholar]
- Peters J. M.; McKay R. M.; McKay J. P.; Graff J. M. Casein kinase I transduces Wnt signals. Nature 1999, 401, 345–350. [DOI] [PubMed] [Google Scholar]
- Knippschild U.; Gocht A.; Wolff S.; Huber N.; Lohler J.; Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell. Signalling 2005, 17, 675–689. [DOI] [PubMed] [Google Scholar]
- Perez D. I.; Gil C.; Martinez A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 2011, 31, 924–954. [DOI] [PubMed] [Google Scholar]
- Kametani F.; Nonaka T.; Suzuki T.; Arai T.; Dohmae N.; Akiyama H.; Hasegawa M. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem. Biophys. Res. Commun. 2009, 382, 405–409. [DOI] [PubMed] [Google Scholar]
- Walsh D. P.; Chang Y. T. Chemical genetics. Chem. Rev. 2006, 106, 2476–2530. [DOI] [PubMed] [Google Scholar]
- Baki A.; Bielik A.; Molnar L.; Szendrei G.; Keseru G. M. A high throughput luminescent assay for glycogen synthase kinase-3beta inhibitors. Assay Drug Dev. Technol. 2007, 5, 75–83. [DOI] [PubMed] [Google Scholar]
- Mashhoon N.; DeMaggio A. J.; Tereshko V.; Bergmeier S. C.; Egli M.; Hoekstra M. F.; Kuret J. Crystal structure of a conformation-selective casein kinase-1 inhibitor. J. Biol. Chem. 2000, 275, 20052–20060. [DOI] [PubMed] [Google Scholar]
- Drewry D. H.; Bamborough P.; Schneider K.; Smith G. K.. The kinome and its impact on medicinal chemistry. In Kinase Drug Discovery; Ward R. A., Goldberg F. W., Eds.; Royal Society of Chemistry: Cambridge, UK, 2012; pp 1–53. [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. A quantitative analysis of kinase inhibitor selectivity. Nature Biotechnol. 2008, 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Iguchi Y.; Katsuno M.; Takagi S.; Ishigaki S.; Niwa J.; Hasegawa M.; Tanaka F.; Sobue G. Oxidative stress induced by glutathione depletion reproduces pathological modifications of TDP-43 linked to TDP-43 proteinopathies. Neurobiol. Dis. 2012, 45, 862–870. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Fan K.; McConnell O. J.; Carter G. T. High throughput artificial membrane permeability assay for blood–brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [DOI] [PubMed] [Google Scholar]
- Crivori P.; Cruciani G.; Carrupt P. A.; Testa B. Predicting blood–brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 2000, 43, 2204–2216. [DOI] [PubMed] [Google Scholar]
- Miguel L.; Frebourg T.; Campion D.; Lecourtois M. Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol. Dis. 2011, 41, 398–406. [DOI] [PubMed] [Google Scholar]
- Romano M.; Feiguin F.; Buratti E.. Drosophila Answers to TDP-43 Proteinopathies. J. Amino Acids 2012, 2012, Article ID 356081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder T.; Yoon K. S.; White B. H.; Keshishian H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 12596–12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemdan M. M.; Fahmy A. F.; Ali N. F.; Hegazi E.; Abd-Elhaleem A. Synthesis of some new heterocycles derived from phenylacetyl isothiocyanate. Chin. J. Chem. 2008, 26, 388–391. [Google Scholar]
- Halgren T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar]
- Stewart J. J. Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SYBYL 8.0; Tripos International: 1699 South Hanley Rd., St., Louis, MO 63144 USA. [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger D.; de Groot B. L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput.-Aided Mol. Des. 2010, 24, 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ADP-Glo Kinase Assay Technical Manual; http://www.promega.com/tbs/.
- Lasbleiz J.; Burgun A.; Marin F.; Rolland Y.; Duvauferrier R. Vertebral trabecular network analysis on CT images. J. Radiol. 2005, 86, 645–649. [DOI] [PubMed] [Google Scholar]
- Yang J. S.; Nam H. J.; Seo M.; Han S. K.; Choi Y.; Nam H. G.; Lee S. J.; Kim S. OASIS: online application for the survival analysis of lifespan assays performed in aging research. PLoS One 2011, 6, e23525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.