

Abstract
Normal hemostasis requires von Willebrand factor (VWF) to support platelet adhesion and aggregation at sites of vascular injury. VWF is a multimeric glycoprotein built from identical subunits that contain binding sites for both platelet glycoprotein receptors and collagen. The adhesive activity of VWF depends on the size of its multimers, which range from 500 to over 10 000 kDa. There is good evidence that the high-molecular-weight multimers (HMWM), which are 5000–10 000 kDa, are the most effective in supporting interaction with collagen and platelet receptors and in facilitating wound healing under conditions of shear stress. Thus, these HMWM of VWF are of particular clinical interest. The unusually large multimers of VWF are, under normal conditions, cleaved by the plasma metalloproteinase ADAMTS13 to smaller, less adhesive multimers. A reduction or lack of HMWM, owing to a multimerization defect of VWF or to an increased susceptibility of VWF for ADAMTS13, leads to a functionally impaired VWF and the particular type 2A of von Willebrand disease. This review considers the biology and function of VWF multimers with a particular focus on the characterization of HMWM – their production, storage, release, degradation, and role in normal physiology. Evidence from basic research and the study of clinical diseases and their management highlight a pivotal role for the HMWM of VWF in hemostasis.
Keywords: ADAMTS13, factor VIII, hemostasis, high-molecular-weight multimers, platelets, von Willebrand disease, von Willebrand factor
Introduction
Under physiological conditions, hemostatic balance is maintained through a complex interplay of procoagulant, anticoagulant, and fibrinolytic factors. A key constituent of the hemostatic system is von Willebrand factor (VWF). Extensive research in recent years has increased our understanding of the structure and function of VWF and the mechanisms underlying its involvement in normal hemostasis and pathological conditions associated with altered coagulation or thrombosis [1].
VWF is a large, highly adhesive, multimeric glycoprotein that is found predominantly in plasma and produced in endothelial cells and megakaryocytes, the precursors of platelets [2–4]. VWF is critical for hemostasis and thrombus formation; it acts as a bridging molecule for normal platelet adhesion and aggregation at sites of vascular injury [1,5]. In addition, VWF is a carrier molecule for procoagulant factor VIII (FVIII), thereby protecting FVIII from rapid clearance, and thus increasing its plasma half-life [6–9]. In this way, VWF is essential to both primary (platelet-mediated) and secondary (coagulation factor-mediated) hemostasis.
VWF is synthesized as a single pre-pro-polypeptide chain. After removal of the signal peptide in the endoplasmic reticulum, VWF is dimerized. VWF then undergoes further maturation in the trans Golgi apparatus and post-Golgi compartments where it is multimerized. The VWF newly synthesized in endothelial cells is either released directly into plasma via the constitutive pathway, or stored in the Weibel–Palade bodies with its propeptide in a 1:1 ratio [10]. Only VWF, and not its propeptide, has a role in platelet adhesion at the endothelial cell surface [11]. In contrast, VWF synthesized in megakaryocytes is stored in platelet α-granules until platelet activation and its subsequent release. This platelet-stored VWF has a high component of high-molecular-weight multimer (HMWM) forms [12]. The relative contribution of VWF from endothelial cells or platelets to hemostasis is currently a subject of investigation [13,14]. It is postulated that platelet-derived VWF can mediate hemostasis but is not required to do so under normal circumstances [12,14]. Indeed, recent evidence suggests that platelet-derived VWF might aggravate thromboinflammatory diseases such as stroke [14].
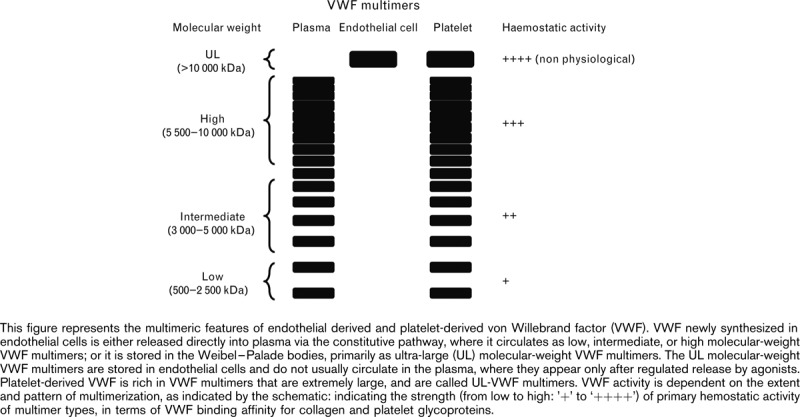
VWF exists in various sizes, referred to as VWF multimers, and include low (L), intermediate (I), high (H), and ultra-large (UL) molecular-weight forms. VWF stored in the Weibel–Palade bodies of endothelial cells or in the α-granules of megakaryocytes is rich in VWF multimers that are extremely large, and are called UL-VWF multimers, whereas the constitutively secreted VWF multimers are shorter, but still of high molecular weight [15–19]. The UL-VWF multimers do not typically circulate in the plasma because of rapid proteolysis that reduces them into smaller multimers soon after their secretion.
Of all the VWF multimers, the HMWM have the greatest activity in terms of hemostasis [binding capacity for collagen and the platelet receptors glycoprotein (GP) Ib and IIb/IIIa, and platelet aggregation under conditions of high fluid shear] [20–25]. Any abnormalities in either the quantity or quality of VWF multimers, particularly of the hemostatically highly active HMWM, can result in defective hemostasis. This article provides a contemporary review of the biology and function of VWF multimers, with a particular focus on the role of HMWM in the physiology of hemostasis.
Biology of von Willebrand factor: synthesis and size distribution of multimers
VWF is a large multimeric glycoprotein present in human plasma as a series of polymers called multimers, consisting of a variable number of subunits linked by disulfide bonds. The number of subunits in each multimer varies, with molecular weights ranging from around 500 kDa for the dimer to over 10 000 kDa for the HMWM (Table 1; Fig. 1) [26,27], thus forming the largest known protein present in human plasma [18]. Each multimeric subunit of VWF has binding sites for the receptor GPIb on nonactivated platelets and the receptor GPIIb/IIIa on activated platelets; this facilitates platelet adhesion and platelet aggregation, respectively, making the VWF HMWM important for normal platelet function.
Table 1.
Multimers of von Willebrand factor and their physiological characteristics
| Multimer | Number of multimers (dimers) | Size (kDa) | Primary distribution | Hemostatic function |
| Low [22] | 1–5 | 500–2500 | Circulating plasma | FVIII carrier only |
| Intermediate [22] | 6–10 | 3000–5000 | Circulating plasma | Low platelet binding affinity; FVIII carrier |
| High (large) [22] | 11–20 | 5500–10 000 | Circulating plasma | High platelet adhesion and aggregation; FVIII carrier |
| Ultra-large [21] | >20 | >10 000 | Uncleaved form of VWF stored in Weibel–Palade bodies and α-granules; rapidly cleaved once released from storage | Cleavage to smaller multimers that are characteristic of the circulating pool of VWF |
VWF, von Willebrand factor. Data from [26,27].
Fig. 1.
No captions available.
The human VWF gene is located at the tip of the short arm of chromosome 12 and consists of 178–180 kilobases and 52 exons [28–32]. The VWF gene encodes a large (240–260 kDa) precursor polypeptide (pre-pro-VWF) made up of a 22-amino-acid signal peptide, a 741-amino acid propeptide (also known as VWF antigen II), and a mature subunit (basic monomer) of 2050 amino acids, and up to 22 carbohydrate side chains [33].
Following its synthesis, precursor VWF is processed to a mature 220-kDa VWF subunit, or monomer. These VWF monomers undergo posttranslational modifications such as glycosylation, dimerization, and subsequently multimerization and propeptide cleavage, resulting in VWF peptides composed of up to 40 identical subunits, which make up the population of circulating VWF multimers, to over 10 000 kDa (ultra-large multimers) for the stored multimers (Fig. 1; Table 1) [26,27].
After synthesis, about 95% of endothelial VWF multimers (L, I, and HMWM) are constitutively secreted into the plasma, and the remainder (ultra-large multimers) are stored in cytoplasmic granules (Weibel–Palade bodies) or in the α-granules of megakaryocytes, as discussed above [15,17]. Understanding of the formation and function of Weibel–Palade bodies of endothelial cells and the controlled release of VWF have been recently reviewed elsewhere [34]. Dimers and the lower molecular-weight multimers of endothelial-derived VWF are not very efficient in initiating platelet adhesion to thrombogenic surfaces under physiological conditions. However, the HMWM are more effective in promoting platelet adhesion, particularly following vessel damage and subsequent high fluid shear stress [35–37].
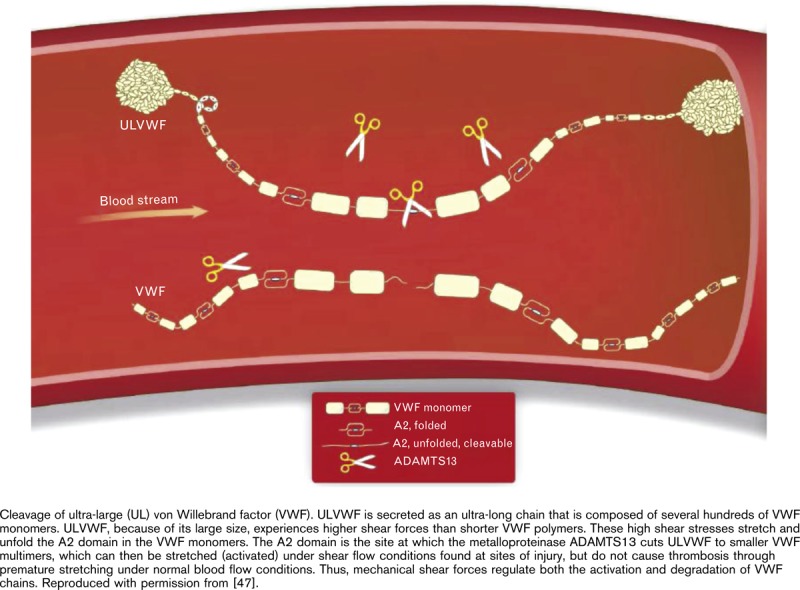
The largest multimeric forms of stored VWF (ultra-large multimers) can be secreted into the plasma via a regulated pathway following stimulation by specific secretagogues [15,18]. Various stimuli for release of these UL-VWF multimers have been identified through the study of cultured cells and platelets, and include exposure to physiological and pharmacological agents such as adrenaline, adenosine diphosphate, collagen, fibrin, histamine, thrombin, complement proteins, and the vasopressin analog desmopressin (DDAVP) [38–42].
UL-VWF multimers released from Weibel–Palade bodies form the strongest bonds to platelets and the extracellular matrix [15,35] (Fig. 1). After release of VWF from storage, some endothelial derived VWF multimers remain anchored to the surface of endothelial cells, forming string-like structures, which, under normal flow, elongate the VWF multimers from a globular to a string-like form, thereby exposing the cleavage site in the A2 domain to the metalloproteinase ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) [37,43,44]. Under conditions of high fluid shear stress, endothelial bound UL-VWF strings are cleaved multiple times by ADAMTS13 to shorter multimers that are still ultra-large in size, suggesting that VWF undergoes further ADAMTS13-mediated proteolysis in circulation [44–46]. This further downstream processing of UL-VWF multimers by ADAMTS13 results in VWF multimers of different sizes that are characteristic of the circulating pool of VWF, ranging from a single dimer to up to 20 dimers (∼10 000 kDa) (Fig. 2) [47–50].
Fig. 2.
No captions available.
The cleavage of VWF by ADAMTS13 is accelerated not only by high shear stress but also by FVIII, platelets, and GPIbα [44,51–53]. Furthermore, the size of multimeric VWF may be regulated by the trimeric glycoprotein thrombospondin-1 (TSP-1), which reduces the average multimer size [54], or by self-association of VWF, either with immobilized VWF or under fluid shear [55,56] or a static condition (interaction with biotinylated VWF in an ELISA system) [57], to form larger fibrillar VWF multimers. Recent evidence shows that, under very high shear stress, plasma VWF multimers undergo a conformational change from a native, inactive state, to a metastable, active state with an increased unfolding barrier, and hence, are harder to cleave by ADAMTS13 [58]. This change in conformation is possibly due to the lateral association of shear-induced VWF multimers into a fibrillar form [58]. In addition, the formation and elongation of UL-VWF multimers under flow conditions have been shown to involve the covalent lateral association of plasma VWF with endothelial-bound UL-VWF, an association that appears to rely on the thiol-disulfide state of UL-VWF [59]. Interestingly, some evidence suggests that ADAMTS13 reduces disulfide bond activity, thereby preventing the covalent lateral association and increased platelet adherence of plasma VWF multimers under high shear stress [60]. However, more recent research demonstrates that reduction of disulfide interactions is not sufficient to alter the hemostatic function of VWF [61]. In that study, increasing concentrations of ADAMTS13 reduced VWF multimer size, resulting in a significant loss of VWF hemostatic function and a rapid clearance of VWF from the circulation. Thus, VWF activity is regulated by ADAMTS13, which cleaves VWF into smaller, less functional VWF molecules [61].
An alternative mechanism for the elongation of UL-VWF multimers has been proposed [46]. Prior to cleavage by ADAMTS13, VWF strings undergo local elongation between adhered platelets on the endothelial surface [46]. The elongation occurs at different sites along the VWF strings and is independent of ADAMTS13, suggesting that this is a general characteristic of most VWF strings under fluid shear flow [46]. Some evidence from patients with subtypes of von Willebrand disease (VWD) suggests that platelet-derived VWF has the potential to play a role in hemostasis [62–64]. For example, patients with type 1 VWD show an inverse relationship between bleeding time and platelet VWF (P <0.001) [62]. Furthermore, a subgroup of patients with type 1 VWD and normal levels of platelet VWF were shown to have almost normal bleeding times in contrast to the prolonged bleeding times found in patients with low platelet VWF concentrations [63]. Normal collagen adhesion ex vivo of platelets derived from ‘platelet normal’ type 1 VWD patients has been observed, thus leading to a suggestion that platelet VWF may contribute to platelet adhesion when plasma VWF is low [64]. Finally, in patients with type 3 VWD, normal bleeding is not always restored following VWF replacement therapy, and thus platelet transfusion may be beneficial in such circumstance; further evidence pointing to a role for platelet-derived VWF in hemostasis [65].
The latest preclinical research also suggests that VWF produced in megakaryocytes/platelets can contribute to hemostasis [13]. Mice expressing VWF only in endothelial cells or only in platelets have been generated to explore this interesting aspect of the biology of VWF [13]. Mice with VWF only of platelet origin showed partially corrected bleeding times, although mice with VWF of endothelial cell origin showed normal duration of bleeding. This study demonstrates the potential ability of VWF released from platelets to contribute to hemostasis, at least in the absence of any endothelial cell-derived VWF [13].
Physiology of von Willebrand factor high-molecular-weight multimers: role in hemostasis and importance of multimer size
The sites of VWF synthesis and its storage offer clues to the role of this glycoprotein in primary hemostasis – a role closely linked to that of platelets [20]. Under normal physiological conditions, circulating multimeric VWF flows through the blood as a loosely coiled protein that does not interact with platelets or with endothelial cell lining, as the GPIb binding domain of VWF is concealed [37,66,67]. However, in response to vascular injury or high shear stress, VWF unwinds, exposing its binding sites within the VWF A1 domain responsible for interaction with platelet GPIbα [67,68].
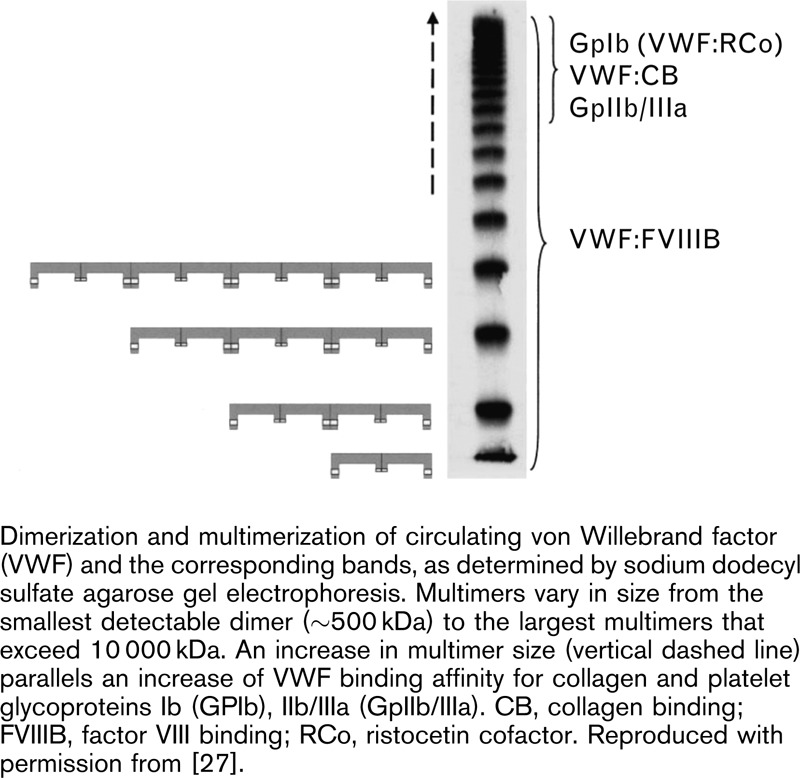
The hemostatic potential of VWF multimers is governed by the multimer size [69]. Studies have shown that as the size of VWF multimers progressively decreases, there is a concomitant loss of VWF function [70]. Specifically, the VWF collagen-binding capacity (VWF:CB) and the functional ability of VWF to bind platelets (the ristocetin cofactor activity; VWF:RCo) decrease as the multimer size of VWF decreases (Fig. 3) [27]. More recently, multimer analysis of a recombinant VWF suggests that there is also a grading of VWF:FVIII binding capacity (FVIIIB) according to multimer size, with a gradual decrease in FVIIIB demonstrated with lower molecular-weight multimers [71]. These effects may be because of fewer binding sites of the smaller multimers, and lower binding affinity between low VWF multimers and platelets, reducing both platelet adhesion and aggregation [72]. Conversely, the VWF HMWM are conformationally more responsive to shear stress, making it easier to unfold the long multimer string and expose their many GPIb binding sites.
Fig. 3.
No captions available.
It should also be noted that a recent cross-laboratory evaluation has been conducted to compare the sensitivity of VWF activity-based assays by exploring their results following progressive depletion of the VWF HMWM [73]. This study showed differences in the sensitivities of the respective tests used: VWF:CB and VWF:RCo assays had higher sensitivity to the loss of VWF HMWM than did the VWF:Act (activity) assay [73].
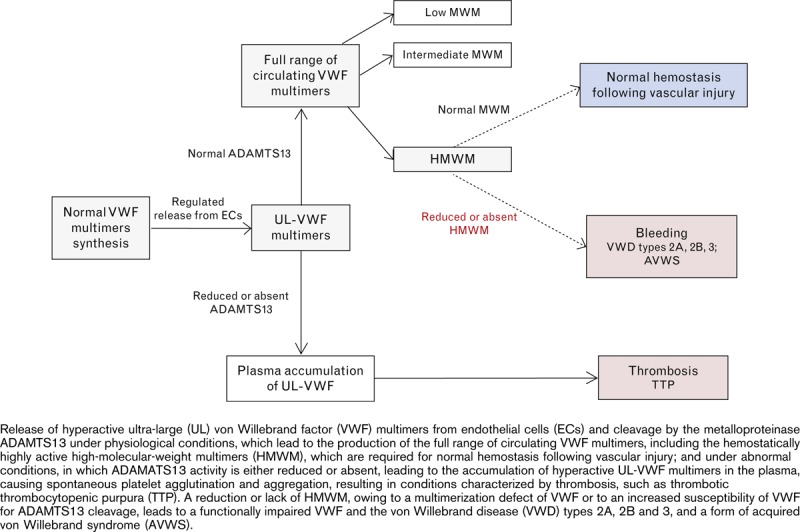
The regulation of VWF multimer size by ADAMTS13 is essential for normal hemostatic function, as evidenced by the pathological states that occur when proteolysis of VWF is defective (Fig. 4): in cases of excessive proteolysis of VWF by ADAMTS13, hemostasis is severely compromised because of the absence of VWF HMWM, resulting in the classical VWD type 2A; conversely, a deficiency of ADAMTS13 results in an abnormal accumulation of the multimers with the largest molecular weight, the UL-VWF multimers, which can cause spontaneous platelet aggregation, leading to the critical condition of thrombotic thrombocytopenic purpura (TTP) [74].
Fig. 4.
No captions available.
In addition to its role in primary hemostasis, VWF also has a role in secondary hemostasis, acting as a carrier protein for FVIII in the plasma. Formation of a VWF/FVIII complex both stabilizes and protects the coagulant activity of FVIII. For more detailed information on VWF/FVIII and its relevance to FVIII activity, readers are referred elsewhere [75,76].
Consequences of reduced or absent von Willebrand factor high-molecular-weight multimers
The importance of VWF HMWM in primary hemostasis is underlined by evidence of a bleeding diathesis when these larger multimers are lacking [5,77–80] (Fig. 4).
Pathophysiological conditions
Congenital von Willebrand disease
In the majority of cases, VWD is a congenital disease that is inherited in an autosomal dominant fashion, affecting men and women with almost the same frequency. The disease results from a qualitative or quantitative deficiency in VWF – with concentration, structure, or function of the larger VWF multimers being affected – and is characterized by an increased risk for bleeding [81,82]. Moreover, the absence of VWF leads to a secondary deficiency of FVIII, causing defects in platelet-plug and fibrin formation. Overall, these defects are reflected by the clinical manifestations of VWD, including excessive and prolonged bleeding following surgery or traumatic injury, mucosal tract hemorrhages such as epistaxis and menorrhagia, and, in more severe forms of the disease, hemophilia-like symptoms such as joint and muscle bleeding [83,84].
VWD has been classified into three major categories (Table 2) [85]: type 1, which is characterized by a partial quantitative deficiency of VWF and accordingly its functional properties; type 2 disease subtypes in which there are qualitative defects affecting VWF function; and type 3 disease in which VWF is totally deficient (Fig. 5) [86]. The distinction between the primary categories of VWF can usually be made by plasma assays for functional activity of VWF, namely, measurement of VWF:RCo/VWF-GPIb-binding and VWF:CB; quantitative assay of VWF antigen (VWF:Ag) and FVIII levels; and qualitative assays of multimers. Specifically, laboratory testing for VWF seeks to distinguish between the disease types with normal or only subtly abnormal multimer distributions (types 1, 2M, and 2N), those characterized by a significant decrease in the proportion of HMWM (types 2A and 2B), and those characterized by a loss of all multimers (type 3) [81,87].
Table 2.
Classification of von Willebrand disease
| Type | Description | Bleeding propensity |
| Type 1 | Partial quantitative deficiency of VWF (structure and distribution of plasma VWF multimers indistinguishable from normal) | Mild-to-moderate |
| Type 2 | Qualitative defects | Variable (usually moderate) |
| 2A | Decreased VWF-dependent platelet adhesion with selective deficiency of HMWM (either from defective multimer assembly or increased sensitivity to ADAMTS13 cleavage) | |
| 2B | Increased affinity for platelet GPIb (due to enhanced interaction of mutant VWF with platelet GPIb) | |
| 2M | Decreased VWF-dependent platelet adhesion without selective deficiency of HMWM, despite normal VWF multimer assembly (results from mutations that disrupt VWF binding to platelets or subendothelium) | |
| 2N | Markedly decreased binding affinity for FVIII (due to mutations that impair FVIII binding capacity) | |
| Type 3 | Virtually complete deficiency of VWF | High (severe bleeding) |
FVIII, factor VIII; HMWM, high-molecular-weight multimer; VWF, von Willebrand factor. Data from [26,81,85].
Fig. 5.
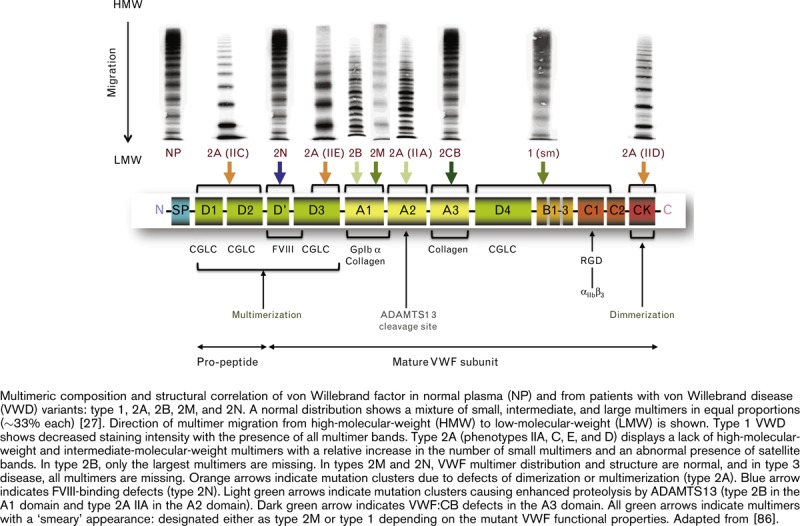
No captions available.
This article, with its focus on the VWF HMWM, reviews the VWD types that are characterized by abnormal VWF multimer profiles, specifically a reduction or absence of the HMWM of VWF, which impact the ability of VWF to bind to platelet GPIb [81,88].
Type 2 von Willebrand disease: qualitative von Willebrand factor defects
In most cases of type 2A VWD, there is a significant relative deficiency of VWF HMWM, which predisposes individuals to bleed. This deficiency of VWF HMWM may be because of impaired biosynthesis of large VWF multimers or an increased sensitivity of plasma VWF multimers to ADAMTS13 cleavage [89,90]. Recent evidence from a combination of molecular dynamic simulations and cleavage experiments suggests that type 2A mutations result from a destabilization of a region in the VWF A2 domain [91]. Such destabilization facilitates exposure of the cleavage site and increases susceptibility to ADAMTS13 cleavage. Type 2A can also be caused by a defective posttranslational processing that includes defects in VWF dimerization or defects in further polymerization of VWF dimers into multimers. The bleeding diathesis may persist even when plasma levels of endogenous VWF are raised by treatment with DDAVP, particularly in those with defects in VWF dimerization or multimerization [42]. Thus, the correction to normal levels of VWF protein is insufficient to support platelet adhesion and aggregation if the level of VWF HMWM is not restored. For these patients, replacement concentrates containing high quantities of VWF HMWM are the treatment of choice [27,87,92,93].
Recently, Haberichter et al. [94] have identified a potential novel pathogenic mechanism for type 2 VWD that involves a defective regulated storage and transportation of the larger multimers of VWF. A resulting loss of large multimers is reflected by decreased VWF–platelet interactions (low VWF:RCo) or low VWF–connective tissue interactions (low VWF:CB) relative to VWF:Ag (<0.6) [83,95].
The clinical expression of type 2A disease is mild-to-moderate mucocutaneous bleeding, which may manifest as easy bruising, epistaxis, prolonged bleeding after injury, during dental surgery, prolonged menses and menorrhagia in women and, in some cases, bleeding from gastrointestinal sites or in the central nervous system, which can be life-threatening [78]. In VWD characterized by reduced VWF HMWM, gastrointestinal bleeding due to angiodysplasia is well recognized, and indeed, such bleeding may relate to a form of acquired von Willebrand syndrome (AVWS) (see below) secondary to cardiovascular disease in some patients [96–99].
Type 2B VWD is characterized by a lack of VWF HMWM due to enhanced affinity of VWF for the GPIb receptor complex on platelets [100]. This enhanced affinity is caused by the presence of mutations in the A1 domain of VWF, resulting in pathological increases in platelet-VWF binding that lead to accelerated, proteolytic degradation by ADAMTS13 of large functional VWF multimers. Some, but not all patients with type 2B disease have a hallmark of thrombocytopenia, at least if it is exacerbated by surgery, pregnancy, or other stress factors such as severe infection [101]. VWF-containing FVIII concentrates are the treatment of choice for patients with type 2B disease, as DDAVP can induce transient thrombocytopenia [102].
Type 3 von Willebrand disease: complete deficiency of von Willebrand factor
In the most severe and rarest form of VWD, all VWF multimers are deficient, and FVIII levels are usually very low. The treatment of choice for this form of VWD is replacement by concentrates containing FVIII with high quantities of the functionally active VWF HMWM [27,87,89,90].
Type 3 VWD is characterized by prolonged and spontaneous bleeding from the nasal, oral, gastrointestinal, and genitourinary mucosa, hypermenorrhagia, and also by joint and muscle bleeding. Experimental studies in murine models of severe VWD clearly illustrate the functional importance of the higher molecular-weight VWF multimers in normal hemostasis [103,104]. In VWF-deficient mice, defects in hemostasis, including prolonged bleeding time and spontaneous bleeding events, are observed. In one study, treatment of VWF-/- mice by gene transfer of wild-type VWF cDNA was able to transiently correct the bleeding diathesis, coupled with a normalization of FVIII levels and the appearance of fully multimerized VWF [103]. In another study, in which the effect of liver-specific gene therapy was examined in a mouse model of severe VWD, treatment with transgenic murine VWF, which contained more than 47% of HMWM of normal murine plasma, restored in-vivo platelet adhesion and aggregation following vascular injury [104]. Moreover, liver-expressed VWF showed the full range of VWF multimers, including the HMWM, and restored FVIII plasma levels [104]. This study is purely a proof-of-concept study showing that VWF can be artificially synthesized in the liver; however, it does raise interesting questions about VWF and its relationship to the liver. Moreover, in the murine gene transfer models, an environment is created in which the liver hepatocytes are stimulated to synthesize VWF, an effect that is not reflective of the normal physiology. Further studies are, therefore, required to explore whether such an approach can be used to induce permanent VWF expression by the liver.
VWF is not synthesized or stored in the liver [105], yet reports have appeared of reversal or alteration of VWD following liver transplantation. For example, one woman experienced a modification of her VWD from type 3 to a moderately severe type 1 following liver transplantation [106]. The authors of that report suggested that the endothelium of the vascular tree transplanted with the liver may be producing sufficient levels of VWF to maintain a normal level of FVIII by protecting it from proteolysis, thereby reducing spontaneous bleeding and the need for replacement therapy. Liver transplantation may, therefore, confer secondary benefits to both hemophilia and VWD type 3 patients [106].
Acquired von Willebrand syndrome
AVWS is a rare bleeding disorder similar to VWD that occurs when there are deficiencies in VWF concentration, structure, or function as a result of acquired conditions, typically lymphoproliferative (e.g. chronic lymphocytic leukemia), myeloproliferative (e.g. thrombocythemia), cardiovascular (e.g. aortic stenosis), immunological (e.g. hypothyroidism) and, rarely autoimmune or neoplastic diseases (e.g. Wilms tumors), or with the use of certain treatments for disease (e.g. anticonvulsants) [98,107,108]. As such, AVWS occurs in patients with no relevant familial history of bleeding [109]. Because of the wide spectrum of underlying conditions that may cause AVWS, it has a heterogeneous clinical presentation, and the syndrome is often not recognized until patients are exposed to major trauma or surgery [103]. Multiple mechanisms have been proposed to explain the pathophysiology of this hemorrhagic condition among individuals with previously normal coagulation [109]. AVWS is characterized by a reduced pool of plasma VWF, where most, although not all forms of AVWS are associated with particular reduction in the HMWM of VWF [109]. Thus, in some forms of AVWS, the multimeric pattern of VWF resembles that of type 2A VWD with an absence or decreased proportion of larger VWF multimers [98,107].
In patients with AVWS and no cure for the underlying disease, treatment with DDAVP or factor concentrates rich in VWF HMWM can be considered a preferred option [110]. However, owing to the diversity of AVWS, different therapeutic options may need to be explored to determine which will provide the best response in individual cases: in many patients with lymphoproliferative disorders, VWF can be normalized for at least 1 week by infusion of high-dose immunoglobulins [111], and in others, by plasma exchange or immunoadsorption.
Consequences of elevated levels of ultra-large von Willebrand factor multimers
Accumulation of UL-VWF multimers, both in the plasma and on the surface of endothelial cells as a result of a deficiency or abnormality in ADAMTS13, can induce platelet aggregation and adhesion to the vascular endothelium. Such accumulation of UL-VWF multimers occurs in association with several conditions, such as thrombotic microangiopathies [74], inflammation and inflammatory responses [112–119], and severe malarial infection [120–122]. There is evidence that some clinical conditions involving microcirculatory thrombosis such as acute myocardial infarction [123], sepsis-induced disseminated intravascular coagulation [124], and diabetic nephropathy [125], may also be associated with increased levels of UL-VWF multimers, possibly related to a deficiency in ADAMTS13 activity.
Conclusion
VWF is an adhesive multimeric glycoprotein with an essential role in normal hemostasis. The multimeric structure of VWF is important, as the HMWM are most effective in supporting platelet adhesion and aggregation at sites of vascular injury and high shear stress, and the presence of these multimers and their hemostatic potential is closely controlled by processes governing the storage, release, and degradation of VWF. VWF has a key role in both physiological and pathophysiological conditions. Acquired or hereditary defects in the synthesis or processing of VWF HMWM result in a hemorrhagic diathesis, as seen in AVWS and type 2A VWD. In contrast, the persistence of UL-VWF due to hereditary or acquired deficiency of the VWF protease ADAMTS13 gives rise to conditions characterized by thromboembolic complications. In conditions in which VWF HMWM are deficient or lacking, replacement concentrates containing high quantities of these hemostatically active multimers are the most effective in restoring hemostasis and in protecting patients against bleeding risks associated with surgical intervention and injury.
Acknowledgements
The authors would like to thank Swiss Medical Press GmbH for editorial assistance.
Conflicts of interest
M.S. is an employee of CSL Behring GmbH. R.S. has received research funding from CSL Behring. U.B. has no conflicts of interest to declare.
References
- 1.Ruggeri ZM. The role of von Willebrand factor in thrombus formation. Thromb Res 2007; 120 Suppl:S5–S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruggeri ZM. Structure and function of von Willebrand factor. Thromb Haemost 1999; 82:576–584 [PubMed] [Google Scholar]
- 3.Jaffe EA, Hoyer LW, Nachman RL. Synthesis of antihemophilic factor antigen by cultured human endothelial cells. J Clin Invest 1973; 52:2757–2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nachman R, Levine R, Jaffe EA. Synthesis of factor VIII antigen by cultured guinea pig megakaryocytes. J Clin Invest 1977; 60:914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem 1998; 67:395–424 [DOI] [PubMed] [Google Scholar]
- 6.Weiss HJ, Sussman II, Hoyer LW. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand's disease. J Clin Invest 1977; 60:390–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoyer LW. The factor VIII complex: structure and function. Blood 1981; 58:1–13 [PubMed] [Google Scholar]
- 8.Rosenberg JB, Foster PA, Kaufman RJ, Vokac EA, Moussalli M, Kroner PA, et al. Intracellular trafficking of factorVIII to von Willebrand factor storage granules. J Clin Invest 1998; 101:613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nogami K, Shima M, Nishiya K, Hosokawa K, Saenko EL, Sakurai Y, et al. A novel mechanism of factor VIII protection by von Willebrand factor from activated protein C-catalyzed inactivation. Blood 2002; 99:3993–3998 [DOI] [PubMed] [Google Scholar]
- 10.Wagner DD, Fay PJ, Sporn LA, Sinha S, Lawrence SO, Marder VJ. Divergent fates of von Willebrand factor and its propolypeptide (von Willebrand antigen II) after secretion from endothelial cells. Proc Natl Acad Sci U S A 1987; 84:1955–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hannah MJ, Skehel P, Erent M, Knipe L, Ogden D, Carter T. Differential kinetics of cell surface loss of von Willebrand factor and its propolypeptide after secretion from Weibel-Palade bodies in living human endothelial cells. J Biol Chem 2005; 280:22827–22830 [DOI] [PubMed] [Google Scholar]
- 12.Blair P, Flaumenhaft R. Platelet α-granules: basic biology and clinical correlates. Blood Rev 2009; 23:177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanaji S, Fahs SA, Shi Q, Haberichter SL, Montgomery RR. Contribution of platelet versus endothelial VWF to platelet adhesion and hemostasis. Thromb Haemost 2012; 10:1646–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verhenne S, Libbrecht S, Vandenbulcke A, Deckmyn H, Vanhoorelbecke K, De Meyer SF. The role of platelet von Willebrand factor in mice. J Thromb Haemost 2013; 11 Suppl 2:14.231 [abstract OC 65.4] [Google Scholar]
- 15.Sporn LA, Marder VH, Wagner D. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell 1986; 46:185–190 [DOI] [PubMed] [Google Scholar]
- 16.Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol 1990; 6:217–246 [DOI] [PubMed] [Google Scholar]
- 17.Wagner DD, Saffaripour S, Bonfanti R, Sadler JE, Cramer EM, Chapman B, et al. Induction of specific storage organelles by von Willebrand factor propolypeptide. Cell 1991; 64:403–413 [DOI] [PubMed] [Google Scholar]
- 18.Ruggeri ZM, Ware J. von Willebrand factor. FASEB J 1993; 7:308–316 [DOI] [PubMed] [Google Scholar]
- 19.Tsai H-M, Nagel RL, Hatcher VB, Sussman II. Multimeric composition of endothelial cell-derived von Willebrand factor. Blood 1989; 73:2074–2076 [PubMed] [Google Scholar]
- 20.Reininger AJ. Function of von Willebrand factor in haemostasis and thrombosis. Haemophilia 2008; 14:11–16 [DOI] [PubMed] [Google Scholar]
- 21.Schneider SW, Nuschele S, Wixforth A, Gorzelanny C, Alexander-Katz A, Netz RR, et al. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci U S A 2007; 104:7899–7903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moake JL, Turner NA, Stathopoulos NA, Nolasco LH, Hellums JD. Involvement of large plasma von Willebrand factor (vWF) multimers and unusually large forms derived from endothelial cells in shear stress-induced platelet aggregation. J Clin Invest 1986; 789:1456–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Federici AB, Bader R, Pagani S, Colibretti ML, De Marco L, Mannucci PM. Binding of von Willebrand factor to glycoproteins Ib and IIb/IIIa complex: affinity is related to multimeric size. Br J Haematol 1989; 73:93–99 [DOI] [PubMed] [Google Scholar]
- 24.Fischer BE, Kramer G, Mitterer A, Grillberger L, Reiter M, Mundt W, et al. Effect of multimerization of human and recombinant von Willebrand factor on platelet aggregation, binding to collagen and binding of coagulation factor VIII. Thromb Res 1996; 84:55–66 [DOI] [PubMed] [Google Scholar]
- 25.Veyradier A, Jumilly AL, Ribba AS, Obert B, Houllier A, Meyer D, et al. New assay for measuring binding of platelet glycoprotein IIb/IIIa to unpurified von Willebrand factor. Thromb Haemost 1999; 82:134–139 [PubMed] [Google Scholar]
- 26.Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev 2010; 24:123–134 [DOI] [PubMed] [Google Scholar]
- 27.Budde U, Pieconka A, Will K, Schneppenheim R. Laboratory testing for von Willebrand disease: contribution of multimer analysis to diagnosis and classification. Semin Thromb Haemost 2006; 32:514–521 [DOI] [PubMed] [Google Scholar]
- 28.Ginsburg D, Handin RI, Bonthron DT, Donlon TA, Bruns GA, Latt SA, et al. Human von Willebrand factor (vWF): isolation of complementary DNA (cDNA) clones and chromosomal localization. Science 1985; 228:1401–1406 [DOI] [PubMed] [Google Scholar]
- 29.Sadler JE, Shelton-Inloes BB, Sorace JM, Harlan JM, Titani K, Davie EW. Cloning and characterization of two cDNAs coding for human von Willebrand factor. Proc Natl Acad Sci U S A 1985; 82:6394–6398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch DC, Zimmerman TS, Collins CJ, Brown M, Morin MJ, Ling EH, et al. Molecular cloning of cDNA for human von Willebrand factor: authentication by a new method. Cell 1985; 41:49–56 [DOI] [PubMed] [Google Scholar]
- 31.Verweij CL, de Vries CJ, Distel B, van Zonneveld AJ, van Kessel AG, van Mourik JA, et al. Construction of cDNA coding for human von Willebrand factor using antibody probes for colony-screening and mapping of the chromosomal gene. Nucleic Acids Res 1985; 13:4699–4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mancuso DJ, Tuley EA, Westfield LA, Worrall NK, Shelton-Inloes BB, Sorace JM, et al. Structure of the gene for human von Willebrand factor. J Biol Chem 1989; 264:19514–19527 [PubMed] [Google Scholar]
- 33.Titani K, Kumar S, Takio K, Ericsson LH, Wade RD, Ashida K, et al. Amino acid sequence of human von Willebrand factor. Biochemistry 1986; 25:3171–3184 [DOI] [PubMed] [Google Scholar]
- 34.Nightingale T, Cutler D. The secretion of von Willebrand factor from endothelial cells: an increasingly complicated story. J Thromb Haemost 2013; 11 Suppl 1:192–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sporn LA, Marder VH, Wagner D. von Willebrand factor released from Weibel-palade bodies binds more avidly to extracellular matrix than that secreted constitutively. Blood 1987; 69:1531–1534 [PubMed] [Google Scholar]
- 36.Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996; 84:289–297 [DOI] [PubMed] [Google Scholar]
- 37.Siediecki CA, Lestini BJ, Kottke-Marchant KK, Eppell SJ, Wilson DL, Marchant RE. Shear dependent changes in the three-dimensional structure of human von Willebrand factor. Blood 1996; 88:2939–2950 [PubMed] [Google Scholar]
- 38.Ribes JA, Francis CW, Wagner DD. Fibrin induces release of von Willebrand factor from endothelial cells. J Clin Invest 1987; 79:117–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamilton KK, Sims PJ. Changes in cytosolic Ca2+ associated with von Willebrand factor release in human endothelial cells exposed to histamine. Study of microcarrier cell monolayers using the fluorescent probe Indo-1. J Clin Invest 1987; 79:600–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine JD, Harlan JM, Harker LA, Joseph ML, Counts RB. Thrombin-mediated release of factor VIII antigen from human umbilical vein endothelial cells in culture. Blood 1982; 60:531–534 [PubMed] [Google Scholar]
- 41.Hattori R, Hamilton KK, McEver RP, Sims PJ. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem 1989; 264:9053–9060 [PubMed] [Google Scholar]
- 42.Ruggeri ZM, Mannucci PM, Lombardi R, Federici AB, Zimmerman TS. Multimeric composition of factor VIII/von Willebrand factor following administration of DDAVP: implications for pathophysiology and therapy of von Willebrand's disease subtypes. Blood 1982; 59:1272–1278 [PubMed] [Google Scholar]
- 43.Zheng XL. Structure-function and regulation of ADMTS-13 protease. J Thromb Haemost 2013; 11 Suppl 1:11–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001; 413:488–494 [DOI] [PubMed] [Google Scholar]
- 45.Jin S-Y, Skipwith CG, Shang D, Zheng XL. von Willebrand factor cleaved from endothelial cells by ADAMTS13 remains ultralarge in size. J Thromb Haemost 2009; 7:1749–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Ceunynck K, Rocha S, Feys HV, De Meyer SF, Uji-i H, Deckmyn H, et al. Local elongation of endothelial cell-anchored von Willebrand factor strings precedes ADAMTS13 protein-mediated proteolysis. J Biol Chem 2011; 286:36361–36367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christof J, Gebhardt M, Rief M. Force signaling in biology. Science 2009; 324:1278–1280 [DOI] [PubMed] [Google Scholar]
- 48.Tsai H-M, Sussman II, Nagel RL. Shear stress enhances the proteolysis of von Willebrand factor in normal plasma. Blood 1994; 83:2171–2179 [PubMed] [Google Scholar]
- 49.Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its confirmation and requires calcium ion. Blood 1996; 87:4235–4244 [PubMed] [Google Scholar]
- 50.Furlan M, Robles R, Affolter D, Meyer D, Baillod P, Lämmle B. Triplet structure of von Willebrand factor reflects proteolytic degradation of high molecular weight multimers. Proc Natl Acad Sci U S A 1993; 90:7503–7507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao W, Krishnaswamy S, Camire RM, Lenting PJ, Zheng XL. Factor VIII accelerates proteolytic cleavage of von Willebrand factor by ADAMTS13. Proc Natl Acad Sci U S A 2008; 105:7416–7421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shim K, Anderson PJ, Tuley EA, Wiswal E, Sadler JE. Platelet-VWF complexes are preferred substrates of ADAMTS13 under fluid shear stress. Blood 2008; 111:651–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Skipwith CG, Cao W, Zheng XL. Factor VIII and platelets synergistically accelerate cleavage of von Willebrand factor by ADAMTS13 under fluid shear stress. J Biol Chem 2010; 285:28596–285603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie L, Chesterman CN, Hogg PJ. Control of von Willebrand factor multimer size by thrombospondin-1. J Exp Med 2001; 193:1341–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Savage B, Sixma JJ, Ruggeri ZM. Functional self-association of von Willebrand factor during platelet adhesion under flow. Proc Natl Acad Sci U S A 2002; 99:425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shankaran H, Alexandridis P, Neelamegham S. Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension. Blood 2003; 101:2637–2645 [DOI] [PubMed] [Google Scholar]
- 57.Ulrichts H, Vanhoorelbeke K, Grima JP, Lenting PJ, Vauterin S, Deckmyn H. The von Willebrand factor self-association is modulated by a multiple domain interaction. J Thromb Haemost 2005; 3:552–561 [DOI] [PubMed] [Google Scholar]
- 58.Wijeratne SS, Botello E, Yeh H-C, Zhou Z, Bergeron A, Frey EW, et al. Mechanical activation of a multimeric adhesive protein through domain conformational change. Phys Rev Lett 2013; 110:108102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y, Choi Z, Nolasco L, Pownall HJ, Voorberg J, Moake JL, et al. Covalent regulation of ULVWF strong formation and elongation on endothelial cells under flow conditions. J Thromb Haemost 2008; 6:1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeh H-C, Zhou Z, Choi H, Tekeoglu S, Way W, III, Wang C, et al. Disulfilde bond reduction of von Willebrand factor by ADAMTS-13. J Thromb Haemost 2010; 8:2778–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shida Y, Brown C, Mewburn J, Sponagle K, Lillicrap D. Impact of ADAMTS13-mediated regulation of von Willebrand factor multimer profile on hemostasis and VWF clearance. J Thromb Haemost 2013; 11 Suppl 2:288.[abstract OC 91.4] [Google Scholar]
- 62.Gralnick HR, Rick ME, McKeown LP, Williams SB, Parker RI, Maisonneuve P, et al. Platelet von Willebrand factor: an important determinant of the bleeding time in type I von Willebrand's disease. Blood 1986; 68:58–61 [PubMed] [Google Scholar]
- 63.Mannucci PM, Lombardi R, Bader R, Vianello L, Federici AB, Solinas S, et al. Heterogeneity of type I von Willebrand disease: evidence for a subgroup with an abnormal von Willebrand factor. Blood 1985; 66:796–802 [PubMed] [Google Scholar]
- 64.d’Alessio P, Zwaginga JJ, de Boer HC, Federici AB, Rodeghiero F, Castaman G, et al. Platelet adhesion to collagen in subtypes of type I von Willebrand's disease is dependent on platelet von Willebrand factor. Thromb Haemost 1990; 64:227–231 [PubMed] [Google Scholar]
- 65.Castillo R, Escolar G, Monteagudo J, Aznar-Salatti J, Reverter JC, Ordinas A. Hemostasis in patients with severe von Willebrand disease improves after normal platelet transfusion and normalizes with further correction of the plasma defect. Transfusion 1997; 37:785–790 [DOI] [PubMed] [Google Scholar]
- 66.Ruggeri ZM. Mechanisms of shear-induced platelet adhesion and aggregation. Thromb Haemost 1993; 70:119–123 [PubMed] [Google Scholar]
- 67.Slayter H, Loscalzo J, Bockenstedt P, Handin RI. Native confirmation of human von Willebrand protein. J Biol Chem 1985; 14:8559–8563 [PubMed] [Google Scholar]
- 68.Auton M, Sowa KE, Behymer M, Cruz MA. N-terminal flanking region of A1 domain in von Willebrand factor stabilizes structure of A1A2A3 complex and modulates platelet activation under shear stress. J Biol Chem 2012; 287:14579–14585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Furlan M. Von Willebrand factor: molecular size and functional activity. Ann Hematol 1996; 72:341–348 [DOI] [PubMed] [Google Scholar]
- 70.Ohmori K, Fretto LJ, Harrison RL, Switzer MEP, Erickson HP, McKee PA. Electron microscopy of human factor VIII/von Willebrand glycoprotein: effect of reducing reagents on structure and function. J Cell Biol 1982; 95:632–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Turecek P, Schrenk G, Rottensteiner H, Varadi K, Bevers E, Lenting P, et al. Structure and function of a recombinant von Willebrand factor drug candidate. Semin Thromb Hemost 2010; 36:510–521 [DOI] [PubMed] [Google Scholar]
- 72.Gralnick HR, Williams SB, Morisato DK. Effect of the multimeric structure of the factor VIII/von Willebrand factor protein on binding to platelets. Blood 1981; 58:387–397 [PubMed] [Google Scholar]
- 73.Favaloro EJ, Bonar R, Chapman K, Meiring M, Funk Adcock D. Differential sensitivity of von Willebrand factor (VWF) ’activity’ assays to large and small VWF molecular weight forms: a cross-laboratory study comparing ristocetin cofactor, collagen-binding and mAb-based assays. J Thromb Haemost 2012; 10:1043–1054 [DOI] [PubMed] [Google Scholar]
- 74.Furlan M, Lämmle B. Haemolytic-uremic syndrome and thrombotic thrombocytopenic purpura: new insights into underlying biochemical mechanisms. Nephrol Dial Transplant 2000; 15:1112–1114 [DOI] [PubMed] [Google Scholar]
- 75.Federici AB. The factor VIII/von Willebrand factor complex: basic and clinical issues. Haematologica 2003; 88 Suppl 9:3–12 [PubMed] [Google Scholar]
- 76.Terraube V, O’Donnell JS, Jenkins PV. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia 2010; 16:3–13 [DOI] [PubMed] [Google Scholar]
- 77.Ruggeri ZM, Zimmerman TS. Variant von Willebrand's disease. Characterization of two subtypes by analysis of multimeric composition of factor VIII/von Willebrand factor in plasma and platelets. J Clin Invest 1980; 65:1318–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Budde U, Drewke E, Mainusch K, Schneppenheim R. Laboratory diagnosis of congenital von Willebrand disease. Semin Thromb Hemost 2002; 28:173–189 [DOI] [PubMed] [Google Scholar]
- 79.Zimmermann TS, Dent JA, Ruggeri ZM, Nannini LH. Subunit composition of plasma von Willebrand factor. J Clin Invest 1986; 77:947–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haselboeck J, Lambers M, Laszkovics C, Ay C, Manner D, Pabinger I, et al. Decreased ADAMTS13 activity levels in patients with a bleeding tendency of unknown origin. Blood 2011; 118:1218 [Google Scholar]
- 81.Sadler JE, Budde U, Eikenboom JCJ, Favaloro EJ, Hill FG, Holmberg L, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the subcommittee on von Willebrand factor. J Thromb Haemost 2006; 4:2103–2114 [DOI] [PubMed] [Google Scholar]
- 82.Federici AB. Classification of inherited von Willebrand disease and implications in clinical practice. Thromb Res 2009; 124 Suppl 1:S2–S6 [DOI] [PubMed] [Google Scholar]
- 83.Schneppenheim R. The evolving classification of von Willebrand disease. Blood Coagul Fibrinolysis 2005; 16 Suppl 1:S3–S10 [DOI] [PubMed] [Google Scholar]
- 84.Budde U. Diagnosis of von Willebrand disease subtypes: implications for treatment. Haemophilia 2008; 14 Suppl 5:27–38 [DOI] [PubMed] [Google Scholar]
- 85.National Heart, Lung, and Blood Institute. U.S. Department of Health and Human Services. National Institutes of Health (NIH) The diagnosis, evaluation, and management of von Willebrand disease. NIH Publication No. 08-5832, December 2007 [Google Scholar]
- 86.Budde U, Schneppenheim R. Michiels JJ. Laboratory diagnostic work-up and influence of DDAVP on VWF mutlimeric pattern: implication for the classification of VWD. Proceedings of the European workshop on von Willebrand factor and von Willebrand disease. Antwerp:University Press Antwerp; 2008. 48–54 [Google Scholar]
- 87.Schneppenheim R. The pathophysiology of von Willebrand disease: therapeutic implications. Thromb Res 2011; 128 Suppl 1:S3–S7 [DOI] [PubMed] [Google Scholar]
- 88.Lillicrap D. von Willebrand disease – phenotype versus genotype: deficiency versus disease. Thromb Res 2007; 120 Suppl 1:S11–S16 [DOI] [PubMed] [Google Scholar]
- 89.Hassenpflug WA, Budde U, Obser T, Angerhaus D, Drewke E, Schneppenheim S, et al. Impact of mutations in the von Willebrand factor A2 domain on ADAMTS13-dependent proteolysis. Blood 2006; 107:2339–2345 [DOI] [PubMed] [Google Scholar]
- 90.Tsai H-M, Sussmann II, Ginsburg D, Lankhof H, Sixma JJ, Nagel RL. Proteolytic cleavage of recombinant type 2A von Willebrand factor mutants R834W and R834Q: inhibition by doxycycline and by monoclonal antibody VP-1. Blood 1997; 89:1954–1962 [PubMed] [Google Scholar]
- 91.Interlandi G, Ling M, Tu AY, Chung DW, Thomas WE. Structural basis of type 2A von Willebrand disease investigated by molecular dynamics simulations and experiments. PLoS One 2012; 7:e45207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Federici AB, Mannucci PM. Optimizing therapy with factor VIII/von Willebrand factor concentrates in von Willebrand disease. Haemophilia 1998; 4 Suppl 3:7–10 [DOI] [PubMed] [Google Scholar]
- 93.Federici AB. Management of von Willebrand disease with factor VIII/von Willebrand factor concentrates: results from current studies and surveys. Blood Coagul Fibrinolysis 2006; 16 Suppl 1:S17–S21 [DOI] [PubMed] [Google Scholar]
- 94.Haberichter S, Budde U, Obser T, Schneppenheim S, Wernes C, Schneppenheim R. The mutation N528S in the von Willebrand factor (VWF) propeptide causes defective multimerization and storage of VWF. Blood 2010; 115:4580–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Favaloro EJ. Appropriate laboratory assessment as a critical facet in the proper diagnosis and classification of von Willebrand disease. Best Pract Res Clin Haematol 2001; 14:299–319 [DOI] [PubMed] [Google Scholar]
- 96.Makris M. Gastrointestinal bleeding in von Willebrand disease. Thromb Res 2006; 118 Suppl 1:S13–S17 [DOI] [PubMed] [Google Scholar]
- 97.Warkentin TE, Moore JC, Anand SS, Lonn EM, Morgan DG. Gastrointestinal bleeding, angiodysplasia, cardiovascular disease, and acquired von Willebrand disease. Transfus Med Rev 2003; 17:272–286 [DOI] [PubMed] [Google Scholar]
- 98.Federici AB, Rand JH, Bucciarelli P, Budde U, van Genderen PJ, Mohri H, et al. Acquired von Willebrand syndrome: data from an international registry. Thromb Haemost 2000; 84:345–349 [PubMed] [Google Scholar]
- 99.Mannucci PM. Treatment of von Willebrand's disease. N Engl J Med 2004; 351:683–694 [DOI] [PubMed] [Google Scholar]
- 100.Ruggeri ZM, Pareti FI, Mannucci PM, Ciavarella N, Zimmerman TS. Heightened interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von Willebrand's disease. N Engl J Med 1980; 302:1047–1051 [DOI] [PubMed] [Google Scholar]
- 101.Federici AB, Mannucci PM, Castaman G, Baronciani L, Bucciarelli P, Canciani MT, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood 2009; 113:526–534 [DOI] [PubMed] [Google Scholar]
- 102.Holmberg L, Nilsson IM, Borge L, Gunnarsson M, Sjörin E. Platelet aggregation induced by 1-desamino-8-D-arginine vasopressin (DDAVP) in type IIB von Willebrand disease. N Engl J Med 1983; 309:816–821 [DOI] [PubMed] [Google Scholar]
- 103.Marx I, Lenting PJ, Adler T, Pendu R, Christophe OD, Denis CV. Correction of bleeding symptoms in von Willebrand factor-deficient mice by liver-expressed von Willebrand factor mutants. Arteriscler Thromb Vasc Biol 2008; 28:419–424 [DOI] [PubMed] [Google Scholar]
- 104.De Meyer SF, Vandeputte N, Pareyn I, Petrus I, Lenting PJ, Chuah MKL, et al. Restoration of plasma von Willebrand factor deficiency is sufficient to correct thrombus formation after gene therapy for severe von Willebrand disease. Arterioscler Thromb Vasc Biol 2008; 28:1621–1626 [DOI] [PubMed] [Google Scholar]
- 105.Senzolo M, Burra P, Cholongitas E, Burroughs AK. New insights into the coagulopathy of liver disease and liver transplantation. World J Gastroenterol 2006; 12:7725–7736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schulman S, Ericzon BG, Eleborg L. Modification of von Willebrand disease after liver transplantation. Thromb Haemost 2001; 86:1588–1589 [PubMed] [Google Scholar]
- 107.Federici AB. Acquired von Willebrand syndrome: is it an extremely rare disorder or do we see only the tip of the iceberg? J Thromb Haemost 2008; 6:565–568 [DOI] [PubMed] [Google Scholar]
- 108.Tiede A, Priesack J, Werwitzke S, Bohlmann K, Oortwijn B, Lenting P, et al. Diagnostic workup of patients with acquired von Willebrand syndrome: a retrospective single-centre cohort study. J Thromb Haemost 2008; 6:569–576 [DOI] [PubMed] [Google Scholar]
- 109.Franchini M, Lippi G. Acquired von Willebrand syndrome: an update. Am J Hematol 2007; 82:368–375 [DOI] [PubMed] [Google Scholar]
- 110.Cuker A, Connors JM, Katz JT, Levy BD, Loscalzo J. A blood mystery. N Engl J Med 2009; 361:1887–1894 [DOI] [PubMed] [Google Scholar]
- 111.Michiels JJ, Berneman Z, Gadisseur A, van der Plancken M, Schroyens W, Budde U, van Vliet HH. Immune mediated etiology of acquired von Willebrand syndrome in systemic lupus erythematosus and in benign monoclonal gammopathy: therapeutic implications. Semin Thromb Hemost 2006; 32:577–588 [DOI] [PubMed] [Google Scholar]
- 112.Chauhan AK, Kisucka J, Brill A, Walsh MT, Scheiflinger F, Wagner DD. ADAMTS13: a new link between thrombosis and inflammation. J Exp Med 2008; 205:2065–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Claus RA, Bockmeyer CL, Sossdorf M, Losche W. The balance between von-Willebrand factor and its cleaving protease ADAMTS13: biomarker in systemic inflammation and development of organ failure? Curr Mol Med 2010; 10:236–248 [DOI] [PubMed] [Google Scholar]
- 114.Bockmeyer CL, Claus RA, Budde U, Kentouche K, Schneppenheim R, Lösche W, et al. Inflammation-associated ADAMTS13 deficiency promotes formation of ultra-large von Willebrand factor. Haematologica 2008; 93:137–140 [DOI] [PubMed] [Google Scholar]
- 115.Banno F, Chauhan AK. The function of ADAMTS13 in thrombogenesis in vivo: insights from mutant mice. Int J Hematol 2010; 91:30–35 [DOI] [PubMed] [Google Scholar]
- 116.Fu X, Chen J, Gallagher R, Zheng Y, Chung DW, Lopez JA. Shear-stress unfolding of VWF accelerates oxidation of key methionine residues in the A1A2A3 region. Blood 2011; 118:5283–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, et al. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood 2009; 114:3329–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fujioka M, Hayakawa K, Mishima K, Kunizawa A, Irie K, Higuchi S, et al. ADAMTS13 gene deletion aggravates ischemic brain damage: a possible neuroprotective role of ADAMTS13 by ameliorating postischemic hypoperfusion. Blood 2010; 115:1650–1653 [DOI] [PubMed] [Google Scholar]
- 119.De Meyer SF, Savchenko AS, Haas MS, Schatzberg D, Carroll MC, Schiviz A, et al. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood 2012; 120:5217–5223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Larkin D, de Laat B, Jenkins V, Bunn J, Craig AG, Terraube V, et al. Severe Plasmodium falciparum malaria is associated with circulating ultra-large von Willebrand factor multimers and ADAMTS13 inhibition. PLoS Pathog 2009; 5:e1000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bridges DJ, Bunn J, van Mourik JA, Grau G, Preston RJ, Molyneux M, et al. Rapid activation of endothelial cells enables Plasmodium falciparum adhesion to platelet-decorated von Willebrand factor strings. Blood 2010; 115:1472–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.López JA. Malignant malaria and microangiopathies: merging mechanisms. Blood 2010; 115:1317–1318 [DOI] [PubMed] [Google Scholar]
- 123.Sakai H, Goto S, Kim JY, Aoki N, Abe S, Ichikawa N, et al. Plasma concentration of von Willebrand factor in acute myocardial infarction. Thromb Haemost 2000; 84:204–209 [PubMed] [Google Scholar]
- 124.Ono T, Mimuro J, Madoiwa S, Soejima K, Kashiwakura Y, Ishiwata A, et al. Severe secondary deficiency of von Willebrand factor-cleaving protease (ADAMTS13) in patients with sepsis-induced disseminated intravascular coagulation: its correlation with development of renal failure. Blood 2006; 107:528–534 [DOI] [PubMed] [Google Scholar]
- 125.Taniguchi S, Hashiguchi T, Ono T, Takenouchi K, Nakayama K, Kawano T, et al. Association between reduced ADAMTS13 and diabetic nephropathy. Thromb Res 2010; 125:310–316 [DOI] [PubMed] [Google Scholar]