

Abstract
Bivalirudin is a direct thrombin inhibitor that is increasingly used in percutaneous coronary intervention (PCI) and has been previously shown to lack inherent platelet activation. Thrombin works through activation of protease activated receptor-1 (PAR1) and PAR4 on human platelets to initiate signaling cascades leading to platelet aggregation. Despite the increasing usage of bivalirudin, the effects on platelet function have not been well defined. Bivalirudin exposure during PCI was therefore assessed for its potential short-term effects on washed platelet function through PAR1 and PAR4. Bivalirudin significantly inhibited low-dose thrombin-mediated platelet aggregation, dense granule secretion, integrin αIIbβ3 activation and Rap1 activation and high dose thrombin-mediated dense granule secretion and Rap1 activation. Exposure to bivalirudin did not alter PAR1 or 4 agonist peptide (PAR1-AP or PAR4-AP) induced aggregation, dense granule secretion, integrin glycoprotein IIbIIIa activation or Rap1 activation. However, exposure to bivalirudin significantly potentiated surface expression of P-selectin following stimulation with high dose thrombin and PAR1-AP, and both low and high dose PAR4-AP. Hence, our data are the first to show that exposure to bivalirudin increased P-selectin expression with certain conditions demonstrating that bivalirudin can increase inherent platelet activity.
Keywords: Thrombin, PCI, Bivalirudin, PAR, P-Selectin
Introduction
Thrombin is the major protease in the coagulation cascade whose pleiotropic actions can ultimately lead to thrombosis and tissue injury. Further, thrombin displays a diverse range of effects in vascular cells that functionally connects tissue damage to both hemostatic and inflammatory responses [1–3]. Many of the cellular effects of thrombin are initiated via activation of the thrombin receptors expressed on the surface of the human platelet, a family of protease-activated receptors (PARs), which are coupled to heterotrimeric G proteins. Thrombin works through activation of PAR1 and PAR4 on human platelets to initiate signaling cascades leading to granule secretion, increases in [Ca]i, trafficking of adhesion molecules to the plasma membrane, and activation of integrins through the small G protein Rap1, all of which lead to subsequent platelet aggregation [4, 5].
Direct thrombin inhibitors, including bivalirudin, have proven to be of benefit in the treatment of patients with cardiovascular disease [6]. The use of bivalirudin is now supported by trials ranging from stable angina to patients presenting with ST segment elevation myocardial infarction (STEMI). The REPLACE-2 demonstrated the long-term efficacy of bivalirudin during PCI in a stable angina patient population [7]. The ACUITY and ISAR-REACT-4 trials have supported bivalirudin monotherapy in patients with diabetes mellitus and Unstable Angina (USA) and Non-ST segment myocardial infarction (NSTEMI) [8, 9]. The HORIZONS AMI trial examined bivalirudin versus heparin plus Glycoprotein (GP) IIbIIIa inhibitors in patients with STEMI [10]. In general, these studies demonstrated that bivalirudin was non-inferior to heparin and a glycoprotein IIbIIIa inhibitor in preventing ischemic events suggesting that bivalirudin provides antiplatelet effects beyond it proven anticoagulation effects. However, a sub-study of the ISAR-REACT-4 study, which stratified patients according to platelet reactivity, found that whereas high platelet reactivity showed similar results in efficacy in the heparin/abciximab arm, the incidence of ischemic events was significantly higher in the high platelet reactivity in the bivalirudin arm compared to the heparin/abciximab arm [11]. This potentially suggests that at least in patients with high platelet reactivity, in the absence of potent platelet inhibition with a GP IIbIIIa inhibitor, bivalirudin may be inferior in preventing ischemic events compared to heparin and a GP IIbIIIa inhibitor.
Despite the increasing usage of bivalirudin, the ex vivo effects of this direct thrombin inhibitor on platelet function have not been fully defined. Unfractionated heparin has a well-described effect that causes increased platelet activation including an increase in aggregation and expression of activated P-selectin on the platelet [12]. Similar studies examining the effects of bivalirudin have not found that pretreatment with bivalirudin increases agonist-induced platelet activation or aggregation. However, these studies have either examined the ex vivo effects of bivalirudin [13] and have limited their investigations to adenosine diphosphate (ADP)-induced platelet activation [13, 14] or have studied relatively small number of patients [15]. In addition, none of these studies have examined PAR1 or PAR4 agonist peptide (AP) stimulation of platelet activation. Although one might predict that a direct thrombin inhibitor would not interfere with PAR-AP-mediated activation, the complexity of PAR-signaling and the precedence of anticoagulants effecting agonist–mediated platelet activation [12] provide a rationale for further study. Just recently, a report of 22 patients undergoing PCI reported that treatment with bivalirudin significantly decreased thrombin and collagen-mediated platelet aggregation in platelet rich plasma [16].
In addition to thrombin’s role in mediating coagulation and inflammation, thrombin also possesses antithrombotic activity at low concentrations [17]. The paradoxical anti-thrombotic activity of thrombin partly stems from the ability of thrombin in the presence of thrombomodulin to activate Protein C, which is a potent anticoagulant that inactivates the coagulation cascade. Activated protein C (APC) also possess anti-inflammatory activity or protective effects as it has been demonstrated to be essential for the maintenance of pregnancy [18] in addition to blocking apoptosis in ischemic brain endothelium [19]. Thus our hypothesis is that inhibition of thrombin by bivalirudin may affect platelet signaling by removing thrombin’s paradoxical anti-thrombotic effect on platelets.
Since both PAR1 and PAR4 are expressed on the platelet surface, it is critical to define the roles of both receptors in several likely clinical settings where PAR antagonists would potentially provide further protection against adverse events. Further, although direct thrombin inhibitors are in common use in interventional cardiology, their potential effects on the signaling state of circulating platelets in pro-thrombotic conditions has not been fully examined.
Materials and methods
Materials
PAR1-AP (SFLLRN) and PAR4-AP (AYPGKF) were purchased from GL Biochem (Shanghai, China). Anti-Rap1 antibody was purchased from Santa Cruz Biotechnology (SanDiego, CA, USA). Blocking buffer and anti-rabbit IRDYE 800 antibody were purchased from LI-COR Biosciences (Lincoln, NE, USA). Protein A Sepharose Beads was purchased from Amersham (Uppsala, Sweden). Lumi-Aggregometer Model 700 and supplies were purchased from Chrono-Log (Havertown, PA, USA). FITC-PAC1 and PE-CD62P (P-selectin) were purchased from BD Pharmingen (San Jose, CA, USA).
Subjects
Study was approved by the Vanderbilt University and Thomas Jefferson University Institutional Review Boards. Informed consent was obtained from all individuals prior to blood donation. Inclusion criteria included patients over 18 years of age (male or female) who had undergone clinically-indicated coronary angiography and subsequent PCI. Patients included those who presented with stable angina and ACS whose subsequent coronary angiography revealed severe stenosis (>70 %) that requires PCI. All patients received one or more coronary stents with the exception of one whose Fractional Flow Reserve revealed a non-hemodynamically significant coronary stenosis. Exclusion criteria included: significant left main coronary artery disease, severely impaired left ventricular systolic function (EF < 35 %), prior treatment with heparin, enoxaparin, bivalirudin (or other thrombin inhibitors), glycoprotein IIbIIIa inhibitors, warfarin, or thrombolytic agents <48 h, prior history of myocardial infarction, CVA, or PCI (<6 months), prior history of severe bleeding, peptic ulcer disease, gastritis, or prior history of hematological abnormalities or HIV/AIDS.
Platelet preparation
To investigate the potential effects of bivalirudin on protease-activated receptor (PAR)-mediated platelet aggregation, platelets which were drawn both prior to and 30 min post-bivalirudin infusion, were stimulated with low and high doses of PAR1-activating peptide (PAR1-AP) or PAR4-AP as determined previously [20]. Agonist concentration was chosen based on the concentration previously shown to result in either partial or full platelet aggregation in washed platelets [20, 21]. Importantly we chose to study the effect of bivalirudin in washed platelets, rather than platelet rich plasma, so we could examine the effect of thrombin stimulation without adding peptide inhibitors that block thrombin-mediated fibrin formation. Human platelets were obtained from blood of patients undergoing PCI in the Vanderbilt University Medical Center Cardiac Catheterization Lab before and 30 min following bolus injection of bivalirudin. Bivalirudin was given as an intravenous bolus dose of 0.75 mg/kg, followed by a continuous intravenous infusion of 1.75 mg/kg/h. For all studies, washed platelets were re-suspended in tyrodes buffer as previously described [21]. All experiments were conducted with a platelet concentration of 2 × 108 platelets/ml. To study the effects of ex vivo bivalirudin on ex vivo thrombin-mediated platelet function, platelets from healthy donors were “spiked” with varying concentrations of bivalirudin (2, 4, 6, 8 and 10 μg) for 30 min, washed either once or twice and resuspended in Tyrode’s buffer to a platelet concentration of 2 × 108 platelets/ml followed by stimulation with 2 nM thrombin.
Platelet aggregation and dense granule secretion
Platelet aggregation and dense granule secretion was measured in washed platelets following stimulation with low and high concentrations of PAR1-AP or PAR4-AP using a lumi-aggregometer (Chronolog Corp.) as previously described [20]. Maximal platelet aggregation was measured as the maximal percentage platelet aggregation following agonist stimulation [22]. Dense granule release was calculated by measuring the maximal ATP release following agonist stimulation with a luminescence detector.
Rap1 activation assay
Rap1 activity was measured using the GST-RalGDS-Rap1-binding domain (RalGDS-RBD) that specifically interacts with activated Rap1 as described elsewhere [21]. Activated Rap1 was detected by immunoblotting with the anti-Rap1 antibody. Rap1 expression was analyzed from total platelet lysate to confirm equal protein loading.
Surface expression of P-selectin and activated glycoprotein IIbIIIa
Human platelet P-selectin and the integrin glycoprotein IIbIIIa were assessed using fluorescence-activated cell sorting (FACS) as previously described [23].
Sample size
Prior data indicate that the difference in the response of matched pairs is normally distributed with a standard deviation of ED50 is 20 [24]. If the true difference in the mean response is 10 %, we calculated that we would need 54 subjects to be able to reject the null hypothesis that this response difference is zero with probability (power) 0.95. The Type I error probability associated with the test of this null hypothesis is 0.05. Sample size calculated with PS Power Version 3.0.
Statistics
Wilcoxon signed rank test was used to detect significant differences between pre and post bivalirudin values. Conclusions were considered significant if the P value ≤ 0.05. Values are expressed as the mean ± SEM.
Results
Bivalirudin effects on thrombin and PAR-induced platelet aggregation
A total of 66 patients were studied that presented with either stable angina, unstable angina or NSTEMI with the demographics represented in Table 1. Low dose stimulation with thrombin resulted in a significant decrease (N = 40, 65 % decrease, P < 0.0001) in the maximal level of platelet aggregation following bivalirudin (Fig. 1a). Low dose stimulation with PAR1-AP showed a minor, but statistically significant decrease in maximal aggregation in the presence of bivalirudin (N = 59, 15 % decrease, P < 0.0001), while the inhibitory effects of PAR4-AP were relatively small (N = 62, 6 % decrease, P = 0.034). When stimulated with high dose of agonist, significant differences were not observed in pre- and post-bivalirudin conditions (Fig. 1a) indicating that a shift in sensitivity to PAR-mediated platelet aggregation may exist following systemic exposure to bivalirudin.
Table 1.
Clinical characteristics of patients undergoing PCI receiving bivalirudin
| Clinical characteristics (n = 66) | Values |
|---|---|
| Age (years, mean, ± SD) | 64.8 ± 9.7 |
| Female (n, %) | 29 (43.9 %) |
| Height (cm ± SD) | 168.0 ± 12.3 |
| Weight (kg ± SD) | 86.0 ± 22.6 |
| Body mass index (BMI) | 30.3 ± 7.0 |
| Systolic blood pressure (mmHG ± SD) | 139.2 ± 19.4 |
| Diastolic blood pressure (mmHG ± SD) | 77.8 ± 12.0 |
| Fasting blood glucose (mg/dl ± SD) | 132 ± 61.3 |
| Diabetes mellitus (n, %) | 17 (25.8 %) |
| Unstable angina/NSTEMI (n, %) | 14 (21.2 %) |
| Smokers (n, %) | 11 (16.7 %) |
| Number of stents per patient (mean + SD) | 1.6 + 0.9 |
| Medications before PCI (n, %) | |
| ASA | 61 (92.4 %) |
| Clopidogrel | 53 (80.3 %) |
| b blocker | 42 (63.6 %) |
| ACE inhibitor | 27 (40.9 %) |
| Angioten sin II receptor blocker | 9 (13.6 %) |
| Ca channel blocker | 21 (31.8 %) |
| Statin | 38 (68.2 %) |
| Lipid lowering (excluding statin) | 18 (27.3 %) |
| Diuretic | 21 (31.8 %) |
| Nitrates | 15 (22.7 %) |
Out of the 14 patients presenting with unstable angina and NSTEMI, two had a NSTEMI. Ethnicity was 89.4 % Caucasian with 10.6 % African American patients
Fig 1.

Panel A bivalirudin effects on PAR-mediated platelet aggregation. Maximal platelet aggregation was measured following stimulation for 10 min with 2 nM thrombin (N = 40; ***P < 0.001), 2.5 μM PAR1-AP (N = 59; ***P < 0.001), or 100 μM PAR4-AP (N = 62; **P < 0.01). Maximal platelet aggregation following stimulation for 10 min with 10 nM thrombin (N = 63), 20 μM PAR1-AP (N = 64) or 200 μM PAR4-AP (N = 62). Panel B bivalirudin effects on dense granule secretion. Stimulation with thrombin at 2 nM (N = 15) or 10 nM (N = 39) resulted in a significant decrease of dense granule secretion (*P < 0.05). There was no significant shift in dense granule secretion following stimulation with low or high concentration PAR4-AP (N = 29 and 30 respectively) and low PAR1-AP (N = 39). There was augmentation with high PAR1-AP (N = 39, P = 0.0008) Panel C bivalirudin alters PAR-mediated alpha granule secretion. Composite fold change in mean fluorescence relative to unstimulated platelets for P-selectin following stimulation with 2 nM thrombin (N = 43; ** P < 0.001), 2.5 μM PAR1-AP (N = 59) or 100 μM PAR4-AP (N = 66; **P < 0.01). Composite fold change for P-selectin following stimulation with 10 nM thrombin (N = 65; **P < 0.005), 20 μM PAR1-AP (N = 66; **P < 0.01) or 200 μM PAR4-AP (N = 66; **P < 0.01). Panel D bivalirudin shifts sensitivity to PAR-mediated activation of glycoprotein IIbIIIa. Composite fold change in mean fluorescence relative to unstimulated platelets for glycoprotein IIbIIIa following stimulation with 2 nM thrombin (N = 43; **P < 0.001), 2.5 μM PAR1-AP (N = 59) or 100 μM PAR4-AP (N = 66). Composite fold change in mean fluorescence relative to unstimulated platelets for αIIbβ3 following stimulation with 10 nM thrombin (N = 65), 20 μM PAR1-AP (N = 66), or 200 μM PAR4-AP (N = 66). Panel E bivalirudin effects on Rap1 activation. Composite fold change in Rap1-GTP following stimulation with 2 nM thrombin (N = 32; ***P < 0.001), 2.5 μM PAR1-AP (N = 39), or 100 μM PAR4-AP (N = 45). Composite fold change in Rap1-GTP levels following stimulation with 10 nM thrombin (N = 46; *P < 0.05), 20 μM PAR1-AP (N = 46) or 200 μM PAR4-AP (N = 45)
To determine if residual bivalirudin was present in the resuspended platelets, which could explain the decrease in stability of platelet aggregates in the presence of low dose thrombin, whole blood from healthy donors was spiked with varying concentrations of bivalirudin, washed either once or twice and resuspended in Tyrode’s buffer followed by stimulation with 2 nM thrombin (Fig. 2, top panel). Washing the platelets once resulted in a dose-dependent inhibition of platelet aggregation which was significantly inhibited with 8 μg/ml bivalirudin and completely inhibited with 10 μg/ml bivalirudin. Washing the spiked platelets twice prior to final resuspension and stimulation resulted in no observable inhibition of platelet aggregation with low dose thrombin, confirming that the decreased aggregate stability observed in the patient samples may be due to residual bivalirudin.
Fig 2.

Top panel residual bivalirudin effects on thrombin-mediated platelet aggregation. Platelet aggregation was measured in platelets pre-treated with varying concentrations of bivalirudin (0–10 μg).F ollowing pre-treatment, platelets were washed either once or twice and resuspended in platelet buffer. Maximal platelet aggregation was measured. Bottom panel comparison of basal levels of PAC-1 and P-selectin before and after treatment with bivalirudin (N = 57; NS)
Bivalirudin modulates PAR-mediated secretion
Platelet activation involves both primary activation through PARs as well as secondary feedback through the ADP receptors P2Y1 and P2Y12 [25]. To identify if bivalirudin alters regulation of platelet activity through inhibition of PAR-induced ATP or ADP secretion, dense granule secretion was measured following stimulation of platelets with thrombin, PAR1-AP or PAR4-AP in pre- and post-bivalirudin conditions (Fig. 1b). Thrombin-induced dense granule secretion was significantly attenuated following treatment with bivalirudin in both low and high thrombin conditions (N = 15, 66 % decrease, P = 0.011 and N = 39, 23 % decrease, P = 0.011, respectively). Dense granule secretion was not, however, significantly decreased post-bivalirudin following stimulation with PAR1-AP or PAR4-AP indicating that the differences in thrombin-mediated dense granule secretion may be due to the direct effect of bivalirudin on thrombin and not on signaling in the platelet.
Alpha granule secretion was also measured to determine if bivalirudin induced differential effects on dense versus alpha granule secretory processes. To measure alpha granule secretion, surface expression of P-selectin was measured by flow cytometry following agonist stimulation (Fig. 1c). At low agonist levels, P-selectin expression was decreased following thrombin stimulation post-bivalirudin (N = 43, 26 % decrease, P = 0.002), whereas there was no difference in PAR1-AP conditions pre or post-bivalirudin and, interestingly, PAR4-AP induced an increase in P-selectin levels post-bivalirudin (N = 66, 25 % increase, P = 0.009). High agonist concentration induced a significant increase in P-selectin levels post-bivalirudin following stimulation with thrombin, PAR1-AP or PAR4-AP (N = 65, 18 % increase, P = 0.002; N = 66 14 % increase, P = 0.003; and N = 66, 20 % increase, P = 0.005, respectively) indicating an increase in the total levels of alpha granule release. Additionally, PAR1 and PAR4 show different levels of sensitivity to bivalirudin with PAR4 being more sensitive at low agonist concentrations to the effects of bivalirudin on alpha granule secretion relative to PAR1. To eliminate the possibility that differences in basal levels of P-selectin and PAC-1 may account for the observed increase in P-selectin secretion following bivalirudin, basal levels of were measured prior to and post-treatment with bivalirudin (Fig. 2, bottom panel) and no significant differences were observed.
Activated glycoprotein IIbIIIa is not significantly altered by treatment with bivalirudin
Following observations in Fig. 1a which showed a significant shift in sensitivity to bivalirudin at low agonist concentration to platelet aggregation, we investigated if this shift may be due to the level of activated glycoprotein IIbIIIa on the platelet surface (Fig. 1d). Similar to what was observed in Fig. 1a, lower concentrations of agonist result in a significant decrease in glycoprotein IIbIIIa activation following thrombin stimulation (N = 43, 17 % decrease, P = 0.004). No significant shift was observed following low or high levels of PAR4-AP. The decrease in glycoprotein IIbIIIa activation observed at low thrombin levels post-bivalirudin was not observed at high thrombin levels supporting a role for bivalirudin in shifting the dose–response curve for agonist-induced glycoprotein IIbIIIa activation to the right.
Thrombin-mediated Rap1 activation is altered by systemic treatment with bivalirudin
Levels and stability of platelet aggregation as well as glyco-protein IIbIIIa activation have been reported in the literature to be partially regulated by the small GTPase Rap1 [21, 22, 26, 27]. To investigate if the observed shift in platelet activation is a consequence of differences in Rap1 activation, the level of Rap1 activation was measured following agonist stimulation both in pre- and post-bivalirudin conditions (Fig. 1e). A significant decrease in Rap1 activation was observed in the post-bivalirudin condition stimulated with either low or high levels of thrombin (N = 32, 25 % decrease, P = 0.0004; N = 46, 20 % decrease, P = 0.021, respectively). However, neither PAR1-AP nor PAR4-AP conditions showed a significant decrease in Rap1 activation. In high PAR1-AP and PAR4-AP conditions however, there is a trend toward a decrease in Rap1 activation in platelets from post-bivalirudin blood.
Platelet function in patients on chronic clopidogrel versus clopidogrel-naïve patients
To determine if chronic treatment with clopidogrel may be a contributing factor to the observed differences in platelet function in pre and post bivalirudin samples, we compared all indices of platelet activation in patients treated with or without clopidogrel (Table 2). In general, no significant differences were observed between the two groups. For example, while low-dose thrombin-mediated platelet aggregation was lower with exposure to bivalirudin (Fig. 3, top panel), this was not statistically significant likely due to the small sample size (n = 13). With respect to alpha granule release, similar trends were observed with high dose thrombin, PAR1-AP and PAR4-AP in the clopidogrel-naïve group, in that post-bivalirudin P-selectin levels were higher, but again did not reach statistical significance (Fig. 4). This trend was observed with glycoprotein IIbIIIa activation (Fig. 5), ATP release and Rap-1 levels (data not shown). To examine the effects of thromboxane inhibition, we also grouped aspirin-naive patients together and compared to patients on chronic aspirin. However, because of our small sample size in the aspirin-naïve group (n = 5), definitive conclusions are difficult to make, although again similar trends are observed (Supplemental data).
Table 2.
Dosing and timing of clopidogrel–naïve patients
| Subject | Clopidogrel dose | Timing of dose in relation to 1st blood draw |
|---|---|---|
| P4-012 | 600 mg | 42 min after |
| P4-022 | 600 mg | 2 min before |
| P4-031 | 600 mg | 173 min after |
| P4-045 | 300 mg | 80 min after |
| P4-046 | 300 mg | 149 min after |
| P4-047 | Unknown dose | Post-procedurea |
| P4-048 | 600 mg | 18 min before |
| P4-050 | 300 mg | 32 min after |
| P4-054 | 600 mg | 6 min before |
| P4-055 | 600 mg | 36 min after |
| P4-056 | 600 mg | 28 min after |
| P4-057 | 600 mg | 31 min after |
| P4-058 | 600 mg | 38 min after |
Eight of the patients were categorized as clopidogrel naïve as they received the loading dose of clopidogrel (either 300 or 600 mg of clopidogrel) at the end of the intervention after the post bivalirudin blood draw. Six of the patients were categorized as clopidogrel-naïve as they received the loading dose within 35 min of the timing of sampling for the first blood draw
Details of the timing or dose of clopidogrel not available, but patient did receive clopidogrel post-procedurally. The patients classified as being on clopidogrel, were on chronic clopidogrel having received clopidogrel daily for at least 7 days prior to presentation
Fig 3.
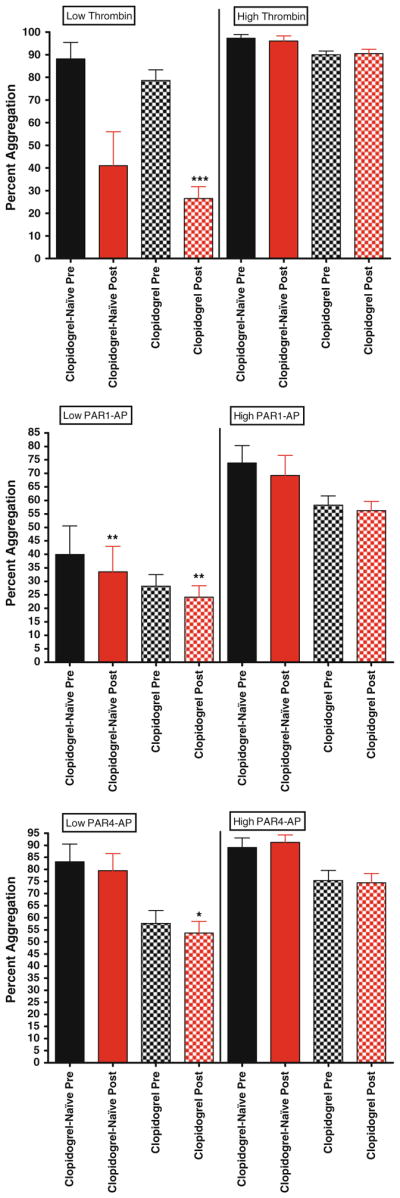
Maximal Platelet aggregation in patients on clopidogrel versus clopidogrel naïve patients. For all graphs shown, black bars depict levels of percent aggregation before bivalirudin administration, while red bars depict levels of percent aggregation post bivalirudin administration. N = 51 for patients on clopidogrel, and n = 13 for clopidogrel-naïve patients. Top panel low and high dose thrombin stimulation. Middle panel low and high dose PAR1-AP stimulation. Bottom panel low and high dose PAR1-AP stimulation
Fig 4.
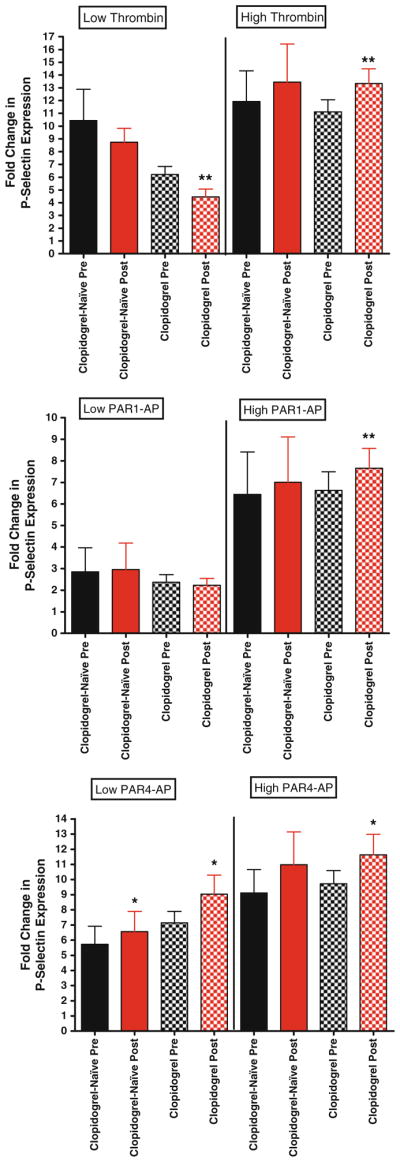
P-selectin expression in clopidogrel versus clopidogrel-naïve patients. For all graphs shown, black bars depict levels of P-selectin expression before bivalirudin administration, while red bars depict post bivalirudin P-selectin expression levels. N = 53 for patients on clopidogrel, and n = 13 for clopidogrel-naïve patients. Top panel low and high dose thrombin stimulation. Middle panel low and high dose PAR1-AP stimulation. Bottom panel low and high dose PAR1-AP stimulation
Fig 5.
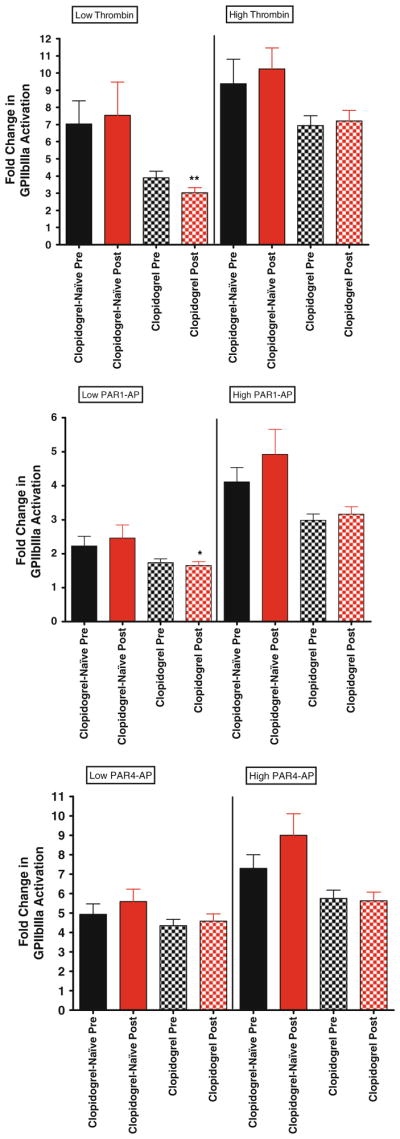
Glycoprotein IIbIIIa expression in clopidogrel versus clopidogrel-naïve patients. For all graphs shown, black bars depict levels of activated glycoprotein IIbIIIa expression before bivalirudin administration, while red bars depict post bivalirudin P-selectin expression levels. N = 53 for patients on clopidogrel, and n = 13 for clopidogrel-naïve patients. Top panel low and high dose thrombin stimulation. Middle panel low and high dose PAR1-AP stimulation. Bottom panel low and high dose PAR1-AP stimulation
Platelet function in patients presenting with acute coronary syndrome compared to patients presenting for elective percutaneous coronary intervention
To determine if bivalirudin effects on platelet function are influenced by clinical presentation, we stratified according to presentation. In general, no significant differences were observed between the two groups. In the ACS group, low-dose thrombin-mediated platelet aggregation and PAR1-AP-mediated glcyoprotein IIbIIIa activation were statistically significantly lower with exposure to bivalirudin (Fig. 6, top panel and Fig. 7 middle panel, respectively). Similar trends were observed with high dose thrombin, PAR1-AP and PAR4-AP-mediated P-selectin expression in clopidogrel-naïve group, in that post-bivalirudin P-selectin level were higher compared to pre-bivalirudin levels, but did not reach statistical significance (Fig. 8) likely due to the small sample size (n = 13).
Fig 6.

Maximal platelet aggregation in elective patients versus patients presenting with ACS. For all graphs shown, black bars depict levels of percent aggregation before bivalirudin administration, while red bars depict levels of percent aggregation post bivalirudin administration. N = 50 for patients undergoing elective PCI, and N = 14 for ACS patients. Top panel low and high dose thrombin stimulation. Middle panel low and high dose PAR1-AP stimulation. Bottom panel low and high dose PAR1-AP stimulation
Fig 7.

Glycoprotein IIbIIIa expression in elective patients versus patients presenting with ACS. For all graphs shown, black bars depict levels of activated glycoprotein IIbIIIa expression before bivalirudin administration, while red bars depict post bivalirudin P-selectin expression levels. N = 52 for patients undergoing elective PCI, and N = 14 for ACS patients. Top panel Low and high dose thrombin stimulation. Middle panel low and high dose PAR1-AP stimulation. Bottom panel low and high dose PAR1-AP stimulation
Fig 8.
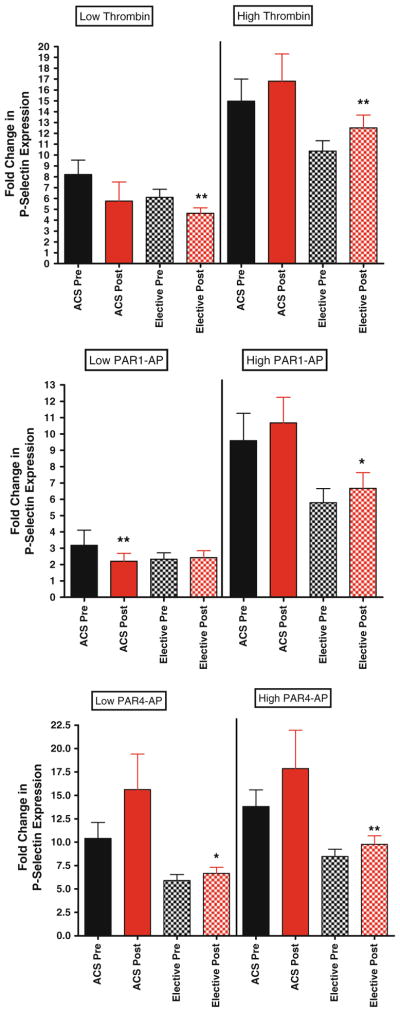
P-selectin expression in elective patients versus patients presenting with ACS. For all graphs shown, black bars depict levels of P-selectin expression before bivalirudin administration, while red bars depict post bivalirudin P-selectin expression levels. N = 52 for patients undergoing elective PCI, and N = 14 for ACS patients. Top panel low and high dose thrombin stimulation. Middle panel low and high dose PAR1-AP stimulation. Bottom panel low and high dose PAR1-AP stimulation
Discussion
Bivalirudin significantly inhibits most aspects of thrombin-mediated platelet activation
Our data in washed platelets demonstrated that exposure to bivalirudin strongly inhibits low and high thrombin mediated activation of platelet aggregation, dense granule secretion, integrin glycoprotein IIbIIIa activation and Rap1 activation. The only facet of platelet activation not inhibited by high dose of thrombin is P-selectin activation (discussed below). Although other studies have reported that exposure to bivalirudin inhibits either platelet aggregation [16] or PAC-1 binding [15] these studies were performed in platelet rich plasma with addition of GPRP (gylcine-L-proyl-L-arginine-L-proline) to platelet rich plasma in order to block fibrin formation permitting measurement of thrombin-mediated platelet aggregation. However, as GPRP has been shown to inhibit ADP-stimulated platelet aggregation, its presence [28] confounds the validity of this method. Nevertheless, our finding confirms these studies and adds further evidence that bivalirudin possess antiplatelet effects that may help to explain its non-inferior status in reducing ischemic events compared to heparin and glycoprotein IIbIIIa inhibitors in patients undergoing PCI [7–10].
PARs may be a viable target for anti-platelet therapy
Current pharmacological therapy for treatment of diseases caused by blood clots, such as heart disease and stroke, often involve the use of antiquated drugs that do not reflect current scientific understanding of these pathologies. However, none of these agents directly block the inflammatory and thrombotic effects mediated by activation of the thrombin receptors PAR1 and PAR4. The fact that national guidelines recommend the use of at least 4 independent agents for the treatment of ACS is testament to the importance of significant inhibition of platelet activation. Our data, which demonstrate that bivalirudin fails to inhibit high doses of PAR1-AP or PAR4-AP activation of platelets, but actually increases PAR1-AP and PAR4-AP-mediated stimulation of P-selectin expression on the platelet surface, gives impetus for the development of additional anti-platelet therapeutics. Indeed, the development of vorapaxar, a PAR1 antagonist that showed promise in Phase II studies in stable patients undergoing PCI [29], disappointingly failed to meet primary ischemic end points in a large Phase III trial and significantly increased intracranial bleeding in ACS patients undergoing PCI [30]. In a secondary prevention trail, vorapaxar did reduce ischemic events in patients at high risk of atherothrombotic events, but did so at the cost of increasing hemorrhagic intracranial bleeding [31].
Recent studies have suggested that PAR1 and PAR4 may form heterodimers and play an essential role in platelet-mediated thrombosis [5]. Addition of bivalirudin to a PAR4 pepducin antagonist inhibited human platelet aggregation and prevented carotid artery occlusion in guinea pigs [32]. Their data suggest that bivalirudin by binding to the hirudin binding site in PAR1, which is in close proximity to the PAR1 agonist peptide sequence, may hinder agonist peptide stimulation of PAR1. Our data demonstrate that exposure to bivalirudin inhibits low dose PAR1-AP stimulation of maximal platelet aggregation and agrees with their findings (Fig. 1a). Because PAR4 does not possess a hirudin binding site, bivalirudin may not hinder PAR4–AP stimulation of platelet activation. In fact, exposure to bivalirudin potentiated PAR4 agonist peptide stimulation of platelet P-selectin expression at both high and low doses. Although the conclusions that PAR1 and PAR4 form heterodimers is not without controversy [32, 33] our data reveal functional differences in PAR-mediated platelet activity with exposure to bivalirudin. Namely, exposure to bivalirudin partially inhibits PAR1 agonist peptide stimulation of platelet aggregation, while potentiating PAR4 agonist peptide stimulation of P-selectin release. This duality of bivalirudin to affect signaling is supportive for a potential role of PAR dimerization in signaling platelet function and further establishes the complexity of platelet signaling both at the platelet surface and the intracellular signaling pathways.
Bivalirudin potentiates PAR-mediated P-selectin expression
Our data are the first to demonstrate that exposure to bivalirudin significantly increases platelet P-selectin expression with high doses of PAR1-AP and with both low and high doses of PAR4-AP. These findings are in contrast to previous studies which indicated that ADP-mediated aggregation and P-selectin expression were not increased in patients with CAD by ex vivo addition (spiking) of bivalirudin [13]. Similar results were observed in another study that examined the effects of in vivo bivalirudin specifically on platelet P-selectin expression and formation of leukocyte aggregates in blood from patients treated with UFH plus eptifibatide compared with bivalirudin [14]. However, neither of these studies examined PAR agonist-mediated platelet activation. In a small group of patients (n = 13), bivalirudin was found to inhibit thrombin-induced platelet reactivity to a greater extent compared to heparin and tirofiban [15]. However, this study also examined the effect of “spiking” bivalirudin in blood obtained from patients undergoing coronary catheterization [15].
Interestingly, Eslam et al. have investigated the effects of bivalirudin on thrombin induction of P-selectin expression in patients undergoing PCI and found that bivalirudin decreased maximal thrombin-mediated P-selectin expression by approximately 100 % compared to pre-bivalirudin levels [34]. Both differences in methodology and patient demographic factors may explain these conflicting results. The Eslam group used whole blood with addition of GPRP when determining thrombin-mediated P-selectin expression by flow cytometry, while we utilized washed platelets to selectively determine the role of bivalirudin exposure on the platelet. Additionally a number of differences exist in the demographics of these two studies including a higher BMI for our subjects (our BMI was 30.3 while their average BMI was 28), more females (we had 44 % females, while they had 35 %), and 14 of our patients presented with acute coronary syndrome while all of their patients had stable angina.
Limitations of the study
One potential limitation of our data is that we cannot control for the effect of PCI, as changes in platelet reactivity could be explained by the act of deploying a coronary stent in a diseased coronary artery, and not by administration of bivalirudin. However, administering bivalirudin to patients with normal coronary arteries or with non-obstructive CAD (i.e., not needing a PCI) for a control group would unnecessarily expose patients to an additional risk of bleeding and potential death. Although the risk of bleeding is decreased with bivalirudin compared to either unfractionated heparin or low-molecular weight heparin, the risk is still present with major bleeding rates ranging from 2.4 % in the REPLACE-2 trial [7], 3.0 % in the ACUITY trial [8] to 4.9 % in the HORIZONS trial [10]. Additionally, our study was limited to treatment with bivalirudin and did not compare the effects of bivalirudin to other modes of anticoagulation, such as heparin, heparin and a glycoprotein IIbIIIa inhibitor or low-molecular weight heparin. Potential future studies will aim at addressing this important question.
Implications of the study
The increase in platelet P-selectin observed after bivalirudin exposure with high doses of PAR1-AP and thrombin and low and high doses of PAR4-AP is a consistent observation in our study. This is intriguing given that bivalirudin caused a small but statistically significant increase in stent thrombosis within 24 h compared to the heparin plus glycoprotein IIbIIIa inhibitor group in the HORIZONS-AMI trial [10]. The simplest explanation for this phenomenon is the short half life of bivalirudin, which most likely accounts for the lower rate of bleeding, but also leaves the patient without adequate anticoagulation before the antiplatelet effects of clopidogrel are fully onboard. Although exposure to bivalirudin decreases PAR1-AP stimulation of maximum platelet aggregation at low doses, the effects of bivalirudin to increase P-selectin expression are significant with high doses of both PAR1- and PAR4-AP. As P-selectin is an inflammatory protein and has been implicated in the development of atherosclerosis [35], there is a body of data demonstrating that platelet P-selectin may play a role in mediating the size and stability of platelet aggregates and thus arterial thrombosis [36–38]. The small, but statistically significant, increase in P-selectin expression observed with exposure to bivalirudin may be overshadowed by the larger inhibitory effect of bivalirudin to inhibit nearly all aspects of thrombin-mediated platelet activation, particularly platelet aggregation. Furthermore, multiple trials have shown significant associations of stent thrombosis with hyporesponsiveness to dual-antiplatelet therapy measured with ADP-induced platelet aggregation, [39, 40] and not thrombin-mediated P-selectin activation.
Overall, the ability of bivalirudin to inhibit thrombin-mediated platelet aggregation support the large number of trials that consistently demonstrate bivalirudin’s equivalence to heparin and glycoprotein IIbIIIa inhibitors in preventing ischemic events.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grants P50-HL081009 to HEH and R00-HL089457 to MH. JHC was supported by the Vanderbilt Clinical & Translational Research Scholars Program.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11239-012-0812-9) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Michael Holinstat, Department of Medicine, Cardeza Foundation for Hematologic Research, Thomas Jefferson University, Philadelphia, PA, USA.
Nancy E. Colowick, Department of Pharmacology, Vanderbilt University Medical Center, Nashville, TN 37232, USA
Willie J. Hudson, Department of Pharmacology, Vanderbilt University Medical Center, Nashville, TN 37232, USA
Dana Blakemore, Department of Biostatistics, Vanderbilt University Medical Center, Nashville, TN, USA.
Qingxia Chen, Department of Biostatistics, Vanderbilt University Medical Center, Nashville, TN, USA.
Heidi E. Hamm, Email: heidi.hamm@vanderbilt.edu, Department of Pharmacology, Vanderbilt University Medical Center, Nashville, TN 37232, USA
John H. Cleator, Email: john.cleator@vanderbilt.edu, Department of Pharmacology, Vanderbilt University Medical Center, Nashville, TN 37232, USA, Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232-6600, USA
References
- 1.Patterson C, Stouffer GA, Madamanchi N, Runge MS. New tricks for old dogs: nonthrombotic effects of thrombin in vessel wall biology. Circ Res. 2001;88:987–997. doi: 10.1161/hh1001.091447. [DOI] [PubMed] [Google Scholar]
- 2.Jamieson GA. Pathophysiology of platelet thrombin receptors. J Thromb Haemostasis. 1997;78:242–246. [PubMed] [Google Scholar]
- 3.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 4.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemostasis. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 5.Leger AJ, Covic L, Kuliopulos A. Protease-activated receptors in cardiovascular diseases. Circulation. 2006;114:1070–1077. doi: 10.1161/CIRCULATIONAHA.105.574830. [DOI] [PubMed] [Google Scholar]
- 6.Eikelboom J, White H, Yusuf S. The evolving role of direct thrombin inhibitors in acute coronary syndromes. J Am Coll Cardiol. 2003;41:70S–78S. doi: 10.1016/s0735-1097(02)02687-6. [DOI] [PubMed] [Google Scholar]
- 7.Lincoff AM, Kleiman NS, Kereiakes DJ, Feit F, Bittl JA, et al. Long-term efficacy of bivalirudin and provisional glycoprotein IIb/IIIa blockade vs heparin and planned glycoprotein IIb/ IIIa blockade during percutaneous coronary revascularization: REPLACE-2 randomized trial. J Am Med Assoc. 2004;292:696–703. doi: 10.1001/jama.292.6.696. [DOI] [PubMed] [Google Scholar]
- 8.Feit F, Manoukian SV, Ebrahimi R, Pollack CV, Ohman EM, et al. Safety and efficacy of bivalirudin monotherapy in patients with diabetes mellitus and acute coronary syndromes: a report from the ACUITY (Acute Catheterization and Urgent Intervention Triage Strategy) trial. J Am Coll Cardiol. 2008;51:1645–1652. doi: 10.1016/j.jacc.2007.11.081. [DOI] [PubMed] [Google Scholar]
- 9.Kastrati A, Neumann FJ, Schulz S, Massberg S, Byrne RA, et al. Abciximab and heparin versus bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med. 2011;365:1980–1989. doi: 10.1056/Nejmoa1109596. [DOI] [PubMed] [Google Scholar]
- 10.Stone GW, Witzenbichler B, Guagliumi G, Peruga JZ, Brodie BR, et al. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med. 2008;358:2218–2230. doi: 10.1056/NEJMoa0708191. [DOI] [PubMed] [Google Scholar]
- 11.Sibbing D, Bernlochner I, Schulz S, Massberg S, Schomig A, et al. Prognostic value of a high on-clopidogrel treatment platelet reactivity in bivalirudin versus abciximab treated non-st-segment elevation myocardial infarction patients: ISAR-REACT- 4 (Intracoronary Stenting and Antithrombotic Regimen: Rapid Early Action for Coronary Treatment-4) platelet substudy. J Am Coll Cardiol. 2012 doi: 10.1016/j.jacc.2012.02.044. [DOI] [PubMed] [Google Scholar]
- 12.Xiao Z, Theroux P. Platelet activation with unfractionated heparin at therapeutic concentrations and comparisons with a low-molecular-weight heparin and with a direct thrombin inhibitor. Circulation. 1998;97:251–256. doi: 10.1161/01.cir.97.3.251. [DOI] [PubMed] [Google Scholar]
- 13.Aggarwal A, Sobel BE, Schneider DJ. Decreased platelet reactivity in blood anticoagulated with bivalirudin or enoxaparin compared with unfractionated heparin: implications for coronary intervention. J Thromb Thrombolysis. 2002;13:161–165. doi: 10.1023/a:1020478923794. [DOI] [PubMed] [Google Scholar]
- 14.Keating FK, Dauerman HL, Whitaker DA, Sobel BE, Schneider DJ. Increased expression of platelet P-selectin and formation of platelet-leukocyte aggregates in blood from patients treated with unfractionated heparin plus eptifibatide compared with bivalirudin. Thromb Res. 2006;118:361–369. doi: 10.1016/j.thromres.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 15.Schneider DJ, Keating F, Sobel BE. Greater inhibitory effects of bivalirudin compared with unfractionated heparin plus eptifibitide on thrombin-induced platelet activation. Coron Artery Dis. 2006;17:471–476. doi: 10.1097/00019501-200608000-00011. [DOI] [PubMed] [Google Scholar]
- 16.Kimmelstiel C, Zhang P, Kapur NK, Weintraub A, Krishnamurthy B, et al. Bivalirudin is a dual inhibitor of thrombin and collagen-dependent platelet activation in patients undergoing percutaneous coronary intervention. Circ Cardiovasc Interv. 2011;4:171–179. doi: 10.1161/CIRCINTERVENTIONS.110.959098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanson SR, Griffin JH, Harker LA, Kelly AB, Esmon CT, et al. Antithrombotic effects of thrombin-induced activation of endogenous protein C in primates. J Clin Investig. 1993;92:2003–2012. doi: 10.1172/JCI116795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isermann B, Sood R, Pawlinski R, Zogg M, Kalloway S, et al. The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat Med. 2003;9:331–337. doi: 10.1038/nm825. [DOI] [PubMed] [Google Scholar]
- 19.Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, et al. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 20.Holinstat M, Voss B, Bilodeau ML, McLaughlin JN, Cleator J, et al. PAR4, but not PAR1, signals human platelet aggregation via Ca2+ mobilization and synergistic P2Y12 receptor activation. J Biol Chem. 2006;281:26665–26674. doi: 10.1074/jbc.M602174200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holinstat M, Voss B, Bilodeau ML, Hamm HE. Protease-activated receptors differentially regulate human platelet activation through a phosphatidic acid-dependent pathway. Mol Pharmacol. 2007;71:686–694. doi: 10.1124/mol.106.029371. [DOI] [PubMed] [Google Scholar]
- 22.Holinstat M, Preininger AM, Milne SB, Hudson WJ, Brown HA, et al. Irreversible platelet activation requires protease-activated receptor 1-mediated signaling to phosphatidylinositol phosphates. Mol Pharmacol. 2009;76:301–313. doi: 10.1124/mol.109.056622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shattil SJ, Cunningham M, Hoxie JA. Detection of activated platelets in whole blood using activation-dependent monoclonal antibodies and flow cytometry. Blood. 1987;70:307–315. [PubMed] [Google Scholar]
- 24.Vericel E, Januel C, Carreras M, Moulin P, Lagarde M. Diabetic patients without vascular complications display enhanced basal platelet activation and decreased antioxidant status. Diabetes. 2004;53:1046–1051. doi: 10.2337/diabetes.53.4.1046. [DOI] [PubMed] [Google Scholar]
- 25.Smyth SS, Woulfe DS, Weitz JI, Gachet C, Conley PB, et al. G-protein-coupled receptors as signaling targets for anti-platelet therapy. Arterioscler Thromb Vasc Biol. 2009;29:449–457. doi: 10.1161/ATVBAHA.108.176388. [DOI] [PubMed] [Google Scholar]
- 26.Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, et al. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10:982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- 27.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC., 2nd Rap1b is required for normal platelet function and hemostasis in mice. J Clin Investig. 2005;115:680–687. doi: 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adelman B, Gennings C, Strony J, Hanners E. Synergistic inhibition of platelet aggregation by fibrinogen-related peptides. Circ Res. 1990;67:941–947. doi: 10.1161/01.res.67.4.941. [DOI] [PubMed] [Google Scholar]
- 29.Becker RC, Moliterno DJ, Jennings LK, Pieper KS, Pei J, et al. Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: a ran-domised, double-blind, placebo-controlled phase II study. Lancet. 2009;373:919–928. doi: 10.1016/S0140-6736(09)60230-0. [DOI] [PubMed] [Google Scholar]
- 30.Tricoci P, Huang Z, Held C, Moliterno DJ, Armstrong PW, et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366:20–33. doi: 10.1056/NEJMoa1109719. [DOI] [PubMed] [Google Scholar]
- 31.Morrow DA, Braunwald E, Bonaca MP, Ameriso SF, Dalby AJ, et al. Vorapaxar in the secondary prevention of athero-thrombotic events. N Engl J Med. 2012;366:1404–1413. doi: 10.1056/NEJMoa1200933. [DOI] [PubMed] [Google Scholar]
- 32.Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, et al. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–1254. doi: 10.1161/CIRCULATIONAHA.105.587758. [DOI] [PubMed] [Google Scholar]
- 33.Nieman MT. Protease-activated receptor 4 uses anionic residues to interact with alpha-thrombin in the absence or presence of protease-activated receptor 1. Biochemistry. 2008;47:13279–13286. doi: 10.1021/bi801334s. [DOI] [PubMed] [Google Scholar]
- 34.Eslam RB, Reiter N, Kaider A, Eichinger S, Lang IM, et al. Regulation of PAR-1 in patients undergoing percutaneous coronary intervention: effects of unfractionated heparin and bivalirudin. Eur Heart J. 2009;30:1831–1836. doi: 10.1093/eurheartj/ehp186. [DOI] [PubMed] [Google Scholar]
- 35.Blann AD, Nadar SK, Lip GY. The adhesion molecule P-selectin and cardiovascular disease. Eur Heart J. 2003;24:2166–2179. doi: 10.1016/j.ehj.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 36.Merten M, Chow T, Hellums JD, Thiagarajan P. A new role for P-selectin in shear-induced platelet aggregation. Circulation. 2000;102:2045–2050. doi: 10.1161/01.cir.102.17.2045. [DOI] [PubMed] [Google Scholar]
- 37.Yokoyama S, Ikeda H, Haramaki N, Yasukawa H, Murohara T, et al. Platelet P-selectin plays an important role in arterial thrombogenesis by forming large stable platelet-leukocyte aggregates. J Am Coll Cardiol. 2005;45:1280–1286. doi: 10.1016/j.jacc.2004.12.071. [DOI] [PubMed] [Google Scholar]
- 38.Merten M, Thiagarajan P. P-selectin in arterial thrombosis. Z Kardiol. 2004;93:855–863. doi: 10.1007/s00392-004-0146-5. [DOI] [PubMed] [Google Scholar]
- 39.Stone GW. Assessment of dual antiplatelet therapy with drug-eluting stents (ADAPT-DES), transcatheter cardiovascular therapeutics meetings; San Franscisco. 7–11 Nov 2011.2011. [Google Scholar]
- 40.Bonello L, Tantry US, Marcucci R, Blindt R, Angiolillo DJ, et al. Consensus and future directions on the definition of high on-treatment platelet reactivity to adenosine diphosphate. J Am Coll Cardiol. 2010;56:919–933. doi: 10.1016/j.jacc.2010.04.047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.