

Abstract
Candidate gene and genome-wide association studies (GWAS) have identified genetic variants that modulate risk for human disease; many of these associations require further study to replicate the results. Here we report the first large-scale application of the phenome-wide association study (PheWAS) paradigm within electronic medical records (EMRs), an unbiased approach to replication and discovery that interrogates relationships between targeted genotypes and multiple phenotypes. We scanned for associations between 3,144 single-nucleotide polymorphisms (previously implicated by GWAS as mediators of human traits) and 1,358 EMR-derived phenotypes in 13,835 individuals of European ancestry. This PheWAS replicated 66% (51/77) of sufficiently powered prior GWAS associations and revealed 63 potentially pleiotropic associations with P < 4.6 × 10−6 (false discovery rate < 0.1); the strongest of these novel associations were replicated in an independent cohort (n = 7,406). These findings validate PheWAS as a tool to allow unbiased interrogation across multiple phenotypes in EMR-based cohorts and to enhance analysis of the genomic basis of human disease.
In recent years, GWAS have provided a powerful systematic method to investigate the impact of common genomic variations on human pathophysiology. Since 2005, more than 1,500 GWAS have identified genomic variants associated with nearly 250 diseases and traits1; a number of the associations had been identified previously by focused genetic studies. These are recorded in the National Human Genome Research Institute’s (NHGRI) web-accessible GWAS catalog (“NHGRI Catalog”)1 (Catalog of Published Genome-Wide Association Studies, http://www.genome.gov/26525384). The majority of GWAS investigate a single disease or trait; the accrual of such a large number of single variant–phenotype associations has led to the serendipitous identification of single loci associated with multiple diseases, or pleiotropy. Notable examples include variants at 9p21.3, which were associated initially with early myocardial infarction2 and subsequently with intracranial aneurysm and abdominal aortic aneurysms3; variants in the human leukocyte antigen (HLA) region and IL23R, which were associated initially with inflammatory bowel disease4 and subsequently with a variety of other autoimmune diseases5,6; and PTPN22 R602W, which was associated initially with lower risk of Crohn’s disease and subsequently with a higher risk of rheumatoid arthritis and other autoimmune diseases7. A recent analysis of the NHGRI catalog noted pleiotropy in 17% of genes and 4.6% of single-nucleotide polymorphisms (SNPs) with reported phenotype associations in the catalog8.
An alternative and complementary approach to query genotype-phenotype associations and to detect pleiotropy is the PheWAS. With PheWAS, associations between a specific genetic variant and a wide range of physiological and/or clinical outcomes and phenotypes can be explored either by using algorithms to parse EMR data9 or by analyzing data collected in observational cohort studies10. Previous small-scale EMR studies have provided initial support for the ability of the EMR-based PheWAS to replicate individual genotype-phenotype associations and to uncover novel associations11–13. However, whether EMR data or PheWAS methods can be used to discover genetic associations with a wide range of phenotypes has not been systematically studied.
Here, we expanded the PheWAS disease classifications to analyze the diverse spectrum of phenotypes in the NHGRI Catalog using EMR data and refined the statistical methods over previous publications9,11–13. We repurposed extant EMR and GWAS data from five institutions in the Electronic Medical Records and Genomics (eMERGE) Network14. We report the results of the largest PheWAS to date, involving 3,144 SNPs in the NHGRI Catalog. Our objectives were to validate PheWAS as a systematic method to detect pleiotropy by replicating known NHGRI Catalog results in EMR-derived data, to discover new associations for all available SNPs in the NHGRI catalog at the time of this study and to establish a comprehensive catalog of phenotypes associated with these SNPs. Our data highlight the value of EMR-based PheWAS as a tool for discovery of genotype- phenotype associations.
RESULTS
Genotype selection and population characteristics
As of April 17, 2012, the NHGRI Catalog contained a total of 6,092 SNPs having 7,486 genomic variant–phenotype associations (including potentially similar phenotypes and nonsignificant associations). A total of 3,144 of these SNPs were present and passed quality control on the Illumina Human660W-Quadv1_A GWAS chip. We studied 13,835 individuals of European descent who were genotyped at one of five different eMERGE sites with EMR-linked DNA biobanks (Supplementary Table 1). Demographics and the most common diagnoses are presented in Supplementary Table 2. The average age was 69.5 years, and 52.6% were female. Subjects had a mean follow-up of 15.7 ± 10.3 years. Our algorithm identified 1,358 unique PheWAS phenotypes, typically diseases and other clinical traits, from 2,080,550 unique dates of interaction with the EMR (e.g., admissions, clinic visits or laboratory tests). Records of individuals were analyzed for replications of existing findings and for new discoveries from the EMR-based PheWAS (Supplementary Fig. 1).
PheWAS replication of NHGRI Catalog associations
To prove the utility of EMR-derived phenotypes and the PheWAS method, we determined whether PheWAS could replicate known genomic variant–disease associations already listed in the NHGRI Catalog. Using the commonly accepted threshold of genome-wide significance, P ≤ 5 × 10−8, we selected NHGRI Catalog associations as candidates for replication. We evaluated 751 SNP-phenotype associations for 673 SNPs that had been identified in at least one prior GWAS using a population of European ancestry (Supplementary Fig. 1 and Supplementary Table 3).
We mapped the NHGRI Catalog phenotypes to 86 unique PheWAS phenotypes (Fig. 1 and Supplementary Fig. 2). Considering all NHGRI Catalog SNP-phenotype associations, including associations inadequately powered (beta < 0.8) in our experiment, PheWAS replicated 210 out of 751 (28%) prior NHGRI Catalog SNP-phenotype associations at P < 0.05 with a consistent direction of effect (Supplementary Fig. 2). The probability of replicating 210 associations out of 751 tests by chance, under the null hypothesis, at the α = 0.05 level is 4 × 10−94. However, when considering NHGRI Catalog binary traits (e.g., diseases) for which an exact match with an adequately powered (beta ≥ 0.8) PheWAS phenotype was found, 51 out of 77 (66%) SNP-phenotype associations were replicated at P < 0.05 (Fig. 1a, binary traits). The probability of replicating 51 out of 77 associations by chance, under the null hypothesis of no association, at the α = 0.05 level is 3 × 10−47. Pruning SNPs in high-linkage disequilibrium in our population (r2 > 0.9) did not alter the replication rates (47/70, or 67%, for adequately powered associations and 200/718, or 28%, for all associations). Similarly, exclusion of the 12 adequately powered associations in the major histocompatibility complex region on chromosome 6 (all of which were replicated) had only a small effect on the replication rate (39/65, or 60%, for adequately powered associations).
Figure 1.

PheWAS replication of NHGRI Catalog SNP-phenotype associations. (a) Each point represents the −log10(P) of a single SNP-phenotype association tested with PheWAS. This study is restricted to SNP-phenotype associations that achieved genome-wide significance (P ≤ 5 × 10−8) in at least one prior GWAS study that included individuals of European ancestry. Numbers in parentheses beside each phenotype represent the sample size within the PheWAS data set. The vertical blue line represents P = 0.05. Binary traits refer to all adequately powered, binary traits in the NHGRI Catalog with exact matches to a PheWAS phenotype. For example, 5/5 catalog SNPs associated with rheumatoid arthritis were replicated at P < 0.05 in PheWAS, and 9/15 SNPs associated with type 2 diabetes were replicated. Continuous traits are those numerically defined traits in the NHGRI Catalog that are related to PheWAS diseases (e.g., “iron deficiency anemia” was the PheWAS trait paired with the “serum iron level” catalog trait). (b) Replication rates of SNP-phenotype associations at different bins of statistical power. Association count refers to the number of SNP-phenotype associations replicated or not replicated at each bin of statistical power (e.g., all tested associations with power <0.1, power 0.1–0.2). The black line represents a linear regression weighted using the number of associations in each bin (y = 0.64×, r2 = 0.96). (c) Replication rate of NHGRI Catalog associations by number of unique publications citing the original SNP-phenotype association. Association count refers to the number of SNP-phenotype associations (among either adequately powered binary or continuous traits) with the corresponding number of publications. (d) Replication rate of NHGRI Catalog associations by discovery P-value. The dashed line indicates P = 5 × 10−8.
The likelihood of our PheWAS replicating an NHGRI Catalog association was directly related to the statistical power of the initial SNP-phenotype association, as demonstrated by the linear regression line (Fig. 1b). Other predictors of the likelihood of replication included the number of times the SNP-phenotype association has been published in the literature (Fig. 1c) and the significance of the original SNP-phenotype association (as measured by P-values, Fig. 1d). The replication rates among continuous traits associated with surrogate PheWAS phenotypes of obesity and musculoskeletal diseases were less than other categories of disease (Supplementary Table 4). However, the replication rate did not differ appreciably for putative functional SNPs compared to intergenic SNPs (Supplementary Table 5). Highly powered associations (beta ≥ 0.8) not replicated in our PheWAS were more likely to have been published only once, have lower effect sizes or represent phenotypes difficult to detect by billing codes (Supplementary Table 6). For example, 11 of the 26 nonreplicated catalog associations were for specific types of diabetes, which can be difficult to accurately distinguish in the EMR without use of more complicated, multicomponent algorithms15,16.
Using a reference standard calculated from phenotypes studied in the NHGRI Catalog, we found that our PheWAS had an area under the receiver operator characteristic curve (ROC) of 0.83 (Supplementary Fig. 3). Subsequent review of highly significant false-positive associations in the ROC analysis revealed evidence from sources outside of the NHGRI Catalog supporting three HLA associations with P < 10−5 in the PheWAS study: rs660895 and rs3135338 with type 1 diabetes17, and rs3135388 with rheumatoid arthritis18. Thus, excluding associations that were erroneous likely due to incorrect differentiation between types 1 and 2 diabetes, all PheWAS associations with P < 0.0002 in the ROC analysis were true positives.
Table 1 presents replicated associations having a P < 4.6 × 10−6 (representing a false discovery rate < 0.1); a full list of tested catalog associations is presented in Supplementary Table 3. Associations replicated by this PheWAS include: Alzheimer’s disease (rs2075650, in linkage disequilibrium with APOE, odds ratio (OR) = 2.41, P = 5.2 × 10−28); diabetes type 1 (HLA-DQB1, OR = 1.42, P = 2.0 × 10−7) and diabetes type 2 (TCF7L2, OR = 1.31, P = 8.3 × 10−16); coronary atherosclerosis and acute myocardial infarction (9p21.3 region, OR = 1.26, P = 1.0 × 10−12 and OR = 1.28, P = 4.0 × 10−8, respectively); gout (ABCG2, OR = 1.72, P = 1.0 × 10−12 and SLC2A9, OR = 0.67, P = 5.1 × 10−8); nonmelanoma skin cancer (EXOC2, OR = 1.32, P = 6.0 × 10−9); prostate cancer (8q24.21, OR = 1.61, P = 2.8 × 10−7); age-related macular degeneration (CFH, OR = 0.51, P = 7.2 × 10−20 and near C2/CFB, OR = 0.57, P = 4.8 × 10−8); and autoimmune diseases such as psoriasis (HLA-C and HCP5) and rheumatoid arthritis (HLA-DRB1 and C6orf10), all P < 2 × 10−6 and OR ≥ 1.5.
Table 1.
NHGRI Catalog associations replicated by PheWAS
| PheWAS phenotype | Cases | Region | Nearest gene | SNP | Odds ratio (95% CI) | P-value | NHGRI Catalog disease(s) | |
|---|---|---|---|---|---|---|---|---|
| Autoimmune | Psoriasis | 327 | 6p21.33 | HLA-C | rs10484554 | 1.71 (1.41, 2.08) | 6.2E-08 | Psoriasis |
| 6p21.33 | HCP5 | rs2395029 | 2.38 (1.74, 3.26) | 2.0E-08 | Psoriasis | |||
| Rheumatoid arthritis | 398 | 6p21.32 | C6orf10 | rs6910071 | 1.50 (1.27, 1.76) | 1.5E-06 | Rheumatoid arthritis | |
| 6p21.32 | HLA-DRB1 | rs660895 | 1.56 (1.33, 1.84) | 6.7E-08 | Rheumatoid arthritis | |||
| Hypothyroidisma | 2,042 | 9q22.33 | FOXE1 | rs7850258 | 0.77 (0.71, 0.83) | 1.1E-11 | Hypothyroidism | |
| Hematologic | Iron metabolism disorder | 40 | 6p22.2 | SLC17A1 | rs17342717 | 6.84 (4.36, 10.7) | 5.3E-17 | Serum ferritin |
| 6p22.2 | HFE | rs1800562 | 12.3 (7.64, 19.7) | 3.4E-25 | Serum transferrin | |||
| 6p22.1 | HIST1H2BJ | rs13194491 | 7.80 (4.76, 12.8) | 3.8E-16 | Serum transferrin | |||
| Neoplastic | Melanoma | 268 | 16q24.3 | MC1R | rs4785763 | 1.52 (1.27, 1.81) | 2.8E-06 | Melanoma |
| Nonmelanoma skin cancer | 1,931 | 6p25.3 | EXOC2 | rs12210050 | 1.32 (1.20, 1.45) | 6.0E-09 | Basal cell carcinoma | |
| Prostate cancer | 848 | 8q24.21 | Intergenic | rs1447295b | 1.61 (1.34, 1.92) | 2.8E-07 | Prostate cancer | |
| Circulatory | Myocardial infarction | 1,382 | 9p21.3 | CDKN2BAS | rs4977574 | 1.28 (1.17, 1.40) | 4.0E-08 | Myocardial infarction |
| Coronary atherosclerosis | 3,499 | 9p21.3 | CDKN2BAS | rs4977574b | 1.26 (1.18, 1.34) | 1.0E-12 | Coronary heart disease | |
| Atrial fibrillation | 1,950 | 4q25 | Intergenic | rs2200733 | 1.52 (1.34, 1.72) | 1.5E-10 | Atrial fibrillation | |
| Endocrine/metabolic | Type 1 diabetes | 615 | 6p21.32 | HLA-DQB1 | rs2647044 | 1.42 (1.24, 1.61) | 2.0E-07 | Type 1 diabetes |
| Type 2 diabetes | 3,122 | 10q25.2 | TCF7L2 | rs7903146b | 1.31 (1.23, 1.40) | 8.3E-16 | Type 2 diabetes | |
| Hypercholesterolemia | 4,518 | 1p13.3 | CELSR2 | rs646776 | 0.77 (0.70, 0.85) | 1.0E-07 | LDL & total cholesterol | |
| 2p24.1 | APOB | rs693 | 0.78 (0.73, 0.85) | 7.4E-10 | LDL & total cholesterol | |||
| 19p13.2 | LDLR | rs6511720 | 0.74 (0.65, 0.84) | 2.5E-06 | LDL cholesterol | |||
| Hyperglyceridemia | 492 | 11q23.3 | APOA5 | rs12272004 | 2.24 (1.70, 2.95) | 7.2E-09 | Triglycerides | |
| 11q23.3 | ZNF259 | rs964184 | 2.22 (1.78, 2.75) | 5.8E-13 | Hypertriglyceridemia | |||
| Gout | 769 | 4p16.1 | SLC2A9 | rs16890979 | 0.67 (0.59, 0.78) | 5.1E-08 | Serum urate | |
| rs13129697b | 0.72 (0.63, 0.81) | 2.4E-07 | Gout, Serum urate | |||||
| 4p16.1 | Intergenic | rs4698036 | 0.68 (0.60, 0.79) | 7.8E-08 | Serum urate | |||
| 4q22.1 | ABCG2 | rs2231142 | 1.72 (1.48, 1.99) | 1.0E-12 | Serum urate | |||
| Hyperbilirubinemia | 46 | 2q37.1 | UGT1A1 | rs887829b | 33.8 (14.5, 78.5) | 3.2E-16 | Serum bilirubin | |
| 2q37.1 | HEATR7B1 | rs2361502 | 7.74 (4.72, 12.7) | 4.2E-16 | Serum bilirubin | |||
| Other | Alzheimer’s disease | 737 | 19q13.32 | TOMM40 | rs157580 | 0.70 (0.62, 0.80) | 8.6E-08 | Alzheimer’s disease |
| rs2075650 | 2.41 (2.06, 2.82) | 5.2E-28 | Alzheimer’s disease | |||||
| Age-related macular degeneration | 749 | 1q31.3 | CFH | rs1329428 | 0.51 (0.45, 0.59) | 7.2E-20 | Age-related macular degeneration | |
| 6p21.33 | SKIV2L/C2/CFB | rs429608 | 0.57 (0.46, 0.70) | 4.8E-08 | Age-related macular degeneration | |||
| Fuchs’ dystrophy | 108 | 18q21.2 | TCF4 | rs613872 | 2.61 (1.90, 3.58) | 2.9E-09 | Fuchs’ dystrophy |
Table limited to phenotypes that exceeded genome-wide significance in previously reported GWAS in individuals of European ancestry and that had a PheWAS P-value < 4.6 × 10−6 (FDR < 0.1). A comprehensive list of replication P-values, odds ratios, minor (reference) alleles and minor allele frequencies can be found in Supplementary Table 3.
This GWAS used a subset of this data set.
PheWAS results include other SNPs in LD with P < 4.6 × 10−6 (Supplementary Tables 3 and 8).
Among the 751 tested SNP-phenotype associations, there were 175 NHGRI Catalog SNP associations with continuous traits for which we selected a surrogate binary disease or trait in PheWAS (e.g., the PheWAS disease “gout” was used as a proxy for the NHGRI Catalog SNP association with “serum uric acid levels”); 68 out of 175 (38.9%) of these continuous traits were replicated (Fig. 1a, continuous traits). Many of the continuous traits reported in the NHGRI Catalog represent associations found with biomarkers, measurements not typically available in the EMR (e.g., Factor VII levels, waist circumference), or values difficult to extract from the EMR (e.g., bone mineral density, which is usually recorded in narrative text)19,20. For these, PheWAS demonstrated associations with clinical diseases related to abnormal values of the measurement, molecule or analyte (Table 1 and Supplementary Table 3). One example is HFE rs1800562 (C282Y), the most common variant found in hereditary hemochromatosis and previously associated with iron levels19; in our PheWAS study this variant was associated with iron metabolism disorders (OR = 12.3, P = 3.4 × 10−25), including hemochromatosis. Moreover, individuals with C282Y were less likely to be diagnosed with iron-deficiency anemia, though the P-value was not significant (OR = 0.72, P = 1.5 × 10−3). Similarly, variants in TMPRSS6, previously associated with decreased serum iron levels, trended toward an association with a risk of iron-deficiency anemia (OR = 1.17, P = 6.2 × 10−4).
Three of the PheWAS phenotypes (hypothyroidism11, Alzheimer’s disease/dementia21 and type 2 diabetes15) have been investigated previously within eMERGE through disease-specific algorithms that used Boolean logic applied to combinations of billing codes, medications, laboratory values and data extracted using natural language processing algorithms and have been validated through comparison to manual review of the EMR by at least two reviewers. We compared performance of the automated PheWAS approach, which used groupings and exclusions of ICD 9 billing codes exclusively, to the eMERGE phenotype algorithms within this set. The replication rate for type 2 diabetes was identical for both methods, but PheWAS was slightly inferior when attempting to replicate Alzheimer’s-associated SNPs (Supplementary Table 7). A comparison with hypothyroidism has been published previously, showing similar results for both methods11.
Novel associations identified by PheWAS
As mentioned above, in our PheWAS study testing each of the 3,144 NHGRI Catalog SNPs for association with 1,358 different diseases and traits (Supplementary Fig. 1), we also searched for novel associations. Using a false discovery rate (FDR)<0.1 (P < 4.6 × 10−6), we detected 202 associations for 102 SNPs and 87 phenotypes; of these, 63 (31%) were judged to be potentially novel associations and 109 (54%) were either replications or associations with phenotypes related to NHGRI Catalog associations. Twenty-eight (14%) were known associations not reported in the NHGRI Catalog. Two (1.0%) were likely the result of erroneous phenotyping (type 2 diabetes patients classified as type 1 diabetes). A full listing is available in Supplementary Table 8. Unique NHGRI Catalog and PheWAS phenotypes associated with these SNPs are presented in Figure 2. An interactive catalog of PheWAS results is available at http://phewascatalog.org/. Both in the NHGRI Catalog and in our PheWAS results, the genomic region with the highest concentration of associations with multiple distinct phenotypes was 6p21.32 in the HLA region; 24 SNPs in this region were associated with 15 distinct disease phenotypes in the NHGRI Catalog, and the PheWAS analysis yielded associations with 12 distinct phenotypes at P < 4.6 × 10−6. Specific new associations and regions of pleiotropy are discussed in more detail below. The Q-Q plot of these results (Supplementary Fig. 4) revealed that the number of observed associations differed from that expected by chance at P-values around 1 × 10−4, which is not surprising as each SNP studied has at least one previously known association from the NHGRI Catalog.
Figure 2.

GWAS and PheWAS associations in the genome. Each diamond represents a unique phenotype association at each SNP. Red diamonds represent associations in the NHGRI Catalog only (including phenotypes not present in the PheWAS catalog), green diamonds represent NHGRI Catalog associations replicated by PheWAS (P < 0.05), and blue diamonds represent new phenotype associations identified by PheWAS (P < 4.6 × 10−6, or a FDR < 0.1). Numbers to the right and left indicate chromosomes.
We then analyzed the novel associations while adjusting for known NHGRI Catalog phenotypes (where possible; phenotypes such as eye color are not present in the EMR) to test whether the novel associations were independent of phenotype comorbidities (Supplementary Table 8). For example, this analysis revealed that the association between “nephritis and nephropathy” with the rs2647044 near HLA-DQB1 is likely mediated by the variant’s association with type 1 diabetes.
The strongest novel associations were with skin phenotypes of actinic keratosis, seborrheic keratosis and nonmelanoma skin cancer (Table 2). IRF4 rs12203592 (Fig. 3a), previously associated with hair and eye color, was strongly associated with actinic keratosis (OR = 1.69, P = 4.1 × 10−26). Variants in or near SLC45A2, EXOC2, HERC2, CDK10 and CDK5RAP1 were also associated with actinic keratosis. Associations with nonmelanoma skin cancer were found for IRF4 rs12203592 (OR = 1.50, P = 3.8 × 10−17) and TYR rs1847134 (OR = 1.28, P = 2.6 × 10−10). The SNP rs2853676 within TERT (Fig. 3b), previously associated with glioma, was associated with seborrheic keratosis (OR = 0.80, P = 1.6 × 10−7). Oral mucosal leukoplakia, a rare phenotype, was also associated with rs2853676, though this finding did not reach significance (44 cases, OR = 2.28, P = 1.6 × 10−4). Of note, rare TERT variants cause dyskeratosis congenita, an autosomal dominant condition that results from defective telomere maintenance and that causes skin hyperpigmentation, hyperkeratosis and leukoplakia22.
Table 2.
Potentially novel associations discovered via PheWAS
| PheWAS phenotype | Cases | Region | Nearest gene | SNP | Odds ratio (95% CI) | P-value | NHGRI catalog disease(s) |
|---|---|---|---|---|---|---|---|
| Actinic keratosis | 2,505 | 5p13.2 | SLC45A2 | rs16891982 | 0.55 (0.44, 0.70) | 6.3E-07 | Hair, eye color, skin pigmentation |
| 6p25.3 | IRF4 | rs12203592 | 1.69 (1.53, 1.86) | 4.1E-26 | Hair, eye color, freckling, PSP | ||
| 6p25.3 | EXOC2 | rs12210050 | 1.32 (1.20, 1.45) | 1.9E-08 | Basal cell carcinoma, tanning | ||
| 15q13.1 | HERC2 | rs12913832 | 0.80 (0.73, 0.87) | 8.7E-07 | Hair, eye color | ||
| 16q24.3 | CDK10 | rs258322 | 1.37 (1.22, 1.55) | 2.1E-07 | Hair color, melanoma | ||
| 20q11.21 | CDK5RAP1 | rs291671 | 1.39 (1.24, 1.57) | 7.0E-08 | Hair color | ||
| Nonmelanoma skin cancer | 1,931 | 6p25.3 | IRF4 | rs12203592a | 1.50 (1.36, 1.64) | 3.8E-17 | Hair, eye color, freckling, PSP |
| 11q14.3 | TYR | rs1847134a | 1.28 (1.18, 1.38) | 2.6E-10 | Eye color | ||
| Seborrheic keratosis | 2,570 | 5p15.33 | TERT | rs2853676 | 0.80 (0.74, 0.87) | 1.6E-07 | Glioma |
| Lipoma | 214 | 3p21.1 | ITIH1 | rs1042779 | 0.70 (0.61, 0.81) | 9.9E-07 | Bipolar disorder |
| Hypercoagulable state | 91 | 1q24 | NME7 | rs16861990b | 3.71 (2.57, 5.34) | 2.0E-12 | D-dimer levels |
| Bronchiectasis | 144 | 1q32.1 | KIF21B | rs2297909 | 1.81 (1.43, 2.29) | 8.7E-07 | Ankylosing spondylitis |
Novel associations with P < 4.6 × 10−6 are reported. For highly related sub-phenotypes (e.g., skin cancer and nonmelanoma skin cancer), only the strongest association is reported. Associations potentially in linkage disequilibrium with known associations are excluded here. All novel associations with P < 4.6 × 10−6 can be found in Supplementary Table 8. PSP, progressive supranuclear palsy.
Other studies have demonstrated associations with squamous cell carcinoma and potentially basal cell carcinoma (P > 1 × 10−4)23,24.
This SNP is in weak LD with the Factor V Leiden mutation (rs6025, r2 = 0.05 per 1,000 genomes).
Figure 3.

PheWAS plots for four SNPs. Each panel represents 1,358 phenotypes tested for association with a particular SNP, using logistic regression assuming an additive genetic model adjusted for age, sex, study site and the first three principal components. Phenotypes are grouped along the x axis by categorization within the PheWAS code hierarchy. The upper red lines indicate P = 4.6 × 10−6 (FDR = 0.1 for entire PheWAS); lower blue lines indicate P = 0.05; dashed lines are a single-SNP Bonferroni correction (P = 0.05/1,358). Diamonds encircling phenotype circles represent known NHGRI Catalog associations. (a) PheWAS associations for rs12203592 in IRF4, previously associated with hair and eye color, freckling and progressive supranuclear palsy. (b) PheWAS associations for rs2853676 in TERT, previously associated with glioma. (c) PheWAS associations for rs4977574 near CDKN2BAS at chr9p21, previously associated with myocardial infarction, and in LD with carotid stenosis. (d) PheWAS associations for rs660895 near HLA-DRB1, previously associated with rheumatoid arthritis. Results and plots for all SNPs included in the present study are available at http://phewascatalog.org/.
The variant rs16861990 near NME7, previously associated with d-dimer levels, was associated with hypercoagulable states (OR = 3.71, P = 2.0 × 10−12); notably, this SNP is in weak linkage disequilibrium (LD) (r2 = 0.05 in the 1000 Genomes CEU (see Online Methods)) with Factor V Leiden (rs6025, not assayed directly on the Illumina 660-Quad). Manual review of a subset of subjects from Vanderbilt for whom complete medical records were available identified 29 cases with the hypercoagulable phenotype, and five of the seven individuals carrying at least one minor allele for rs16861990 were found to have the Factor V Leiden mutation through prior testing as part of clinical care. Thus, the NME7 association may represent an association with Factor V Leiden.
Pleiotropy revealed by PheWAS analysis
We investigated SNPs associated with multiple phenotypes as a measure of possible pleiotropy within the PheWAS results. Pleiotropic effects of variants at the 9p21.3 locus near CDKN2BAS (Fig. 3c) were noted; the same SNPs were associated not only with coronary atherosclerosis (OR = 1.26, P = 1 × 10−12), acute myocardial infarction (OR = 1.28, P = 4 × 10−8) and abdominal aortic aneurysm (OR = 1.29, P = 0.001), consistent with prior publications3, but also with other near-significant “vascular” phenotypes such as unstable angina, carotid stenosis and hemorrhoids. Associations with hemorrhoids, abdominal aortic aneurysms and carotid stenosis all persisted when the regression model was adjusted for coronary atherosclerosis or myocardial infarction as a comorbidity.
Our study replicated the association between rheumatoid arthritis and rs660895 near HLA-DRB1 (Fig. 3d; OR = 1.56, P = 6.7 × 10−8). This SNP was also strongly associated with type 1 diabetes (OR = 1.44, P = 7.1 × 10−8) and potentially associated with inflammatory arthritides (OR = 1.64, P = 3.1 × 10−5), a parent phenotype of giant cell arteritis (OR = 1.94, P = 6.3 × 10−5). Both of these associations persisted when adjusting for rheumatoid arthritis (P = 1.8 × 10−7 for type 1 diabetes and P = 2.3 × 10−5 for inflammatory arthritides). Polymyalgia rheumatica, which often co-occurs with giant cell arteritis, was not associated with this variant (P = 0.064), whereas it may be associated with other variants in the HLA-region (rs10484561; OR = 1.59, P = 8.5 × 10−5); however, rs10484561 was not associated with giant cell arteritis (P = 0.21), suggesting that different HLA variants may be involved in polymyalgia rheumatica and giant cell arteritis. rs2647044 near HLA-DQB1 was the most pleiotropic SNP identified, being associated with nine phenotypes (P < 4.6 × 10−6), including a variety of autoimmune disorders such as celiac disease (OR = 2.60, P = 6.3 × 10−7), type 1 diabetes (OR = 1.42, P = 2.0 × 10−7) and lupus (OR = 2.60, P = 3.3 × 10−6).
Because a number of the new SNP-phenotype associations discovered in this PheWAS were skin phenotypes, we compared the patterns of phenotype associations for these SNPs (Fig. 4). IRF4 was uniquely associated with sunburns and other phenotypes influenced by sun exposure: actinic keratosis, seborrheic keratosis, basal cell carcinoma and eye neoplasms. However, the other SNPs associated with nonmelanoma skin cancer (TYR, EXOC2, MC1R, CDK10) and melanoma (MC1R) were not associated with sunburns. The TERT variant was not associated with skin phenotypes other than seborrheic keratosis.
Figure 4.
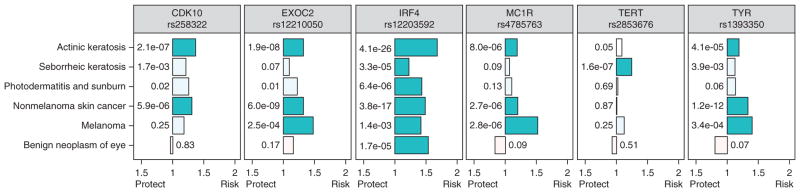
Risk variants for skin phenotypes have different pleiotropy patterns. Association odds ratios are graphed on the x axis and P-values (numbers next to the bars) are from the PheWAS analysis for that SNP. All SNPs use the minor allele as the coded allele, except rs2853676 (TERT). Darker colored bars represent significant associations, calculated as P = 0.05 divided by the number of associations displayed, or 0.05/(6 phenotypes*6 SNPs) = 1.4 × 10−3. Tests for heterogeneity revealed significant heterogeneity among the six phenotypes (I2 = 59–94%, all P < 0.05) and among the six SNPs (I2 = 23–83%, all P < 0.05). Bars oriented leftward toward “protect” represent SNPs in which the coded allele favors decreased prevalence of disease, and bars oriented rightward toward “risk” represent coded alleles favoring increased prevalence of disease.
Independent replication of PheWAS associations
We selected three phenotypes (actinic keratosis, seborrheic keratosis and nonmelanoma skin cancer) with novel associations discovered by our PheWAS to analyze in a separate EMR-linked GWAS population (n = 7,406) for replication of these results; we used a cutoff of P < 0.05. For these replications, we developed and validated algorithms using natural language processing to find each phenotype in the EMR in order to verify the accuracy of both the phenotype and the SNP-phenotype association discovered in our PheWAS. Physician review gave high positive predictive values (>98%) for each phenotype algorithm. Associations between IRF4 and actinic keratosis were replicated (OR = 1.60, 95% CI 1.26–2.04, P = 1.2 × 10−4), as were those between seborrheic keratosis and TERT (OR = 0.72, 95% CI 0.56–0.95, P = 0.02). Associations between nonmelanoma skin cancer and IRF4 (OR = 1.50, 95% CI 1.23–1.83, P = 4.8 × 10−5) and TYR (OR = 1.32, 95% CI 1.11–1.57, P = 1.5 × 10−3) were replicated. Despite being underpowered, two additional associations with actinic keratosis were replicated: HERC2 (OR = 0.76, 95% CI 0.60–0.97, P = 0.03) and CDK10 (OR = 1.42, 95% CI 1.03–1.96, P = 0.03). TYR and IRF4 variants also were associated with the more specific phenotypes of basal cell carcinoma and squamous cell carcinoma, which was possible to evaluate using the natural language processing algorithms deployed during this phase of analysis. Prior studies have demonstrated of this IRF4 variant23 and related TYR variants24 with squamous cell carcinoma and, more weakly, with basal cell carcinoma; this study suggests that IRF4 and TYR variants are associated with both skin cancers. Full results are available in Supplementary Table 9.
DISCUSSION
Here, we demonstrate that extant EMR-linked genetic data can be used in an unbiased search across large numbers of phenotypes to broadly replicate known GWAS associations in real-world, practice-based populations. We replicated 66% (51/77) of the prior NHGRI Catalog associations for which our analysis was adequately powered. When we included associations that were underpowered for replication, PheWAS analysis replicated 210 of all 751 (28%) testable NHGRI Catalog associations. PheWAS further discovered 63 potentially new associations (at P < 4.6 × 10−6), some of which were pleiotropic associations. Associations with actinic keratosis, seborrheic keratosis and nonmelanoma skin cancer discovered through PheWAS were replicated in a separate population using biopsy-confirmed diagnoses, thus validating not only the genetic association but also the quality of the EMR phenotype used in the PheWAS. Our findings therefore reinforce the utility of phenome scanning as a tool for not only replicating genotype-phenotype associations but also in providing a comprehensive catalog of human diseases associated with published variants.
In these EMR-based populations, associations for 42 phenotypes surpassed Bonferroni significance, including replications of SNPs associated with diverse phenotypes such as Alzheimer’s disease, type 2 diabetes, atrial fibrillation, gout, bilirubin metabolism, age-related macular degeneration, Fuchs’ corneal dystrophy, hypothyroidism, iron metabolism disorders (a code used for hemochromatosis), hypercholesterolemia, hypertriglyceridemia and coronary atherosclerosis. Notably, PheWAS replication rates of previously described associations were significantly higher for variants that had multiple published associations and for associations that had more significant P-values in previous publications. These results suggest that some of the lower-significance, so far nonreplicated associations in the NHGRI Catalog may require further investigation to evaluate the possibility of false positives.
Pleiotropy has been previously identified for a number of loci and is particularly prevalent among autoimmune phenotypes in the NHGRI Catalog. Although the sample size in this study does not permit a robust examination of pleiotropy for all SNPs considered here, most of the SNPs in our study did not show association for more than one phenotype. However, although only 102 of the 3,144 (3.2%) SNPs tested had at least one association with P < 4.6 × 10−6, 44 of these (43%) were associated with more than one phenotype. Fourteen SNPs (0.45%) were associated with more than three phenotypes at P < 4.6 × 10−6 (Supplementary Table 8). Thus, with a larger EMR population, one might suspect that pleiotropy would be more common than suggested by previous estimations of pleiotropy for 4.6% of SNPs in the NHGRI Catalog8.
Most of the highest-significance associations observed in this study were known, which is expected, given that the SNPs tested were included because they had been found in prior association studies. The strongest of the potential novel associations were with phenotypes not yet studied by GWAS, such as actinic and seborrheic keratoses. Some of these potentially novel pleiotropic associations were in fact SNPs in LD with known associations (e.g., MSH5 SNPs and type 1 diabetes, as noted in Table 2). The ability of PheWAS to demonstrate these pleiotropic associations using a single study population contrasts with current labor-intensive methods required to recognize pleiotropy, which depend on integration of multiple studies, genotyping platforms and LD maps.
By simultaneously evaluating associations with a broad range of phenotypes, PheWAS can highlight pleiotropic differences between multiple SNPs related to a common disease, some of which may suggest a mechanism of action. For example, the collection of sun exposure–related phenotypes associated with IRF4, as well as its known associations with hair and eye color, suggest its role in such risks may involve sun sensitivity. However, SNPs in other genes such as rs4785763 in MC1R (associated with melanoma) and TERT (a novel association with seborrheic keratosis) did not have an association with sunburns and other sun-exposed phenotypes, suggesting that the risk they confer may not be explained by sun sensitivity. In support of this hypothesis, MC1R variants predict melanoma risk not captured by skin pigmentation and sun exposure questionnaires25. Combining PheWAS-type methods with large-scale EMR data mining methods, which can identify phenotype comorbidities and exposure risks26–28, may identify compound phenotypes for genetic study and may elucidate the mechanism of action behind risk variants.
This analysis was performed on data from 13,835 individuals; as a result, the study is underpowered to make many discoveries, given that the power to detect a finding in PheWAS is determined by the minor allele frequency, the effect size and the prevalence of a phenotype within the population. We envision a future with much larger genotyped populations linked to longitudinal EMRs. Such a population will facilitate detection of associations between rare diseases and dissection of genetic influences on prognosis, responses to medication and comorbidity risk. In addition to the samples available in the eMERGE Network, large-scale EMR-linked genomic efforts are underway at Kaiser Permanente29, the Million Veterans Program and the UK Biobank30. Collectively, these efforts will soon involve >1 million patients with dense genotype data. Significant challenges to analyzing the associated EMR data remain; such challenges can be met in part through enhanced data collection within the EMR, as mandated through the “meaningful use standards” enacted by the Health Information Technology for Economic and Clinical Health (HITECH) Act31, and through improved mining of EMR data, including use of natural language processing32,33.
The NHGRI Catalog is composed of SNP-phenotype associations that were reported in research studies that employed a wide variety of study designs, including observational cohorts and controlled trials. Given the nature of this study, we were not able to take into account the nuances of the phenotype definition underlying a particular SNP-phenotype association. Many research studies use narrowly defined phenotypes, with strict inclusion criteria for both cases and controls. That we failed to replicate some of these associations with data derived from the heterogeneous environment of clinically indicated testing and disease coding is not surprising. Many of the phenotypes represented in this PheWAS were diseases that are surrogates for a physiological phenotype; as such, the PheWAS phenotypes are a stand-in for health outcomes sufficiently poor to trigger healthcare interventions. These surrogates include morbid obesity (PheWAS) for BMI (GWAS) and osteoporosis (PheWAS) for decreased bone mineral density (GWAS).
Limitations counsel caution in interpretation of this study. First, subsequent work is needed to verify our potentially novel associations beyond the skin findings, as some may be false positives. Given the number of known findings in the top 202 new associations (137), the false-positive rate could be estimated as high as 29%. Second, the current PheWAS efforts considered pleiotropy as multiple clinically apparent diseases and traits, which could classify as pleiotropic two traits that share underlying mechanisms. Detection of true pleiotropy for these SNPs would require larger data sets, statistical analyses of independence and potentially biological validation. Regardless, our analyses highlight phenotypes with common genetic underpinnings, and tests of independence demonstrate that our strongest associations were not a result of known comorbid associations. Third, PheWAS phenotypes were defined using billing codes, which have imperfect sensitivity and positive predictive value, owing to inherent variations in the coding scheme itself (i.e., how broadly or narrowly a code defines disease) and variation in how codes are assigned to patients. Such inaccuracies typically bias results toward the null hypothesis by reducing the magnitude of association. Our replication population for the novel skin associations, however, used a gold standard of pathologist-reviewed biopsy specimens. Methods leveraging multiple modalities of information in the EMR allow for more accurate distinction between phenotypes, such as types 1 and 2 diabetes15, and detection of rare phenotypes34 not defined by billing codes. However, these results demonstrate that large data sets can provide robustness to some phenotype misclassification using just billing codes, with many replicated associations for types 1 and 2 diabetes. Fourth, because our study used GWAS data, it shares limitations inherent to GWAS, such as the inability to pinpoint causal alleles and the general lack of inclusion of rare alleles. When calculating our power to detect an association, we used the strongest odds ratio reported in the NHGRI Catalog, which may overestimate the true effect size and thus falsely inflate our power to replicate. Indeed, the odds ratios for PheWAS associations were typically less than those found in the NHGRI Catalog (Supplementary Fig. 5), which likely represents both some degree of phenotype misclassification as well as a result of the “winner’s curse,” such that the GWAS in which the association was discovered often overestimates the true effect size35. Finally, we did not test SNPs in the NHGRI Catalog that would have required imputation, as that would have introduced another potential source of error into interpreting replication (or lack thereof) of a known association.
Another consideration when interpreting these data is that because the eMERGE population was selected from clinical populations, they were, in general, older and sicker than the general population (e.g., 66% had hypertension with a mean age of 69.5 years), due in part to the fact that the primary phenotypes that established eligibility for the cohort included diseases associated with advancing age, including Alzheimer’s disease, cataracts and peripheral vascular disease. It may be helpful to take into account age of disease onset when selecting control groups in future PheWAS analyses. Furthermore, the prevalence of disease is enriched relative to the general population at some eMERGE sites because of the individuals’ presentation for health care at academic medical centers.
An EMR-based, phenome-wide catalog of phenotypic associations for discovered genetic variants may increase the speed and efficiency of genetic exploration and may uncover pleiotropy, thereby aiding discovery of biological underpinnings of disease phenotypes and highlighting new research directions. In addition, EMR-linked genetic data may add clinical context to our growing knowledge of genomic diversity.
ONLINE METHODS
Study design
This study was performed in the eMERGE Network, a project sponsored by the National Human Genome Research Institute comprising five institutions (Group Health Cooperative, Marshfield Clinic, Mayo Clinic, Northwestern University and Vanderbilt University Medical Center) that each have DNA biorepositories linked to their EMR. Details of these biobanks and of the eMERGE Network have been published elsewhere14,36,37. All studies were approved by local Institutional Review Boards. Patients gave consent as part of the DNA biobanks at Group Health Cooperative, Marshfield Clinic, Mayo Clinic, Northwestern University; Vanderbilt uses an opt-out model as previously described and evaluated36,38. Biobank recruitment is nonrandom and based on clinic populations. Individuals with DNA linked to EMR records were selected for genome-wide genotyping based on being a case or control for one of five phenotypes (Supplementary Table 1); these data served as the genetic data for the initial GWAS. For the analyses reported here, 13,835 individuals passed quality control (QC) whose race was classified as “White” or “Caucasian” in the EMR or from self-reported data. The SNPs analyzed by PheWAS included all SNPs in the NHGRI Catalog, first downloaded on January 16, 2011. After development of the methods and phenotype categorization for performing PheWAS using billing and demographic data from the EMR, the catalog was updated to the April 17, 2012 version and the analysis rerun. PheWAS phenotypes that matched or nearly matched with NHGRI Catalog phenotypes were considered replications. Catalog phenotypes that were continuous measures (e.g., weight) were matched to the nearest PheWAS phenotype when a suitable match was available (e.g., obesity). Following alignment of PheWAS phenotypes with NHGRI Catalog phenotypes, PheWAS analysis was performed for all 3,144 SNPs available for testing (Supplementary Fig. 1).
Genotyping
Genotyping was performed at the Center for Genotyping and Analysis at the Broad Institute (for two eMERGE sites) and the Center for Inherited Disease Research at Johns Hopkins University (for the remaining three eMERGE sites) using the Human660W-Quadv1_A genotyping platform, consisting of 561,490 SNPs and 95,876 intensity-only probes on a total of 13,835 EMR-identified European-American subjects across each of the five eMERGE sites. Genotypes were merged across the five sites and cleaned using the QC pipeline developed by the eMERGE Genomics Working Group39. This process includes evaluation of sample and marker call rate, gender mismatch and anomalies, duplicate and HapMap concordance, batch effects, Hardy-Weinberg equilibrium (HWE), sample relatedness and population stratification (using STRUCTURE40 and EIGENSTRAT41). Relatedness was determined based on identity by descent (IBD) estimates generated from the genome-wide genotype data in PLINK.
522,164 SNPs passed the following QC criteria: SNP call rate >95%, sample call rate >99%, minor allele frequency >0.01, 99.99% concordance rate in duplicates, unrelated samples only, and individuals of European-descent only (based on STRUCTURE analysis of >90% probability of being in the CEU cluster, which was derived from Utah residents with ancestry from northern and western Europe). We flagged all markers with HWE P < 1 × 10−4 for further evaluation post-analysis using standard criteria. Finally, we selected SNPs that were also in the NHGRI Catalog (as of April 17, 2012), which yielded 3,144 SNPs (at any P-value); these were used for PheWAS analysis (described below). The QC and data analysis were performed using a combination of PLINK, PLATO and the R statistical package.
Organization of billing codes for PheWAS
In this study, we revised and expanded our earlier PheWAS phenotype categorization to a total of 1,645 phenotypes identified from International Classification of Disease, Ninth revision, Clinical Modification (ICD9) codes. (Our initial PheWAS phenotype categorization included 744 phenotypes9.) The ICD9 coding system is divided into four components: diseases, signs and symptoms (“three digit” codes, 001–999), external causes of injury (“E” codes), procedures (“two digit” codes 00.0–99.9) and supplemental classifications (“V” codes). The prior PheWAS code groupings included only diseases, signs and symptoms (three digit) ICD9 codes9. We revised and expanded the PheWAS phenotypes by (i) adding V codes (commonly used to record personal histories of given diseases) and E codes (which refer to external causes of injury) to the PheWAS code mapping, (ii) redesigning the code system to be hierarchical, such that one phenotype could be a parent of another subphenotype (e.g., cardiac arrhythmias is a parent of atrial fibrillation, atrial flutter and other arrhythmias), and (iii) including more granular phenotypes into the coding system (e.g., “type 1 diabetes with ketoacidosis”). Creation of hierarchical phenotypes included creation of phenotypes not present in the ICD9 billing hierarchy, such as “inflammatory bowel disease” as the parent phenotype for “Crohn’s disease” and “ulcerative colitis.” In this process, we were guided by the hierarchical organization of the Clinical Classifications Software (CCS) produced by the Agency for Healthcare Research and Quality42; the 2011 version of the CCS contains 727 phenotypes. The resulting PheWAS code group currently contains 1,645 phenotypes, 1,358 of which had at least 25 cases (a prevalence of 0.18% in our data set) in the eMERGE cohort, our threshold for these analyses. The current version of the PheWAS codes, with ICD9 mappings and control groups, is available from http://knowledgemap.mc.vanderbilt.edu/research/content/phewas.
Replication of NHGRI Catalog associations using PheWAS phenotypes
The NHGRI Catalog was downloaded and parsed into a local database. We selected all SNP-phenotype associations that contained (i) SNPs that were present and passed QC in our genotyping platform and (ii) phenotypes that can be represented in the PheWAS phenotypes, and attempted to replicate these by PheWAS analysis. We did not analyze phenotypes occurring in fewer than 25 eMERGE patients, and we did not impute the genetic data before aligning with catalog SNPs.
Two authors (L.B. and J.C.D.), working together, mapped each NHGRI Catalog phenotype to a PheWAS phenotype using review of the original papers and data supplements. NHGRI Catalog phenotypes were divided into continuous (e.g., weight, height, laboratory values) and binary traits (e.g., disease status). Because many phenotypes studied in the NHGRI Catalog are traits (such as height or C-reactive protein levels) that are not represented by the current PheWAS phenotype definition, NHGRI Catalog traits were mapped into similar corresponding traits and classified into one of four match types: (i) exact match (binary traits that match a PheWAS disease); (ii) PheWAS phenotypes related to a catalog continuous trait (e.g., obesity for catalog traits “body mass index” or “waist circumference”; iron-deficiency anemia for catalog trait “serum iron levels”); (iii) PheWAS phenotypes that were either broader or narrower than their NHGRI Catalog counterparts (e.g., Alzheimer’s disease for Alzheimer’s subtypes); (iv) catalog phenotypes that lack corresponding PheWAS phenotypes (e.g., hair and eye color, height, medication response phenotypes, age at menopause). The complete listing of NHGRI Catalog diseases and their associated PheWAS phenotypes are found in Supplementary Table 3.
For replication analysis, we considered only those SNPs that were associated with at least one phenotype at P ≤ 5 × 10−8 in the NHGRI Catalog, were tested in a population that contained individuals of European ancestry, and had phenotypes testable in the list of PheWAS phenotypes (e.g., height is not a PheWAS phenotype at the current time as it is not a billable diagnosis; see above); 673 SNPs met these criteria. Each SNP-phenotype association test was run independently with PLINK43 using logistic regression adjusted for age, gender, site (e.g., Vanderbilt, Marshfield Clinic, etc.), and the first three principal components as calculated by EIGENSTRAT, using ancestry informative markers chosen from the entire set of GWAS SNPs41. Analysis was performed assuming an additive genetic model. These data were aggregated and analyzed using Perl scripts and the R statistical package. To count as a replication, a phenotype had to have P ≤ 0.05 and a consistent direction of effect. Directional consistency was tested for using the listed allele in the NHGRI Catalog and review of the original papers for those associations for which an allele was not listed or was unclear in the catalog. The direction of effect was obtained from the primary analysis in all but 5 of the 215 possible replicated associations (those with P < 0.05); we considered these 5 associations, with unknown directions of effect, replicated.
Phenome-wide analysis to detect novel associations
We combined the entire cohort of self-identified European American individuals identified across the five eMERGE sites (n = 13,835 individuals) into one analysis. To define diseases, we queried all ICD9 codes from the respective EMRs from the five eMERGE sites. The PheWAS software then used these ICD9 codes to classify each person as having one of the 1,358 possible clinical phenotypes belonging to >25 patients in the populations (as noted above). For each disease, the PheWAS code defined relevant control groups for each disease or finding, such that patients with related diseases do not serve as controls for that disease (e.g., a patient with Graves disease cannot serve as a control for an analysis of thyroiditis).
We have previously found that the positive predictive value for some algorithms to establish a diagnosis from EMR data is improved by requiring the presence of multiple instances of disease-associated ICD9 codes44. For example, to be considered a case for tuberculosis, a patient is required to have at least two ICD9 codes in the ranges of 10–18 (tuberculosis infections of different sites), 137 (late effects of tuberculosis) or V12.01 (personal history of tuberculosis). Accordingly, for the present study, we used a threshold of relevant ICD9 codes on two distinct days to establish that person as a “case” for a given phenotype. Controls are patients without any ICD9 codes in the corresponding control range; thus, patients with a single ICD9 case code are excluded for the analysis as neither a case nor a control. Each SNP-phenotype association test was run independently with PLINK43, using logistic regression adjusted for age, gender, site (e.g., Vanderbilt, Marshfield Clinic), and the first three principal components as calculated by EIGENSTRAT, using ancestry informative markers as above41. Analysis was performed assuming an additive genetic model. These data were aggregated and analyzed using Perl scripts and the R statistical package.
Categorization of PheWAS results
All PheWAS associations at P < 4.6 × 10−6 not in the NHGRI Catalog were manually reviewed by three authors (J.R.F., L.B. and J.C.D.) to categorize them as: (i) a catalog replication (“replicated”), (ii) a known finding not in the NHGRI Catalog or in LD with a SNP known to be associated with that disease (“known”), (iii) a phenotype association related to a known finding (“related”; for example, “disorders of lipoid metabolism” is a parent phenotype of “hyperlipidemia”) or (iv) a previously unreported finding. Categorizing related findings is particularly useful because of the hierarchical nature of the PheWAS phenotypes: if a ‘child’ phenotype (e.g., “atrial fibrillation”) is associated with a SNP, the ‘parent’ phenotype (“cardiac dysrhythmias”) may also be associated. To look for known associations, we reviewed the NHGRI Catalog and queried for the SNP in PubMed. We also generated a list of SNPs in LD using SNAP45 and searched for these. SNP-phenotype associations not found in any of these steps were considered previously unreported and reviewed by three authors (L.B., J.R.F. and J.D.).
Replication analysis for actinic keratosis, seborrheic keratosis and nonmelanoma skin cancer
We sought to replicate seven SNP-phenotype associations identified by PheWAS in an independent population with previous GWAS-level genotyping; this population consisted of 7,406 individuals derived from the Vanderbilt EMR-linked DNA biobank (BioVU) with data from Illumina OMNI 1M or 5M BeadChips. There is no overlap between these subjects and those included in the Vanderbilt eMERGE data set used for the primary analysis. QC on this set was performed as above. For analysis of variants in IRF4, HERC2, CDK10, CDK5RAP1 and TYR, all 7,406 were eligible. TERT rs2853676 is not assayed on the OMNI 5M platform, and no suitable SNPs in strong LD passed QC; for this reason, 5,515 patients (all genotyped on the OMNI 1M) were used for the TERT analysis with seborrheic keratosis.
Natural language processing was used to identify cases through extraction of pathology results for each diagnosis and recognition of these diagnoses from the physician-maintained problem list, which is unstructured text. Phenotype algorithms based on natural language processing allowed us to divide the nonmelanoma skin cancer phenotype into the more specific phenotypes and squamous cell carcinoma; ICD9 codes did not distinguish between basal cell carcinoma and squamous cell carcinoma until 2012. Controls were selected among the population of genotyped patients who did not have a diagnosis, by ICD9 code, for actinic keratosis, seborrheic keratosis, basal cell carcinoma or squamous cell carcinoma. To evaluate algorithm-positive predictive value, 50 randomly selected cases and 50 randomly selected controls were reviewed, in a random order, by a physician not associated with algorithm development to validate accuracy of the determinations. The physician was blinded to the algorithmic determination. Review criteria to certify a true case required presence of pathology results, a dermatologist assertion of the diagnosis, or a primary care physician asserting the diagnosis with treatment and date data (indicative of past or outside dermatologist involvement). The genetic association analysis was performed using logistic regression assuming an additive genetic model adjusted for age and sex.
ROC curve analysis
Using all SNPs with at least one adequately powered association (beta > 0.8), we created a reference standard of associations to perform ROC analysis (Supplementary Fig. 3). We restricted the list of phenotypes to those PheWAS phenotypes also studied in the NHGRI Catalog at the time of this study. True positives were any phenotype association with that SNP that was either directly mentioned in the NHGRI Catalog, or was in LD (r2 > 0.8) with another SNP in the Catalog that was associated with the phenotype. We selected as true negatives any PheWAS phenotype studied in the NHGRI Catalog and not associated with the SNP or a SNP in LD. PheWAS phenotypes not studied in the NHGRI Catalog were considered neither a true positive nor true negative, as there was no reference standard by which to evaluate them. This resulted in a total of 2,146 SNP phenotype associations evaluated for 29 SNPs. An ROC curve was generated using the −log(P-value) as the threshold variable.
Statistical analysis
The primary outcome for this analysis was the extent of replication of known findings in the NHGRI Catalog that achieved genome-wide significance (P < 5 × 10−8) in a prior GWAS and for which we were adequately powered to detect a difference. For this analysis, we used P = 0.05 to determine significance of a replication, and tested for directional consistency with the original trait (see above). For binary traits recorded in the NHGRI Catalog, we determined our power to replicate a known association based on the minor allele frequency taken from the eMERGE population, the expected effect size (i.e., odds ratio) taken from the largest effect size for that association in the NHGRI Catalog, and the number of cases of that phenotype in the eMERGE population (using the PheWAS-defined phenotypes). All sample size calculations were based on 80% power. We set alpha at 0.05, given that each of the tested replications has been previously established at genome-wide significance in the NHGRI Catalog. Power was calculated using Quanto46.
To test the probability of replicating X out of Y tested NHGRI Catalog associations at alpha = 0.05, we calculated based on the probability of drawing P-values randomly from a normal distribution with at least X of them having P ≤ 0.05 (X being the number of replicated associations). Thus, the probability of getting X SNP-phenotype associations replicated (P ≤ 0.05) out of Y tested SNP-phenotype associations is:
where P = 0.05 and C(Y,X) represents the number of combinations among Y items selecting X. The calculation was performed using the R pbinom method.
Our second outcome was identification of new phenotype associations. The threshold for significance to determine new associations was established using an FDR of 0.10, calculated using the Benjamini & Hochberg47 method using the R p.adjust method. (A Bonferroni correction for all tested associations would be P = 0.05/1,358/3,144 = 1.2 × 10−8.) All tests of association were performed with PLINK as indicated above using logistic regression adjusted for age, sex and the first three principal components. All reported P-values for both discovery and replications assumed a two-sided analysis.
To test for heterogeneity among SNPs and phenotypes with similar associations (in Fig. 4), we calculated the variation across SNPs and across each phenotype as I2 using METAL48. We compared both the different phenotypes across the same SNP (to test whether SNPs demonstrated differential effects on different phenotypes) and the same phenotype across different SNPs (to test whether different SNPs had different influences on a phenotype). I2 is calculated using the following formula:
Where Q is Cochran’s heterogeneity statistic and df is the degrees of freedom.
Supplementary Material
Acknowledgments
This work was supported by the eMERGE Network, initiated and funded by the National Human Genome Research Institute (NHGRI), with additional funding from the National Institute of General Medical Sciences (NIGMS), through the following grants: U01-HG004610 and U01-HG006375 (Group Health Cooperative/University of Washington); U01-HG004608 (Marshfield Clinic); U01-HG004599 and U01-HG006379 (Mayo Clinic); U01-HG004609 and U01-HG006388 (Northwestern University); U01-HG006389 (Essentia Institute of Rural Health/Marshfield Clinic); U01-HG004603 and U01-HG006378 (Vanderbilt University); and U01-HG006385 (Vanderbilt University serving as the Coordinating Center). Funding support for eMERGE genotyping was provided by NHGRI through the grants: U01-HG004424 (The Broad Institute) and U01-HG004438 (Johns Hopkins University, Center for Inherited Disease Research). Replication genotypes were derived from a pharmacogenomics resource supported by NIGMS RC2-GM092318. Development of the PheWAS method is also supported by R01-LM010685 from the National Library of Medicine. BioVU received and continues to receive support through the National Center for Research Resources UL1 RR024975, which is now the National Center for Advancing Translational Sciences, 2 UL1 TR000445. Additional support for this work at the University of Washington was partially provided by the National Center for Advancing Translational Sciences grant UL1TR000427.
Footnotes
Accession codes. dbGaP: Phs000360.
AUTHOR CONTRIBUTIONS
The experiment was conceived by J.C.D., L.B., D.R.M., and D.M.R. J.C.D. and L.B. designed the final PheWAS algorithm, phenotype classification and matching to NHGRI Catalog phenotypes. L.B. performed the PheWAS. Statistical analysis was performed by J.C.D., L.B., R.J.C. and J.D.M. eMERGE Phenotype algorithms were developed primarily by D.S.C., A.N.K. and J.C.D. Novel phenotype algorithms for skin phenotypes were generated and executed by J.C.D., L.B. and R.Z. and evaluated by J.D.M. S.A.P. performed power calculations. J.R.F., J.C.D. and L.B. reviewed the literature for previous publications for each SNP. Genetic quality control and the merged data set were performed by M.D.R. with input from D.C.C., D.R.C. and J.L.H. Data were provided by D.S.C., P.L.P., A.N.K., J.A.P., L.V.R., D.R.C., P.K.C., J.P., S.J.B. and M.A.B. J.C.D., L.B. and D.M.R. drafted the manuscript, with substantial revision and direction by D.R.M., J.L.H., D.C.C., M.D.R. and J.R.F. Guidance and critical revision were provided by T.A.M. and L.A.H. All authors edited the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Hindorff LA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helgadottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 3.Helgadottir A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–224. doi: 10.1038/ng.72. [DOI] [PubMed] [Google Scholar]
- 4.Lees CW, Barrett JC, Parkes M, Satsangi J. New IBD genetics: common pathways with other diseases. Gut. 2011;60:1739–1753. doi: 10.1136/gut.2009.199679. [DOI] [PubMed] [Google Scholar]
- 5.Cotsapas C, et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. 2011;7:e1002254. doi: 10.1371/journal.pgen.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramos PS, et al. A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PLoS Genet. 2011;7:e1002406. doi: 10.1371/journal.pgen.1002406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10:241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- 8.Sivakumaran S, et al. Abundant pleiotropy in human complex diseases and traits. Am J Hum Genet. 2011;89:607–618. doi: 10.1016/j.ajhg.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denny JC, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–1210. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pendergrass SA, et al. Phenome-wide association study (PheWAS) for detection of pleiotropy within the Population Architecture using Genomics and Epidemiology (PAGE) Network. PLoS Genet. 2013;9:e1003087. doi: 10.1371/journal.pgen.1003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denny JC, et al. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenome-wide studies. Am J Hum Genet. 2011;89:529–542. doi: 10.1016/j.ajhg.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ritchie MD, et al. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127:1377–1385. doi: 10.1161/CIRCULATIONAHA.112.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hebbring SJ, et al. A PheWAS approach in studying HLA-DRB1*1501. Genes Immun. 2013;14:187–191. doi: 10.1038/gene.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCarty CA, et al. The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics. 2011;4:13. doi: 10.1186/1755-8794-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kho AN, et al. Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome-wide association study. J Am Med Inform Assoc. 2012;19:212–218. doi: 10.1136/amiajnl-2011-000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klompas M, et al. Automated detection and classification of type 1 versus type 2 diabetes using electronic health record data. Diabetes Care. 2012 doi: 10.2337/dc12-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He C, et al. MHC fine mapping of human type 1 diabetes using the T1DGC data. Diabetes Obes Metab. 2009;11 (suppl 1):53–59. doi: 10.1111/j.1463-1326.2008.01003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plenge RM, et al. TRAF1–C5 as a risk locus for rheumatoid arthritis–a genomewide study. N Engl J Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka T, et al. A genome-wide association analysis of serum iron concentrations. Blood. 2010;115:94–96. doi: 10.1182/blood-2009-07-232496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kolz M, et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5:e1000504. doi: 10.1371/journal.pgen.1000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDavid A, et al. Enhancing the power of genetic association studies through the use of silver standard cases derived from electronic medical records. PLoS ONE. 2013;8:e63481. doi: 10.1371/journal.pone.0063481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dokal I. Dyskeratosis congenita. Hematology (Am Soc Hematol Educ Program) 2011;2011:480–486. doi: 10.1182/asheducation-2011.1.480. [DOI] [PubMed] [Google Scholar]
- 23.Han J, et al. A germline variant in the interferon regulatory factor 4 gene as a novel skin cancer risk locus. Cancer Res. 2011;71:1533–1539. doi: 10.1158/0008-5472.CAN-10-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gudbjartsson DF, et al. ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nat Genet. 2008;40:886–891. doi: 10.1038/ng.161. [DOI] [PubMed] [Google Scholar]
- 25.Kanetsky PA, et al. Does MC1R genotype convey information about melanoma risk beyond risk phenotypes? Cancer. 2010;116:2416–2428. doi: 10.1002/cncr.24994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanauer DA, Rhodes DR, Chinnaiyan AM. Exploring clinical associations using ‘-omics’ based enrichment analyses. PLoS ONE. 2009;4:e5203. doi: 10.1371/journal.pone.0005203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roque FS, et al. Using electronic patient records to discover disease correlations and stratify patient cohorts. PLoS Comput Biol. 2011;7:e1002141. doi: 10.1371/journal.pcbi.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rzhetsky A, Wajngurt D, Park N, Zheng T. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci USA. 2007;104:11694–11699. doi: 10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffmann TJ, et al. Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics. 2011;98:79–89. doi: 10.1016/j.ygeno.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collins R. What makes UK Biobank special? Lancet. 2012;379:1173–1174. doi: 10.1016/S0140-6736(12)60404-8. [DOI] [PubMed] [Google Scholar]
- 31.Blumenthal D, Tavenner M. The ‘meaningful use’ regulation for electronic health records. N Engl J Med. 2010;363:501–504. doi: 10.1056/NEJMp1006114. [DOI] [PubMed] [Google Scholar]
- 32.Friedman C, Shagina L, Lussier Y, Hripcsak G. Automated encoding of clinical documents based on natural language processing. J Am Med Inform Assoc. 2004;11:392–402. doi: 10.1197/jamia.M1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilke RA, et al. The emerging role of electronic medical records in pharmacogenomics. Clin Pharmacol Ther. 2011;89:379–386. doi: 10.1038/clpt.2010.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denny JC, Arndt FV, Dupont WD, Neilson EG. Increased hospital mortality in patients with bedside hippus. Am J Med. 2008;121:239–245. doi: 10.1016/j.amjmed.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 35.Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- 36.Roden DM, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84:362–369. doi: 10.1038/clpt.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarty CA, Wilke RA, Giampietro PF, Wesbrook SD, Caldwell MD. Marshfield Clinic Personalized Medicine Research Project (PMRP): design, methods and recruitment for a large population-based biobank. Pers Med. 2005;2:49–79. doi: 10.1517/17410541.2.1.49. [DOI] [PubMed] [Google Scholar]
- 38.Pulley J, Clayton E, Bernard GR, Roden DM, Masys DR. Principles of human subjects protections applied in an opt-out, de-identified biobank. Clin Transl Sci. 2010;3:42–48. doi: 10.1111/j.1752-8062.2010.00175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turner S, et al. Quality control procedures for genome-wide association studies. Curr Protoc Hum Genet. 2011;68:1.19. doi: 10.1002/0471142905.hg0119s68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 42.Cowen ME, et al. Casemix adjustment of managed care claims data using the clinical classification for health policy research method. Med Care. 1998;36:1108–1113. doi: 10.1097/00005650-199807000-00016. [DOI] [PubMed] [Google Scholar]
- 43.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ritchie MD, et al. Robust replication of genotype-phenotype associations across multiple diseases in an electronic medical record. Am J Hum Genet. 2010;86:560–572. doi: 10.1016/j.ajhg.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson AD, et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gauderman WJ. Sample size requirements for association studies of gene-gene interaction. Am J Epidemiol. 2002;155:478–484. doi: 10.1093/aje/155.5.478. [DOI] [PubMed] [Google Scholar]
- 47.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;125:289–300. [Google Scholar]
- 48.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.