

Abstract
Oxygen deprivation followed by reoxygenation causes pathological responses in many disorders, including ischemic stroke, heart attacks and reperfusion injury. Key aspects of ischemia-reperfusion can be modeled by a C. elegans behavior, the O2-ON response, which is suppressed by hypoxic preconditioning or inactivation of the O2-sensing HIF (hypoxia-inducible-factor) hydroxylase EGL-9. From a genetic screen, we found that the cytochrome P450 oxygenase CYP-13A12 acts in response to the EGL-9/HIF-1 pathway to facilitate the O2-ON response. CYP-13A12 promotes oxidation of polyunsaturated fatty acids into eicosanoids, signaling molecules that can strongly affect inflammatory pain and ischemia-reperfusion injury responses in mammals. We propose that roles of the EGL-9/HIF-1 pathway and cytochrome P450 in controlling responses to anoxia-reoxygenation are evolutionarily conserved.
Ischemia-reperfusion-related disorders, such as strokes and heart attacks, are the most common causes of adult deaths worldwide (1). Blood delivers O2 and nutrients to target tissues, and ischemia results when the blood supply is interrupted. The restoration of O2 from blood flow after ischemia, known as reperfusion, can exacerbate tissue damage (2). How organisms prevent ischemia-reperfusion injury is poorly understood. Studies of the nematode C. elegans led to discovery of an evolutionarily conserved family of O2-dependent enzymes (EGL-9 in C. elegans and EGLN2 in mammals) that hydroxylate the HIF transcription factor and link hypoxia to HIF-mediated physiological responses (3–7). Exposure to chronic low concentrations of O2 (hypoxic preconditioning) or direct inhibition of EGLN2 strongly protects mammals from stroke and ischemia-reperfusion injury (2, 8, 9). Similarly, EGL-9 inactivation in C. elegans blocks a behavioral response to reoxygenation, the O2-ON response (characterized by a rapidly increased locomotion speed triggered by reoxygenation after anoxia) (10, 11), which is similar to mammalian tissue responses to ischemia-reperfusion: (i) reoxygenation drives the O2-ON response and is the major pathological driver of reperfusion injury, (ii) hypoxic preconditioning can suppress both processes, and (iii) the central regulators (EGL-9/HIF) of both processes are evolutionarily conserved. How the EGL-9/HIF-1 and EGLN2/HIF pathways control the O2-ON response and ischemia-reperfusion injury, respectively, is largely unknown.
To seek EGL-9/HIF-1 effectors important in the O2-ON response, we performed an egl-9 suppressor screen for mutations that can restore the defective O2-ON response in egl-9 mutants (fig. S1A). We identified new alleles of hif-1 in this screen; because EGL-9 inhibits HIF-1, hif-1 mutations suppress the effects of egl-9 mutations (10). We also identified mutations that are not alleles of hif-1 (Figs. 1A–1C and fig. S1B). hif-1 mutations recessively suppressed three defects of egl-9 mutants: the defective O2-ON response, defects in egg-laying and the ectopic expression of the HIF-1 target gene cysl-2 (previously called K10H10.2) (fig. S1C) (10, 12). By contrast, one mutation, n5590, dominantly suppressed the O2-ON defect but did not suppress the egg-laying defect or the ectopic expression of cysl-2∷GFP (Figs. 1D, 1E and fig. S2). n5590 restored the sustained phase (starting at 30s post-reoxygenation) better than it did the initial phase (within 30s post-reoxygenation) (Figs. 1A–1C). egl-9; hif-1; n5590 triple mutants displayed a normal O2-ON response, just like the wild type and egl-9; hif-1 double mutants (fig. S1D). Thus, n5590 specifically suppresses the egl-9 defect in the sustained phase of the O2-ON response.
Fig. 1. n5590 suppresses the defect of egl-9 mutants in the O2-ON response.
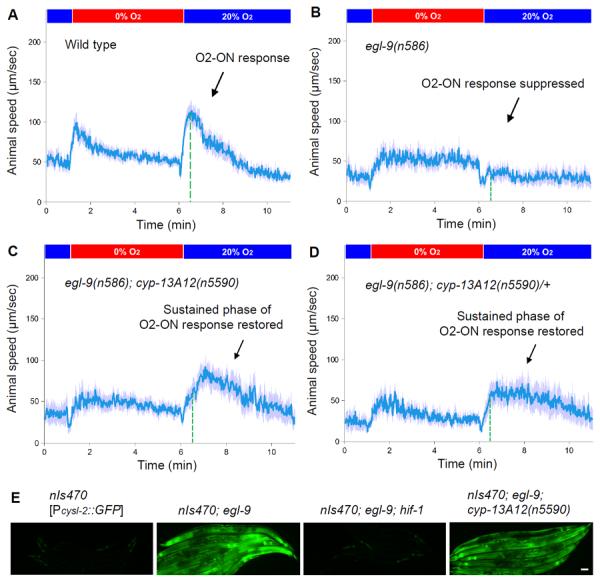
(A) Speed graph of wild-type animals, showing a normal O2-ON response. Average speed values ± 2 SEMs (blue) of animals (n > 50) are shown with step changes of O2 between 20% and 0% at the indicated times. The mean speed within 0–120 s after O2 restoration is increased compared with that before O2 restoration (p < 0.01, one-sided unpaired t-test). The dashed green line indicates the approximate boundary (30s post-reoxygenation) between the initial and sustained phases of the O2-ON response. (B) Speed graph of egl-9(n586) mutants, showing a defective O2-ON response. (C) Speed graph of egl-9(n586); cyp-13A12(n5590) mutants, showing a restored O2-ON response mainly in the sustained phase (right of the dashed green line). The mean speed within 30–120 s after O2 restoration was significantly higher than that of egl-9(n586) mutants (p <0.01). (D) Speed graph of egl-9(n586); cyp-13A12(n5590)/+ mutants, showing a restored O2-ON response in the sustained phase. (E) hif-1 but not cyp-13A12(n5590) suppressed the expression of cysl-2∷GFP by egl-9(n586) mutants. GFP fluorescence micrographs of 5–7 worms aligned side by side carrying the transgene nIs470 [Pcysl-2∷GFP] are shown. Scale bar, 50 μm.
We genetically mapped n5590 and identified an M46I missense mutation in the gene cyp-13A12 by whole-genome sequencing (Fig. 2A, fig. S3A and Table S1A). Decreased wild-type cyp-13A12 gene dosage in animals heterozygous for a wild-type allele and the splice acceptor null mutation gk733685, which truncates the majority of the protein, did not recapitulate the dominant effect of n5590 (Fig. 2B). gk733685 homozygous mutants similarly did not recapitulate the effect of n5590 (Fig. 2C). Thus, n5590 does not cause a loss of gene function. By contrast, increasing wild-type cyp-13A12 gene dosage by overexpression restored the sustained phase of the O2-ON response (Fig. 2D), and RNAi against cyp-13A12 abolished the effect of n5590 (Fig. 2E). We conclude that n5590 is a gain-of-function allele of cyp-13A12.
Fig. 2. n5590 is a gain-of-function allele of cyp-13A12.
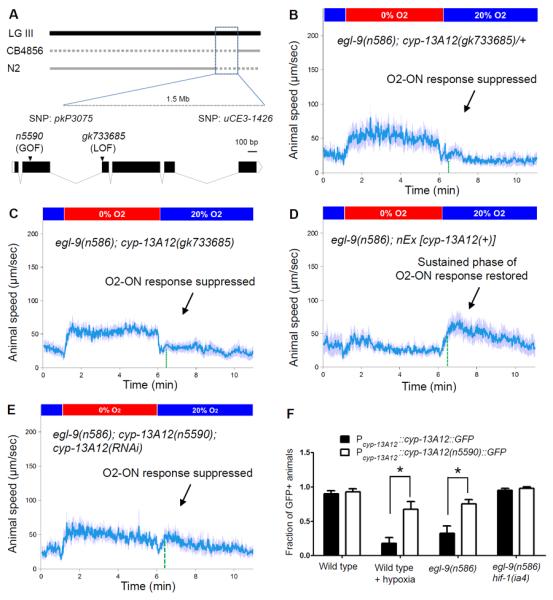
(A) Genetic mapping positioned n5590 between the SNPs pkP3075 and uCE3-1426. Solid grey lines indicate genomic regions for which recombinants exhibited a defective O2-ON response, thus excluding n5590 from those regions. The locations of n5590 and gk733685 are indicated in the gene diagram of cyp-13A12. (B) Speed graph of egl-9(n586); cyp-13A12(gk733685)/+ animals, showing a defective O2-ON response. (C) Speed graph of egl-9(n586); cyp-13A12(gk733685) mutants, showing a defective O2-ON response. (D) Speed graph of egl-9(n586); nEx [cyp-13A12(+)] animals, showing a restored O2-ON response in the sustained phase (right of the dashed green line). (E) Speed graph of egl-9(n586); cyp-13A12(n5590); cyp-13A12(RNAi) animals, showing a suppressed O2-ON response. (F) Fractions of animals expressing CYP-13A12∷GFP or CYP-13A12(n5590)∷GFP (* p<0.01, two-way ANOVA with Bonferroni's test, n=4).
cyp-13A12 encodes a cytochrome P450 oxygenase (CYP). CYPs can oxidize diverse substrates (13–15). The C. elegans genome contains 82 CYP genes, at least two of which are polyunsaturated fatty acid (PUFA) oxygenases that generate eicosanoid signaling molecules (fig. S3B) (16, 17). The closest human homolog of CYP-13A12 based on BLASTP scores is CYP3A4 (fig. S4). We aligned the protein sequences of CYP-13A12 and CYP3A4 and found that n5590 converts methionine 46 to an isoleucine, the residue in the corresponding position of normal human CYP3A4 (fig. S4). Methionines can be oxidized by free radicals, which are produced in the CYP enzymatic cycle, rendering CYPs prone to degradation (18, 19). Using transcriptional and translational GFP-based reporters, we identified the pharyngeal marginal cells as the major site of expression of cyp-13A12 (fig. S5) and observed that the abundance of CYP-13A12∷GFP protein was decreased by prolonged hypoxic preconditioning and also decreased in egl-9 but not in egl-9; hif-1 mutants (Fig. 2F and fig. S5). The n5590 mutation prevented the decrease in CYP-13A12∷GFP abundance by hypoxia or egl-9. Thus, n5590 acts, at least in part, by restoring the normal abundance of CYP-13A12, which then promotes the O2-ON response in egl-9 mutants.
We tested whether CYP-13A12 was normally required for the O2-ON response in wild-type animals. The cyp-13A12 null allele gk733685 abolished the sustained phase of the O2-ON response; the initial phase of the O2-ON response was unaffected (Fig. 3A). A wild-type cyp-13A12 transgene fully rescued this defect (Fig. 3B). A primary role of CYP-13A12 in the sustained phase of the O2-ON response explains the incomplete rescue of the defective O2-ON response of egl-9 mutants by n5590 during the initial phase (Fig. 1C). The activity of most and possibly all C. elegans CYPs requires EMB-8, a CYP reductase that transfers electrons to CYPs (20). No non-CYP EMB-8 targets are known. emb-8(hc69) causes a temperature-sensitive embryonic lethal phenotype. We grew emb-8(hc69) mutants at the permissive temperature to the young-adult stage. A shift to the non-permissive temperature simultaneously with E. coli-feeding RNAi against emb-8 nearly abolished the O2-ON response (Figs. 3C and 3D) (Both the hc69 mutation and RNAi against emb-8 were required to substantially reduce the level of EMB-8 (17).) CYP-13A12 is thus required for the sustained phase of the O2-ON response, and one or more other CYPs likely act with CYP-13A12 to control both phases of the O2-ON response.
Fig. 3. Requirement of CYP-13A12 for a normal O2-ON response.

(A) Speed graph of cyp-13A12(gk733685) loss-of-function mutants, showing an O2-ON response with a normal initial phase but a diminished sustained phase (left and right, respectively, of the dashed green line). (B) Speed graph of cyp-13A12(gk733685) mutants with a rescuing wild-type cyp-13A12 transgene, showing the O2-ON response with a normal initial phase and sustained phase. The mean speed within 30–120 s after O2 restoration was higher than that of cyp-13A12(gk733685) mutants (p <0.01, one-sided unpaired t-test, n >50). (C) Speed graph of emb-8(hc69) mutants growing at the permissive temperature of 15°C with simultaneous E. coli feeding RNAi against emb-8, showing a normal O2-ON response. (D) Speed graph of emb-8(hc69) mutants growing post-embryonically at the restrictive temperature of 25°C with simultaneous E. coli feeding-RNAi against emb-8, showing a reduced O2-ON response.
CYP oxygenases define one of three enzyme families that can convert PUFAs to eicosanoids, signaling molecules that affect inflammatory pain and ischemia-reperfusion responses of mammals (15, 21–23); the other two families, cyclooxygenases and lipoxygenases, do not appear to be present in C. elegans (17, 24). To test whether eicosanoids are regulated by EGL-9 and CYP-13A12, we used high-performance liquid chromatography (HPLC) coupled with mass spectrometry (MS) to profile steady-state amounts of 21 endogenous eicosanoid species from cell extracts of wild-type, egl-9(n586) and egl-9(n586); cyp-13A12(n5590) strains. Only free eicosanoids have potential signaling roles (21, 22, 24), so we focused on free eicosanoids. The egl-9 mutation caused a markedly decreased overall amount of free eicosanoids, while the total amount of eicosanoid, including both free and membrane-bound fractions, was unaltered (Fig. 4A and fig. S6). Among the eicosanoids profiled, 17,18-DiHEQ (17,18-diolhydroxyeicosatetraenoic acid) was the most abundant species (fig. S6B). 17,18-DiHEQ is the catabolic hydrolase product of 17,18-EEQ (17,18-epoxyeicosatetraenoic acid), an epoxide active in eicosanoid signaling (25). Free cytosolic 17,18-EEQ and 19-hydroxyeicosatetraenoic acid (19-HETE) were present in the wild type but undetectable in egl-9 mutants (Figs. 4C–4F). egl-9(n586); cyp-13A12(n5590) mutants exhibited partially restored free overall eicosanoid levels as well as restored levels of 17,18-EEQ and 19-HETE (Figs. 4A–4F and fig. S6B). Thus, both EGL-9 and CYP-13A12 regulate amounts of free cytosolic eicosanoids.
Fig. 4. Modulation of eicosanoid concentrations by EGL-9 and CYP-13A12.

(A) Overall levels of free eicosanoids, calculated by adding the values of the profiled 21 eicosanoids in the wild type and egl-9(n586) and egl-9(n586); cyp-13A12(n5590) strains. (B) Schematic illustrating the conversion of arachidonic acid (AA, 20:4n-6) to 19-HETE and of EPA (20:5n-3) to 17,18-EEQ by CYPs. (C) Quantification of 19-HETE and 17,18-EEQ concentrations in the wild type and egl-9(n586); cyp-13A12(n5590) and egl-9(n586) mutant strains. Amounts of free (membrane unbound) forms of 17,18-EEQ and 19-HETE from extracts of age-synchronized young adult hermaphrodites are shown. p < 0.01, one-way ANOVA post hoc test, n = 3. Error bars are SEMs. (D–F) Representative HPLC-MS traces indicating free 17,18-EEQ levels based on the spectrograms of three MS samples: (D) wild type, (E) egl-9(n586), and (F) egl-9(n586); cyp-13A12(n5590). Peaks of 17,18-EEQ at its transition m/z (mass-to-charge ratio) were measured and extracted (MassHunter). The x-axis shows the retention time (minutes); the y-axis shows the abundance (counts), with specific integral values over individual peaks indicated above each peak. (G) Speed graph of fat-2 mutants, showing a defective O2-ON response. Animals were supplemented with the solvents used in (H) as a control. (H) Speed graph of fat-2 mutants, showing the O2-ON response rescued by C20 PUFA (AA) supplementation. (I–J) Model of how EGL-9 and CYPs control the O2-ON response under (I) normoxic conditions and (J) conditions of hypoxic preconditioning or in egl-9 mutants (see text for details). The light blue indicates low protein activity, low amounts of eicosanoids or a defective O2-ON response.
We tested whether the O2-ON response requires PUFAs, which are CYP substrates and eicosanoid precursors. PUFA-deficient fat-2 and fat-3 mutants (26) exhibited a complete lack of the O2-ON response, although the acceleration in response to anoxia preceding the O2-ON response was normal (Fig. 4G and figs. S7A–S7C). The defective O2-ON response of fat-2 mutants was restored by feeding animals the C20 PUFA arachidonic acid (Fig. 4H) but not oleate, a C18 monounsaturated fatty acid that is processed by FAT-2 to generate C20 PUFAs (fig. S7D). These results demonstrate an essential role of PUFAs for the O2-ON response.
We suggest a model in which CYPs, which are strictly O2-dependent (27, 28), generate eicosanoids to drive the O2-ON response (Figs. 4I and fig. S8). In this model, EGL-9 acts as a chronic O2-sensor, so that during hypoxic preconditioning, the O2-dependent activity of EGL-9 is inhibited, HIF-1 is activated and unknown HIF-1 up-regulated targets decrease CYP protein abundance. The low abundance of CYPs defines the hypoxic preconditioned state. Without hypoxic preconditioning, CYPs generate eicosanoids, which drive the O2-ON response. By contrast, with hypoxic preconditioning or in egl-9 mutants, the CYP amounts are insufficient to generate eicosanoids and the O2-ON response is not triggered. Neither C20 PUFAs nor overexpression of CYP-29A3 restored the defective O2-ON response of egl-9 mutants (figs. S9 and S10), indicating that this defect is unlikely caused by a general deficiency in C20 PUFAs or CYPs. Since the O2-ON response requires EMB-8, a general CYP reductase, but only the sustained phase requires CYP-13A12, we propose that CYP-13A12 and other CYPs act as acute O2 sensors and produce eicosanoids, which are short-lived and act locally (22) during reoxygenation to signal nearby sensory circuits that drive the O2-ON response.
In humans, a low uptake of PUFAs or an imbalanced ratio of ω3/ω6 PUFAs is associated with elevated risk of stroke, cardiovascular disease and cancer (21, 23, 29, 30). Cytochrome P450s and eicosanoid production also have been implicated in mammalian ischemia-reperfusion (15, 21). Nonetheless, the causal relationships among and mechanisms relating O2 and PUFA homeostasis, CYP and PUFA-mediated cell signaling and organismal susceptibility to oxidative disorders are poorly understood. We identify a novel pathway in which EGL-9/HIF-1 regulates CYP-eicosanoid signaling, demonstrate that PUFAs confer a rapid response to reoxygenation via CYP-generated eicosanoids and provide direct causal links among CYPs, PUFA-derived eicosanoids, and an animal behavioral response to reoxygenation. As molecular mechanisms of O2 and PUFA homeostasis are fundamentally similar and evolutionarily conserved between nematodes and mammals (7, 11, 26), we suggest that the C. elegans O2-ON response is analogous to the mammalian tissue/cellular response to ischemia-reperfusion injury and that the principle of CYP-mediated regulation and the molecular pathway including EGL-9/HIF-1 and CYPs in controlling responses to anoxia-reoxygenation are evolutionarily conserved.
Supplementary Material
Acknowledgments
We thank C. Bargmann, A. Fire, A. Hart, Y. Iino, J. Powell-Coffman and C. Rongo for reagents; the Caenorhabditis Genetics Center and the Million Mutation Project for strains. H.R.H. is an Investigator of the Howard Hughes Medical Institute and the David H. Koch Professor of Biology at MIT. Supported by NIH grant GM24663 (H.R.H), German Research Foundation grant ME2056/3–1 (R.M.), NSF Graduate Research Fellowship (N.B.), MIT Undergraduate Research Opportunities Program (S.Z.) and a Helen Hay Whitney Foundation postdoctoral fellowship (D.K.M.).
References and Notes
- 1.Go AS, et al. Circulation. 2012 Dec 12; [Google Scholar]
- 2.Eltzschig HK, Eckle T. Nat Med. 2011;17:1391. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epstein AC, et al. Cell. 2001 Oct 5;107:43. [Google Scholar]
- 4.Kaelin WG, Jr., Ratcliffe PJ. Mol Cell. 2008 May 23;30:393. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Trent C, Tsung N, Horvitz HR. Genetics. 1983 Aug;104:619. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semenza GL. Cell. 2012 Feb 3;148:399. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powell-Coffman JA. Trends Endocrinol Metab. 2010 Jul;21:435. doi: 10.1016/j.tem.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 8.Semenza GL. Biochim Biophys Acta. 2011 Jul;1813:1263. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aragones J, et al. Nat Genet. 2008 Feb;40:170. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- 10.Ma DK, Vozdek R, Bhatla N, Horvitz HR. Neuron. 2012 Mar 8;73:925. doi: 10.1016/j.neuron.2011.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma DK, Ringstad N. Front Biol. 2012 Jun;7:246. doi: 10.1007/s11515-012-1219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Budde MW, Roth MB. Genetics. 2011 Oct;189:521. doi: 10.1534/genetics.111.129841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson DR, et al. Pharmacogenetics. 1996 Feb;6:1. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Gotoh O. Mol Biol Evol. 1998 Nov;15:1447. doi: 10.1093/oxfordjournals.molbev.a025872. [DOI] [PubMed] [Google Scholar]
- 15.Gottlieb RA. Arch Biochem Biophys. 2003 Dec 15;420:262. doi: 10.1016/j.abb.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Kosel M, et al. Biochem J. 2011 May 1;435:689. doi: 10.1042/BJ20101942. [DOI] [PubMed] [Google Scholar]
- 17.Kulas J, Schmidt C, Rothe M, Schunck WH, Menzel R. Arch Biochem Biophys. 2008 Apr 1;472:65. doi: 10.1016/j.abb.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Berlett BS, Stadtman ER. J Biol Chem. 1997 Aug 15;272:20313. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 19.Zangar RC, Davydov DR, Verma S. Toxicol Appl Pharmacol. 2004 Sep 15;199:316. doi: 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 20.Rappleye CA, Tagawa A, Le Bot N, Ahringer J, Aroian RV. BMC developmental biology. 2003 Oct 3;3:3. doi: 10.1186/1471-213X-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szefel J, et al. Curr Mol Med. 2011 Feb;11:13. doi: 10.2174/156652411794474374. [DOI] [PubMed] [Google Scholar]
- 22.Wymann MP, Schneiter R. Nat Rev Mol Cell Biol. 2008 Feb;9:162. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- 23.Chapkin RS, Kim W, Lupton JR, McMurray DN. Prostaglandins Leukot Essent Fatty Acids. 2009 Aug-Sep;81:187. doi: 10.1016/j.plefa.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panigrahy D, Kaipainen A, Greene ER, Huang S. Cancer Metastasis Rev. 2010 Dec;29:723. doi: 10.1007/s10555-010-9264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold C, et al. J Biol Chem. 2010 Oct 22;285:32720. doi: 10.1074/jbc.M110.118406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watts JL. Trends Endocrinol Metab. 2009 Mar;20:58. doi: 10.1016/j.tem.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harder DR, et al. Circ Res. 1996 Jul;79:54. doi: 10.1161/01.res.79.1.54. [DOI] [PubMed] [Google Scholar]
- 28.Ward JP. Biochim Biophys Acta. 2008 Jan;1777:1. doi: 10.1016/j.bbabio.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Gerber M. Br J Nutr. 2012 Jun;107(Suppl 2):S228. doi: 10.1017/S0007114512001614. [DOI] [PubMed] [Google Scholar]
- 30.Mozaffarian D, Wu JH. J Am Coll Cardiol. 2011 Nov 8;58:2047. doi: 10.1016/j.jacc.2011.06.063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.