

Abstract
Certain amphipathic α-helical peptides can functionally mimic many of the properties of full-length apolipoproteins, thereby offering an approach to modulate high-density lipoprotein (HDL) for combating atherosclerosis. In this Perspective, we summarize the key findings and advances over the past 25 years in the development of peptides that mimic apolipoproteins, especially apolipoprotein A-I (apoA-I). This assemblage of information provides a reasonably clear picture of the state of the art in the apolipoprotein mimetic field, an appreciation of the potential for such agents in pharmacotherapy, and a sense of the opportunities for optimizing the functional properties of HDL.
INTRODUCTION
Atherosclerosis is a disease that involves the hardening of arteries due to the accumulation of plaque on the inside of blood vessel walls. This chronic process of plaque deposition, which ultimately interferes with or blocks the flow of blood, thereby inducing tissue ischemia, is responsible across the globe for most cases of heart disease and a high incidence of human deaths.1 To treat atherosclerosis, the general standard of care calls for a low-fat, low-cholesterol diet along with drugs that lower serum cholesterol levels.2 The “statins”, which inhibit HMG-CoA reductase, a key enzyme in the cholesterol biosynthesis pathway, are widely prescribed to treat hypercholesterolemia, particularly elevated serum low-density lipoprotein-cholesterol (LDL-C), to decrease the risk of heart attack or stroke.3 While statin therapy can reduce LDL-C by up to 50–60%, some people are resistant to the positive effects.3–5 Since most patients on statins will take them for life, the side effects may prove troublesome, as statins can have the following tolerability issues: muscle pain and damage, liver problems, digestive problems, rash or flushing, blood glucose elevation, and memory loss or confusion.3,5 Other agents in medical use are fibrates, niacin, bile acid resins, and ezetimibe.6–8
An alternative strategy for combating atherosclerosis is the modulation of high-density lipoprotein (HDL),8–14 to increase its plasma levels and/or its ability to transport cholesterol. The process of reverse cholesterol transport (RCT), which removes excess cholesterol from peripheral tissues and delivers it to the liver for elimination, is greatly facilitated by HDL.15–17 In addition, HDL exhibits atheroprotective properties due to its antioxidant and anti-inflammatory activity.18,19 Experimental studies have consistently shown that administration of HDL or apolipoprotein A-I (apoA-I), the major protein component of HDL, significantly reduces atherosclerosis in animal models.20–40 In humans, positive results have been observed in clinical studies involving intravenous administration of human apoA-I or its Milano variant.41–51
Although HDL-targeted therapies have attracted considerable attention lately, questions remain about how best to harness the potential of HDL for medical applications. For example, recent clinical trials with niacin and cholesterol ester-transfer protein (CETP) inhibitors (dalcetrapib and torcetrapib) failed to show cardiovascular benefits, despite an increase in total plasma HDL levels.52–54 Also, a recent meta-analysis challenged previous epidemiological findings that higher total plasma HDL levels lower the risks for cardiovascular disease.55 These findings and others56–58 suggest that simply raising HDL levels is not sufficient to protect against atherosclerosis. Rather, it would seem that HDL functional properties have to be seriously taken into account,54,56–58 such that specific subtypes of HDL particles or specific HDL functions may be more important than a high level of total plasma HDL (i.e., quality vs. quantity).9,15,59 In this sense, boosting RCT activity ought to be a key factor in enhancing the atheroprotection of HDL.60–66 Altogether, several mechanisms are responsible for the antiatherogenic properties of HDL, including promotion of cholesterol efflux from cells, antioxidant properties, and anti-inflammatory effects.15–19
In seeking agents that modulate HDL function, a salient consideration is the heterogeneity of HDL particles, which vary in size (diam. = 7–13 nm), shape (discoidal or spherical), and density (ρ = 1.06–1.20 g/mL).15,59,67–69 These nanoparticles exist in constant dynamic flux as part of a complex “lipoproteostasis” network, in which they undergo a remodeling process that encompasses the influx, efflux, or modification of constituent lipids, cholesterol, and small molecules, mediated in some instances by specific proteins and enzymes (Figure 1).10,13 Five distinct HDL particle sizes have been identified by nondenaturing gradient gel electrophoresis (NDGGE): HDL2b (diam. = 9.7–13.0 nm), HDL2a (8.8–9.7 nm), HDL3a (8.2–8.8 nm), HDL3b (7.8–8.2 nm), and HDL3c (7.2–7.8 nm).70 Through remodeling, the specific populations of HDL species across the HDL size spectrum are defined, and the diverse small molecules and proteins contained within the HDL particles are altered. Presently, the differences in function for discrete HDL subtypes are poorly understood; however, it is clear that the smallest, most dense HDL particles (called lipid-poor or pre-beta HDL) are crucial for combating atherosclerosis. These “guardian angels of the arterial wall”71 are preferred substrates for certain key enzymes, thus predisposing them to absorb cholesterol and transport it away from peripheral tissues.62,72 Consequently, the remodeling of mature, large HDLs into lipid-poor, small HDLs, along with the promotion of RCT, would constitute a central feature of promising atherosclerosis therapies.
Figure 1.

Schematic depicting HDL species, HDL remodeling, cholesterol flux and reverse cholesterol transport, and the influence of apoA-I mimetic peptides (indicated by the blue arrows). ApoA-I is secreted by the liver and intestine in the form of a lipid-free or lipid-poor protein. Lipid-poor apoA-I is also formed by the fragmentation of HDL or LDL particles. Lipid-free and lipid-poor apoA-I bind phospholipids and cholesterol from cellular membranes in a process mediated by ABCA1. The resulting small, lipid-poor pre-β HDL particles gradually increase in size as the result of cholesterol uptake and esterification, catalyzed by LCAT, leading to the formation of a cholesterol ester-rich lipid core. The larger, spherical HDL species can accept phospholipids and free cholesterol from peripheral tissues via ABCG1. Ultimately, cholesterol esters are offloaded from HDL particles into hepatocytes via scavenger receptor B1 (SR-B1), or are transferred to LDL in a process mediated by CETP. The actions of PLTP and various lipases lead to plasma HDL remodeling by promoting particle fusion or fragmentation. Abbreviations: ABCA1/ABCG1, ATP-binding cassette transporter protein A1/G1; CETP, cholesterol ester transfer protein; LDL, low-density lipoproteins; LCAT, lecithin–cholesterol acyltransferase; LDLR, low-density lipoprotein receptor; PLTP, phospholipid transfer protein; SR-B1, scavenger receptor B1.
The primary protein component (~70%) of HDL particles is apoA-I, a highly lipophilic, 243-aminoacid protein (~28 kDa) with a secondary structure consisting of 10 conserved amphipathic α-helices, eight 22-mers and two 11-mers.10,73 The strong interaction between apoA-I and phospholipids is promoted by the amphipathic α-helix structures in apoA-I, which possess a hydrophilic face and a hydrophobic face (vide infra).74–76 These helices are stabilized by contact with themselves and/or with lipids, and the structure of lipid-associated apoA-I in the discoidal or spherical HDL particles reflects such stabilizing interactions.77 Most of the eight 22-mer segments display a “class A” α-helical motif, which is defined by a specific distribution of nonpolar and charged/polar amino acid residues (vide infra).78,79
Endogenously, apoA-I is secreted in the form of a lipid-free or lipid-poor (pre-β) protein. Lipid-poor apoA-I is also formed by the remodeling of circulating HDL particles. Lipid-deficient apoA-I accepts phospholipids and cholesterol from peripheral tissues in a process mediated by the ATP-binding cassette transporter protein ABCA1.10,13 The resulting discoidal HDL particles, originally ~7 nm in size, become enlarged due to cholesterol uptake that is mediated by ABCA1 or ABCG1. HDL cholesterol is esterified by lecithin-cholesterol acyltransferase (LCAT) leading to larger, spherical particles with a cholesterol ester-rich lipid core. Ultimately, cholesterol is off-loaded from HDL particles into hepatocytes via scavenger receptor B-1 (SR-B1), or into LDL particles under mediation by CETP. Thus, in the overall RCT process cholesterol is mobilized and transported from peripheral tissues to the liver (Figure 1).10,13
ApoA-I plays a key role in HDL biogenesis, function, and structural dynamics. Compelling evidence for the antiatherogenicity of apoA-I derives from observations that i.v. infusions of apoA-I or reconstituted HDL particles, or over-expression of apoA-I, confer protective effects.20–51 Unfortunately, the use of apoA-I itself as a therapeutic agent has faced serious challenges stemming from its high cost of manufacture and its lack of oral bioavailability. Fortunately, there may be useful alternatives in the form of apoA-I mimetic peptides.
Functional apoA-I mimetic peptides adopt a class A, amphipathic α-helical structure75,78,80 that mimics apoA-I by modulating the properties of HDL. Helical peptides of this type exhibit many biologically useful functions, particularly strong lipid-associating ability,81–83 activation of enzymes involved in HDL remodeling,84–86 promotion of cholesterol efflux,83,86–88 binding of oxidized lipids,89 anti-inflammatory effects,90 and inhibition of atherosclerosis in mice91 (vide infra). Studies with various peptides have shown that a balance between peptide–peptide and peptide–lipid interactions is needed for optimal biological activity,92 with antioxidant and anti-inflammatory properties also being important.92 A diversity of peptide sequences have shown efficacy as apolipoprotein mimetics,92–104 including those with no homology to natural apolipoproteins and peptides composed of D-amino acids (vide infra). Five small, human clinical trials involving mimetic peptides have been described: two with a lipid formulation of the 22-mer H-PVLDLFRELLNELLEALKQKLK-OH (1; ETC-642),105 derived from the apoA-I sequence, two with the 18-mer 4F,106 and one with D4F (enantiomer of 4F).107 These studies were principally aimed at characterizing safety, pharmacokinetics (PK), and pharmacodynamics, although a significant decrease in HDL inflammatory index was reported with D4F.107 Thus, short, synthetic, apolipoprotein mimetic peptides with amphipathic α-helical structures, which are potentially less costly to produce than full-length proteins, can recapitulate many of the protective functions associated with apoA-I.108–114
It is important to gain an appreciation for the common physicochemical and biological assays that are used to characterize the biophysical and functional properties of apoA-I analogues and peptide mimetics. A gauge of the lipid-associating propensity for a specific molecular entity can be obtained by measuring the rate and extent of lipid clearance when incubating a turbid suspension of lipid vesicles with the test molecule.115 The degree of association is related to clearance of the cloudy sample, whereby the suspended vesicles are dispersed by formation of smaller protein–lipid or peptide–lipid nanoparticles. To determine the capacity to promote cholesterol efflux, macrophage cells are loaded with radiolabeled or fluorescently labeled116 cholesterol and treated with the agent of interest (protein, peptide, or serum from a dosed animal). Then, the levels of cholesterol in the cells and the media are separately measured after an incubation period.88,117 This assay can be carried out with or without prior activation of ABCA1, to determine if the efflux is mediated by this transporter. Rader et al. developed an in vivo extension of the cholesterol efflux assay, in which 3H-cholesterol-laden macrophages are injected intraperitoneally into mice, followed by measurement of radioactivity in the plasma, liver, bile salts, and other tissues.118 One mechanism for promoting RCT involves increasing the levels of lipid-poor HDL (i.e., pre-βHDL) in the plasma (a process called HDL remodeling), since these small nanoparticles serve as the primary acceptor for free cholesterol from ABCA1 in an early step of RCT. The typical HDL remodeling assay entails treating plasma with the molecule of interest (in vitro or in vivo), after which the plasma sample is subjected to NDGGE to separate the HDL subspecies, followed by immunoblotting for apoA-I.119 More recently, a monoclonal antibody that is specific for the conformation of apoA-I in pre-β HDL has been employed in an ELISA method that provides a more quantitative measure of pre-β HDL concentrations.119 Cell-free and cell-based assays have been used to assess the antioxidant and anti-inflammatory properties of HDLs in compound-treated plasma samples (in vitro or in vivo). The cell-free assay for antioxidant activity monitors the ability of HDLs to either prevent the formation of or inactivate oxidized lipids, by measuring the conversion of dichlorofluorescin to dichlorofluorescein.120 Anti-inflammatory activity has been assessed by the cell-based monocyte chemotaxis assay (MCA), which involves a coculture of aortic endothelial cells and smooth muscle cells in the presence of human sera and LDL. Reactive oxygen species (ROS) induce the production of monocyte chemotactic protein 1 (MCP-1), leading to migration of monocytes into the subendothelial space (by chemotaxis), which is inhibited by inclusion of HDL (or other antioxidants).121 Finally, compounds can be assayed for their efficacy in preventing the development or progression of atherosclerotic lesions in animal models. Mice genetically predisposed to develop atherosclerosis, such as apoE-null or LDL receptor (LDLr)-null animals, are commonly used, being dosed for an extended time period, such as 6–10 weeks. At the end of this regime, the mice are sacrificed and the area and/or volume of atherosclerotic lesions in the aortic region are evaluated.
Given the high degree of pharmaceutical interest in HDL-modulating drugs, and the dearth of exposure to apolipoprotein mimetic peptides in the medicinal chemistry community, the time is ripe for presenting a Perspective on this field. In this review we summarize the key findings and advances over the past 25 years and establish the current state of knowledge. Additionally, we present some research results from our own laboratory at Scripps. This assemblage of information should provide a reasonably clear picture of the state of the art in the apolipoprotein mimetic field, as well as a sense of the potential of such agents in pharmacotherapy.
APOLIPOPROTEIN A-I AND ITS PEPTIDE FRAGMENTS
Apolipoprotein A-I, as the major protein component of HDL particles, accounts for about 70% of the HDL protein mass.10,73 Also, it is the most abundant apolipoprotein in humans, with a high plasma concentration of about 130 mg/dL (ca. 45 μM).10 In HDL particles, apoA-I serves key structural and functional roles, especially in mediating the formation of HDL species to transport lipids, as part of the RCT pathway. ApoA-I directs the evolution of HDL particles for off-loading and eliminating excess cholesterol via the liver, which prevents the chronic accumulation of cholesterol in the arteries. Agents that mimic the functional properties of apoA-I could therefore furnish useful therapeutics for treating atherosclerosis. In this section, we review the nature of apoA-I in some detail, along with structure–function relationships of apoA-I-derived peptides.
The mature 28-kDa human protein, containing 243 amino acids, has a secondary structure defined by 10 conserved amphipathic α-helices: eight 22-mers and two 11-mers (vide infra).10,73 The plasma half-life of apoA-I is around four days,10 with the main sites of catabolism and elimination being the liver and kidney. Lipid-rich apoA-I can be removed from plasma, along with mature HDL or LDL particles, by hepatic HDL holoparticle receptors or LDL receptors, respectively, whereas lipid-poor apoA-I can be removed via glomerular filtration in the kidneys. Given the complexity of the HDL life cycle, apoA-I metabolism is influenced by many receptors, lipid-transfer proteins, and enzymes, such that its plasma stability and concentration are greatly affected by apoA-I gene mutations, as well as by various proteins associated with the evolution of HDL particles. Generally, any mutations that prevent the lipidation of HDL particles will reduce plasma levels of apoA-I, since the kidneys readily filter the small, lipid-free protein.122
ApoA-I and its peptide fragments (as well as apoA-I mimetic peptides) can be “reconstituted” into HDL-like nanoparticles, called rHDL.123 The most common method for preparing rHDL particles is the cholate dialysis method, in which phospholipid vesicles, apolipoproteins or mimetic peptides, and cholate (as detergent) are mixed, followed by extensive dialysis to remove most of the cholate from the assembled rHDLs. Alternatively, some molecules that possess inherent detergent-like properties, such as certain apoA-I fragments and mimetic peptides, can generate rHDL particles simply by being mixed with phospholipid vesicles (no cholate necessary). The resultant nanoparticles are often purified by using size-exclusion chromatography (SEM) or ultracentrifugation, and are typically characterized by using transmission electron microscopy (TEM).
Secondary and Tertiary Structure of ApoA-I
The secondary structure of apoA-I, like that of apoA-II, apoA-IV, apoC-I, apoC-II, and apoE, is predominantly α-helical.10 In 1977, three groups independently reported that the apoA-I sequence contains multiple, amphipathic α-helical segments,124–126 of which the minimal subunit is 11 residues. Human apoA-I contains two 11-residue and eight 22-residue segments, the latter having each arisen from tandem duplication of two 11-residue subunits. The repetitive, 11-mer subunits have been rationalized on the basis of 3.6 amino acids per turn in a standard α-helix, since 11 residues completes three turns. By the same token, tandem duplication to 22-mer subunits provides six complete helical turns with little twisting between the adjacent helices.78–80 The helical segments are often interrupted by a proline, which is thought to permit the conformational flexibility needed to develop the morphology of the various HDL particles. In human apoA-I, the ten helical segments constitute residues 44–65, 66–87, 88–98, 99–120, 121–142, 143–164, 165–186, 187–208, 209–219, and 220–241 (Figure 2).73
Figure 2.

Primary amino acid sequence of apoA-I and selected apoA-I mimetic peptides. The sequence of apoA-I is broken up by helical segments, with Pro residues highlighted.
Analysis of protein sequences for many species73 indicates that helix 7, located in the center of apoA-I and involved in interactions with ABCA1 and LCAT, is the most conserved subunit, while helix 10 is the least conserved. Deletion of helix 10, the subunit with the highest lipid affinity,127 from human apoA-I is very impactful in that this abolishes ABCA1-mediated cholesterol efflux128 and impairs the binding to lipids.129,130 However, it is interesting that apoA-I in two monkey species, orangutan and cottontop tamarin, is missing helices 9 and 10.73 Since apoA-I does not contain cysteines, it cannot form intramolecular or intermolecular disulfide bonds; so, self-dimerization is not an issue. In addition, there are no known post-translational modifications, such as glycosylation or phosphorylation. Three of ten prolines and three of seven tyrosines are conserved across 31 species, probably to maintain important structural, lipid binding, and antioxidant features.73
Most of the α-helical segments in apoA-I (and other apolipoproteins) have a class A structural motif, as first described in 1974 by Segrest and coworkers.131 Class A α-helices are defined by an amphipathic structure in which the cationic residues are clustered at the polar/nonpolar interface and the anionic residues are located near the center of the polar region (Figure 3).80 In particular, on the polar face there is a distinct cluster of positively charged amino acids [e.g., Lys (K) and Arg (R)] at the polar/nonpolar boundary of the helix and negatively charged amino acids [e.g., Asp (D) and Glu (E)] at the center of the polar face.78–80 Studies involving model peptides have shown that the amphipathic α-helix is the minimal lipid-associating domain of apolipoproteins and that those with a class A topology bind to lipids with high affinity.80,132 The strong lipid affinity has been ascribed to the cationic Lys and Arg residues being able to bury the hydrophobic portion of their side chains into the membrane while extending the terminal charged groups into the aqueous phase, proximate to phospholipid head groups (a process called “snorkeling”).80 Four of the helices in apoA-I (3, 4, 9, 10) are class Y helices, which exhibit high lipid affinity and are similar to class A helices, but possess an additional cluster of positive residues at the center of the polar face.
Figure 3.

Comparative representations of 18-residue apoA-I mimetic peptides 2F and 4F, and the 22-residue apoA-I consensus peptide. Top panel: Edmunson helical wheel diagrams with the amino acids (single letter code) numbered starting at the N-terminus with “1” (hydrophobic, acid, basic, and uncharged polar residues are denoted by gray, blue, red, and green, respectively). The helices exhibit a class A topology, having basic residues at the polar–nonpolar interface (positions 4/15/9/13 in the 18-residue peptides) and acidic residues in the central region of the polar face (positions 8/1/12/16). Positions 3 and 14 change in going from 2F to 4F (L→F). Middle panel: Electrostatic surface maps of the polar faces of the peptides, again showing the class A helix topology consisting of basic residues in the interfacial region and acidic residues at the center of the polar face. Bottom panel: Space-filled molecular models of the hydrophobic face of the peptides. The highlighted residues 3 and 14 change in going from 2F to 4F, while substitution of position 13 in the consensus peptide with a hydrophic residue affords improved lipid binding affinity and cholesterol efflux efficiency (see text for details). Color code: red, polar acidic; blue, polar basic; green, polar uncharged; white, nonpolar.
The tertiary structure of human apoA-I is defined by two domains: an N-terminal helical bundle (residues 1–180), and a less ordered C-terminal lipid-associating segment (residues 181–243). Details of the tertiary structure are somewhat ill-defined, owing to the conformational flexibility of the globular, lipid-free protein. It is known that the hydrophobic C-terminal domain is required to initiate lipid binding,130 with subsequent opening of the four-helix bundle allowing lipids to be enclosed in the nascent HDL particle. The N-terminal domain is more highly conserved across 31 species than the C-terminal domain.73 Mouse apoA-I, which shares 65% identity with the human protein, also folds into a similar, two-domain tertiary structure.133 A crystal structure for full-length, lipid-free, human apoA-I was reported in 2006,134 but that structure was cast into doubt in 2010 by allegations of fabricated data.135,136 Crystal structures of two truncated apoA-I variants have been reported.137,138
ApoA-I is fascinating due to its ability to exist in solution in a wide range of lipid-free to lipid-bound states, with varied conformations. In the lipid-free state, apoA-I aggregates at concentrations higher than ca. 0.1 mg/mL (ca. 0.3 μM), which may stabilize the structure.139,140 Chemical and thermal denaturation experiments indicated that the protein denatures with a relatively low free energy of 2.2–2.7 kcal/mol, consistent with sedimentation velocity studies,139 which point to major conformational heterogeneity and flexibility. The disparate structures that can be adopted by apoA-I underlie its function within HDL particles amidst lipoproteostasis and RCT (vide infra). For example, in the lipid-free/lipid-poor states, the hydrophobic regions of the amphipathic α-helices in apoA-I interact with each other to form a globular bundle, which is the primary substrate for ABCA1, thus transfering phospholipids and free cholesterol to apoA-I to generate nascent HDL (first step of RCT).141 On the other hand, LCAT interacts preferentially with apoA-I in discoidal HDL, rather than in lipid-free or mature HDL, to esterify HDL-associated cholesterol (second step of RCT).142 Considering this complex picture, it might seem highly improbable that short, α-helical, apolipoprotein mimetic peptides could reproduce the characteristics of native apoA-I. However, it is important to consider that mimetic peptides may act in part by enhancing or modulating the activities of native apoA-I, such as by increasing the levels of lipid-poor apoA-I or by improving the function of apoA-I.143,144
HDL Structure and Dynamics
HDL nanoparticles are highly heterogeneous, dynamic complexes, making them resistant to structure determination by X-ray crystallography or NMR. Accordingly, alternative techniques have been applied, often with reconstituted HDL (rHDL) particles that contain apoA-I as the sole protein. A variety of approaches have been used to elucidate the structural disposition of helices in rHDLs, including chemical cross-linking/mass spectrometry, fluorescence resonance-energy transfer (FRET), small-angle neutron scattering, electron paramagnetic resonance spectroscopy, and molecular dynamics.77,145–151 Despite some residual debate, these complementary approaches have led to a generally accepted notion that two or more apoA-I molecules wrap like a double belt around the edge of a phospholipid bilayer in an antiparallel fashion to form and stabilize discoidal lipid particles.152 The conformationally flexible apoA-I is thought to adjust to different particles sizes by rearranging to form a trefoil scaffold that maintains the underlying double-belt architecture.147 The HDL structures are characterized by rapidly interchanging, coexisting conformations of apoA-I, depending on the particle size and composition. The interested reader is directed to more comprehensive, recent discussions of HDL structure.77,145–151
As discussed in the Introduction, there is a broad spectrum of HDL particles, which undergo dynamic interchange as part of RCT (Figure 1). HDL nanoparticles have different structures and functions that are dictated not just by apoA-I, but by the various small-molecule and protein constituents. Another important apolipoprotein component is apoA-II. The proteomics of HDLs is very complex, with more than 100 proteins having been identified in HDL particles, although many are at relatively low levels compared to apoA-I.153–164 Thus, HDL particles are in dynamic flux in a complex lipoproteostasis network, in which remodeling defines the specific populations of HDL species across the HDL spectrum, as noted earlier (vide supra).10,13
While the differences in function of each HDL subspecies are not fully understood, it is clear that the lipid-poor particles (pre-β HDL) are crucial to combating atherosclerosis.71 Even though only 5–10% of circulating apoA-I exists in a lipid-poor/free state, this small transient population is thought to be particularly cardioprotective because it serves as the primary acceptor for free cholesterol from ABCA1 in the early, rate-limiting step of RCT.62,72 In fact, in a study of serum samples from 263 patients, de la Llera-Moya et al. found that the capacity to promote cholesterol efflux, a useful clinical metric of HDL function in vivo,61 correlated better with the concentration of lipid-poor HDL than with HDL-cholesterol levels.62 Many HDL-targeted therapies, including apoA-I mimetic peptides, seek to increase the plasma level of lipid-poor apoA-I. Nevertheless, it should be kept in mind that shifting the HDL distribution toward lipid-poor particles may have the unintended consequence of lowering the overall plasma concentration of apoA-I, by concomitantly favoring proteolysis of apoA-I and its removal by the kidneys.
Structure–Function Studies of ApoA-I Mutants and Fragments
There have been numerous studies aimed at determining structure–function relationships for the different regions of apoA-I, by using both apoA-I mutants and peptide fragments corresponding to segments of apoA-I.139,165 This subsection summarizes the findings of selected efforts in this vein, which may be instructive for the design apoA-I mimetic peptides. An important caveat for studies with apoA-I peptide fragments is that they may behave quite differently from the full-length protein, due to differences in aggregation state, intramolecular and intermolecular interactions, and absence of folding cooperativity. One notable aspect is that ABCA1, LCAT, and SR-B1 exhibit rather low protein–protein recognition specificity for apoA-I; instead, their effects are driven mainly by an amphipathic α-helix topology, as opposed to a specific amino acid sequence (vide infra). This lack of specificity is probably a key reason that peptides with diverse sequences can effectively mimic many of the functional properties of apoA-I.
Lipid binding
The two terminal apoA-I helices bind lipids with higher affinity than any of the central helices (Table 1, entries 1–8).81,127,166,167 From examining peptides that correspond to each of the 22-residue segments in apoA-I, only N-terminal helix 1, apoA-I(44–65), and C-terminal helix 10, apoA-I(220–241), were effective in a standard lipid-clearance assay.127 The N- and C-terminal peptides also proved to be the most membrane active by several other measures, including partitioning into (R)-(+)-1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC) liposomes and the exclusion pressure for penetrating an egg yolk phosphatidylcholine monolayer. In a follow-up study, Mishra et al. compared the lipid-associating properties of 22-mer peptides to a panel of 33-mer, 44-mer, and 55-mer peptides.81 Again, the most terminal, tandem helix segments, apoA-I(44–87) and apoA-I(209–241), were best able to bind to lipid (Table 1, entries 9–18). However, the full-length protein was more effective than any of the peptides in reducing the enthalpy of the lipid gel-to-liquid crystal phase transition. Thus, it was suggested that cooperative intermolecular and intramolecular interactions between apoA-I molecules and lipids stabilize the overall HDL structure.81 Considering that all of the apoA-I helices are similar amphipathic class A or class Y helices, it is surprising that only the peptides derived from the terminal helices show such high affinity for lipids. Nevertheless, these data are bolstered by other studies with synthetic peptides166,168 and by CNBr-fragmentation studies of apoA-I, which revealed that apoA-I(1–86) and apoA-I(149–243) associated with (R)-(+)-1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC) to form discoidal nanoparticles, but fragments apoA-I(87–112) and apoA-I(113–148) did not.169 Apparently, the central helices of apoA-I have evolved to bind to lipids more weakly, possibly to allow them more freedom to interact with each other or with proteins involved in HDL metabolism.81
Table 1.
Peptide Fragments Derived from ApoA-I
| entry | apoA-I helix | apoA-I peptides | AA’sa | DMPC MLV clearanceb | % chol effluxc |
|---|---|---|---|---|---|
| 1 | 1 | apoA-I(44–65) | 22 | 89% | 46 (27) |
| 2 | 2 | apoA-I(66–87) | 22 | <5% | 5 (0) |
| 3 | 4 | apoA-I(99–120) | 22 | <5% | 0 (0) |
| 4 | 5 | apoA-I(121–142) | 22 | <5% | 0 (0) |
| 5 | 6 | apoA-I(143–164) | 22 | <5% | 0 (0) |
| 6 | 7 | apoA-I(165–186) | 22 | <5% | 0 (0) |
| 7 | 8 | apoA-I(187–208) | 22 | <5% | 0 (0) |
| 8 | 10 | apoA-I(220–241) | 22 | 94% | 0 (2) |
| 9 | 1,2 | apoA-I(44–87) | 44 | 95% | 13 (9) |
| 10 | 2,3 | apoA-I(66–98) | 33 | <5% | 0 |
| 11 | 2,3,4 | apoA-I(66–120) | 55 | <5% | ND |
| 12 | 3,4 | apoA-I(88–120) | 33 | <5% | 0 |
| 13 | 4,5 | apoA-I(99–142) | 44 | <5% | 0 |
| 14 | 5,6 | apoA-I(121–164) | 44 | <5% | 0 |
| 15 | 6,7 | apoA-I(143–186) | 44 | <5% | 0 |
| 16 | 7,8 | apoA-I(165–208) | 44 | <5% | 0 |
| 17 | 8,9 | apoA-I(187–219) | 33 | <5% | 0 |
| 18 | 9,10 | apoA-I(209–241) | 33 | 95% | 66 (61) |
| 19 | WT protein | apoA-I(1–243) | 243 | 89%, 90% | 100 (100) |
Studies involving deletion variants of apoA-I and chimeric proteins further support the concept that the C-terminal helices are crucial for lipid association. Burgess et al. showed that the apoA-I mutant lacking residues 187–243 could not associate with cellular lipids, form lipoprotein particles, or promote cholesterol efflux.170 In rabbits, deletion of apoA-I residues 201–243, 217–243, or 226–243171 and 190–243129 resulted in higher rates of apoA-I catabolism and lower levels of mature HDL, consistent with impaired lipid binding. Furthermore, weak lipid association was observed for a chimeric protein in which residues 190–243 of apoA-I were replaced with helices from apoA-II (residues 12–77), even though apoA-II has a higher lipid-binding affinity than apoA-I.129 Engineered apoA-I mutants with modified hydrophobic residues in the last helix exhibited impaired binding to phospholipids compared to the wild-type protein.172 Thus, C-terminal helix 10, apoA-I(220–241), is critical for the initial association of apoA-I with lipids and the early formation of HDL particles.
ABCA1-mediated cholesterol efflux
Lipid-free and lipid-poor apoA-I species receive cholesterol and lipids from the ATP-binding cassette transporter ABCA1 in the initial, rate-limiting step of RCT.141,173 ABCA1 can transfer cholesterol with broad specificity to multiple HDL apolipoproteins, including apo A-I, A-II, C, E-3, and E-4.167,174 However, truncation mutants of apoA-I lacking helix 10 did not promote cholesterol efflux.128,175 Comprehensive studies with peptides corresponding to each of the apoA-I helical segments showed that, in general, the same apoA-I helical segments capable of lipid binding could also promote cholesterol efflux, albeit at 50–70% of the level of the full-length protein (Table 1).167 For a series of 33-mer peptides in which different helices from apoA-I were combined, Natarajan et al. found that ABCA1-dependent cholesterol efflux was best stimulated by a structure containing a linear array of negatively charged amino acids spanning the polar faces of two amphipathic α-helices.176 In other studies, single, class A amphipathic α-helices with no direct sequence relation to apoA-I promoted cholesterol efflux as well as apoA-I did.88,100 Interestingly, a peptide synthesized from all D-amino acids was as effective as the L-isomer in promoting cholesterol efflux, ruling out stereoselective requirements for cholesterol efflux.177 Thus, a lipid-binding, amphipathic α-helix is the major structural element required for promotion of cholesterol efflux by ABCA1.
LCAT-mediated cholesterol esterification
ApoA-I central helices 5, 6, and 7 have been implicated as the site of interaction between apoA-I and LCAT.178–184 Removing either helix 6 or 7 (residues 143–164) reduced both LCAT binding to rHDL and the kinetics of cholesterol esterification, with overall LCAT activity decreased by 50–95%.178–181 Minnich et al. found a near-complete impairment of esterification activity for an apoA-I deletion of helix 7 plus part of helix 6 (residues 148–186).182 Likewise, reversing the orientation of helix 6 by mutagenesis reduced LCAT activity, and replacing helix 6 with helix 10 somewhat restored LCAT activity, but with impaired binding and kinetics.183 Another study implicated apoA-I helix 6 (residues 140–150) as being essential for apoA-I to activate LCAT.184 Reduced LCAT activity was observed for apoA-I variants that exhibited impaired lipid binding, such as mutants of the C-terminal region, since the lipid-associated apoA-I conformation in discoidal HDL is a preferred LCAT substrate.74,182 Apolipoproteins other than apoA-I can activate LCAT, but only to about 30% of apoA-I.85,185 In a study of 22-mer and 44-mer peptide variants of the apoA-I consensus sequence, some peptides were as effective as the full-length protein in activating LCAT.85 A model 20-mer peptide lacking sequence homology to apoA-I was 65% as effective as apoA-I in LCAT activation.186 Thus, a specific amino acid sequence is not necessary for activation of LCAT; rather, an amphipathic α-helix with certain topological properties is sufficient. However, 22-mer peptides derived from the apoA-I consensus sequence that efficiently activated LCAT and associated with lipids did not significantly inhibit atherosclerosis in C57Bl/6J mice on a high-fat diet, indicating that these two features are not sufficient for in vivo antiatherosclerotic efficacy.187
SR-B1 and cholesterol off-loading
Scavenger receptor B1 (SR-B1) is the primary receptor for cholesterol off-loading from mature HDL particles to the liver (final step of RCT). SR-B1 binds HDLs with high affinity and then mediates the transfer of lipids from the nanoparticle to the cell membrane.188 SR-B1 is known to bind and off-load cholesterol from a variety of sources, including HDL, LDL, VLDL, and rHDL.188,189 Indeed, binding and cross-linking studies showed that SR-B1 interacts with a number of the apoA-I helices, as well as with a model class A, amphipathic peptide.190 ApoA-I variants with mutations in helix 4 (D102A/D103A) or helix 6 (R160V/H162A) could bind SR-B1 with affinity similar to wild-type apoA-I, but off-loaded cholesterol with less than 50% efficiency,191 leading to the idea that SR-B1 ligand binding and cholesterol off-loading are partially independent functions. Surprisingly, rHDL particles containing apoA-I mimetic peptide D4F off-loaded cholesteryl esters 20-fold more efficiently than rHDL containing wild-type apoA-I.192 This finding indicates a lack of sequence-dependent requirements (and lack of stereoselective demands) in the promotion of cholesteryl ester off-loading by SR-B1. Like ABCA1 and LCAT, SR-B1 does not require strict sequence dependence for interaction partners; rather it is activated by an amphipathic α-helix with certain topological properties.
Naturally Occurring Mutants and Dysfunctions of ApoA-I
Understanding the properties of natural apoA-I mutants can assist in the design of apoA-I mimetic peptides by teaching how the protein sequence relates to its functions. Likewise, by appreciating the various ways in which the protein can become dysfunctional in vivo via covalent modification, it may be possible to design improved mimetic peptides that evade these natural mechanisms of impairment.
Many naturally occurring apoA-I variants with mutations in the N-terminal helix-bundle domain are known. These mutations can be roughly categorized based on their position within the protein sequence: mutations in the first 100 amino acids are typically associated with amyloid formation, whereas mutations within the central region comprising helices 5–7 (residues 140–180) are mostly associated with defective interactions with LCAT. A recent X-ray crystal structure of a truncated apoA-I variant, Δ(185–243)apoA-I,137 provides a structural basis to rationalize apoA-I amyloidosis, since the sites of known amyloidogenic mutations corresponded to positions that stabilize the helix bundle.145 Fewer natural variants have been described having mutations in the disordered C-terminal domain, probably because this domain is more tolerant of mutations, as evidenced by its lower overall degree of sequence conservation.73 The interested reader may wish to consult some comprehensive reviews on naturally occurring apoA-I mutants.13,73,139
The best-known natural variant of apoA-I is apoA-I Milano (apoA-IM), which was described in 1980 after its discovery in the population of a small town in northern Italy.193 This variant corresponds to an R173C point mutation, located in helix 7 of the protein. Carriers of apoA-IM exhibit substantially reduced plasma levels of HDL, but nonetheless enjoy a lower risk for cardiovascular disease compared to those with wild-type apoA-I.194 Likewise, the apoA-I Paris mutant, which corresponds to an R151C point mutation in helix 6, is cardioprotective despite reduced plasma HDL levels.195 Intriguingly, the Paris and Milano mutations, which exhibit similar cardioprotective effects despite reduced HDL levels, are Arg→Cys substitutions located at the same position of a helical segment, but differing in which helix is affected. The apoA-IM variant undergoes faster proteolysis compared to normal apoA-I,194,196 which can explain the lower plasma HDL levels. On the other hand, the source of the cardioprotective properties of these variants is not well understood, although the introduction of cysteine imparts improved antioxidant properties and allows formation of disulfide homodimers or heterodimers with other apolipoproteins.197–199 ApoA-IM has been the subject of numerous in vivo studies, including human clinical trials, as detailed below. To our knowledge, there is no example of a mimetic peptide designed with Cys residues, in an effort to specifically recapitulate the beneficial properties of the apoA-I Milano or Paris mutants.
Great interest has developed for defining the function of various subspecies of HDL particles, given that improved HDL quality (i.e., function) may be more important than simply increased HDL quantity. As a corollary, it is important to have a sound grasp of HDL dysfunction, one source of which derives from chemical modification.200,201 The best-understood source of apoA-I modification is myeloperoxidase (MPO), a heme enzyme secreted by monocytes and artery wall macrophages that uses H2O2 to generate diverse oxidant species involved in the innate immune system.201 However, being abundant in atherosclerotic lesions, MPO also leads to the inadvertent oxidation of apoA-I. It is striking that apoA-I from cardiovascular patient plasmas or atherosclerotic plaques is enriched up to 500-fold in MPO-specific oxidation products, such as 3-chlorotyrosine and 3-nitrotyrosine.202 Of the seven Tyr residues in apoA-I, two are substantially modified by MPO: primarily Tyr192 in helix 8, and secondarily Tyr166.203 MPO was suggested to directly bind to apoA-I helix 8,202 but later studies indicated that a specific YXXK amino acid motif in apoA-I is responsible for the site specificity of the Tyr modifications.204,205 Importantly, the degree of modification at Tyr192 was strongly correlated with impaired ABCA1-mediated cholesterol efflux.203,206,207 The amino acids Met, Trp, Lys, and His are also known to undergo oxidation by MPO, but the functional outcomes here are less clear.201 Replacement of all four Trp residues in apoA-I with Phe yielded a fully functional protein that was resistant to MPO inactivation of ABCA1-dependent cholesterol efflux.208 The MPO oxidation of Met148 in apoA-I to the sulfoxide (Figure 4) resulted in an 85% decrease in LCAT activity, but this oxidative impairment was absent in apoA-I(M148L) mutant. These findings provide a strong impetus for efforts to design apoA-I mimetics resistant to oxidative modification, through judicious choice of the amino acid components (Figure 4).
Figure 4.

Chemical modifications that can lead to apoA-I dysfunction, and unnatural amino acids that could be used to evade such modifications in apoA-I mimetic peptides. (a) Oxidation of Met residues, such as Met148, by MPO to the sulfoxide derivative causes impaired apoA-I function. Norleucine bears a side chain that is structurally similar to that of Met, but not susceptible to oxidation. (b) Covalent modification of Lys residues by acrolein or malondialdehyde impairs the ability of apoA-I to promote cholesterol efflux. Replacement of Lys with the dimethyl-Lys analogue would render the amino acid unreactive with such reactive carbonyls.
Another type of apoA-I modification occurs via reactive carbonyl electrophiles, which originate from lipid peroxidation or oxidized carbohydrates. Modification of Lys residues in the C-terminal region of apoA-I, especially Lys226, by acrolein impaired ABCA1-mediated cholesterol efflux (Figure 4).209 An EXXK amino acid motif was shown to be responsible for the site-selective modification of Lys residues by acrolein in a model amphipathic α-helical peptide.210 Modification of Lys residues by malondialdehyde, primarily in C-terminal apoA-I helices 7–10, was found to prevent effective cholesterol efflux (Figure 4).211 Glycation of Lys residues in apoA-I, a condition prevalent in diabetics, is known to impair the ability of apoA-I to associate with lipids and thereby mediate anti-inflammatory and antioxidant activity.212,213
In Vivo Studies of ApoA-I and ApoA-I Milano
Compelling evidence for the antiatherogenicity of apoA-I derives from numerous observations that i.v. infusions of apoA-I or rHDL particles, or over-expression of apoA-I, confer protective effects in animal models and humans.214 The most common experimental animals used to characterize antiatherosclerotic agents are mice and rabbits.215,216 Wild-type mice are resistant to atherosclerosis, owing to their high levels of HDL and low levels of LDL/VLDL, and thus require genetic modification to become a suitable animal model. Two common mouse models, with mutations that lead to hypercholesterolemia and atherosclerosis development, are apoE-null (apoE−/−) and LDLr-null (LDLr−/−) strains. An important distinction between mice and humans is that mice lack CETP, which converts HDLs into larger lipoproteins in humans.215 On the other hand, wild-type rabbits carry CETP and develop hypercholesterolemia and atherosclerosis when fed a cholesterol-rich diet.216
In cholesterol-fed rabbits, weekly infusions of purified rabbit HDL36,37 or wild-type apoA-I32 slowed the progression of atherosclerosis and even regressed established lesions. Likewise, infusion of apoA-IM reduced arterial thickening and macrophage content after vascular injury in rabbits.33,34 In apoE-null mice, introduction of a gene for human apoA-I reduced the progression of atherosclerosis by more than 80%.40 Similarly, lesions were suppressed following infection of apoE-null or LDLr-null mice with a virus encoding human apoA-I or apoA-IM.28–30 Remarkably, a single infusion of apoA-IM to apoE-null mice was sufficient to reverse endothelial dysfunction and prevent the progression of aortic atherosclerotic lesions.25,27 An important caveat for the above studies is that atherosclerotic plaque in humans develops over decades and has a more complex pathophysiology compared to plaques in these animal models; so, human lesions may not respond in the same manner to the infusion of apoA-I, apoA-IM, or HDLs.
In the first clinical trial involving HDL infusion, sponsored by Esperion Therapeutics, recombinant apoA-IM formulated with POPC (ETC-216) was administered weekly for 5 weeks at two doses, beginning within 2 weeks of an acute coronary event.48 Among the 47 patients completing the study, the treatment reduced atheroma volume by 4.2% (combined treatment groups), relative to placebo. Esperion was purchased by Pfizer for $1.3 billion in 2003,217 but Pfizer discontinued all activities on Esperion product candidates four years later.218 In 2008, the company was sold to an investor syndicate for $23 million and relaunched,218 and Esperion now has an oxidation-resistant apoA-I in preclinical development.219 ApoA-IM was licensed from Pfizer by The Medicines Company in late 2009.220
In the Effect of rHDL on Atherosclerosis–Safety and Efficacy (ERASE) clinical trial, conducted by CSL Limited, 183 patients with a recent acute coronary event were given four weekly infusions of placebo or purified human apoA-I combined with soybean phosphatidylcholine (CSL-111).45 Of the two doses used, the higher dose (80 mg/kg) was discontinued due to hepatic enzyme elevation, while the lower dose (40 mg/kg) was well tolerated.45 Atheroma volumes and secondary endpoints tied to plaque characterization indices and coronary angiographic changes improved significantly in the treated group, but not in the placebo group. These data suggest that infusions of HDL or apoA-I may reduce events, particularly among patients with acute coronary syndrome (ACS), and that even short-term treatment can impart clinical benefits. In fact, the magnitude of change in the coronary angiography after the four weekly infusions was similar to that typically seen in 2-year trials with statins.214 Studies were discontinued because of liver toxicity,45 but a new formulation of apoA-I, a novel rHDL complex (CSL-112),221 completed two Phase 1 clinical trials, and entered a small Phase 2a trial in February 2012 to study safety, tolerability, and pharmacokinetics (clinicaltrials.gov identifier NCT01499420).
A third clinical trial, reported in 2010, involved a group of 28 ACS patients who received seven weekly infusions of their own HDLs that had been de-lipidated with an experimental device developed by Lipid Sciences.222 The reinfusion process was well tolerated and dramatically increased the 6% baseline level of lipid-poor, pre-β HDL in patients to 79%. Total atheroma volumes decreased, as compared to an increase in the control group.
An “HDL mimetic” based on human apoA-I from Cerenis Therpeutics (CER-001)223 has been studied as an infusion therapy in patients with ACS. It showed preclinical and clinical efficacy in mobilizing cholesterol and promoting RCT.223 Clinical studies were intended to eliminate atherosclerotic plaque and thereby reduce the risk of cardiac events. A single, rising-dose Phase 1 study of 32 healthy dyslipidemic volunteers, reported in May 2010, showed that the treatment was safe and well tolerated at dosages up to 45 mg/kg, and mobilizes cholesterol at 2 mg/kg and up.223 A Phase 2 safety and efficacy study, initiated in March 2011, is assessing the regression of coronary atherosclerotic plaque, as measured by intravascular ultrasound (IVUS). The study, involving at least 500 patients at centers in the USA, Canada, and Europe, will examine three different dose levels administered in six weekly i.v. infusions.223
While the antiatherogenic properties of apoA-I, and the potential of apoA-I as a therapeutic agent, have been documented in numerous studies, including several, preliminary clinical trials, the use of apoA-I as a therapeutic faces serious challenges. The large amounts of protein (3–5 g/single infusion) required, coupled with laborious production and purification methods, can be cost prohibitive unless simpler methods for its production are devised. Additionally, apoA-I is not orally bioavailable, so a parenteral route of administration would limit its widespread chronic use in the management of atherosclerosis. The possibility that short, synthetic, orally deliverable peptides could serve as alternatives to apoA-I for HDL-targeted therapy has spurred considerable interest in the discovery and development of apoA-I mimetic peptides.
APOLIPOPROTEIN MIMETIC PEPTIDES
In the Introduction, we mentioned that synthetic peptides with a class A amphipathic α-helical structure75,78,80 can mimic apoA-I and thereby modulate the properties of HDL. As the field developed, physicochemical and in vitro biological studies evolved into studies of atherogenesis in mouse models, ultimately with clinical aspirations. Indeed, it is noteworthy that the apoA-I mimetic peptides 4F,106 D4F,107 and 1105 were advanced into human clinical trials.
Early studies in this area were concerned with explaining the interactions between proteins and lipids within plasma lipoproteins based on the “amphipathic helix hypothesis” that was proposed in the mid-1970’s.131,224 A model peptide with the 18-amino-acid sequence DWLKAFYDKVAEKLKEAF (“18A”) was designed to mimic apoA-I by virtue of certain structural features relating to a class A, amphipathic α-helix [ref] (see Figure 3). The 18A peptide has hydrophobic amino acids (W, L, A, F, Y, V) on the nonpolar face of the α-helix and hydrophilic amino acids (D, E, K, R) on the polar face, with positively charged lysines at the polar/nonpolar interface and negatively charged D or E at the center of the polar face. By mimicking an apoA-I α-helix, 18A exhibits appropriate biological functionality, despite constituting just a single, isolated helix with a much different peptide sequence.225,226 For example, 18A was found to associate strongly with liposomes,132 and was able to displace apoA-I from native HDL and apoE from native VLDL.227 To interact effectively with lipids, the distribution of the charged, polar residues was found to be very important, as analogue KWLDAFYKDVAKELEKAF (“18R”; italics denote charge-altered polar residues) had weak lipid affinity.84,225 An end-capped version of 18A, Ac-18A-NH2 (aka 2F; Figure 3), in which the charged groups at the N- and C-termini were neutralized, had higher helical content and better lipid affinity than 18A (Table 2, entry 2).86 As expected, 2F also had better lipid affinity than Ac-18R-NH2.228 For activating LCAT, 2F was better than 18A, and essentially comparable to apo A-I.86 This information provided a basis for the idea that a simple, model, class A, amphipathic helix can replicate the lipid-binding, and other, properties of apoA-I.
Table 2.
ApoA-I Mimetic Peptides: Single Helices
| entry | peptidea | AA’sb | EPC dissolnc | monocyte chemotaxd | chol effluxe | mouse antiatherof |
|---|---|---|---|---|---|---|
| 1 | DWLKAFYDKVAEKLKEAF (18A) | 18 | 20%g | 7800h | ||
| 2 | Ac-DWLKAFYDKVAEKLKEAF-NH2 (2F) | 18 | 70%i | 45%i | 2600h | 0%i |
| 3 | Ac-DWFKAFYDKVAEKFKEAF-NH2 (4F) | 18 | 85%i | 70%i | 2000j | 60%k; 0%l |
| 4 | Ac-DWFKAFYDKVAEKLKEAF-NH2 (3F3) | 18 | 45%i | 10%i | ||
| 5 | Ac-DWLKAFYDKVAEKFKEAF-NH2 (3F14) | 18 | 45%i | −15%i | 0%m | |
| 6 | Ac-DWLKAFYDKVFEKFKEFF-NH2 (5F) | 18 | 80%i | 60%i | 40%i,n | |
| 7 | Ac-DWLKAFYDKFFEKFKEFF-NH2 (6F) | 18 | 70%i | 60%i | ||
| 8 | Ac-DWFKAFYDKFFEKFKEFF-NH2 (7F) | 18 | 70%i | 25%i | ||
| 9 | Ac-FWLKAFYDKVAEKLKEAF-NH2 (3F-1) | 18 | 65%o | 70%p | ||
| 10 | Ac-DFLKAFYDKVAEKLKEAF-NH2 (3F-2) | 18 | 80%o | 75%p | 20%m | |
| 11 | Ac-DWFRAFYDKVAEKFREAF-NH2 (4F-R)q | 18 | 55%k | |||
| 12 | Ac-DWFKAFYDRVAERFKEAF-NH2 (4F-R′)q | 18 | 20%k | |||
| 13 | Ac-DWLXAFYDXVAEXLXEAF-NH2 (2F′) | 18 | 45%r |
For entries 1–12, boldface F’s are provided for clarity in comparison. For entries 11 and 12, italicized R’s are for clarity in comparison. For entry 13, X = L-2,4-diaminobutyric acid (Lys with two methylenes removed from the side chain), with X italicized for clarity.
Number of amino acids.
Dissolution of egg PC vesicles, reported as percent clarification of turbidity at 30 min (0% at t = 0), unless otherwise noted.
LDL-induced monocyte chemotaxis in human artery wall cell cocultures, reported as percent inhibition (HDL = 75%), unless otherwise noted. A negative value indicates stimulation.
Efflux of cholesterol from cholesterol-laden mouse macrophages (EC50 values in nM units; apoA-I = 53 nM in ref 88; 100 nM in ref 98).
Inhibition of atherosclerotic lesion formation in mice fed a high-fat diet, reported as percent reduction of mean aortic lesion area relative to vehicle baseline, unless otherwise noted.
Ref 86.
Ref 88.
Ref 115.
This value was measured for D4F (ref 98).
Ref 231. Inhibition of atherosclerotic lesion size in chow-fed, 14-week-old, apoE-null mice with daily i.p. administration; mean aortic lesion area in en face preparations after 16 weeks of treatment.
Ref 230. Inhibition of arterial atherosclerotic lesion size in chow-fed, 20-week-old, apoE-null mice after 8 weeks of i.p. administration every other day.
Ref 236. Inhibition of atherosclerotic lesion formation in chow-fed, 4-week-old, apoE-null mice; mean lesion cross-sectional area after 6 weeks of daily i.p. administration.
Ref 91.
Ref 121. Dissolution of POPC multilamellar vesicles, reported as percent clarification of turbidity at 30 min (0% at t = 0). 4F gave 85%, as with egg PC, but 3F3 and 3F14 were much more effective than with egg PC (95% and 100%, respectively).
Ref 121. Oxidized lipid-induced monocyte chemotaxis in human artery wall cell cocultures, reported as percent inhibition (HDL = 55%). Peptide 4F gave 90%; 3F3 and 3F14 were much weaker (30% and 20%, respectively).
4F-R and 4F-R′ are terms used herein for peptides with Lys→Arg substitutions at positions 4,15 and 9,13, respectively (ref 231).
Ref 228.
To test the idea that cooperativity between helical domains might benefit the function of apoA-I mimetic molecules, peptides containing two amphipathic helical regions separated by a short linker were subsequently studied. In complexes between certain peptides and DMPC by multiple techniques, the proline-linked dimer of 18A, DWLKAFYDKVAEKLKEAF-P-DWLKAFYDKVAEKLKEAF (“37pA”), exhibited better lipid affinity than 18A.225,226 In cholesterol efflux assays with mouse J774 macrophages, 2F and 37pA were 3-fold and 18-fold better than 18A, respectively; however, 37pA remained 8-fold poorer than apoA-I (Table 2, entries 1 and 2; Table 3, entries 1 and 2).88,229 The end-capped 37pA and IHS 43-mer (two 4F helices linked by apoA-I interhelical sequence KVEPLRA; Table 2, entries 10 and 12) exhibited about the same effectiveness in promoting cellular cholesterol efflux, which was better than that of 4F by a factor of 3–4.101 The same rank ordering of these peptides was observed for displacement of apoA-I from HDL.226 Modified HDL with 40% of its apoA-I displaced by 18A behaved like unmodified HDL as a substrate for LCAT, indicating that 18A had just a minor effect. By contrast, incubation of 37pA with egg phosphatidylcholine (PC) vesicles produced discoidal nanoparticles that powerfully activated LCAT, exceeding the activity achieved with apoA-I by 40%. A comparative study showed that 37pA binds much better to DMPC than 18A-A-18A (“37aA”) and 18A-18A (“36A”) did (decreasing lipid affinity: 37pA > 37aA > 36A); 37pA also bound more effectively than 36A to egg PC monolayers.103 In a DMPC dissolution study, end-capped versions of 37pA and 37aA were dramatically different, with 60% and 0% clarification of turbidity, respectively (Table 3, entries 10 and 11), supporting the idea that a local turn conformation introduced by the proline between each amphipathic helix is preferable over alanine for enhanced lipid affinity.101 The IHS 43-mer had the same high level of lipid binding as end-capped 37pA (Table 3, entries 10 and 12).101 The collection of results from these and other229 studies with bihelical peptides supports the idea that multivalency of class A amphipathic helices can lead to enhanced apoA-I mimetic properties, presumably caused by cooperativity between the two helices.
Table 3.
ApoA-I Mimetic Peptides: Multiple Helices
| entry | peptidea | AA’sb | DMPC dissolnc | chol effluxd | mouse antiatheroe |
|---|---|---|---|---|---|
| 1 | DWLKAFYDKVAEKLKEAF-P- DWLKAFYDKVAEKLKEAF (37pA) | 37 | 55%f | 440g; +h,i | |
| 2 | DWLKAFYDKVAEKLKEAF-A- DWLKAFYDKVAEKLKEAF (37aA) | 37 | +i | ||
| 3 | DWLKAFYDKVAEKLKEAF- DWLKAFYDKVAEKLKEAF (36A) | 36 | |||
| 4 | DWLKAFYDKVAEKLKEAF-P- DWLKAFYDKVAEKAKEAF (1A) | 37 | 75%f | +h | |
| 5 | DWLKAFYDKVAEKLKEAF-P- DWLKAFYDKVAEKAKEAA (2A) | 37 | 65%f | +h | |
| 6 | DWLKAFYDKVAEKLKEAF-P- DWFKAFYDKAAEKAKEAA (3A) | 37 | 70%f | +h | |
| 7 | DWLKAFYDKVAEKLKEAF-P- DWFKAAYDKAAEKAKEAA (4A) | 37 | 70%f | +h | |
| 8 | DWLKAFYDKVAEKLKEAF-P- DWAKAAYDKAAEKAKEAA (5A) | 37 | 80%f | 3400j; +h | |
| 9 | DWAKAAYDKAAEKAKEAA -P- DWAKAAYDKAAEKAKEAA (10A) | 37 | 14%f | −h | |
| 10 | Ac-DWFKAFYDKVAEKFKEAF-P- DWFKAFYDKVAEKFKEAF-NH2k (4F-P-4F) | 37 | 60%l | +l | 0%m |
| 11 | Ac-DWFKAFYDKVAEKFKEAF-A- DWFKAFYDKVAEKFKEAF-NH2n (4F-A-4F) | 37 | 0%l | ||
| 12 | Ac-DWFKAFYDKVAEKFKEAF-KVEPLRA- DWFKAFYDKVAEKFKEAF-NH2o (4F-IHS-4F) | 43 | 66%l | +l |
Interhelix linkers: -P-, -A-, -KVEPLRA-. For entries 4–9, italicized A’s are for clarity in comparison.
Number of amino acids.
Dissolution of DMPC; reported as percent decrease in turbidity, from relative absorbance or optical density, at 30 min (0% at t = 0), unless otherwise noted.
Efflux of cholesterol from cholesterol-laden mouse macrophages (EC50 values in nM units; apoA-I = 53 nM in ref 88; 100 nM in ref 98), unless otherwise noted. A plus sign (+) signifies activity, but without quantification (ref 101, 229).
Inhibition of atherosclerotic lesion formation in mice fed a high-fat diet, reported as percent reduction of mean aortic lesion area relative to vehicle baseline, unless otherwise noted.
Ref 102.
Ref 88.
Efflux of cholesterol from ABCA1-transfected HeLa cells. A positive sign (+) signifies activity, but without quantification; a negative sign (−) signifies inactivity (ref 102).
Ref 229.
Ref 98.
Tandem dimer of 4F linked by Pro (ref 101).
Ref 101. Clarification of turbidity of a DMPC suspension as measured by light scattering (value for 4F in this work was 90%).
Ref 230. Inhibition of arterial atherosclerotic lesion size in chow-fed, 20-week-old, apoE-null mice after 8 weeks of i.p. administration (100 μg) every other day.
Tandem dimer of 4F linked by Ala (ref 101).
Tandem dimer of 4F linked by KVEPLRA, the 7-amino acid, interhelical segment between helices 4 and 5 of human apoA-I (“HIS”) (ref 101).
The fact that apoA-I is composed of multiple amphipathic helices attracted further interest in bihelical peptides.100,102,177 Both 37pA and its enantiomer, D37pA, strongly promoted cholesterol efflux from ABCA1-transfected HeLa cells, to nearly the same degree.177 Unlike apoA-I, these peptides were able to efflux cholesterol by a passive, energy-independent mechanism (in control HeLa cells). Sethi et al. examined systematic substitution of hydrophobic residues by alanine in the C-terminal helix (Table 3, entries 4–8).102 In lipid dissolution with DMPC vesicles, 5A was the most effective peptide (80%) and it exceeded 37A (55%); however, in the case of vesicles formed with a mixture of phospholipids, 37A was somewhat better than 5A. In cholesterol efflux studies with ABCA1-transfected HeLa cells, 10A (Table 3, entry 9) was essentially inactive, whereas 1A–5A were very effective.102 On the other hand, in efflux studies with control HeLa cells 37A and 1A were very effective, 2A and 3A were moderately effective, 4A and 5A were weak, and 10A was inactive.102 Clearly, the loss of overall hydrophobicity caused by replacing 10 greasy residues with Ala, as in 10A, was detrimental to the functional properties. Whereas 37pA exhibited frank cytotoxicity in hemolysis of red blood cells, 5A was rather benign. In a study involving a series of 22 bihelical peptides, largely proline-linked 37-mers, the effectiveness of the different peptides in several antiatherogenic assays, such as cholesterol efflux, anti-inflammatory properties, and antioxidant activity, was rather variable.100 EKLKELLEKVAEKLKELL-P-EKLKELLEKVAEKLKELL (“ELK-2A2K2E”) and EKFKELLEKFLEKFKELL-P-EKFKELLEKFLEKFKELL (“ELK-2F”) were the most efficient in cholesterol efflux from THP-1 cells, better than 5A and about the same as apoA-I, and EKLKALLEKLLAKLKELL-P-EKLKALLEKLLAKLKELL (“ELK-2A”) was one of the least efficient peptides. However, ELK-2A was as anti-inflammatory as apoA-I in inhibiting VCAM-1 expression in a mouse endothelial cell line. Bihelical peptides containing cysteine or histidine were among the best antioxidants in an LDL oxidation assay. Although structure–function correlations were inconsistent across various bioassays, D’Souza et al. outlined a set of parameters that relate to structural features associated with optimal activity for different criteria.100
Structure–Function Studies of 4F-Type ApoA-I Mimetic Peptides
In 2001, Datta et al. expanded on improved, end-capped 2F with structural modifications that increased hydrophobicity, specifically by replacing certain nonpolar residues with phenylalanines, in an effort to optimize apoA-I mimetic peptides based on physicochemical and biological properties (Table 2, entries 3–8).115 Various physicochemical parameters were measured, including hydrophobicity, α-helicity, and solubility.115 Interaction of the peptides with phospholipid monolayers was explored by determining the monolayer exclusion pressure, that is, the surface pressure at which a peptide would no longer penetrate an egg PC monolayer.115 ApoA-I and 18A measured 34 and 30 dyn/cm, respectively; 2F, 3F3, 3F14, and 4F measured 38–40 dyn/cm; 5F, 6F, and 7F measured 45–46 dyn/cm; and 37A measured 41 dyn/cm, which is consistent overall with their hydrophobicity. With the apparent absence of sensitivity here, two distinct sets of peptides were defined, one containing 2F, 3F3, 3F14, and 4F, and one containing 5F, 6F, and 7F. Right-angle light scattering was used to assess the dissolution of egg PC multilamellar vesicles mediated by the peptides over time, and the response values at 30 min were: 45% for 3F3 and 3F14; 70% for 2F, 6F, and 7F; 80% for 5F; and 85% for 4F (Table 2).115 Reducing the length of each of the Lys side chains in 2F by two methylenes decreased the dissolution value to 45% (2F′; Table 2, entry 13).228 Not only was 4F the most effective peptide in this series, but it also gave the most rapid rate of dissolution.
The peptides were further evaluated as activators of LCAT by determining their effect on the initial rate of reaction of LCAT with egg PC/cholesterol vesicles.115 Peptides 5F and 6F were the best activators at levels of 80% and 70% relative to apoA-I, respectively, whereas 2F, 3F, 4F, and 7F were much weaker (ca. 40% of apoA-I),115 and 18A was very weak.86 Certain peptides potently inhibited LDL-induced monocyte chemotaxis, with 4F, 5F, and 6F being the most effective, in the realm of HDL; 2F was moderately effective; 7F was weak; and 3F3 and 3F14 were essentially inactive (Table 2).115 The results of the chemotaxis experiments suggested that the more effective peptides better sequestered the oxidized lipids (“seeding molecules”) present in LDL (i.e., ox-LDL), which are responsible for promoting monocyte chemotaxis via the secretion of MCP-1 and macrophage colony-stimulating factor (M-CSF). In the final analysis, there was not a clear-cut correlation between the effectiveness of peptides in monocyte chemotaxis, their physicochemical properties, or their ability to activate LCAT.115 However, 4F was among the best performers in egg PC dissolution and monocyte chemotaxis, while possessing favorably moderate hydrophobicity in this family of peptides.
The weak-to-nonexistent chemotactic activity for 3F3 and 3F14 spurred the examination of related peptides 3F-1 and 3F-2 (Table 2, entries 9 and 10), in which the surface of the nonpolar face of the α-helix was modified.121 In dissolution of POPC multilamellar vesicles (different protocol than discussed earlier), 3F-2 was very effective (80% clarification in 30 min) and essentially equal to 4F (85%), whereas 3F-1 was moderately effective (65%). The inhibitory activity of 3F-1 and 3F-2 in oxidized lipid-induced monocyte chemotaxis in human artery wall cell cocultures (different protocol than discussed earlier) was 70% and 75%, respectively, with 4F being more potent (90%) and 3F3 and 3F14 being much weaker (30% and 20%, respectively), as noted for LDL-induced chemotaxis. These results, and other experiments, indicate that the surface topography (shape) of the nonpolar face of the α-helix, influenced here by the arrangement of aromatic groups, as well as hydrophobicity, play a role in antichemotactic activity.121
The antiatherogenic activity of 5F was studied by i.p. administration (20 μg/day; ca. 1 mg/kg) over 16 weeks in wild-type (C57BL/6J) mice fed a high-fat diet.91 The treated mice had significantly less aortic lesions (mean lesion area = 20.1 × 103 μm2) compared with mice in the untreated PBS group (mean lesion area = 35.7 × 103 μm2), for a 40–45% reduction, without alteration of the lipoprotein profile (Table 2). HDL isolated from the treated mice was more effective in inhibiting LDL oxidation and LDL-induced monocyte chemotaxis than HDL from untreated mice,91 whereas peptide 2F, by comparison, was much less active in this mouse chemotaxis model.115 The in vivo results with 5F constituted the first demonstration of antiatherogenic activity for an apoA-I mimetic peptide.
The 4F peptide inhibited atherosclerotic lesion formation in chow-fed, 10-week-old, apoE-null mice 4 weeks after i.p. administration (50 μg/mouse, or ca. 2.5 mg/kg, every other day; ~80% reduction vs. PBS control), but not in 20-week-old mice after 8 weeks of dosing, whereas 4F-P-4F (100 μg/mouse, or ca. 5 mg/kg, every other day) did not inhibit atherosclerotic lesion size in either situation (Table 2, entry 3; Table 3, entry 10).230 Thus, 4F worked better in early-stage atherogenesis, as compared to more mature arterial lesions. In another study involving chow-fed, 14-week-old, apoE-null mice with daily i.p. administration (100 μg/mouse), the mean aortic lesion area in en face preparations after 16 weeks of treatment was reduced by 60% (Table 2, entry 3).231 The enantiomer of 4F, D4F, which is discussed in some detail later in this section, exhibited frank antiatherogenic activity in vivo.232 In a p.o.-dosing study with 10-week-old LDLr-null mice on a high-fat diet, D4F decreased aortic lesions by 79% (2.5 mg of D4F in 100 μL of liposomes; twice daily for 6 weeks), while administration of D4F in the drinking water of apoE-null mice (0.05 mg/mL) decreased lesions by 75%.232 Notably, these results took place without changes in total plasma cholesterol or HDL-cholesterol, suggesting that the ability of HDL to prevent LDL oxidation may be more important than HDL levels in determining the development of lesions.91,232–234 Differential effects on evolving and established plaque were also recorded with D4F (in apoE-null mice).235 Oral and i.p. administration of D4F reduced evolving atherosclerotic lesions, plaque lipids, and macrophage activity in vein grafts, but had no impact on established lesions in aortic sinus.
Peptides 3F-2 and 3F14 were studied for inhibition of atherosclerotic lesion formation in chow-fed, 4-week-old, apoE-null mice with daily i.p. administration (20 μg/mouse).236 Measurement of the mean lesion area after 6 weeks of treatment showed no effect for 3F14 and a modest 20% decrease for 3F-2 (Table 2, entries 5 and 10). Analogues of 4F in which two lysines were replaced by arginines were studied to test the importance of the location of interfacial Arg residues on the polar face of the α-helix to biological properties (Table 2, entries 11 and 12).231 In chow-fed, 14-week-old, apoE-null mice with daily i.p. administration (100 μg/mouse) after 16 weeks of treatment, 4F-R and 4F-R′ showed 55% and 20% reduction of mean aortic lesion areas in en face preparations, respectively (Table 2, entries 11 and 12). A determination of plasma levels for 4F, 4F-R, and 4F-R′ indicated that the AUC for 4F-R′ was about half that of 4F and 4F-R, which could account for its weaker antiatherogenic activity.231
In general, apoA-I mimetic peptides remodel HDL in plasma, which results in the migration of apoA-I from larger to smaller nanoparticles and consequently the production of pre-β HDL. This process is often monitored by gel electrophoresis (such as NDGGE), but this method offers just a qualitative, or semi-quantitative, measure for comparative purposes. An effective quantitative method, based on ELISA with a monoclonal antibody (mAb 55201) that recognizes the apoA-I conformation present in pre-β HDL, was described.98,119 Troutt et al. established a correlation between the dose of apoA-I mimetic peptides and pre-β1 HDL formation in human plasma.119 For D4F, the increase in pre-β1 HDL with increasing dose in the ELISA assay was nicely mirrored in the gel band density for pre-β1 HDL (Western blotting). These results with D4F agreed reasonably well with an earlier report that used two-dimensional electrophoresis (agarose/native PAGE).237 Carballo-Jane et al. were able to generate useful dose-response curves for a series of different 22-mer, α-helical apoA-I-derived peptides.98 In the supporting information of their manuscript, the authors reported that D4F was very potent in remodeling HDL in human plasma in vitro.98 Peptides 4F, 4F-P-4F, and 4F-A-4F at 5 μM substantially remodeled human HDL,101 and 4F was more effective than 4F-R, which was better than 4F-R′.231 Interestingly, the importance of pre-β HDL formation to the antiatherogenic activity of apoA-I mimetic peptides has been drawn into question, since the concentration of peptide required to remodel HDLs is higher than that required to reduce the development of atherosclerotic lesions.238,239
A curious 4F-containing “peptibody”, consisting of a mouse IgG Fc fragment fused to two copies of 4F to give a tetrameric display of 4F (Fc is a dimer; Figure 4) was recently described.240 The peptibody was more efficient than 4F in promoting cholesterol efflux, and nanoparticles generated from it were larger than those from apoA-I, owing to the large size of the Fc fragment (50 kDa). When mice were treated with a single dose of the peptibody, levels of total plasma cholesterol and HDL-cholesterol were increased (4-h time point), which was ascribed to enhanced RCT. A co-immunoprecipitation analysis of HDL particles from this experiment offered direct evidence for apoA-I mimetic peptides and apoA-I coexisting in the same lipoprotein particles after in vivo administration of test peptide.
Focus on D4F
To address the expected plasma instability of 4F and to try to enable oral dosing, D4F (enantiomer of 4F consisting of D-amino acids) was explored.232,237 The point here is that having all D-amino acids would lead to a more robust peptide for in vivo studies. In the case of 4F, the residence time in mouse plasma after i.p. administration was rather short, with t1/2 = ca. 2 h, such that there would be limited coverage accompanying once-daily dosing.231 Four hours after administration by oral gavage to LDLr-null mice, 4F was essentially depleted, while D4F was present at a substantial level.232 Nevertheless, the oral bioavailability of D4F was actually less than 1%.237 For example, after administration of 500 μg of D4F by oral gavage to apoE-null mice, the highest observed plasma concentration was 140 nM (320 ng/mL). This low plasma level begs the question about how D4F could exert its bioactivity in mice despite a high plasma level of apoA-I of ca. 35 μM. A possible answer was revealed in a subsequent investigation with apoE-null mice, wherein the intestine was suggested as a major site of action for D4F during oral or parenteral administration; the authors posited a mechanism of action for 4F involving binding and inactivation of oxidized lipids in the intestine, resulting in reduced systemic inflammation.234,241 Another point pertains to the antioxidant and anti-inflammatory mechanisms of action for D4F, and the fact that D4F must work in conjunction with apoA-I (as discussed below with respect to studies in LDLr-null mice lacking apoA-I143,144).
As mentioned earlier, D4F markedly reduced atherosclerotic lesions in LDLr-null and apoE-null mice after oral administration,232 although there were differential effects for evolving vs. established lesions.235 Whereas oral and i.p. administration of D4F reduced evolving atherosclerotic lesions, plaque lipids, and macrophage activity in vein grafts, there was virtually no effect on established lesions in aortic sinus.235 Navab et al. found that a daily p.o. dose of 50 μg/mouse did not prevent lesion formation nor alter HDL inflammatory properties. However, when this dose regimen was paired with a subtherapeutic p.o. dose of pravastatin (HMG-CoA reductase inhibitor) in apoE-null mice, there was a strong synergistic response.242 It is noteworthy that lesion formation was prevented in young mice, and established lesions were reduced in older mice. Interestingly, the retro-inverso variant of D4F, reverse-D4F, was found to decrease aortic root atherosclerotic lesions and lesion macrophage content in chow-fed, 4-week-old apoE-null mice, when dosed in drinking water at 0.4 mg/mL (ca. 1.6 mg/day) over 6 weeks93 (the 4F peptide was ineffective under this protocol93). Plasma total cholesterol and HDL-cholesterol were unaffected by reverse-D4F treatment.
After dosing apoE-null mice with 500 μg of D4F by oral gavage, small lipid particles (7–8 nm) with pre-β mobility were generated in plasma and the HDL became anti-inflammatory.237 In vitro, D4F promoted HDL-mediated cholesterol efflux from macrophages237 and bound avidly to pro-inflammatory oxidized lipids (with a much higher affinity than apoA-I).89 In apoE-null mice and wild-type mice, oral D4F increased pre-β HDL, decreased lipid hydroperoxides in HDL, and increased lipid hydroperoxides in pre-β HDL.237 In LDLr-null mice lacking apoA-I, D4F improved arterial vasoreactivity, but not HDL inflammatory properties, nor did it decrease arterial wall thickening, indicating that D4F works best in conjunction with apoA-I.143,144 Mechanistically, D4F appears to combat atherogenesis by a combination of effects, including: (1) inhibition of inflammatory and oxidative events at the endothelium, such as by limiting LDL oxidation so as to reduce proinflammatory LDL and by inhibiting monocyte adhesion; and (2) improving HDL function, such as increasing pre-β HDL, decreasing lipid hydroperoxides in HDL, and enhancing the anti-inflammatory properties of HDL.
Treatment of cholesterol-fed rabbits (New Zealand White) with daily s.c. injections of D4F (20 mg/mL; 10 mg/kg/day) for one month resulted in a significant reduction of aortic lesions without altering levels of HDL-C.243 Biomarkers, such as HDL inflammatory index and serum amyloid A (SAA), proved useful as predictors of antiatherogenic activity. In monkey studies, administration of D4F (1.7 mg/kg, p.o., single dose) reduced lipid hydroperoxides and improved the anti-inflammatory activity of HDL.244 In addition, D4F enhanced monkey HDL-mediated cholesterol efflux from human macrophages. A single, 10-mg/kg oral dose increased paraoxonase activity in monkeys and produced pre-β HDL (at 6 h).92
Addition of D4F (250 ng/mL) to human plasma induced pre-β HDL formation, reduced HDL lipid peroxides, and increased paraoxonase activity, as observed with mice and monkeys in vivo.92 As noted earlier, the quantitative ELISA method for pre-β HDL particles found that D4F is very potent in vitro at remodeling HDL to pre-β HDL in human plasma.98,119 Such positive results, in conjunction with the collection of favorable antiatherogenic data for D4F in test animals, led to human clinical trials. A rising-single-dose, placebo-controlled study (30, 100, 300, or 500 mg, p.o.; n = 8 per dose) was performed with unformulated D4F in patients with coronary heart disease or equivalent risk under fasting conditions.107 No safety events occurred and D4F was detected in plasma at all doses, with tmax values of 0.5, 1.0, and 2 h for doses of 30, 100, and 300/500 mg, respectively. The maximal plasma concentration was ca. 4 nM and the AUC at the 500 mg dose was 55 ng/h/mL, indicating low oral bioavailability. The HDL inflammatory index was improved at 300 and 500 mg, 4 hours after dosing. In addition to this clinical probe with D4F, two human clinical studies were performed with 4F, administered parenterally, in patients with coronary heart disease or equivalent risk, and on a statin: a randomized, double-blind, placebo-controlled i.v. study with ascending multiple doses (3, 10, 30, or 100 mg), as a daily 2-h infusion over seven days; and a parallel, randomized, double-blind, placebo-controlled s.c. study with multiple doses, as daily injections for 28 days.106 No safety issues arose and the mean maximal plasma concentration reached 2900 ng/mL for the i.v. study (t1/2 of ca. 1.5 h) and 400 ng/mL for the s.c. (t1/2 of ca. 3 h) study. Although the exposures matched levels that were effective in animal models, the biomarkers for HDL function, such as inflammatory index and paraoxonase activity, did not improve.
Mimetic Peptides Derived from the ApoA-I Sequence
Several studies have examined the in vivo effects of peptides directly derived from apoA-I. Esperion Therapeutics advanced a 22-mer peptide that differs from the apoA-I consensus sequence (Figure 2) by three mutations: Glu5→Leu, Lys9→Leu, and Glu13→Leu. A formulation of this peptide with sphingomyelin and (R)-(+)-1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC), 1, was the focus of two human clinical studies.105 In the first Phase 1 study, a single i.v. infusion of 1 at 3 mg/kg was well-tolerated and promoted cholesterol mobilization in vivo; in a second Phase 1 trial, single i.v. infusions of 10–30 mg/kg were well-tolerated.105 In 2011, three studies involving 1 reported on the anti-inflammatory and antiatherosclerotic effects of the complex.95,245,246 Two infusions of 1 reduced inflammation in rabbits to a similar degree as infusions of rHDL, as indicated by a dose-dependent reduction in the levels of cell-adhesion molecules.95,245 Twice weekly infusions of 1 at a 50-mg/kg dose for 12 weeks in rabbits inhibited the progression of aortic atherosclerosis, as determined by vessel, lumen, and plaque volumes (imaged by IVUS).246
In 2010, two related studies were published on the development of apoA-I mimetics originating from the 22-residue apoA-I consensus sequence. In the first study, the authors took a peptide “stapling” approach, in which two side chain functional groups in the hydrophobic face were replaced with olefinic amino acids that could be covalently linked via olefin metathesis.96 Such stapling strategies are known to stabilize helical peptide conformations and improve in vivo stability.247 In this case, introduction of the staple increased the α-helical content of the peptide and yielded sequences that promoted cholesterol efflux, with EC50 values around 1 μM (WT apoA-I EC50 = 0.1 μM), and the formation of pre-β HDL. However, the most efficient peptide contained the olefin residues not coupled by metathesis, suggesting that the introduction of the hydrophobic side chains, rather than stapling, might be responsible for the improved activities. A follow-up study described a series of variants of the apoA-I consensus sequence in which one or two hydrophobic amino acids containing alkyl chains of 5–8 carbons were introduced at various positions of the peptide chain.98 The optimal compounds were superior to D4F and similarly efficient as native apoA-I in promoting cholesterol efflux; they also induced pre-β HDL formation in vitro and in vivo. Interestingly, installing a hydrophobic residue in place of Glu13 of the apoA-I consensus peptide had the most dramatic effect in improving cholesterol efflux efficiency, consistent with the stapling study,96 with the Glu13→Leu substitution present in 1, and with the improved lipid affinity of the Glu13→Ala consensus sequence variant.85 Thus, as would be expected, replacing a polar amino acid with a hydrophobic one in the nonpolar face of an amphiphilic helical peptide appears to benefit lipid association and cholesterol efflux (Figure 3).
Cerenis Therpeutics has advanced to a Phase 1 study a peptide analogue of apoA-I (CER-522; structure not disclosed) for treating aortic valve stenosis.248
Mimetic Peptides Inspired by Apolipoproteins Other Than ApoA-I
Besides apoA-I, there are numerous apolipoproteins that associate with HDL, such as apoA-II, apoIV, apoV, apoC-I, apoC-II, apoC-III, apoD, apoE, apoJ, and apoM,249 which could be candidates for discovering useful mimetic peptides. Apolipoproteins apoE and apoJ, in particular, have served as a basis for the design of medium-size peptides with antiatherogenic activity.239,249,250 The crux of the mimicry involved the use of key segments from the full-length apolipoproteins.
ApoE is a 299-amino-acid protein present in larger lipoproteins, including intermediate-density lipoproteins (IDLs), LDLs, VLDLs, and chylomicrons that transport cholesterol and lipids in plasma.250 The presence of multiple apoE molecules within these lipoproteins enables high-affinity binding to the LDLr. ApoE is composed of four amphipathic α-helices that are organized into a bundle topology at the N-terminus, containing the LDLr-binding domain (residues 1–191), and a lengthy, amphipathic α-helical segment at the C-terminus (lipid-associating domain; residues 203–266), which are linked together by a so-called hinge region.250 The sequence LRKLRLRLLR (“hE”) within human apoE, from 141–150, is a heparin-binding domain that is important for binding to the LDLr. This 10-residue apoE fragment was attached to the class A, amphipathic, α-helical peptide 18A, and end-capped, to obtain Ac-LRKLRLRLLR-18A-NH2 (Ac-hE18A-NH2), in order to afford strong lipid-binding properties.251 It was found that this dual-domain peptide markedly enhances LDL uptake and degradation by fibroblasts.
Intravenous administration of Ac-hE18A-NH2 to wild-type mice on an atherogenic diet, apoE-null mice, or apoE-null/LDLr-null mice (100 μg/mouse) rapidly reduced serum cholesterol levels, with the effect lasting for more than 6 hours.252 Administration of this peptide to Watanabe hyperlipidemic rabbits (15 mg/kg, i.v. bolus) reduced plasma cholesterol levels (by ca. 50% at 18 h) and improved arterial endothelial function, such as by reducing plasma lipid hydroperoxide content.253 A single dose of this peptide in apoE-null mice (100 μg/mouse) lowered serum cholesterol by 90% at 5 hours, and administration (100 μg/mouse/day, 3 days/week) for six weeks produced a moderate reduction in aortic sinus lesions.254 The mice that were treated for 6 weeks showed reduced plasma cholesterol and triglyceride levels, as well as an increase in PON-1 activity. By contrast, related peptide Ac-nhE18A-NH2, in which nhE is the nonheparin-binding domain DADDLQKRLA (apoE 151–160), did not decrease plasma cholesterol, nor increase PON-1 activity, nor inhibit atherosclerosis in apoE-null mice.254 In fact, Ac-nhE18A-NH2 displayed proinflammatory and proatherogenic effects. A head-to-head comparison of Ac-hE18A-NH2 and 4F was conducted in apoE-null mice under different parenteral administration protocols.255 Both peptides were effective in reducing aortic lesions by en face assessment; however, at a similar dose regime Ac-hE18A-NH2 had a more pronounced effect. Thus, attachment of 18A to the peptide sequence apoE(141–150), which is responsible for high-affinity LDLr binding, gave a dual-domain peptide, Ac-hE18A-NH2, with antiatherogenic and anti-inflammatory properties.252–256
Bielicki et al. described a different type of peptide that is based on the C-terminal helix of apoE (segment 238–266): Ac-EVRSKLEEWFAAFREFAEEFLARLKS-NH2 (ATI-5261).99 This 26-mer, amphipathic, α-helical peptide with a class A arrangement of hydrophobic and hydrophilic groups was potent in stimulating cholesterol efflux in vitro and in vivo. When old LDLr-null mice fed a high-fat Western diet were treated with daily i.p. injections (30 mg/kg) for 6 weeks, the aortic lesion area was reduced by 30%. When old apoE-null mice fed a high-fat Western diet were treated every other day with i.p. injections for 6 weeks, the atherosclerosis was reduced by 45%. Similar antiatherogenic results were obtained with a POPC formulation. Later, it was determined that this peptide readily forms oligomers by self-association, similar to the behavior of native apolipoproteins.257
Handattu et al. studied Ac-GFRRFLGSWARIYRAFVG-NH2 (“mR18L”), a class L, cationic, amphipathic, α-helical peptide, which interacted strongly with lipids and promoted LDL uptake by HepG2 cells.97 Intravenous administration of mR18L to apoE-null mice (75 μg/mouse) caused a dramatic decrease in plasma cholesterol levels. Despite the low oral bioavailability of mR18L, oral administration to 4-week-old apoE-null mice (1 mg per 4 g of chow) prevented aortic sinus lesions to the extent of ca. 30%, after six weeks of treatment, although only a minor effect on cholesterol levels was observed. In a study of Ac-hE18A-NH2 and mR18L with LDLr-null mice fed a high-fat Western diet (retro-orbital dosing, 100 μg/mouse, twice weekly for 8 weeks),258 a similar reduction in plasma cholesterol occurred and both peptides reduced aortic plaque burden, with Ac-hE18A-NH2 showing a greater effect.
Human apoJ (aka clusterin) is a 427-amino-acid protein found in plasma and associated with HDL.259 Navab et al. synthesized and examined amphipathic peptides representing putative class G* α-helical segments in apoJ.260 Seven of the 17 potential helical segments were tested in the artery wall cell culture assay and six were found to decrease LDL-induced monocyte chemotactic activity. The two peptides that were equipotent with full-length apoJ, apoJ(113–122) and apoJ(336–357), were resynthesized from D-amino acids and tested preliminarily for antiatherogenic activity in apoE-null mice with oral administration. The 10-residue peptide D-apoJ(113–122), lvgrqleefl (lower case for D-amino acids), was found to inhibit lesion formation. Although D4F can readily remodel apoA-I to form pre-β particles, D-apoJ(113–122) did not. An oral dose of D-apoJ(113–122) was cleared from plasma in mice much more slowly than D4F. Treatment of apoE-null mice with D-apoJ(113–122) orally (125 μg/mouse/day) sharply reduced atherosclerosis (70% reduction of aortic sinus lesions). This compound enhanced cholesterol efflux and improved HDL anti-inflammatory properties in mice and monkeys. Contrary to the behavior of D4F, D-apoJ(113–122) was hardly α-helical in the absence of a lipid environment. It was suggested260 that a common mechanism for the effectiveness of D4F and D-apoJ(113–122) may be binding and sequetration of pro-inflammatory oxidized lipids, leading to the activation of antioxidant enzymes, such as PON-1.
Serum amyloid A (SAA) proteins constitute a family of apolipoproteins (isoforms) that are associated with HDL in plasma and are produced in response to inflammatory stimuli.261,262 As such, SAA can serve as an early-phase biomarker for inflammation. Peptide fragments from the sequence of SAA (isoform 2.1) in liposomes were studied for effects on cholesterol homeostasis.262 SAA(1–20) inhibited ACAT activity and SAA(74–103) stimulated cholesterol ester hydrolase activity, which makes cholesterol available for transport and elimination; also, each peptide promoted cholesterol efflux from macrophages in vitro and in vivo (mice).262 Consequently, SAA(1–20) and SAA(74–103) were investigated as liposomal formulations for antiatherosclerotic activity in apoE-null mice on a high-fat diet, dosed every fourth day by i.v. injection.263 Each peptide (15 μg/mouse) prevented atherosclerosis, as judged by a 40–45% decrease in the amount of aortic lesions, and coadministration (7.5 μg/mouse for each) gave a remarkable 70% reduction. A more challenging plaque regression protocol also furnished positive results, especially for the combination of peptides.
EXPLORATORY WORK FROM OUR LABORATORY264
Earlier in this Perspective, we described studies with dimeric or bihelical mimetics of apoA-I that were largely based of a pair of 18A, 2F, or 4F units coupled by a short linker (Table 3; Figure 5) (vide supra). The first examples of multivalent constructs, where more than two segments of apoA-I are attached to a scaffold, appeared in the 1990’s.265,266 At the time of these studies, it was thought that apoA-I-derived peptides containing a single amphiphilic α-helix or two tandem helices would not bind to lipids with high affinity.267 Demoor and colleagues hypothesized that cooperativity among the multiple helices of apolipoproteins was essential for optimal lipid association and that branched molecules displaying several amphiphilic α-helices would better mimic the lipid-binding properties of a full-length apolipoprotein. As a consequence, molecules containing three or four copies of the 39-residue apoA-I(145–183) were synthesized on a dendrimeric, lysine-based peptide core, thereby presenting a total of six or eight amphipathic α-helices, respectively. Compared to the 39-mer peptide, the multivalent constructs were more effective at clearing DMPC vesicles: monomer, 10% clearance; trimer, 80% clearance; tetramer, 75% clearance (relative to apoA-I after 16 h).265 Additionally, compared to apoA-I(145–183), the multivalent peptides generated nanoparticles that were more like those from apoA-I in size and homogeneity.265 The lipid complex formed from the trimeric species stimulated cholesterol efflux to a similar degree as the complex formed from apoA-I.266 Despite these positive findings, the authors noted an important concern about the structural integrity of the materials involved, which derives from the parallel-synthesis approach that was used. Since three or four 39-mers were synthesized in parallel on the core peptide, there was a distinct likelihood that the constructs would contain one, or a few, amino acid deletions (“micro-heterogeneities”), even after purification. Around the same time, the first studies showing that short 22-mer peptides corresponding to apoA-I N-terminal or C-terminal helices bind to lipids with high affinity appeared,81,127 indicating that multiple helices are not functionally necessary. No further reports of multivalent apoA-I mimetics appeared in the literature for the next 15 years, as researchers in the field focused intensely on peptides consisting of single or tandem amphipathic α-helices.
Figure 5.
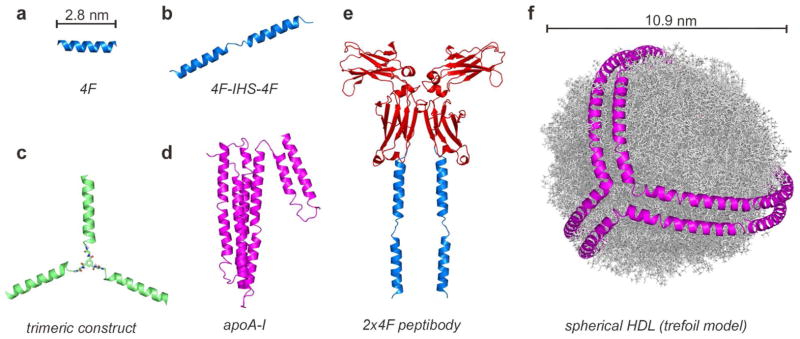
Structural models of several apoA-I mimetic molecules, along with lipid-free apoA-I and spherical HDL. The models are drawn to scale. (a) The 18-amino acid 4F peptide. (b) A tandem apoA-I mimetic, consisting of two 4F segments connected by a seven-residue interhelical sequence (IHS) peptide linker.101 (c) A branched, trimeric apoA-I mimetic construct consisting of three copies of a 23-amino acid apoA-I helix 10-derived peptide attached to a small scaffold.264 (d) Lipid-free apoA-I.134–136 (e) A 4F “peptibody”, consisting of a mouse IgG Fc fragment fused to two copies of 4F to give a tetrameric display of 4F (Fc is a dimer).240 (f) The trefoil model of spherical HDL, showing lipid molecules as gray sticks.147 The following protein structures were used to create the models: PDB# 2A01, lipid-free apoA-I and helical segments; PDB# 3HKF, murine IgG Fc fragment; PMDB# PM0075240, spherical HDL.
The highly dynamic HDL structure is characterized by many rapidly interchanging, coexisting conformations of apoA-I, which are related to the particle size and composition. ApoA-I exhibits a high degree of conformationally plasticity in forming the various particle sizes and morphologies amidst HDL maturation. We thought that branched, multimeric α-helical constructs, although not found in nature, might be better suited than monomeric helices for lipid nanoparticle scaffolding and conformational adaptability, thereby better replicating the dynamic HDL remodeling process. Thus, we carried out a systematic study of chemically homogeneous, branched apoA-I mimetics containing different numbers of attached helices.264 We synthesized and characterized two families of monomer, dimer, trimer, and tetramer apoA-I mimetic constructs (see Figure 5 for a model of a trimeric construct). A convergent synthetic strategy employing native chemical ligation (NCL)268 furnished the multivalent apoA-I mimetic peptides with good synthetic efficiency (ca. 50% isolated yield for the trimer). The two families of constructs differed in the length of the helical peptide; the first series employed a 23-mer peptide sequence largely based on human apoA-I helix 10 (residues 222–243), and the second series employed a truncated 16-mer variant. The 23-mer peptide CGVLESFKASFLSALEEWTKKLQ-NH2 has a cysteine at the N-terminus to effect NCL and an adjacent glycine to act as a spacer. Flexible, uncharged scaffolds were employed to avoid influencing the functional properties of the constructs.
For both series of constructs, the multivalent apoA-I mimetics were more effective than the monomer in effluxing cholesterol from mouse macrophages (cholesterol efflux EC50 values (μM): monomer, 5.8 ± 1.2; dimer, 0.4 ± 0.1; trimer, 1.4 ± 0.4; tetramer, 0.8 ± 0.1; 4F, 6.2 ± 1.3; apoA-I, 0.2 ± 0.1). The multivalent constructs were also found to be remarkably stable to proteolysis and to have a protracted half-life in mouse plasma in vivo (PK AUC values (μM•h): monomer23, 110 ± 10; dimer23 1570 ± 130μ trimer23, 500 ± 40; monomer16, <10 μM•h; dimer16, 340 ± 30 μM•h; trimer16, 180 ± 30 μM•h; and tetramer16, 260 ± 30 μM•h, respectively). Moreover, the 23-mer trimer construct, despite being synthesized from all L-amino acids, was resistant to degradation by pepsin, making oral dosing a possibility. Indeed, administration of 23-mer trimer-DMPC nanoparticles ad libitum in drinking water over 10 weeks to LDLr-null mice on a high-fat diet (50 mg/kg/day, n = 7) reduced aortic atherosclerotic lesions by 58% compared to control animals given DMPC alone (n = 7; p = 2 × 10−7, unpaired two-tailed Student’s t-test). Intramolecular association of the α-helices in the multivalent constructs might confer the high proteolytic stabilities.269 The enhanced stabilities observed for the multivalent peptides, together with their improved activities in functional assays, appear to afford a practical advantage over monomeric apoA-I mimetics. The multivalent architecture provides an additional layer of manipulation, beyond the primary sequence, over the functional properties of the apoA-I mimetic entities.
To gain mechanistic understanding, we investigated how the multivalents constructs interact with native lipoproteins in vitro. The 23-mer family of peptides was alkylated on the Cys side chain with a fluorescein moiety, and each of the peptides was then separately assembled into lipid nanoparticles with DMPC containing 1% of a rhodamine-labeled DMPC analogue. The labeled nanoparticles were incubated with human plasma, which was then subjected to NDGGE. Fluorescent imaging and Western blotting (for apoA-I) of the gel were used to visualize the fluorescently-labeled components and HDL species, respectively, as the peptide and lipid constituents of the nanoparticles interacted with native lipoproteins in the sample. Basically, the labeled peptides and the lipids rapidly associated with the native lipoproteins. Fluorescence from the multivalent peptides was overlaid with the apoA-I-containing HDL bands (7.5–11 nm), but the monomer was largely bound to albumin, which is consistent with a report that monomeric 4F only weakly associates with HDL and is mainly bound to plasma proteins.104 By contrast, the tandem peptide 4F-P-4F binds to lipoproteins more selectively.104
CARDIOVASCULAR PROTECTIVE EFFECTS
Throughout this Perspective, we have referred to the atheroprotective effects of certain HDL species, apoA-I itself, and apoA-I mimetic peptides. The purpose of this section is to summarize the associated mechanisms in one place, along with additional details that will shed more light on them.
The widespread use of drugs that reduce LDL-C (the “bad cholesterol”) has had a major, positive impact on the prevalence of coronary heart disease. However, there are numerous patients who do not respond well to the current standards of care, or who experience unacceptable side effects, which call for additional pharmaceutical intervention. Thus, a keen interest developed for elevating levels of HDL-C (the “good cholesterol”), although this approach has yet to bear fruit. In human studies, the unimpressive results with agents that elevate HDL-C have brought doubts to the surface and have spurred cogent thoughts about the benefits of HDL quality over HDL quantity.54,57,270,271 Indeed, with respect to the atheroprotective effects of HDL, it is becoming increasingly evident that function trumps quantity. The composition and functional effects of HDL particle subpopulations can define whether HDL is atheroprotective or not (or even proatherogenic).272
From a historical standpoint, population studies have indicated that the main risk factors for cardiovascular disease include elevated plasma LDL-C and plasma triglycerides, along with low HDL-C.273 Whereas the rate of cardiovascular events has markedly declined with current therapies that lower LDL-C, risk assessment based on LDL-C alone has failed to identify a substantial portion of the at-risk patients. Alternatively, low HDL-C has been viewed as a significant risk factor for the development of cardiovascular disease, particularly atherosclerosis.274–276 Relative to HDL elevation, in human clinical trials an increase in plasma HDL levels by just 1 mg/dL resulted in a 2–3% decrease in cardiovascular risk,276 which is a larger effect than that seen with a similar decrease in plasma LDL-C, such as via statins. In this vein, human epidemiological studies have shown that elevated plasma levels of HDL correlate inversely with the development of coronary heart disease.274,277 Although a recent meta-analysis has challenged these epidemiological findings,55 numerous experimental studies in humans and animals have indicated that HDL and/or apoA-I protect against the development of atherosclerosis (vide supra).
While HDL particles have several identified functions in vivo, the precise mechanisms by which they prevent atherosclerosis are not completely understood. Among the various subpopulations in the HDL fraction from human plasma, those responsible for the atheroprotective properties of HDL have not been completely established, although the focus had been on the smaller HDL particles and lipid-free apoA-I. Key mechanisms in atheroprotection include efflux of cholesterol from cells via RCT,60–66 especially removal of macrophage cholesterol from atherosclerotic plaques, and exerting anti-inflammatory and antioxidant properties.18,19 Besides these mechanisms, other functions of HDLs could be important to their cardioprotective properties, as underscored by recent proteomic analyses that have identified an unexpectedly wide variety of proteins associated with HDL particles.153–164 The putative functions of HDLs suggested by these studies include roles in hemostasis and thrombosis, the complement pathway, host defense, and the acute-phase response.153
Reverse Cholesterol Transport and Lipoproteostasis
HDL plays a key role in the process of RCT, which removes excess cholesterol from peripheral tissues and delivers it to the liver for elimination,15–17 as illustrated in the basic cartoon in Figure 1. It is important to appreciate that specific subpopulations within the realm of HDL particles are effective for effluxing cholesterol, particularly the small, lipid-poor particles, known as the pre-β fraction. In fact, cholesterol efflux capacity better correlates with pre-β serum concentration than with HDL-C or apoA-I levels.62 These pre-β particles are mainly composed of phospholipids and apoA-I, although their exact composition is debated. Besides the lipid-poor pre-β particles, lipid-free apoA-I also promotes efflux as an early step of RCT. Under normal circumstances, the atheroprotective pre-β particles are a minor subpopulation, accounting for only 5–15% of the heterogeneous HDL particles.278 Certain α-type particles can contribute to cholesterol efflux via ABCG1 (Figure 1), as well, and thus exert atheroprotective effects.279
Pre-β particles accumulate phospholipids and cholesterol, which passes across cell membranes under the influence of ABCA1. The cholesterol in these nascent particles is esterified by LCAT, and the resulting cholesterol esters accumulate to form the core of larger, spherical HDL particles, in which substantial triglycerides are also present. The cholesterol esters are largely off-loaded by CETP for hepatic disposal via apoB-containing particles, with concomitant replacement by triglycerides, or through the agency of SR-B1. The HDL triglyceride and phospholipid content is depleted by lipases (hepatic and endothelial)280,281 and PLTP to form smaller HDL particles and restart the cycle. Interference with the key steps of RCT have caused accelerated atherosclerosis, and the overexpression of key factors in the cycle have provided atheroprotection.282
The dynamic flux of such nanolipid particles defines a complex “lipoproteostasis” network, in which HDL is remodeled with the influx, efflux, or modification of diverse constituents (phospholipids, lipids, and cholesterol; proteins, peptides, and small molecules).153–164 Addition of apoA-I to the system will shift the balance toward pre-β particles and thus be antiatherogenic. ApoA-I mimetic peptides have a similar effect by inducing remodeling of larger HDL particles to smaller ones.92 Since it is unlikely that single, amphipathic, α-helical peptides can serve in the same structural role as full-length apoA-I, perhaps the mimetics work by cooperating with native apoA-I and somehow amplifying its functional behavior.143,144 These peptides could enter the larger particles to coexist with apoA-I,234 displace some apoA-I which would be able to form nascent discoidal particles, and favorably influence the function of the apoA-I that remains in the particles. Our in vitro fluorescence-imaging studies with labeled dimer and trimer constructs indicated that these mimetic peptides rapidly transferred from synthetic nanolipid particles to HDL particles in human plasma, thereby coexisting with apoA-I in native HDLs.264
Assuming a primary role of HDL in atherogenesis due to RCT, increasing the plasma levels of HDL became of major interest to pharmaceutical companies. However, the CETP inhibitors torcetrapib and dalcetrapib failed to show efficacy in reducing cardiovascular events in Phase 3 trials, as did niacin, despite having significantly increased HDL levels in all cases. This outcome led to serious concerns about the usefulness of promoting RCT and the relevance of boosting plasma HDL-C levels to cardiovascular disease.54,57,266,267 In any event, the pending results from human clinical studies with newer CETP inhibitors, anacetrapib and evacetrapib,271 should help clarify this issue.
HDL Function: Anti-inflammatory and Antioxidant Activity
While RCT is the highly recognized function of HDL, the atheroprotective effects of HDL are closely linked to anti-inflammatory19–22,283 and antioxidant actions,18 as well. Earlier in this Perspective, we also noted that apoA-I mimetic peptides frequently manifest anti-inflammatory and antioxidant properties, which may partially emanate from their cooperation with apoA-I within HDL particles. In two studies with D4F in mice, there was a substantial reduction in atherosclerotic lesions without a significant alteration in plasma cholesterol; rather, the HDL particles had improved anti-inflammatory properties in cellular assays.89,232,237
Atherosclerosis is a chronic inflammatory condition, the hallmark of which is compositionally complex plaque deposits on the inner lining of arteries. HDL subtypes protect endothelial cells in blood vessels by decreasing cytokine-dependent adhesion molecules and monocyte adhesion.284,285 For example, HDL inhibits the expression of VCAM-1 in endothelial cells that is caused by inflammatory cytokines, such as interleukin-1 (IL-1) and tissue necrosis factor-α (TNF-α). HDLs also interfere with the production of ROS, which induce production of MCP-1 and the migration of monocytes into the endothelium. Consequently, anti-inflammatory activity for apoA-I and apoA-I mimetic peptides has been assessed by the cell-based monocyte chemotaxis assay (MCA), involving a coculture of endothelial cells and smooth muscle cells amidst human sera and LDL. HDL stimulates endothelial nitric oxide synthase (eNOS), which generates NO in blood vessels to regulate vascular tone and relax.278 This action combats endothelial dysfunction due to adverse events, such as damage from ROS and leukocyte infiltration. Interestingly, lipid-free apoA-I does not induce eNOS.278
Oxidative processes and inflammation are inextricably entwined, in that oxidative stress is a key source of inflammation.284 Lipid oxidation produces lipid hydroperoxides and oxidized LDL (ox-LDL), which promote inflammatory sequelae, but is counteracted by HDL, apoA-I, and apoA-I mimetics. The antioxidative properties of HDL rely on certain partners, such as PON1, lipoprotein-associated phospholipase A2, and LCAT, with PON1 being especially effective.286 PON1 is an HDL-associated enzyme that can prevent lipid oxidation and inactivate oxidized lipids.13,56,110 Proteomics studies have provided a connection between HDL particle subclasses and antioxidative function,154 as well as biomarkers to understand HDL functionality.155,157 It is notable that 4F has been reported to bind oxidized lipids with much higher affinity than apoA-I,89 and that the binding of oxidized lipids in the intestine has been proposed as the main mechanism of action of the 4F peptide.234
CONCLUSION AND OUTLOOK
Molecules that mimic apoA-I hold great promise for combating atherosclerosis, and possibly other diseases associated with inflammation. Studies in animal models have demonstrated that apolipoprotein mimetic peptides can enhance many facets of the RCT pathway, such as increasing lipid-poor HDL, promoting cholesterol efflux, activating LCAT, and stimulating cholesterol off-loading. The peptides can recapitulate other beneficial functions associated with HDLs, such as anti-inflammatory and antioxidant effects. These characteristics have resulted in clinically relevant improvements in animal models of atherosclerosis, most notably inhibition of the development or progression of atherosclerotic lesions. Significant challenges remain in advancing apoA-I mimetic peptides to approved drugs. Among the hurdles are high production costs and the possibility that parenteral dosing would be required, although there is precedence for synthetic peptides as marketed drugs.287,288 More problematic, however, are the considerable duration and cost of the clinical studies that would be required for apoA-I mimetics, and the lack of robust predictive clinical biomarkers relevant to reduction of cardiovascular events.
The diverse sequences of apoA-I mimetic peptides, along with in vitro and in vivo effects in a variety of disease models, suggest that they may exert their biological properties through a manifold of different mechanisms. Until the mechanisms of action are better understood, the design of improved agents will continue to require much trial and error. However, impacting cholesterol homeostasis may be a common mechanism underlying many of the disparate functions of apoA-I mimetic peptides, since cellular cholesterol content is critical in regulating the functions of diverse cells types.109 The best-understood mechanisms of action to date are the role of mimetic peptides in effluxing cholesterol from cells and their antioxidant/anti-inflammatory activity.
More detailed structure–function studies could lead to a deeper understanding of how to design improved agents. In this regard, a critical limitation is the lack of knowledge as to which in vitro assays best correlate with in vivo antiatherogenic efficacy. The development of reasonably high-throughput assays with high accuracy and good predictive power would allow novel synthetic peptides to be rapidly surveyed, perhaps even by using combinatorial peptide libraries. With improved SAR, it may become possible to design mimetics with a specific function, rather than multiple functions, with optimized potency. This would enable a more rational design of mimetic peptides and would provide useful research tools to dissect the complex functions of HDLs. Further studies will likely reveal that single, short peptides do not have the versatility to reproduce all the beneficial effects of the full-length native protein, so judicious choices will need to be made in deciding which functions to target and optimize.
There are several attractive, but underexplored, avenues by which the in vivo functions of apoA-I mimetic agents might be improved. First, by analogy to apoA-I, the in vivo half-life of a given apoA-I mimetic peptide could be lengthened by modifying its structure to increase inherent stability in vivo and its partitioning from plasma into lipoproteins. Second, replacing residue types in the peptides known to be covalently modified in apoA-I, such as Trp, Tyr, Met, or Lys, with less susceptible residue types may prolong the function of the peptide in vivo, especially at the sites of atherosclerotic plaques (Figure 4). In this regard, it remains to be seen whether it is possible to construct an efficacious apoA-I mimetic peptide without the current Trp, Tyr, and Lys residues. Also, there has been limited use of nonproteinogenic amino acids in the development of SAR for mimetic peptides, such as for the 2F series. For example, one might wonder about the effect of replacing Phe or Trp residues with naphthylalanine or biphenylalanine. Third, introduction of Cys or His residues should be more fully explored for improving the antioxidant properties of a given peptide. What’s more, Cys residues would provide for the formation of disulfide homodimers or heterodimers, making possible the mimicry of the cardioprotective functions of apoA-IM and potentially increasing the peptide’s biological half-life by attachment to a larger apolipoprotein that cannot be removed by glomerular filtration. The incorporation into apoA-I mimetics of nonproteinogenic amino acids that would confer antioxidant activity is also an attractive idea.
A complicating factor in the development of apoA-I mimetic compounds to treat atherosclerosis is the role of lipidation. Lipid-free apoA-I has different functional properties than apoA-I in HDLs, and the same would point apply to lipid-free vs. lipidated peptides. Some open questions pertain to how the bound lipids change the functional properties of apoA-I mimetic peptides, or how one lipid will affect the properties of a peptide compared to a different lipid. Mimetic peptides are often studied in vitro as lipid-free materials, but it is likely that most of these amphipathic peptides rapidly bind lipids and/or combine with endogenous lipoproteins when introduced into plasma. Furthermore, apoA-I mimetic peptides that are lipidated and characterized as such complexes will likely undergo rapid remodeling on exposure to plasma, resulting in the exchange of peptides, lipids, and proteins with native lipoproteins in vivo.
Given the diversity of peptide sequences that have been reported as mimetics of apoA-I, it is somewhat surprising that all such agents to date have employed the α-helical peptide architecture. It stands to reason that other helical scaffolds, such as foldamers derived from β-, α-/β-, or γ-peptides,289,290 could also be functional as apolipoprotein mimetics. More broadly, it is likely that other amphipathic molecules, such as amphipathic polymers, peptoids, dendrimers, etc.,289–294 could function like apoA-I mimetic peptides for some mechanistic aspects. The time is ripe for more broadly exploring the chemical sequence space of molecules that mimic apoA-I, to go beyond the constraints imposed by the backbones and amino acid side chains available from Nature.
In the final analysis, there is substantial opportunity to impact pharmacotherapy for atherosclerosis by advancing improved apoA-I mimetic agents. Considerable information that has been collected to date is quite supportive of the potential here. However, it is still necessary to devise a suitable drug candidate for clinical development.
Chart 1. Glossary of Terms.
- Apolipoprotein (apo):
Specialized protein that binds to lipids (e.g., fats, cholesterol) to form lipoproteins.
- ATP-binding cassette transporter protein (ABC):
Member of a large family of proteins involved in membrane transport that use the energy from ATP to move a substrate across a cell membrane.
- Atherosclerosis:
A chronic disease involving the buildup of plaques on the interior of the arteries, leading to hardening and thickening of the vessel walls.
- Cholesterol (C):
A fatty substance produced in the human body or ingested from animal products, which is a major component of cell membranes.
- Cholesteryl ester (CE):
Molecule that forms the core of a lipoprotein particle, and converts nascent HDL to mature HDL, generated by esterification of cholesterol with a fatty acid.
- Cholesteryl ester transfer protein (CETP):
An enzyme involved in the movement of cholesterol from peripheral tissues to the liver by mediating the transfer from HDL to VLDL or LDL of cholesteryl esters, which are then eliminated by the liver. A CETP deficiency causes markedly higher plasma levels of HDL and apoA-I.
- High-density lipoprotein (HDL):
The smallest and densest of the plasma lipoprotein particles. “Nascent” discoidal particles contain phospholipids in a bilayer configuration encircled by apolipoproteins; “mature” spherical particles have a cholesteryl ester core, some triglycerides and unesterified cholesterol, and a coating of phospholipids, free cholesterol, and apolipoproteins.
- Hypercholesterolemia:
Condition of elevated cholesterol in blood plasma.
- Lecithin–cholesterol acyltransferase (LCAT):
An enzyme that converts free cholesterol into cholesteryl esters and thereby mediates conversion of nascent HDL to mature HDL.
- Lipoproteins:
Soluble lipid-protein complexes that transport lipids in the blood stream.
- Low-density lipoprotein (LDL):
Larger, less dense plasma lipoproteins, composed of greater amounts of lipid than protein. As the primary cholesterol-containing particles, high levels of LDL-C (so-called “bad cholesterol”) increase the risk of atherosclerosis.
- Oxidized low-density lipoprotein (ox-LDL):
A form of LDL that has been chemically transformed by reactive oxygen species (ROS) such that the particles are enriched in oxidized lipid molecules. Ox-LDL promotes atherosclerosis by attracting leukocytes to the site on the arterial wall, causing inflammation and promoting cholesterol build up.
- Paraoxonase 1 (PON1):
An aryl esterase that is a major antiatherosclerotic component of HDL.
- Phospholipid:
Lipid molecules that are composed mainly of fatty acids, a phosphate group, and a small organic backbone, such as glycerol, which are the main components of cell membranes.
- Plaque:
A fatty deposit containing other substances and cells that accumulates on the inner lining of arteries. Fragmentation of an unstable plaque deposit can cause a heart attack or a stroke.
- Reactive oxygen species (ROS):
Energetic oxygen-containing molecules, such as superoxide anion radical, hydroxyl radical, and hydroperoxide, produced from oxygen-consuming metabolic processes. While ROS are important in cell signaling at normal levels, at elevated levels from cellular “oxidative stress” they cause adverse physiology, such as inflammatory sequelae.
- Reverse cholesterol transport (RCT):
A multistep process in plasma that results in the movement of cholesterol from peripheral tissues to the liver for metabolism or elimination.
- Scavenger receptor B1 (SR-B1):
An integral membrane protein that functions as a receptor for HDL, facilitating the uptake of cholesteryl esters from HDL for their elimination in the liver.
- Statin:
A drug that inhibits HMG-CoA reductase and reduces LDL-cholesterol levels.
- Triglyceride:
A lipid derived from food or produced endogenously that is comprised of glycerol triply esterified with fatty acids.
Acknowledgments
We are grateful to NIH NHLBI for funding our research efforts in the area of apoA-I mimetic peptides, Dr. Linda K. Curtiss and Dr. Yannan Zhao for helpful discussions, and Mr. Ryan Troseth for assistance with manuscript preparation.
ABBREVIATIONS USED
- ABC
ATP-binding cassette transporter protein
- ACS
acute coronary syndrome
- apo
apolipoprotein
- AUC
area under the curve
- CETP
cholesteryl ester transfer protein
- DMPC
(R)-(+)-1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine
- DPPC
(R)-(+)-1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine
- ELISA
enzyme-linked immunosorbent assay
- Fc
fragment crystallizable region
- HDL
high-density lipoprotein
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- IgG
immonoglobulin G
- IHS
interhelical sequence
- IVUS
intravascular ultrasound
- LCAT
lecithin–cholesterol acyltransferase
- LDL
low-density lipoprotein
- LDLr
low-density lipoprotein receptor
- MCP
monocyte chemotactic protein
- M-CSF
macrophage colony-stimulating factor
- MPO
myeloperoxidase
- NCL
native chemical ligation
- NDGGE
nondenaturing gradient gel electrophoresis
- ox-LDL
oxidized low-density lipoprotein
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate-buffered saline
- PC
phosphatidylcholine
- PK
pharmacokinetics
- PLTP
phospholipid transfer protein
- PON
paraoxonase
- POPC
(R)-(+)-1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine
- RCT
reverse cholesterol transport
- ROS
reactive oxygen species
- SAA
serum amyloid A
- SR-B1
scavenger receptor B-1
- TG
triglyceride
- VCAM
vascular cell adhesion molecule
- VLDL
very low-density lipoprotein
- WT
wild-type
- 4F
Ac-DWFKAFYDKVAEKFKEAF-NH2
Biographies
Luke J. Leman earned a B.A. degree (2001) summa cum laude from the University of Colorado at Boulder, and a Ph.D. degree (2006) from The Scripps Research Institute (TSRI) in La Jolla, California. His doctoral research involved the development of catalysts for acyl transfer reactions in water. He is currently Assistant Professor of Chemistry at TSRI (La Jolla), where his broad interest is in engineering new peptide-based molecules and materials (peptides, peptide nucleic acids, and peptidomimetics) that can interact with biomolecules in useful ways.
Bruce E. Maryanoff earned B.S. (1969) and Ph.D. (1972) degrees from Drexel University, and conducted postdoctoral studies at Princeton University. He joined McNeil Laboratories (Johnson & Johnson company) and advanced to Distinguished Research Fellow, the highest scientific position. He discovered TOPAMAX® topiramate, a blockbuster drug for treating epilepsy and migraine. In industry, he pursued drugs for CNS, cardiovascular, metabolic, and pulmonary disorders. He has published 275 scientific papers and is an inventor on 70 issued U.S. patents. Dr. Maryanoff received three ACS national awards: Heroes of Chemistry Award (2000); Award in Industrial Chemistry (2003); Hershberg Award (2013). He was inducted into the ACS Medicinal Chemistry Hall of Fame (2008), received the Smissman Award (2009); he is a Fellow of the ACS, RSC, and AAAS.
M. Reza Ghadiri is Professor of Chemistry and Molecular Biology, and Member of the Skaggs Institute for Chemical Biology at The Scripps Research Institute (TSRI). He conducts a broad multidisciplinary research program that encompass the de novo design of synthetic peptides and catalysts; design of intrasterically regulated semi-synthetic enzymes, self-replicating molecular systems and complex networks; adaptive biomaterials; design of antimicrobial and bioactive agents; single-molecule stochastic sensing; molecular computation; and prebiotic chemistry. Prof. Ghadiri has received a number of awards, including the Searle Scholars Award (1991), ACS Award in Pure Chemistry (1995), Feynman Prize in Nanotechnology (1998), Arthur C. Cope Scholar Award (1999), and Vincent du Vigneaud Award (2010); he is a Fellow of the AAAS.
Footnotes
The authors declare no competing financial interest.
References
- 1. [accessed in January 2013]; http://www.who.int/mediacentre/factsheets/fs310/en/index.html.
- 2. [accessed in January 2013]; http://www.heart.org/HEARTORG/Conditions/Cholesterol/PreventionTreatmentofHighCholesterol/Prevention-and-Treatment-of-High-Cholesterol_UCM_001215_Article.jsp.
- 3. [accessed in January 2013]; http://en.wikipedia.org/wiki/Statin.
- 4. [accessed in January 2013]; http://www.drugsupdate.com/news/view/2/530.
- 5. [accessed in January 2013]; http://www.mayoclinic.com/health/statins/CL00010.
- 6.Jain KS, Kathiravan MK, Somani RS, Shishoo CJ. The biology and chemistry of hyperlipidemia. Bioorg Med Chem. 2007;15:4674–4699. doi: 10.1016/j.bmc.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 7.Lee SD, Gershkovich P, Darlington JW, Wasan KM. Inhibition of cholesterol absorption: targeting the intestine. Pharm Res. 2012;29:3235–3250. doi: 10.1007/s11095-012-0858-6. [DOI] [PubMed] [Google Scholar]
- 8.Joy TR. Novel HDL-based therapeutic agents. Pharmacol Ther. 2012;135:18–30. doi: 10.1016/j.pharmthera.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Redondo S, Martinez-Gonzalez J, Urraca C, Tejerina T. Emerging therapeutic strategies to enhance HDL function. Lipids Health Dis. 2011;10:175. doi: 10.1186/1476-511X-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kontush A, Chapman MJ. High-density lipoproteins. Structure, metabolism, function and therapeutics. Wiley; Hoboken, N. J: 2012. [Google Scholar]
- 11.deGoma EM, Rader DJ. Novel HDL-directed pharmacotherapeutic strategies. Nat Rev Cardiol. 2011;8:266–277. doi: 10.1038/nrcardio.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy AJ, Remaley AT, Sviridov D. HDL therapy: two kinds of right? Curr Pharm Des. 2010;16:4134–4147. doi: 10.2174/138161210794519228. [DOI] [PubMed] [Google Scholar]
- 13.Schaefer EJ. High density lipoproteins, dyslipidemia, and coronary heart disease. Springer; New York, N.Y: 2010. [Google Scholar]
- 14.Linsel-Nitschke P, Tall AR. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov. 2005;4:193–205. doi: 10.1038/nrd1658. [DOI] [PubMed] [Google Scholar]
- 15.Rothblat GH, Phillips MC. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol. 2010;21:229–238. doi: 10.1097/mol.0b013e328338472d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009 Apr;(Suppl):S189–S194. doi: 10.1194/jlr.R800088-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.von Eckardstein A, Nofer JR, Assmann G. High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2001;21:13–27. doi: 10.1161/01.atv.21.1.13. [DOI] [PubMed] [Google Scholar]
- 18.Bandeali S, Farmer J. High-density lipoprotein and atherosclerosis: the role of antioxidant activity. Curr Atheroscler Rep. 2012;14:101–107. doi: 10.1007/s11883-012-0235-2. [DOI] [PubMed] [Google Scholar]
- 19.Murphy AJ, Woollard KJ. High-density lipoprotein: a potent inhibitor of inflammation. Clin Exp Pharmacol Physiol. 2010;37:710–718. doi: 10.1111/j.1440-1681.2009.05338.x. [DOI] [PubMed] [Google Scholar]
- 20.Patel S, Di Bartolo BA, Nakhla S, Heather AK, Mitchell TW, Jessup W, Celermajer DS, Barter PJ, Rye KA. Anti-inflammatory effects of apolipoprotein A-I in the rabbit. Atherosclerosis. 2010;212:392–397. doi: 10.1016/j.atherosclerosis.2010.05.035. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Dong JB, Wu MP. Human apoA-I overexpression diminishes LPS-induced systemic inflammation and multiple organ damage in mice. Eur J Pharmacol. 2008;590:417–422. doi: 10.1016/j.ejphar.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 22.Puranik R, Bao S, Nobecourt E, Nicholls SJ, Dusting GJ, Barter PJ, Celermajer DS, Rye KA. Low dose apolipoprotein A-I rescues carotid arteries from inflammation in vivo. Atherosclerosis. 2008;196:240–247. doi: 10.1016/j.atherosclerosis.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Valenta DT, Bulgrien JJ, Banka CL, Curtiss LK. Overexpression of human apoA-I transgene provides long-term atheroprotection in LDL receptor-deficient mice. Atherosclerosis. 2006;189:255–263. doi: 10.1016/j.atherosclerosis.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 24.Choudhury RP, Rong JX, Trogan E, Elmalem VI, Dansky HM, Breslow JL, Witztum JL, Fallon JT, Fisher EA. High-density lipoproteins retard the progression of atherosclerosis and favorably remodel lesions without suppressing indices of inflammation or oxidation. Arterioscler Thromb Vasc Biol. 2004;24:1904–1909. doi: 10.1161/01.ATV.0000142808.34602.25. [DOI] [PubMed] [Google Scholar]
- 25.Kaul S, Coin B, Hedayiti A, Yano J, Cercek B, Chyu KY, Shah PK. Rapid reversal of endothelial dysfunction in hypercholesterolemic apolipoprotein E-null mice by recombinant apolipoprotein A-I Milano-phospholipid complex. J Am Coll Cardiol. 2004;44:1311–1319. doi: 10.1016/j.jacc.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 26.Chiesa G, Monteggia E, Marchesi M, Lorenzon P, Laucello M, Lorusso V, Di Mario C, Karvouni E, Newton RS, Bisgaier CL, Franceschini G, Sirtori CR. Recombinant apolipoprotein A-I Milano infusion into rabbit carotid artery rapidly removes lipid from fatty streaks. Circ Res. 2002;90:974–980. doi: 10.1161/01.res.0000018422.31717.ee. [DOI] [PubMed] [Google Scholar]
- 27.Shah PK, Yano J, Reyes O, Chyu KY, Kaul S, Bisgaier CL, Drake S, Cercek B. High-dose recombinant apolipoprotein A-I Milano mobilizes tissue cholesterol and rapidly reduces plaque lipid and macrophage content in apolipoprotein E-deficient mice. Potential implications for acute plaque stabilization. Circulation. 2001;103:3047–3050. doi: 10.1161/hc2501.092494. [DOI] [PubMed] [Google Scholar]
- 28.Benoit P, Emmanuel F, Caillaud JM, Bassinet L, Castro G, Gallix P, Fruchart JC, Branellec D, Denefle P, Duverger N. Somatic gene transfer of human apoA-I inhibits atherosclerosis progression in mouse models. Circulation. 1999;99:105–110. doi: 10.1161/01.cir.99.1.105. [DOI] [PubMed] [Google Scholar]
- 29.Van Craeyveld E, Gordts SC, Nefyodova E, Jacobs F, De Geest B. Regression and stabilization of advanced murine atherosclerotic lesions: a comparison of LDL lowering and HDL raising gene transfer strategies. J Mol Med. 2011;89:555–567. doi: 10.1007/s00109-011-0722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lebherz C, Sanmiguel J, Wilson JM, Rader DJ. Gene transfer of wild-type apoA-I and apoA-I Milano reduce atherosclerosis to a similar extent. Cardiovasc Diabetol. 2007;6:15. doi: 10.1186/1475-2840-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shah PK, Nilsson J, Kaul S, Fishbein MC, Ageland H, Hamsten A, Johansson J, Karpe F, Cercek B. Effects of recombinant apolipoprotein A-I Milano on aortic atherosclerosis in apolipoprotein E-deficient mice. Circulation. 1998;97:780–785. doi: 10.1161/01.cir.97.8.780. [DOI] [PubMed] [Google Scholar]
- 32.Miyazaki A, Sakuma S, Morikawa W, Takiue T, Miake F, Terano T, Sakai M, Hakamata H, Sakamoto Y, Natio M, Ruan Y, Takahashio K, Ohta T, Horiuchi S. Intravenous injection of rabbit apolipoprotein A-I inhibits the progression of atherosclerosis in cholesterol-fed rabbits. Arterioscler Thromb Vasc Biol. 1995;15:1882–1888. doi: 10.1161/01.atv.15.11.1882. [DOI] [PubMed] [Google Scholar]
- 33.Soma MR, Donetta E, Parolini C, Sirtori CR, Fumagalli R, Franceschini G. Recombinant apolipoprotein A-I Milano dimer inhibits carotid intimal thickening induced by perivascular manipulation in rabbits. Circ Res. 1995;76:405–411. doi: 10.1161/01.res.76.3.405. [DOI] [PubMed] [Google Scholar]
- 34.Ameli S, Hultgardh-Nilsson A, Cercek B, Shah PK, Forrester JS, Ageland H, Nilsson J. Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation. 1994;90:1935–1941. doi: 10.1161/01.cir.90.4.1935. [DOI] [PubMed] [Google Scholar]
- 35.Rubin EM, Krauss RM, Spangler EA, Verstuyft JG, Clift SM. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein A-I. Nature. 1991;353:265–267. doi: 10.1038/353265a0. [DOI] [PubMed] [Google Scholar]
- 36.Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85:1234–1241. doi: 10.1172/JCI114558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Badimon JJ, Badimon L, Galvez A, Dische R, Fuster V. High density lipoprotein plasma fractions inhibit aortic fatty streaks in cholesterol-fed rabbits. Lab Invest. 1989;60:455–461. [PubMed] [Google Scholar]
- 38.Belalcazar LM, Merched A, Carr B, Oka K, Chen KH, Pastore L, Beaudet A, Chan L. Long-term stable expression of human apolipoprotein A-I mediated by helper-dependent adenovirus gene transfer inhibits atherosclerosis progression and remodels atherosclerotic plaques in a mouse model of familial hypercholesterolemia. Circulation. 2003;107:2726–2732. doi: 10.1161/01.CIR.0000066913.69844.B2. [DOI] [PubMed] [Google Scholar]
- 39.Tsukamoto K, Tangirala R, Chun SH, Pure E, Rader DJ. Rapid regression of atherosclerosis induced by liver-directed gene transfer of ApoE in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:2162–2170. doi: 10.1161/01.atv.19.9.2162. [DOI] [PubMed] [Google Scholar]
- 40.Plump AS, Scott CJ, Breslow JL. Human apolipoprotein A-I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci US A. 1994;91:9607–9611. doi: 10.1073/pnas.91.20.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drew BG, Carey AL, Natoli AK, Formosa MF, Vizi D, Reddy-Luthmoodoo M, Weir J, Barlow CK, van Hall G, Meikle PJ, Duffy SJ, Kingwell BA. Reconstituted high-density lipoprotein infusion modulates fatty acid metabolism in patients with type 2 diabetes mellitus. J Lipid Res. 2011;52:572–581. doi: 10.1194/jlr.P012518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tardif JC, Heinonen T, Noble S. High-density lipoprotein/apolipoprotein A-I infusion therapy. Curr Atheroscler Rep. 2009;11:58–63. doi: 10.1007/s11883-009-0009-7. [DOI] [PubMed] [Google Scholar]
- 43.Patel S, Drew BG, Nakhla S, Duffy SJ, Murphy AJ, Barter PJ, Rye KA, Chin-Dusting J, Hoang A, Sviridov D, Celermajer DS, Kingwell BA. Reconstituted high-density lipoprotein increases plasma high-density lipoprotein anti-inflammatory properties and cholesterol efflux capacity in patients with type 2 diabetes. J Am Coll Cardiol. 2009;53:962–971. doi: 10.1016/j.jacc.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 44.Shaw JA, Bobik A, Murphy A, Kanellakis P, Blombery P, Mukhamedova N, Woollard K, Lyon S, Sviridov D, Dart AM. Infusion of reconstituted high-density lipoprotein leads to acute changes in human atherosclerotic plaque. Circ Res. 2008;103:1084–1091. doi: 10.1161/CIRCRESAHA.108.182063. [DOI] [PubMed] [Google Scholar]
- 45.Tardif JC, Gregoire J, L’Allier P, Ibrahim R, Lesperance J, Heinonen TM, Kouz S, Berry C, Basser R, Lavoie MA, Guerin M, Rodes-Cabau J. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis. J Am Med Assoc. 2007;297:1675–1682. doi: 10.1001/jama.297.15.jpc70004. [DOI] [PubMed] [Google Scholar]
- 46.Nicholls SJ, Tuzcu EM, Sipahi I, Schoenhagen P, Crowe T, Kapadia S, Nissen SE. Relationship between atheroma regression and change in lumen size after infusion of apolipoprotein A-I Milano. J Am Coll Cardiol. 2006;47:992–997. doi: 10.1016/j.jacc.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 47.Kujiraoka T, Nanjee MN, Oka T, Ito M, Nagano M, Cooke CJ, Takahashi S, Olszewski WL, Wong JS, Stepanova IP, Hamilton RL, Egashira T, Hattori H, Miller NE. Effects of intravenous apolipoprotein A-I/phosphatidylcholine discs on LCAT, PLTP, and CETP in plasma and peripheral lymph in humans. Arterioscler Thromb Vasc Biol. 2003;23:1653–1659. doi: 10.1161/01.ATV.0000089328.23279.3F. [DOI] [PubMed] [Google Scholar]
- 48.Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant apoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes. Randomized controlled trial. J Am Med Assoc. 2003;290:2292–2300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- 49.Spieker LE, Sudano I, Hurlimann D, Lerch PG, Lang MG, Binggeli C, Corti R, Ruschitzka F, Luscher TF, Noll G. High-density lipoprotein restores endothelial function in hypercholesterolemic men. Circulation. 2002;105:1399–1402. doi: 10.1161/01.cir.0000013424.28206.8f. [DOI] [PubMed] [Google Scholar]
- 50.Nanjee MN, Cooke CJ, Garvin R, Semeria F, Lewis G, Olszewski WL, Miller NE. Intravenous apoA-I/lecithin discs increase pre-β-HDL concentration in tissue fluid and stimulate reverse cholesterol transport in humans. J Lipid Res. 2001;42:1586–1593. [PubMed] [Google Scholar]
- 51.Eriksson M, Carlson LA, Miettinen TA, Angelin B. Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A-I: Potential reverse cholesterol transport in humans. Circulation. 1999;100:594–598. doi: 10.1161/01.cir.100.6.594. [DOI] [PubMed] [Google Scholar]
- 52.Nicholls SJ. Is niacin ineffective? Or did AIM-HIGH miss its target? Cleve Clin J Med. 2012;79:38–43. doi: 10.3949/ccjm.79a.11166. [DOI] [PubMed] [Google Scholar]
- 53.Joy TR, Hegele RA. The failure of torcetrapib: what have we learned? Br J Pharmacol. 2008;154:1379–1381. doi: 10.1038/bjp.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 55.Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Hólm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart AFR, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PIW, Klungel OH, Maitland-van der Zee AH, Peters BJM, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VHM, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WMM, Boer JMA, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, König IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schäfer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O’Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soran H, Hama S, Yadav R, Durrington PN. HDL functionality. Curr Opin Lipidol. 2012;23:353–366. doi: 10.1097/MOL.0b013e328355ca25. [DOI] [PubMed] [Google Scholar]
- 57.Heinecke JW. A new era for quantifying HDL and cardiovascular risk? Nat Med. 2012;18:1346–1347. doi: 10.1038/nm.2930. [DOI] [PubMed] [Google Scholar]
- 58.Besler C, Luscher TF, Landmesser U. Molecular mechanisms of vascular effects of high-density lipoprotein: alterations in cardiovascular disease. EMBO Mol Med. 2012;4:251–268. doi: 10.1002/emmm.201200224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Camont L, Chapman MJ, Kontush A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol Med. 2011;17:594–603. doi: 10.1016/j.molmed.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 60.Hoang A, Drew BG, Low H, Remaley AT, Nestel P, Kingwell BA, Sviridov D. Mechanism of cholesterol efflux in humans after infusion of reconstituted high-density lipoprotein. Eur Heart J. 2012;33:657–665. doi: 10.1093/eurheartj/ehr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. New Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de la Llera-Moya M, Drazul-Schrader D, Asztalos BF, Cuchel M, Rader DJ, Rothblat GH. The ability to promote efflux via ABCA1 determines the capacity of serum specimens with similar high-density lipoprotein cholesterol to remove cholesterol from macrophages. Arterioscler Thromb Vasc Biol. 2010;30:796–801. doi: 10.1161/ATVBAHA.109.199158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oram JF, Yokoyama S. Apolipoprotein-mediated removal of cellular cholesterol and phospholipids. J Lipid Res. 1996;37:2473–2491. [PubMed] [Google Scholar]
- 64.Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211–228. [PubMed] [Google Scholar]
- 65.Jonas A. Lecithin-cholesterol acyltransferase in the metabolism of high-density lipoproteins. Biochim Biophys Acta, Lipids Lipid Metab. 1991;1084:205–220. doi: 10.1016/0005-2760(91)90062-m. [DOI] [PubMed] [Google Scholar]
- 66.Johnson WJ, Mahlberg FH, Rothblat GH, Phillips MC. Cholesterol transport between cells and high-density lipoproteins. Biochim Biophys Acta. 1991;1085:273–298. doi: 10.1016/0005-2760(91)90132-2. [DOI] [PubMed] [Google Scholar]
- 67.Cavigiolio G, Shao B, Geier EG, Ren G, Heinecke JW, Oda MN. The interplay between size, morphology, stability, and functionality of high-density lipoprotein subclasses. Biochemistry. 2008;47:4770–4779. doi: 10.1021/bi7023354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Asztalos BF, Tani M, Schaefer EJ. Metabolic and functional relevance of HDL subspecies. Curr Opin Lipidol. 2011;22:176–185. doi: 10.1097/MOL.0b013e3283468061. [DOI] [PubMed] [Google Scholar]
- 69.Rosenson RS, Brewer HB, Jr, Chapman MJ, Fazio S, Hussain MM, Kontush A, Krauss RM, Otvos JD, Remaley AT, Schaefer EJ. HDL measures particle heterogeneity proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin Chem. 2011;57:392–410. doi: 10.1373/clinchem.2010.155333. [DOI] [PubMed] [Google Scholar]
- 70.Warnick GR, McNamara JR, Boggess CN, Clendenen F, Williams PT, Landolt CC. Polyacrylamide gradient gel electrophoresis of lipoprotein subclasses. Clin Lab Med. 2006;26:803–846. doi: 10.1016/j.cll.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 71.Kontush A, Chapman MJ. Antiatherogenic small, dense HDL--guardian angel of the arterial wall? Nat Clin Pract Cardiovasc Med. 2006;3:144–153. doi: 10.1038/ncpcardio0500. [DOI] [PubMed] [Google Scholar]
- 72.Kane JP, Malloy MJ. Prebeta-1 HDL and coronary heart disease. Curr Opin Lipidol. 2012;23:367–371. doi: 10.1097/MOL.0b013e328353eef1. [DOI] [PubMed] [Google Scholar]
- 73.Bashtovyy D, Jones MK, Anantharamaiah GM, Segrest JP. Sequence conservation of apolipoprotein A-I affords novel insights into HDL structure-function. J Lipid Res. 2011;52:435–450. doi: 10.1194/jlr.R012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein A-I: a critical analysis and review. Biochim Biophys Acta. 2001;1531:4–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- 75.Segrest JP, Garber DW, Brouillette CG, Harvey SC, Anantharamaiah GM. The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv Protein Chem. 1994;45:303–369. doi: 10.1016/s0065-3233(08)60643-9. [DOI] [PubMed] [Google Scholar]
- 76.Brouillette CG, Anantharamaiah GM. Structural models of human apolipoprotein A-I. Biochim Biophys Acta. 1995;1256:103–129. doi: 10.1016/0005-2760(95)00018-8. [DOI] [PubMed] [Google Scholar]
- 77.Phillips MC. New insights into the determination of HDL structure by apolipoproteins. J Lipid Res. 2012 doi: 10.1194/jlr.R034025. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Anantharamaiah GM, Jones MK, Segrest JP. An atlas of the amphipathic helical domains of human exchangeable plasma apolipoproteins. In: Epand RM, editor. Amphipathic Helix. CRC Press; Boca Raton, Florida: 1993. pp. 109–142. [Google Scholar]
- 79.Segrest JP, De Loof H, Dohlman JG, Brouillette CG, Anantharamaiah GM. Amphipathic helix motif: classes and properties. Proteins: Struct Funct Genetics. 1990;8:103–117. doi: 10.1002/prot.340080202. [DOI] [PubMed] [Google Scholar]
- 80.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 81.Mishra VK, Palgulachari MN, Datta G, Phillips MC, Lund-Katz S, Adeyeye SO, Segrest JP, Anantharamaiah GM. Studies of synthetic peptides of human apolipoprotein A-I containing tandem amphipathic α-helices. Biochemistry. 1998;37:10313–10324. doi: 10.1021/bi980042o. [DOI] [PubMed] [Google Scholar]
- 82.Corijn J, Deleys R, Labeur C, Vanloo B, Lins L, Brasseur R, Baert J, Ruysschaert JM, Rosseneu M. Synthetic model peptides for apolipoproteins. II. Characterization of the discoidal complexes generated between phospholipids and synthetic model peptides for apolipoproteins. Biochim Biophys Acta, Lipids Lipid Metab. 1993;1170:8–16. doi: 10.1016/0005-2760(93)90169-a. [DOI] [PubMed] [Google Scholar]
- 83.Labeur C, Lins L, Vanloo B, Baert J, Brasseur R, Rosseneu M. Design of a new class of amphipathic helical peptides for the plasma apolipoproteins that promote cellular cholesterol efflux but do not activate LCAT. Arterioscler Thromb Vasc Biol. 1997;17:580–588. doi: 10.1161/01.atv.17.3.580. [DOI] [PubMed] [Google Scholar]
- 84.Epand RM, Gawish A, Iqbal M, Gupta KB, Chen CH, Segrest JP, Anantharamaiah GM. Studies of synthetic peptide analogs of the amphipathic helix. Effect of charge distribution, hydrophobicity, and secondary structure on lipid association and lecithin:cholesterol acyltransferase activation. J Biol Chem. 1987;262:9389–9396. [PubMed] [Google Scholar]
- 85.Anantharamaiah GM, Venkatachalapathi YV, Brouillette CG, Segrest JP. Use of synthetic peptide analogues to localize lecithin:cholesterol acyltransferase activating domain in apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 1990;10:95–105. doi: 10.1161/01.atv.10.1.95. [DOI] [PubMed] [Google Scholar]
- 86.Venkatachalapathi YV, Phillips MC, Epand RM, Epand RF, Tytler EM, Segrest JP, Anantharamaiah GM. Effect of end group blockage on the properties of a class A amphipathic helical peptide. Proteins: Struct Funct Genetics. 1993;15:349–359. doi: 10.1002/prot.340150403. [DOI] [PubMed] [Google Scholar]
- 87.Davidson WS, Lund-Katz S, Johnson WJ, Anantharamaiah GM, Palgunachari MN, Segrest JP, Rothblat GH, Phillips MC. The influence of apolipoprotein structure on the efflux of cellular free cholesterol to high density lipoprotein. J Biol Chem. 1994;269:22975–22982. [PubMed] [Google Scholar]
- 88.Yancey PG, Bielicki JK, Johnson WJ, Lund-Katz S, Palgunachari MN, Anantharamaiah GM, Segrest JP, Phillips MC, Rothblat GH. Efflux of cellular cholesterol and phospholipid to lipid-free apolipoproteins and class A amphipathic peptides. Biochemistry. 1995;34:7955–7965. doi: 10.1021/bi00024a021. [DOI] [PubMed] [Google Scholar]
- 89.Van Lenten BJ, Wagner AC, Jung CL, Ruchala P, Waring AJ, Lehrer RI, Watson AD, Hama S, Navab M, Anantharamaiah GM, Fogelman AM. Anti-inflammatory apoA-I-mimetic peptides bind oxidized lipids with much higher affinity than human apoA-I. J Lipid Res. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Navab M, Anantharamaiah GM, Reddy ST, Fogelman AM. ApoA-I mimetic peptides as anti-inflammatory agents. Curr Med Chem: Immunol Endocr Metab Agents. 2005;5:335–338. [Google Scholar]
- 91.Garber DW, Datta G, Chaddha M, Palgunachari MN, Hama SY, Navab M, Fogelman AM, Segrest JP, Anantharamaiah GM. A new synthetic class A amphipathic peptide analogue protects mice from diet-induced atherosclerosis. J Lipid Res. 2001;42:545–552. [PubMed] [Google Scholar]
- 92.Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Yu N, Ansell BJ, Datta G, Garber DW, Fogelman AM. Apolipoprotein A-I mimetic peptides. Arterioscler Thromb Vasc Biol. 2005;25:1325–1331. doi: 10.1161/01.ATV.0000165694.39518.95. [DOI] [PubMed] [Google Scholar]
- 93.Qin S, Kamanna VS, Lai JH, Liu T, Ganji SH, Zhang L, Bachovchin WW, Kashyap ML. Reverse D4F, an apolipoprotein-AI mimetic peptide, inhibits atherosclerosis in apoE-null mice. J Cardiovasc Pharmacol Ther. 2012;17:334–343. doi: 10.1177/1074248411434598. [DOI] [PubMed] [Google Scholar]
- 94.Sviridov DO, Ikpot IZ, Stonik J, Drake SK, Amar M, Osei-Hwedieh DO, Piszczek G, Turner S, Remaley AT. Helix stabilization of amphipathic peptides by hydrocarbon stapling increases cholesterol efflux by the ABCA1 transporter. Biochem Biophys Res Commun. 2011;410:446–451. doi: 10.1016/j.bbrc.2011.05.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Di Bartolo BA, Vanags LZ, Tan JTM, Bao S, Rye KA, Barter PJ, Bursill CA. The apolipoprotein A-I mimetic peptide, ETC-642, reduces chronic vascular inflammation in the rabbit. Lipids Health Dis. 2011;10:224. doi: 10.1186/1476-511X-10-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ingenito R, Burton C, Langella A, Chen X, Zytko K, Pessi A, Wang J, Bianchi E. Novel potent apoA-I peptide mimetics that stimulate cholesterol efflux and pre-beta particle formation in vitro. Bioorg Med Chem Lett. 2010;20:236–239. doi: 10.1016/j.bmcl.2009.10.128. [DOI] [PubMed] [Google Scholar]
- 97.Handattu SP, Datta G, Epand RM, Epand RF, Palgunachari MN, Mishra VK, Monroe CE, Keenum TD, Chaddha M, Anantharamaiah GM, Garber DW. Oral administration of L-mR18L, a single domain cationic amphipathic helical peptide, inhibits lesion formation in ApoE null mice. J Lipid Res. 2010;51:3491–3499. doi: 10.1194/jlr.M006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carballo-Jane E, Chen Z, O’Neill E, Wang J, Burton C, Chang CH, Chen X, Eveland S, Frantz-Wattley B, Gagen K, Hubbard B, Ichetovkin M, Luell S, Meurer R, Song X, Strack A, Langella A, Cianetti S, Rech F, Capito E, Bufali S, Veneziano M, Verdirame M, Bonelli F, Monteagudo E, Pessi A, Ingenito R, Bianchi E. ApoA-I mimetic peptides promote pre-beta HDL formation in vivo causing remodeling of HDL and triglyceride accumulation at higher dose. Bioorg Med Chem. 2010;18:8669–8678. doi: 10.1016/j.bmc.2010.09.074. [DOI] [PubMed] [Google Scholar]
- 99.Bielicki JK, Zhang H, Cortez Y, Zheng Y, Narayanaswami V, Patel A, Johansson J, Azhar S. A new HDL mimetic peptide that stimulates cellular cholesterol efflux with high efficiency greatly reduces atherosclerosis in mice. J Lipid Res. 2010;51:1496–1503. doi: 10.1194/jlr.M003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.D’Souza W, Stonik JA, Murphy A, Demosky SJ, Sethi AA, Moore XL, Chin-Dusting J, Remaley AT, Sviridov D. Structure/function relationships of apolipoprotein A-I mimetic peptides: implications for antiatherogenic activities of high-density lipoprotein. Circ Res. 2010;107:217–227. doi: 10.1161/CIRCRESAHA.110.216507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wool GD, Reardon CA, Getz GS. Apolipoprotein A-I mimetic peptide helix number and helix linker influence potentially anti-atherogenic properties. J Lipid Res. 2008;49:1268–1283. doi: 10.1194/jlr.M700552-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sethi AA, Stonik JA, Thomas F, Demosky SJ, Amar M, Neufeld E, Brewer HB, Davidson WS, D’Souza W, Sviridov D, Remaley AT. Asymmetry in the lipid affinity of bihelical amphipathic peptides: a structural determinant for the specificity of ABCA1-dependent cholesterol efflux by peptides. J Biol Chem. 2008;283:32273–32282. doi: 10.1074/jbc.M804461200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mishra VK, Palgunachari MN, Lund-Katz S, Phillips MC, Segrest JP, Anantharamaiah GM. Effect of the arrangement of tandem repeating units of class A amphipathic alpha-helixes on lipid interaction. J Biol Chem. 1995;270:1602–1611. doi: 10.1074/jbc.270.4.1602. [DOI] [PubMed] [Google Scholar]
- 104.Wool GD, Vaisar T, Reardon CA, Getz GS. An apoA-I mimetic peptide containing a proline residue has greater in vivo HDL binding and anti-inflammatory ability than the 4F peptide. J Lipid Res. 2009;50:1889–1900. doi: 10.1194/jlr.M900151-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. [accessed in February 2013]; http://www.prnewswire.co.uk/news-releases/esperion-reports-positive-results-for-second-phase-1-study-of-etc-642-155462665.html.
- 106.Watson CE, Weissbach N, Kjems L, Ayalasomayajula S, Zhang Y, Chang I, Navab M, Hama S, Hough G, Reddy ST, Soffer D, Rader DJ, Fogelman AM, Schecter A. Treatment of patients with cardiovascular disease with L-4F, an apo-A1 mimetic, did not improve select biomarkers of HDL function. J Lipid Res. 2011;52:361–373. doi: 10.1194/jlr.M011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bloedon LT, Dunbar R, Duffy D, Pinell-Salles P, Norris R, DeGroot BJ, Movva R, Navab M, Fogelman AM, Rader DJ. Safety, pharmacokinetics, and pharmacodynamics of oral apoA-I mimetic peptide D-4F in high-risk cardiovascular patients. J Lipid Res. 2008;49:1344–1352. doi: 10.1194/jlr.P800003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gordon SM, Davidson WS. Apolipoprotein A-I mimetics and high-density lipoprotein function. Curr Opin Endocrinol Diabetes Obes. 2012;19:109–114. doi: 10.1097/MED.0b013e32835056d4. [DOI] [PubMed] [Google Scholar]
- 109.Osei-Hwedieh DO, Amar M, Sviridov D, Remaley AT. Apolipoprotein mimetic peptides: mechanisms of action as anti-atherogenic agents. Pharmacol Ther. 2011;130:83–91. doi: 10.1016/j.pharmthera.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Imaizumi S, Navab M, Morgantini C, Charles-Schoeman C, Su F, Gao F, Kwon M, Ganapathy E, Meriwether D, Farias-Eisner R, Fogelman AM, Reddy ST. Dysfunctional high-density lipoprotein and the potential of apolipoprotein A-1 mimetic peptides to normalize the composition and function of lipoproteins. Circ J. 2011;75:1533–1538. doi: 10.1253/circj.cj-11-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Getz GS, Reardon CA. Apolipoprotein A-I and A-I mimetic peptides: a role in atherosclerosis. J Inflamm Res. 2011;4:83–92. doi: 10.2147/JIR.S12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Navab M, Shechter I, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Fogelman AM. Structure and function of HDL mimetics. Arterioscler Thromb Vasc Biol. 2010;30:164–168. doi: 10.1161/ATVBAHA.109.187518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hovingh G, Bochem AE, Kastelein JJ. Apolipoprotein A-I mimetic peptides. Curr Opin Lipidol. 2010;21:481–486. doi: 10.1097/MOL.0b013e3283404507. [DOI] [PubMed] [Google Scholar]
- 114.Getz GS, Wool GD, Reardon CA. Biological properties of apolipoprotein A-I mimetic peptides. Curr Atheroscler Rep. 2010;12:96–104. doi: 10.1007/s11883-010-0097-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Datta G, Chaddha M, Hama S, Navab M, Fogelman A, Garber DW, Mishra VK, Epand RM, Epand RF, Lund-Katz S, Phillips MC, Segrest JP, Anantharamaiah GM. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096–1004. [PubMed] [Google Scholar]
- 116.Sankaranarayanan S, Kellner-Weibel G, de la Llera-Moya M, Phillips MC, Asztalos BF, Bittman R, Rothblat GH. A sensitive assay for ABCA1-mediated cholesterol efflux using BODIPY-cholesterol. J Lipid Res. 2011;52:2332–2340. doi: 10.1194/jlr.D018051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rothblat GH, de la Llera-Moya M, Favari E, Yancey PG, Kellner-Weibel G. Cellular cholesterol flux studies: methodological considerations. Atherosclerosis. 2002;163:1–8. doi: 10.1016/s0021-9150(01)00713-4. [DOI] [PubMed] [Google Scholar]
- 118.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 119.Troutt JS, Alborn WE, Mosior MK, Dai J, Murphy AT, Beyer TP, Zhang Y, Cao G, Konrad RJ. An apolipoprotein A-I mimetic dose-dependently increases the formation of pre-beta1 HDL in human plasma. J Lipid Res. 2008;49:581–587. doi: 10.1194/jlr.M700385-JLR200. [DOI] [PubMed] [Google Scholar]
- 120.Kelesidis T, Currier JS, Huynh D, Meriwether D, Charles-Schoeman C, Reddy ST, Fogelman AM, Navab M, Yang OO. A biochemical fluorometric method for assessing the oxidative properties of HDL. J Lipid Res. 2011;52:2341–2351. doi: 10.1194/jlr.D018937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Datta G, Epand RF, Epand RM, Chaddha M, Kirksey MA, Garber DW, Lund-Katz S, Phillips MC, Hama S, Navab M, Fogelman AM, Palgunachari MN, Segrest JP, Anantharamaiah GM. Aromatic residue position on the nonpolar face of class A amphipathic helical peptides determines biological activity. J Biol Chem. 2004;279:26509–26517. doi: 10.1074/jbc.M314276200. [DOI] [PubMed] [Google Scholar]
- 122.Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest. 2006;116:3090–3100. doi: 10.1172/JCI30163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bricarello DA, Smilowitz JT, Zivkovic AM, German JB, Parikh AN. Reconstituted lipoprotein: a versatile class of biologically-inspired nanostructures. ACS Nano. 2011;5:42–57. doi: 10.1021/nn103098m. [DOI] [PubMed] [Google Scholar]
- 124.McLachlan AD. Repeated helical pattern in apolipoprotein A-I. Nature. 1977;267:465–466. doi: 10.1038/267465a0. [DOI] [PubMed] [Google Scholar]
- 125.Barker WC, Dayhoff MO. Evolution of lipoproteins deduced from protein sequence data. Comp Biochem Physiol. 1977;57:309–315. doi: 10.1016/0305-0491(77)90060-8. [DOI] [PubMed] [Google Scholar]
- 126.Fitch WM. Phylogenies constrained by the crossover process as illustrated by human hemoglobins and a thirteen-cycle, eleven-amino-acid repeat in human apolipoprotein A-I. Genetics. 1977;86:623–644. doi: 10.1093/genetics/86.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Palgunachari MN, Mishra VK, Lund-Katz S, Phillips MC, Adeyeye SO, Alluri S, Anantharamaiah GM, Segrest JP. Only the two end helixes of eight tandem amphipathic helical domains of human apo A-I have significant lipid affinity; implications for HDL assembly. Arterioscler Thromb Vasc Biol. 1996;16:328–338. doi: 10.1161/01.atv.16.2.328. [DOI] [PubMed] [Google Scholar]
- 128.Panagotopulos SE, Witting SR, Horace EM, Hui DY, Maiorano JN, Davidson WS. The role of apolipoprotein A-I helix 10 in apolipoprotein-mediated cholesterol efflux via the ATP-binding cassette transporter ABCA1. J Biol Chem. 2002;277:39477–39484. doi: 10.1074/jbc.M207005200. [DOI] [PubMed] [Google Scholar]
- 129.Holvoet P, Zhao Z, Deridder E, Dhoest A, Collen D. Effects of deletion of the carboxyl-terminal domain of apoA-I or of its substitution with helices of apoA-II on in vitro and in vivo lipoprotein association. J Biol Chem. 1996;271:19395–19401. doi: 10.1074/jbc.271.32.19395. [DOI] [PubMed] [Google Scholar]
- 130.Rogers DP, Roberts LM, Lebowitz J, Datta G, Anantharamaiah GM, Engler JA, Brouillette CG. The lipid-free structure of apolipoprotein A-I: effects of amino-terminal deletions. Biochemistry. 1998;37:11714–11725. doi: 10.1021/bi973112k. [DOI] [PubMed] [Google Scholar]
- 131.Segrest JP, Jackson RL, Morrisett JD, Gotto AM., Jr A molecular theory of lipid–protein interactions in the plasma lipoproteins. FEBS Lett. 1974;38:247–253. doi: 10.1016/0014-5793(74)80064-5. [DOI] [PubMed] [Google Scholar]
- 132.Kanellis P, Romans AY, Johnson BJ, Kercret H, Chiovetti R, Jr, Allen TM, Segrest JP. Studies of synthetic peptide analogs of the amphipathic helix. Effect of charged amino acid residue topography on lipid affinity. J Biol Chem. 1980;255:11464–11472. [PubMed] [Google Scholar]
- 133.Tanaka M, Koyama M, Dhanasekaran P, Nguyen D, Nickel M, Lund-Katz S, Saito H, Phillips MC. Influence of tertiary structure domain properties on the functionality of apolipoprotein A-I. Biochemistry. 2008;47:2172–2180. doi: 10.1021/bi702332b. [DOI] [PubMed] [Google Scholar]
- 134.Ajees AA, Anantharamaiah GM, Mishra VK, Hussain MM, Murthy HM. Crystal structure of human apolipoprotein A-I: insights into its protective effect against cardiovascular diseases. Proc Natl Acad Sci US A. 2006;103:2126–2131. doi: 10.1073/pnas.0506877103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 135.Schekman R. Editorial expression of concern for multiple articles. Proc Natl Acad Sci US A. 2010;107:6551–6551. doi: 10.1073/pnas.1003210107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Borrell B. Fraud rocks protein community. Nature. 2009;462:970. doi: 10.1038/462970a. [DOI] [PubMed] [Google Scholar]
- 137.Mei X, Atkinson D. Crystal structure of c-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem. 2011;286:38570–38582. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci US A. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Frank PG, Marcel YL. Apolipoprotein A-I: structure-function relationships. J Lipid Res. 2000;41:853–872. [PubMed] [Google Scholar]
- 140.Vitello LB, Scanu AM. Studies on human serum high density lipoproteins. Self-association of apolipoprotein A-I in aqueous solutions. J Biol Chem. 1976;251:1131–1136. [PubMed] [Google Scholar]
- 141.Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- 142.Kunnen S, Van Eck M. Lecithin-cholesterol acyltransferase: old friend or foe in atherosclerosis? J Lipid Res. 2012;53:1783–1799. doi: 10.1194/jlr.R024513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Navab M, Anantharamaiah GM, Fogelman AM. An apolipoprotein A-I mimetic works best in the presence of apolipoprotein A-I. Circ Res. 2005;97:1085–1086. doi: 10.1161/01.RES.0000194558.86099.ba. [DOI] [PubMed] [Google Scholar]
- 144.Ou J, Wang J, Xu H, Ou Z, Sorci-Thomas MG, Jones DW, Signorino P, Densmore JC, Kaul S, Oldham KT, Pritchard KA., Jr Effects of D-4F on vasodilation and vessel wall thickness in hypercholesterolemic LDL receptor-null and LDL receptor/apolipoprotein A-I double-knockout mice on Western diet. Circ Res. 2005;97:1190–1197. doi: 10.1161/01.RES.0000190634.60042.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gursky O, Mei X, Atkinson D. The crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on amyloid formation by the N-terminal fragment. Biochemistry. 2012;51:10–18. doi: 10.1021/bi2017014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Segrest JP, Jones MK, Catte A, Thirumuruganandham SP. Validation of previous computer models and MD simulations of discoidal HDL by a recent crystal structure of apoA-I. J Lipid Res. 2012;53:1851–1863. doi: 10.1194/jlr.M026229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Huang R, Silva RA, Jerome WG, Kontush A, Chapman MJ, Curtiss LK, Hodges TJ, Davidson WS. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol. 2011;18:416–422. doi: 10.1038/nsmb.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gogonea V, Gerstenecker GS, Wu Z, Lee X, Topbas C, Wagner MA, Tallant TC, Smith JD, Callow P, Pipich V, Malet H, Schoehn G, Didonato JA, Hazen SL. The low resolution structure of nascent high density lipoprotein reconstituted with DMPC with and without cholesterol reveals a mechanism for particle expansion. J Lipid Res. 2013 doi: 10.1194/jlr.M032763. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jones MK, Zhang L, Catte A, Li L, Oda MN, Ren G, Segrest JP. Assessment of the validity of the double superhelix model for reconstituted high density lipoproteins: a combined computational-experimental approach. J Biol Chem. 2010;285:41161–41171. doi: 10.1074/jbc.M110.187799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gogonea V, Wu Z, Lee X, Pipich V, Li XM, Ioffe AI, Didonato JA, Hazen SL. Congruency between biophysical data from multiple platforms and molecular dynamics simulation of the double-super helix model of nascent high-density lipoprotein. Biochemistry. 2010;49:7323–7343. doi: 10.1021/bi100588a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wu Z, Gogonea V, Lee X, May RP, Pipich V, Wagner MA, Undurti A, Tallant TC, Baleanu-Gogonea C, Charlton F, Ioffe A, DiDonato JA, Rye KA, Hazen SL. The low resolution structure of ApoA1 in spherical high density lipoprotein revealed by small angle neutron scattering. J Biol Chem. 2011;286:12495–12508. doi: 10.1074/jbc.M110.209130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Segrest JP, Jones MK, Klon AE, Sheldahl CJ, Hellinger M, De Loof H, Harvey SC. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 153.Shah AS, Tan L, Lu Long J, Davidson WS. The proteomic diversity of high density lipoproteins: our emerging understanding of its importance in lipid transport and beyond. J Lipid Res. 2013 doi: 10.1194/jlr.R035725. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Davidson WS, Silva RA, Chantepie S, Lagor WR, Chapman MJ, Kontush A. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler Thromb Vasc Biol. 2009;29:870–876. doi: 10.1161/ATVBAHA.109.186031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Heinecke JW. The HDL proteome: a marker--and perhaps mediator--of coronary artery disease. J Lipid Res. 2009;50(Suppl):S167–171. doi: 10.1194/jlr.R800097-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Davidsson P, Hulthe J, Fagerberg B, Camejo G. Proteomics of apolipoproteins and associated proteins from plasma high-density lipoproteins. Arterioscler Thromb Vasc Biol. 2010;30:156–163. doi: 10.1161/ATVBAHA.108.179317. [DOI] [PubMed] [Google Scholar]
- 157.Gordon S, Durairaj A, Lu JL, Davidson WS. High-density lipoprotein proteomics: identifying new drug targets and biomarkers by understanding functionality. Curr Cardiovasc Risk Rep. 2010;4:1–8. doi: 10.1007/s12170-009-0069-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gordon SM, Deng J, Lu JL, Davidson WS. Proteomic characterization of human plasma high density lipoprotein fractionated by gel filtration chromatography. J Prot Res. 2010;9:5239–5249. doi: 10.1021/pr100520x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Vaisar T, Mayer P, Nilsson E, Zhao XQ, Knopp R, Prazen BJ. HDL in humans with cardiovascular disease exhibits a proteomic signature. Clin Chim Acta. 2010;411:972–979. doi: 10.1016/j.cca.2010.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Alwaili K, Bailey D, Awan Z, Bailey SD, Ruel I, Hafiane A, Krimbou L, Laboissiere S, Genest J. The HDL proteome in acute coronary syndromes shifts to an inflammatory profile. Biochim Biophys Acta. 2012;1821:405–415. doi: 10.1016/j.bbalip.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 161.Rezaee F, Casetta B, Levels JH, Speijer D, Meijers JC. Proteomic analysis of high-density lipoprotein. Proteomics. 2006;6:721–730. doi: 10.1002/pmic.200500191. [DOI] [PubMed] [Google Scholar]
- 162.Karlsson H, Leanderson P, Tagesson C, Lindahl M. Lipoproteomics II: mapping of proteins in high-density lipoprotein using two-dimensional gel electrophoresis and mass spectrometry. Proteomics. 2005;5:1431–1445. doi: 10.1002/pmic.200401010. [DOI] [PubMed] [Google Scholar]
- 163.Levels JH, Geurts P, Karlsson H, Maree R, Ljunggren S, Fornander L, Wehenkel L, Lindahl M, Stroes ES, Kuivenhoven JA, Meijers JC. High-density lipoprotein proteome dynamics in human endotoxemia. Proteome Sci. 2011;9:34. doi: 10.1186/1477-5956-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hoofnagle AN, Heinecke JW. Lipoproteomics: using mass spectrometry-based proteomics to explore the assembly, structure, and function of lipoproteins. J Lipid Res. 2009;50:1967–1975. doi: 10.1194/jlr.R900015-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 166.Sparrow JT, Gotto AM., Jr Phospholipid binding studies with synthetic apolipoprotein fragments. Ann NY Acad Sci. 1980;348:187–211. doi: 10.1111/j.1749-6632.1980.tb21300.x. [DOI] [PubMed] [Google Scholar]
- 167.Gillotte KL, Zaiou M, Lund-Katz S, Anantharamaiah GM, Holvoet P, Dhoest A, Palgunachari MN, Segrest JP, Weisgraber KH, Rothblat GH, Phillips MC. Apolipoprotein-mediated plasma membrane microsolubilization. J Biol Chem. 1999;274:2021–2028. doi: 10.1074/jbc.274.4.2021. [DOI] [PubMed] [Google Scholar]
- 168.Srinivas RV, Rui Z, Owens RJ, Compans RW, Venkatachalapathi YV, Gupta KB, Srinivas SK, Anatharamaiah GM, Segrest JP. Inhibition of virus-induced cell fusion by apolipoprotein A-I and its amphipathic peptide analogs. J Cell Biochem. 1991;45:224–237. doi: 10.1002/jcb.240450214. [DOI] [PubMed] [Google Scholar]
- 169.Vanloo B, Morrison J, Fidge N, Lorent G, Lins L, Brasseur R, Ruysschaert JM, Baert J, Rosseneu M. Characterization of the discoidal complexes formed between apoA-I-CNBr fragments and phosphatidylcholine. J Lipid Res. 1991;32:1253–1264. [PubMed] [Google Scholar]
- 170.Burgess JW, Frank PG, Franklin V, Liang P, McManus DC, Desforges M, Rassart E, Marcel YL. Deletion of the C-terminal domain of apolipoprotein A-I impairs cell surface binding and lipid efflux in macrophage. Biochemistry. 1999;38:14524–14533. doi: 10.1021/bi990930z. [DOI] [PubMed] [Google Scholar]
- 171.Schmidt HHJ, Remaley AT, Stonik JA, Ronan R, Wellmann A, Thomas F, Zech LA, Brewer HB, Jr, Hoeg JM. Carboxyl-terminal domain truncation alters apolipoprotein A-I in vivo catabolism. J Biol Chem. 1995;270:5469–5475. doi: 10.1074/jbc.270.10.5469. [DOI] [PubMed] [Google Scholar]
- 172.Laccotripe M, Makrides SC, Jonas A, Zannis VI. The carboxyl-terminal hydrophobic residues of apolipoprotein A-I affect its rate of phospholipid binding and its association with high density lipoprotein. J Biol Chem. 1997;272:17511–17522. doi: 10.1074/jbc.272.28.17511. [DOI] [PubMed] [Google Scholar]
- 173.Fu Y. Rate-limiting factors of cholesterol efflux in reverse cholesterol transport: acceptors and donors. Clin Exp Pharmacol Physiol. 2010;37:703–709. doi: 10.1111/j.1440-1681.2010.05386.x. [DOI] [PubMed] [Google Scholar]
- 174.Fitzgerald ML, Morris AL, Chroni A, Mendez AJ, Zannis VI, Freeman MW. ABCA1 and amphipathic apolipoproteins form high-affinity molecular complexes required for cholesterol efflux. J Lipid Res. 2004;45:287–294. doi: 10.1194/jlr.M300355-JLR200. [DOI] [PubMed] [Google Scholar]
- 175.Vedhachalam C, Liu L, Nickel M, Dhanasekaran P, Anantharamaiah GM, Lund-Katz S, Rothblat GH, Phillips MC. Influence of ApoA-I structure on the ABCA1-mediated efflux of cellular lipids. J Biol Chem. 2004;279:49931–49939. doi: 10.1074/jbc.M406924200. [DOI] [PubMed] [Google Scholar]
- 176.Natarajan P, Forte TM, Chu B, Phillips MC, Oram JF, Bielicki JK. Identification of an apolipoprotein A-I structural element that mediates cellular cholesterol efflux and stabilizes ATP binding cassette transporter A1. J Biol Chem. 2004;279:24044–24052. doi: 10.1074/jbc.M400561200. [DOI] [PubMed] [Google Scholar]
- 177.Remaley AT, Thomas F, Stonik JA, Demosky SJ, Bark SE, Neufeld EB, Bocharov AV, Vishnyakova TG, Patterson AP, Eggerman TL, Santamarina-Fojo S, Brewer HB. Synthetic amphipathic helical peptides promote lipid efflux from cells by an ABCA1-dependent and an ABCA1-independent pathway. J Lipid Res. 2003;44:828–836. doi: 10.1194/jlr.M200475-JLR200. [DOI] [PubMed] [Google Scholar]
- 178.Sorci-Thomas MG, Kearns MW, Lee JP. Apolipoprotein A-I domains involved in lecithin-cholesterol acyltransferase activation. Structure–function relationships. J Biol Chem. 1993;268:21403–21409. [PubMed] [Google Scholar]
- 179.Sorci-Thomas MG, Thomas MJ, Curtiss LK, Landrum M. Single repeat deletion in apoA-I blocks cholesterol esterification and results in rapid catabolism of delta6 and wild-type ApoA-I in transgenic mice. J Biol Chem. 2000;275:12156–12163. doi: 10.1074/jbc.275.16.12156. [DOI] [PubMed] [Google Scholar]
- 180.Sorci-Thomas MG, Thomas MJ. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121–128. doi: 10.1016/s1050-1738(01)00163-3. [DOI] [PubMed] [Google Scholar]
- 181.Sorci-Thomas MG, Bhat S, Thomas MJ. Activation of lecithin:cholesterol acyltransferase by HDL apoA-I central helices. Clin Lipidol. 2009;4:113–124. doi: 10.2217/17584299.4.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Minnich A, Collet X, Roghani A, Cladaras C, Hamilton RL, Fielding CJ, Zannis VI. Site-directed mutagenesis and structure-function analysis of the human apolipoprotein A-I. Relation between lecithin-cholesterol acyltransferase activation and lipid binding. J Biol Chem. 1992;267:16553–16560. [PubMed] [Google Scholar]
- 183.Sorci-Thomas MG, Curtiss LK, Parks JS, Thomas MJ, Kearns MW, Landrum M. The hydrophobic face orientation of apolipoprotein A-I amphipathic helix domain 143–164 regulates lecithin: cholesterol acyltransferase activation. J Biol Chem. 1998;273:11776–11782. doi: 10.1074/jbc.273.19.11776. [DOI] [PubMed] [Google Scholar]
- 184.Sviridov D, Hoang A, Sawyer WH, Fidge NH. Identification of a sequence of apolipoprotein A-I associated with the activation of lecithin: cholesterol acyltransferase. J Biol Chem. 2000;275:19707–19712. doi: 10.1074/jbc.M000962200. [DOI] [PubMed] [Google Scholar]
- 185.Chen CH, Albers JJ. Activation of lecithin: cholesterol acyltransferase by apolipoproteins E-2, E-3, and A-IV isolated from human plasma. Biochim Biophys Acta. 1985;836:279–285. doi: 10.1016/0005-2760(85)90131-6. [DOI] [PubMed] [Google Scholar]
- 186.Pownall HJ, Hu A, Gotto AM, Jr, Albers JJ, Sparrow JT. Activation of lecithin: cholesterol acyltransferase by a synthetic model lipid-associating peptide. Proc Natl Acad Sci USA. 1980;77:3154–3158. doi: 10.1073/pnas.77.6.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Anantharamaiah GM, Mishra VK, Garber DW, Datta G, Handattu SP, Palgunachari MN, Chaddha M, Navab M, Reddy ST, Segrest JP, Fogelman AM. Structural requirements for antioxidative and anti-inflammatory properties of apolipoprotein A-I mimetic peptides. J Lipid Res. 2007;48:1915–1923. doi: 10.1194/jlr.R700010-JLR200. [DOI] [PubMed] [Google Scholar]
- 188.Krieger M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J Clin Invest. 2001;108:793–797. doi: 10.1172/JCI14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Liadaki KN, Liu T, Xu S, Ishida BY, Duchateaux PN, Krieger JP, Kane J, Krieger M, Zannis VI. Binding of high density lipoprotein (HDL) and discoidal reconstituted HDL to the HDL receptor scavenger receptor class B type I. Effect of lipid association and APOA-I mutations on receptor binding. J Biol Chem. 2000;275:21262–21271. doi: 10.1074/jbc.M002310200. [DOI] [PubMed] [Google Scholar]
- 190.Williams DL, de La Llera-Moya M, Thuahnai ST, Lund-Katz S, Connelly MA, Azhar S, Anantharamaiah GM, Phillips MC. Binding and cross-linking studies show that scavenger receptor BI interacts with multiple sites in apolipoprotein A-I and identify the class A amphipathic alpha-helix as a recognition motif. J Biol Chem. 2000;275:18897–18904. doi: 10.1074/jbc.M002411200. [DOI] [PubMed] [Google Scholar]
- 191.Liu T, Krieger M, Kan HY, Zannis VI. The effects of mutations in helices 4 and 6 of ApoA-I on scavenger receptor class B type I (SR-BI)-mediated cholesterol efflux suggest that formation of a productive complex between reconstituted high density lipoprotein and SR-BI is required for efficient lipid transport. J Biol Chem. 2002;277:21576–21584. doi: 10.1074/jbc.M112103200. [DOI] [PubMed] [Google Scholar]
- 192.Song X, Fischer P, Chen X, Burton C, Wang J. An apoA-I mimetic peptide facilitates offloading cholesterol from HDL to liver cells through scavenger receptor BI. Int J Biol Sci. 2009;5:637–646. doi: 10.7150/ijbs.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Franceschini G, Sirtori CR, Capurso A, 2nd, Weisgraber KH, Mahley RW. A-I Milano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J Clin Invest. 1980;66:892–900. doi: 10.1172/JCI109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Eberini I, Gianazza E, Calabresi L, Sirtori CR. ApoA-I Milano from structure to clinical application. Ann Med. 2008;40:48–56. [Google Scholar]
- 195.Bruckert E, von Eckardstein A, Funke H, Beucler I, Wiebusch H, Turpin G, Assmann G. The replacement of arginine by cysteine at residue 151 in apolipoprotein A-I produces a phenotype similar to that of apolipoprotein A-I Milano. Atherosclerosis. 1997:121–128. doi: 10.1016/s0021-9150(96)05982-5. [DOI] [PubMed] [Google Scholar]
- 196.Rocco AG, Sensi C, Gianazza E, Calabresi L, Franceschini G, Sirtori CR, Eberini I. Structural and dynamic features of apolipoprotein A-I cysteine mutants, Milano and Paris, in synthetic HDL. J Mol Graph Model. 2010;29:406–414. doi: 10.1016/j.jmgm.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 197.Favari E, Gomaraschi M, Zanotti I, Bernini F, Lee-Rueckert M, Kovanen PT, Sirtori CR, Franceschini G, Calabresi L. A unique protease-sensitive high density lipoprotein particle containing the apolipoprotein A-I (Milano) dimer effectively promotes ATP-binding Cassette A1-mediated cell cholesterol efflux. J Biol Chem. 2007;282:5125–5132. doi: 10.1074/jbc.M609336200. [DOI] [PubMed] [Google Scholar]
- 198.Bielicki JK, Oda MN. Apolipoprotein A-I Milano and apolipoprotein A-I Paris exhibit an antioxidant activity distinct from that of wild-type apolipoprotein A-I. Biochemistry. 2002;41:2089–2096. doi: 10.1021/bi011716p. [DOI] [PubMed] [Google Scholar]
- 199.Ibanez B, Giannarelli C, Cimmino G, Santos-Gallego CG, Alique M, Pinero A, Vilahur G, Fuster V, Badimon L, Badimon JJ. Recombinant HDL Milano exerts greater anti-inflammatory and plaque stabilizing properties than HDL wild-type. Atherosclerosis. 2012;220:72–77. doi: 10.1016/j.atherosclerosis.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 200.Fisher EA, Feig JE, Hewing B, Hazen SL, Smith JD. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2012;32:2813–2820. doi: 10.1161/ATVBAHA.112.300133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shao B. Site-specific oxidation of apolipoprotein A-I impairs cholesterol export by ABCA1, a key cardioprotective function of HDL. Biochim Biophys Acta. 2012;1821:490–501. doi: 10.1016/j.bbalip.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zheng L, Nukuna B, Brennan ML, Sun M, Goormastic M, Settle M, Schmitt D, Fu X, Thomson L, Fox PL, Ischiropoulos H, Smith JD, Kinter M, Hazen S. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J Clin Invest. 2004;114:529–541. doi: 10.1172/JCI21109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zheng L, Settle M, Brubaker G, Schmitt D, Hazen SL, Smith JD, Kinter M. Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. J Biol Chem. 2005;280:38–47. doi: 10.1074/jbc.M407019200. [DOI] [PubMed] [Google Scholar]
- 204.Bergt C, Fu X, Huq NP, Kao J, Heinecke JW. Lysine residues direct the chlorination of tyrosines in YXXK motifs of apolipoprotein A-I when hypochlorous acid oxidizes high density lipoprotein. J Biol Chem. 2004;279:7856–7866. doi: 10.1074/jbc.M309046200. [DOI] [PubMed] [Google Scholar]
- 205.Shao B, Oda MN, Bergt C, Fu X, Green PS, Brot N, Oram JF, Heinecke JW. Myeloperoxidase impairs ABCA1-dependent cholesterol efflux through methionine oxidation and site-specific tyrosine chlorination of apolipoprotein A-I. J Biol Chem. 2006;281:9001–9004. doi: 10.1074/jbc.C600011200. [DOI] [PubMed] [Google Scholar]
- 206.Bergt C, Pennathur S, Fu X, Byun J, O’Brien K, McDonald TO, Singh P, Anantharamaiah GM, Chait A, Brunzell J, Geary RL, Oram JF, Heinecke JW. The myeloperoxidase product hypochlorous acid oxidizes HDL in the human artery wall and impairs ABCA1-dependent cholesterol transport. Proc Natl Acad Sci US A. 2004;101:13032–13037. doi: 10.1073/pnas.0405292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Shao B, Bergt C, Fu X, Green P, Voss JC, Oda MN, Oram JF, Heinecke JW. Tyrosine 192 in apolipoprotein A-I is the major site of nitration and chlorination by myeloperoxidase, but only chlorination markedly impairs ABCA1-dependent cholesterol transport. J Biol Chem. 2005;280:5983–5993. doi: 10.1074/jbc.M411484200. [DOI] [PubMed] [Google Scholar]
- 208.Peng DQ, Brubaker G, Wu Z, Zheng L, Willard B, Kinter M, Hazen SL, Smith JD. Apolipoprotein A-I tryptophan substitution leads to resistance to myeloperoxidase-mediated loss of function. Arterioscler Thromb Vasc Biol. 2008;28:2063–2070. doi: 10.1161/ATVBAHA.108.173815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Shao B, Fu X, McDonald TO, Green PS, Uchida K, O’Brien KD, Oram JF, Heinecke JW. Acrolein impairs ATP binding cassette transporter A1-dependent cholesterol export from cells through site-specific modification of apolipoprotein A-I. J Biol Chem. 2005;280:36386–36396. doi: 10.1074/jbc.M508169200. [DOI] [PubMed] [Google Scholar]
- 210.Zheng Y, Kim SH, Patel AB, Narayanaswami V, Iavarone AT, Hura GL, Bielicki JK. Positional specificity of EXXK motifs within an amphipathic alpha-helix dictates preferential lysine modification by acrolein: Implications for the design of HDL mimetic peptides. Biochemistry. 2012;51:6400–6412. doi: 10.1021/bi300626g. [DOI] [PubMed] [Google Scholar]
- 211.Shao B, Pennathur S, Pagani I, Oda MN, Witztum JL, Oram JF, Heinecke JW. Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J Biol Chem. 2010;285:18473–18484. doi: 10.1074/jbc.M110.118182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hedrick CC, Thorper SR, Fu MX, Harper CM, Yoo J, Kim SM, Wong H, Peters AL. Glycation impairs high-density lipoprotein function. Diabetologia. 2000;43:312–320. doi: 10.1007/s001250050049. [DOI] [PubMed] [Google Scholar]
- 213.Calvo C, Verdugo C. Association in vivo of glycated apolipoprotein A-I with high density lipoproteins. Eur J Clin Chem Clin Biochem. 1992;30:3–5. doi: 10.1515/cclm.1992.30.1.3. [DOI] [PubMed] [Google Scholar]
- 214.Tardif JC. Emerging high-density lipoprotein infusion therapies: fulfilling the promise of epidemiology? J Clin Lipidol. 2010;4:399–404. doi: 10.1016/j.jacl.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 215.Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn J, Princen HM, Kooistra T. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27:1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- 216.Yanni AE. The laboratory rabbit: an animal model of atherosclerosis research. Lab Animals. 2004:38. doi: 10.1258/002367704323133628. [DOI] [PubMed] [Google Scholar]
- 217. [accessed in February 2013]; http://www.cnn.com/2003/BUSINESS/12/21/us.pfizer.reut.
- 218. [accessed in February 2013]; http://www.drugdevelopment-technology.com/projects/etc.
- 219. [accessed in January 2013]; http://www.esperion.com.
- 220. [accessed in February 2013]; http://www.themedicinescompany.com.
- 221.Diditchenko S, Gille A, Pragst I, Stadler D, Waelchli M, Hamilton R, Leis A, Wright Samuel D. Novel formulation of a reconstituted high-density lipoprotein (CSL112) dramatically enhances ABCA1-dependent cholesterol efflux. Arterioscler Thromb Vasc Biol. 2013;33:2202–2211. doi: 10.1161/ATVBAHA.113.301981. [DOI] [PubMed] [Google Scholar]
- 222.Waksman R, Torguson R, Kent KM, Pichard AD, Suddath WO, Satler LF, Martin BD, Perlman TJ, Maltais JA, Weissman NJ, Fitzgerald PJ, Brewer HB., Jr A first-in-man randomized placebo-controlled study to evaluate the safety and feasibility of autologous delipidated high-density lipoprotein plasma infusions in patients with acute coronary syndrome. J Am Coll Cardiol. 2010;55:2727–2735. doi: 10.1016/j.jacc.2009.12.067. [DOI] [PubMed] [Google Scholar]
- 223. [accessed in February 2013]; http://www.cerenis.com/pipeline_cer001.asp.
- 224.Jackson RL, Morrisett JD, Gotto AM, Jr, Segrest JP. Mechanism of lipid-binding by plasma lipoproteins. Mol Cell Biochem. 1975;6:43–50. doi: 10.1007/BF01731865. [DOI] [PubMed] [Google Scholar]
- 225.Anantharamaiah GM, Jones JL, Brouillette CG, Schmidt CF, Chung BH, Hughes TA, Bhown AS, Segrest JP. Studies of synthetic peptide analogs of the amphipathic helix. J Biol Chem. 1985;18:10248–10255. [PubMed] [Google Scholar]
- 226.Chung BH, Anatharamaiah GM, Brouillette CG, Nishida T, Segrest JP. Studies of synthetic peptide analogs of the amphipathic helix. J Biol Chem. 1985;260:10256–10262. [PubMed] [Google Scholar]
- 227.Segrest JP, Chung BH, Brouillette CG, Kanellis P, McGahan R. Studies of synthetic peptide analogs of the amphipathic helix. Competitive displacement of exchangeable apolipoproteins from native lipoproteins. J Biol Chem. 1983;258:2290–2295. [PubMed] [Google Scholar]
- 228.Mishra VK, Palgulachari MN, Segrest JP, Anatharamaiah GM. Interactions of synthetic peptide analogs of the class A amphipathic helix with lipids. J Biol Chem. 1994;269:7185–7191. [PubMed] [Google Scholar]
- 229.Mendez AJ, Anantharamaiah GM, Segrest JP, Oram JF. Synthetic amphipathic helical peptides that mimic apolipoprotein A-I in clearing cellular cholesterol. J Clin Invest. 1994;94:1698–1705. doi: 10.1172/JCI117515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Wool GD, Cabana VG, Lukens J, Shaw PX, Binder CJ, Witztum JL, Reardon CA, Getz GS. 4F Peptide reduces nascent atherosclerosis and induces natural antibody production in apolipoprotein E-null mice. FASEB J. 2011;25:290–300. doi: 10.1096/fj.10-165670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Nayyar G, Mishra VK, Handattu SP, Palgunachari MN, Shin R, McPherson DT, Deivanayagam CC, Garber DW, Segrest JP, Anantharamaiah GM. Sidedness of interfacial arginine residues and anti-atherogenicity of apolipoprotein A-I mimetic peptides. J Lipid Res. 2012;53:849–858. doi: 10.1194/jlr.M019844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Navab M. Oral administration of an apo A-I mimetic peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–292. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- 233.Navab M, Berliner JA, Subbanagounder G, Hamas S, Lusis AJ, Castellani LW, Reddy S, Shih D, Shi W, Watson AD, Van Lenten BJ, Vora D, Fogelman AM. HDL and the inflammatory response induced by LDL-derived oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2001;21:481–488. doi: 10.1161/01.atv.21.4.481. [DOI] [PubMed] [Google Scholar]
- 234.Navab M, Reddy ST, Van Lenten BJ, Buga GM, Hough G, Wagner AC, Fogelman AM. High-density lipoprotein and 4F peptide reduce systemic inflammation by modulating intestinal oxidized lipid metabolism: novel hypotheses and review of literature. Arterioscler Thromb Vasc Biol. 2012;32:2553–2560. doi: 10.1161/ATVBAHA.112.300282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Li X, Chyu KY, Faria Neto JR, Yano J, Nathwani N, Ferreira C, Dimayuga PC, Cercek B, Kaul S, Shah PK. Differential effects of apolipoprotein A-I-mimetic peptide on evolving and established atherosclerosis in apolipoprotein E-null mice. Circulation. 2004;110:1701–1705. doi: 10.1161/01.CIR.0000142857.79401.69. [DOI] [PubMed] [Google Scholar]
- 236.Handattu SP, Garber DW, Horn DC, Hughes DW, Berno B, Bain AD, Mishra VK, Palgunachari MN, Datta G, Anantharamaiah GM, Epand RM. ApoA-I mimetic peptides with differing ability to inhibit atherosclerosis also exhibit differences in their interactions with membrane bilayers. J Biol Chem. 2007;282:1980–1988. doi: 10.1074/jbc.M606231200. [DOI] [PubMed] [Google Scholar]
- 237.Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, Wagner AC, Frank JS, Datta G, Garber D, Fogelman AM. Oral D-4F causes formation of pre-beta high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation. 2004;109:3215–3220. doi: 10.1161/01.CIR.0000134275.90823.87. [DOI] [PubMed] [Google Scholar]
- 238.Getz GS, Wool GD, Reardon CA. Apoprotein A-I mimetic peptides and their potential anti-atherogenic mechanisms of action. Curr Opin Lipidol. 2009;20:171–175. doi: 10.1097/MOL.0b013e32832ac051. [DOI] [PubMed] [Google Scholar]
- 239.Getz GS, Wool GD, Reardon CA. HDL apolipoprotein-related peptides in the treatment of atherosclerosis and other inflammatory disorders. Curr Pharm Des. 2010;16:3173–3184. doi: 10.2174/138161210793292492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Lu SC, Atangan L, Won Kim K, Chen MM, Komorowski R, Chu C, Han J, Hu S, Gu W, Veniant M, Wang M. An apoA-I mimetic peptibody generates HDL-like particles and increases alpha-1 HDL subfraction in mice. J Lipid Res. 2012;53:643–652. doi: 10.1194/jlr.M020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Navab M, Reddy ST, Anantharamaiah GM, Imaizumi S, Hough G, Hama S, Fogelman AM. Intestine may be a major site of action for the apoA-I mimetic peptide 4F whether administered subcutaneously or orally. J Lipid Res. 2011;52:1200–1210. doi: 10.1194/jlr.M013144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Navab M, Anantharamaiah GM, Hama S, Hough G, Reddy ST, Frank JS, Garber DW, Handattu S, Fogelman AM. D-4F and statins synergize to render HDL antiinflammatory in mice and monkeys and cause lesion regression in old apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1426–1432. doi: 10.1161/01.ATV.0000167412.98221.1a. [DOI] [PubMed] [Google Scholar]
- 243.Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hama S, Reddy ST, Fogelman AM. Lipoprotein inflammatory properties and serum amyloid A levels but not cholesterol levels predict lesion area in cholesterol-fed rabbits. J Lipid Res. 2007;48:2344–2353. doi: 10.1194/jlr.M700138-JLR200. [DOI] [PubMed] [Google Scholar]
- 244.Navab M, Ananthramaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fonarow GC, Vahabzadeh K, Hama S, Hough G, Kamranpour N, Berliner JA, Lusis AJ, Fogelman AM. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res. 2004;45:993–1007. doi: 10.1194/jlr.R400001-JLR200. [DOI] [PubMed] [Google Scholar]
- 245.Di Bartolo BA, Nicholls SJ, Bao S, Rye KA, Heather AK, Barter PJ, Bursill C. The apolipoprotein A-I mimetic peptide ETC-642 exhibits anti-inflammatory properties that are comparable to high density lipoproteins. Atherosclerosis. 2011;217:395–400. doi: 10.1016/j.atherosclerosis.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 246.Iwata A, Miura S, Zhang B, Imaizumi S, Uehara Y, Shiomi M, Saku K. Antiatherogenic effects of newly developed apolipoprotein A-I mimetic peptide/phospholipid complexes against aortic plaque burden in Watanabe-heritable hyperlipidemic rabbits. Atherosclerosis. 2011;218:300–307. doi: 10.1016/j.atherosclerosis.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 247.Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Meth Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- 248. [accessed in February 2013]; http://www.cerenis.com/pipeline_cer522.asp.
- 249.Petraki MP, Mantani PT, Tselepis AD. Recent advances on the antiatherogenic effects of HDL-derived proteins and mimetic peptides. Curr Pharm Des. 2009;15:3146–3166. doi: 10.2174/138161209789057977. [DOI] [PubMed] [Google Scholar]
- 250.Sharifov OF, Nayyar G, Garber DW, Handattu SP, Mishra VK, Goldberg D, Anantharamaiah GM, Gupta H. Apolipoprotein E mimetics and cholesterol-lowering properties. Am J Cardiovasc Drugs. 2011;11:371–381. doi: 10.2165/11594190-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 251.Datta G, Chaddha M, Garber DW, Chung BH, Tytler EM, Dashti N, Bradley WA, Gianturco SH, Anantharamaiah GM. The receptor binding domain of apolipoprotein E, linked to a model class A amphipathic helix, enhances internalization and degradation of LDL by fibroblasts. Biochemistry. 2000;39:213–220. doi: 10.1021/bi991209w. [DOI] [PubMed] [Google Scholar]
- 252.Garber DW, Handattu S, Aslan I, Datta G, Chaddha M, Anantharamaiah GM. Effect of an arginine-rich amphipathic helical peptide on plasma cholesterol in dyslipidemic mice. Atherosclerosis. 2003;168:229–237. doi: 10.1016/s0021-9150(03)00101-1. [DOI] [PubMed] [Google Scholar]
- 253.Gupta H, White CR, Handattu S, Garber DW, Datta G, Chaddha M, Dai L, Gianturco SH, Bradley WA, Anantharamaiah GM. Apolipoprotein E mimetic peptide dramatically lowers plasma cholesterol and restores endothelial function in Watanabe heritable hyperlipidemic rabbits. Circulation. 2005;111:3112–3118. doi: 10.1161/CIRCULATIONAHA.104.497107. [DOI] [PubMed] [Google Scholar]
- 254.Nayyar G, Handattu SP, Monroe CE, Chaddha M, Datta G, Mishra VK, Keenum TD, Palgunachari MN, Garber DW, Anantharamaiah GM. Two adjacent domains (141–150 and 151–160) of apoE covalently linked to a class A amphipathic helical peptide exhibit opposite atherogenic effects. Atherosclerosis. 2010;213:449–457. doi: 10.1016/j.atherosclerosis.2010.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Nayyar G, Garber DW, Palgunachari MN, Monroe CE, Keenum TD, Handattu SP, Mishra VK, Anantharamaiah GM. Apolipoprotein E mimetic is more effective than apolipoprotein A-I mimetic in reducing lesion formation in older female apo E null mice. Atherosclerosis. 2012;224:326–331. doi: 10.1016/j.atherosclerosis.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Datta G, White CR, Dashti N, Chaddha M, Palgunachari MN, Gupta H, Handattu SP, Garber DW, Anantharamaiah GM. Anti-inflammatory and recycling properties of an apolipoprotein mimetic peptide, Ac-hE18A-NH2. Atherosclerosis. 2010;208:134–141. doi: 10.1016/j.atherosclerosis.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zheng Y, Patel AB, Narayanaswami V, Hura GL, Hang B, Bielicki JK. HDL mimetic peptide ATI-5261 forms an oligomeric assembly in solution that dissociates to monomers upon dilution. Biochemistry. 2011;50:4068–4076. doi: 10.1021/bi2002955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Handattu SP, Nayyar G, Garber DW, Palgunachari MN, Monroe CE, Keenum TD, Mishra VK, Datta G, Anantharamaiah GM. Two apolipoprotein E mimetic peptides with similar cholesterol reducing properties exhibit differential atheroprotective effects in LDL-R null mice. Atherosclerosis. 2013;227:58–64. doi: 10.1016/j.atherosclerosis.2012.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.De Silva HV, Harmony JAK, Stuart WD, Gil CM, Robbins J. Apolipoprotein J: structure and tissue distribution. Biochemistry. 1990;29:5380–5389. doi: 10.1021/bi00474a025. [DOI] [PubMed] [Google Scholar]
- 260.Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Wagner AC, Hama S, Hough G, Bachini E, Garber DW, Mishra VK, Palgunachari MN, Fogelman AM. An oral apoJ peptide renders HDL antiinflammatory in mice and monkeys and dramatically reduces atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1932–1937. doi: 10.1161/01.ATV.0000174589.70190.e2. [DOI] [PubMed] [Google Scholar]
- 261.De Beer MC, De Beer FC, Beach CM, Carreras I, Sipe JD. Mouse serum amyloid A protein. Complete amino acid sequence and mRNA analysis of a new isoform. Biochem J. 1992;283:673–678. doi: 10.1042/bj2830673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kisilevsky R, Tam SP. Macrophage cholesterol efflux and the active domains of serum amyloid A 2.1. J Lipid Res. 2003;44:2257–2269. doi: 10.1194/jlr.M300133-JLR200. [DOI] [PubMed] [Google Scholar]
- 263.Tam SP, Ancsin JB, Tan R, Kisilevsky R. Peptides derived from serum amyloid A prevent, and reverse, aortic lipid lesions in apoE−/− mice. J Lipid Res. 2005;46:2091–2101. doi: 10.1194/jlr.M500191-JLR200. [DOI] [PubMed] [Google Scholar]
- 264.Zhao Y, Imura T, Leman LJ, Curtiss LK, Maryanoff BE, Ghadiri MR. Mimicry of high-density lipoprotein: functional peptide-lipid nanoparticles based on multivalent peptide constructs. J Am Chem Soc. 2013 doi: 10.1021/ja404714a. in press. http://dx.doi.org/10.1021/ja404714a. [DOI] [PMC free article] [PubMed]
- 265.Demoor L, Boutillon C, Fievet C, Vanloo B, Baert J, Rosseneu M, Fruchart JC, Tartar A. Branched synthetic constructs that mimic the physicochemical properties of apolipoprotein A-I in reconstituted high-density lipoproteins. Eur J Biochem. 1996;239:74–84. doi: 10.1111/j.1432-1033.1996.0074u.x. [DOI] [PubMed] [Google Scholar]
- 266.Nion S, Demoor L, Boutillon C, Luchoomun J, Vanloo B, Fievet C, Castro G, Rosseneu M, Fruchart JC, Tartar A, Clavey V. Branched synthetic peptide constructs mimic cellular binding and efflux of apolipoprotein A-I in reconstituted high density lipoproteins. Atherosclerosis. 1998;141:227–235. doi: 10.1016/s0021-9150(98)00176-2. [DOI] [PubMed] [Google Scholar]
- 267.Vanloo B, Demoor L, Boutillon C, Lins L, Brasseur R, Baert J, Fruchart JC, Tartar A, Rosseneu M. Association of synthetic peptide fragments of human apolipoprotein A-I with phospholipids. J Lipid Res. 1995;36:1686–1696. [PubMed] [Google Scholar]
- 268.Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Synthesis of proteins by native chemical ligation. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 269.Pini A, Falciani C, Bracci L. Branched peptides as therapeutics. Curr Protein Pept Sci. 2008;9:468–477. doi: 10.2174/138920308785915227. [DOI] [PubMed] [Google Scholar]
- 270.Kolata G. Doubt cast on the ‘good’ in ‘good cholesterol’. [accessed in March 2013];The New York Times. 2012 May 16; http://www.nytimes.com/2012/05/17/health/research/hdl-good-cholesterol-found-not-to-cut-heart-risk.html?_r=0.
- 271.Kypreos KE, Gkizas S, Rallidis LS, Karagiannides I. HDL particle functionality as a primary pharmacological target for HDL-based therapies. Biochem Pharmacol. 2013 doi: 10.1016/j.bcp.2013.03.004. epub ahead of print, ( http://dx.doi.org/10.1016/j.bcp.2013.1003.1004) [DOI] [PubMed]
- 272.Tsompanidi EM, Brinkmeier MS, Fotiadou EH, Giakoumi SM, Kypreos KE. HDL biogenesis and functions: role of HDL quality and quantity in atherosclerosis. Atherosclerosis. 2010;208:3–9. doi: 10.1016/j.atherosclerosis.2009.05.034. [DOI] [PubMed] [Google Scholar]
- 273.Assmann G. Pro and con: high-density lipoprotein, triglycerides, and other lipid subfractions are the future of lipid management. Am J Cardiol. 2001;87:2B–7B. doi: 10.1016/s0002-9149(01)01448-5. [DOI] [PubMed] [Google Scholar]
- 274.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham study. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 275.Tall AR. Plasma high density lipoproteins. Metabolism and relationship to atherogenesis. J Clin Invest. 1990;86:379–384. doi: 10.1172/JCI114722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Gordon DJ, Probstfield JL, Garrison RJ, Neaton JD, Castelli WP, Knoke JD, Jacobs DR, Jr, Bangdiwala S, Tyroler HA. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79:8–15. doi: 10.1161/01.cir.79.1.8. [DOI] [PubMed] [Google Scholar]
- 277.Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham study. J Am Med Assoc. 1986;256:2835–2838. [PubMed] [Google Scholar]
- 278.Assmann G, Gotto Antonio M., Jr HDL cholesterol and protective factors in atherosclerosis. Circulation. 2004;109(Suppl III):8–14. doi: 10.1161/01.CIR.0000131512.50667.46. [DOI] [PubMed] [Google Scholar]
- 279.Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, Frank J, Francone OL, Edwards PA. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–131. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 280.Badellino KO, Rader DJ. The role of endothelial lipase in high-density lipoprotein metabolism. Curr Opin Cardiol. 2004;19:392–395. doi: 10.1097/01.hco.0000130161.89169.02. [DOI] [PubMed] [Google Scholar]
- 281.Santamarina-Fojo S, Gonzalez-Navarro H, Freeman L, Wagner E, Nong Z. Hepatic lipase, lipoprotein metabolism, and atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1750–1754. doi: 10.1161/01.ATV.0000140818.00570.2d. [DOI] [PubMed] [Google Scholar]
- 282.von Eckardstein A, Nofer JR, Assmann G. Acceleration of reverse cholesterol transport. Curr Opin Cardiol. 2000;15:348–354. doi: 10.1097/00001573-200009000-00007. [DOI] [PubMed] [Google Scholar]
- 283.Navab M, Anantharamaiah GM, Fogelman AM. The role of high-density lipoprotein in inflammation. Trends Cardiovasc Med. 2005;15:158–161. doi: 10.1016/j.tcm.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 284.Marsche G, Saemann Marcus D, Heinemann A, Holzer M. Inflammation alters HDL composition and function: implications for HDL-raising therapies. Pharmacol Ther. 2013;137:341–351. doi: 10.1016/j.pharmthera.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 285.Prosser HC, Ng MK, Bursill CA. The role of cholesterol efflux in mechanisms of endothelial protection by HDL. Curr Opin Lipidol. 2012;23:182–189. doi: 10.1097/MOL.0b013e328352c4dd. [DOI] [PubMed] [Google Scholar]
- 286.Moren X, Deakin S, Liu ML, Taskinen MR, James RW. HDL subfraction distribution of paraoxonase-1 and its relevance to enzyme activity and resistance to oxidative stress. J Lipid Res. 2008;49:1246–1253. doi: 10.1194/jlr.M700439-JLR200. [DOI] [PubMed] [Google Scholar]
- 287.Nestor JJ., Jr The medicinal chemistry of peptides. Curr Med Chem. 2009;16:4399–4418. doi: 10.2174/092986709789712907. [DOI] [PubMed] [Google Scholar]
- 288.Bray BL. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat Rev Drug Discov. 2003;2:587–593. doi: 10.1038/nrd1133. [DOI] [PubMed] [Google Scholar]
- 289.Horne WS. Peptide and peptoid foldamers in medicinal chemistry. Expert Opin Drug Discov. 2011;6:1247–1262. doi: 10.1517/17460441.2011.632002. [DOI] [PubMed] [Google Scholar]
- 290.Martinek TA, Fueloep F. Peptidic foldamers: ramping up diversity. Chem Soc Rev. 2012;41:687–702. doi: 10.1039/c1cs15097a. [DOI] [PubMed] [Google Scholar]
- 291.Zuckermann RN, Kodadek T. Peptoids as potential therapeutics. Curr Opin Mol Ther. 2009;11:299–307. [PubMed] [Google Scholar]
- 292.Olsen CA. Peptoid-peptide hybrid backbone architectures. ChemBioChem. 2010;11:152–160. doi: 10.1002/cbic.200900618. [DOI] [PubMed] [Google Scholar]
- 293.Palmer LC, Stupp SI. Molecular self-assembly into one-dimensional nanostructures. Acc Chem Res. 2008;41:1674–1684. doi: 10.1021/ar8000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Wang Y, Grayson SM. Approaches for the preparation of non-linear amphiphilic polymers and their applications to drug delivery. Adv Drug Delivery Rev. 2012;64:852–865. doi: 10.1016/j.addr.2012.03.011. [DOI] [PubMed] [Google Scholar]