

Abstract
Physiologically based pharmacokinetic (PBPK) modeling has been broadly used to facilitate drug development, hereby we developed a PBPK model to systematically investigate the underlying mechanisms of the observed positive food effect of compound X (cpd X) and to strategically explore the feasible approaches to mitigate the food effect. Cpd X is a weak base with pH-dependent solubility; the compound displays significant and dose-dependent food effect in humans, leading to a nonadherence of drug administration. A GastroPlus Opt logD Model was selected for pharmacokinetic simulation under both fasted and fed conditions, where the biopharmaceutic parameters (e.g., solubility and permeability) for cpd X were determined in vitro, and human pharmacokinetic disposition properties were predicted from preclinical data and then optimized with clinical pharmacokinetic data. A parameter sensitivity analysis was performed to evaluate the effect of particle size on the cpd X absorption. A PBPK model was successfully developed for cpd X; its pharmacokinetic parameters (e.g., Cmax, AUCinf, and tmax) predicted at different oral doses were within ±25% of the observed mean values. The in vivo solubility (in duodenum) and mean precipitation time under fed conditions were estimated to be 7.4- and 3.4-fold higher than those under fasted conditions, respectively. The PBPK modeling analysis provided a reasonable explanation for the underlying mechanism for the observed positive food effect of the cpd X in humans. Oral absorption of the cpd X can be increased by reducing the particle size (<100 nm) of an active pharmaceutical ingredient under fasted conditions and therefore, reduce the cpd X food effect correspondingly.
KEY WORDS: GastroPlus, particle size, physiologically based pharmacokinetics, precipitation, solubility
INTRODUCTION
Physiologically based pharmacokinetic (PBPK) models establish a physiological environment to estimate drug absorption and distribution using a series of mathematical equations. GastroPlus™ (Simulation Plus, Inc., Lancaster, CA) is a commercial PBPK modeling tool based on compartmental absorption and transit (CAT) model originally proposed by Amidon et al. (1), which was later expanded to the advanced CAT or ACAT model (2). The model contains nine compartments in sequence representing anatomic segments of the gastrointestinal (GI) tract, namely stomach, duodenum, jejunum (two compartments), ileum (three compartments), caecum, and ascending colon. Default physiological conditions such as pH values and transit time for each compartment are incorporated in the model. The absorption processes of compounds throughout GI tract can be estimated based on specific physicochemical properties such as pKa, partition coefficient, and particle size of the compounds, as well as related dosing regimen. Transmembrane passage of the compound in each GI segment is estimated based on the permeability and concentration gradient across the membrane in that GI compartment. The absorption in each segment following first-pass metabolism is subsequently estimated, accumulated, and convoluted with disposition kinetics to generate drug pharmacokinetic profiles.
Compound X (Cpd X) is a lipophilic compound displaying a low, pH-dependent aqueous solubility and moderate intestinal permeability. The compound demonstrates properties consistent with the Biopharmaceutics Classification System (BCS) class II (or IV) category (3). The absorption of cpd X in humans has been investigated in a mass-balance study following a single oral dose of 400 mg C14-labeled cpd X. Majority of the dose (∼70%) was recovered in feces as parent drug with large individual variability, suggesting a low oral absorption under fasted conditions. Metabolism occurs primarily in the liver via hydroxylation and oxidation mediated by the CYP3A4 enzyme. Unchanged cpd X form is the main circulating drug component in the serum and primarily responsible for the drug’s pharmacologic activities.
The cpd X is currently marketed as an immediate release (IR) capsule formulation. The systemic exposure of the cpd X is increased when given with food. Compared to the fasted state, the mean systemic exposure of cpd X (400 mg single dose) measured from an area under the plasma concentration–time curve (AUC) and maximum plasma concentration (Cmax) were increased by 29% and 55%, respectively, when dosed 30 min after a light meal, and increased by 82% and 112%, respectively, after a high fat meal. While absolute oral bioavailability of cpd X has not been determined due to the lack of a suitable intravenous formulation, it has been estimated to be less than 30%.
Food may interact with drug absorption via mechanisms including delay in gastric emptying, change in GI pH and bile excretion. The relationship of food effect with physicochemical properties of compounds has been reported by Fleisher et al. (4), lipophilic compounds with poor aqueous solubility mostly exhibit a positive food effect (increase in exposure after food intake) because of improved solubilization due to high bile salt concentration (5). These findings were confirmed by Gu et al. who investigated food effects of 92 compounds which correlated with physicochemical properties (6); nearly 71% of the BCS class II compounds (low solubility and high permeability) displayed a positive food effect.
Physiologically based pharmacokinetic models have been used to quantitatively predict oral drug absorption and food effects for poorly soluble drugs, based on in vitro biorelevant solubility and dissolution results, as well as investigation in animal models (7–9). The current study applied a similar PBPK approach to understand the underlying mechanism of food effects on the absorption of a low solubility compound and to explore potential formulation solutions to overcome the food effects.
MATERIALS AND METHODS
Clinical Food Effect Studies of Compound X
Pharmacokinetic data from four Novartis clinical studies conducted from 2005 to 2012 were analyzed. Healthy adult male and sterile or postmenopausal female subjects with ages from 18 to 65 years participated in these studies. All study participants were required to provide written informed consent prior to study initiation. Marketed capsule formulation was selected to investigate drug absorption with or without meal. Fasted state was defined as overnight fasting for 10 h before dosing and fed state as dosing within 30 min following ingestion of high fat meal (approximately 1,000 calories with 50% from fat content). Study details and demographic information of participants are shown in Table I.
Table I.
Clinical Studies of Cpd X at Various Doses
| Study number | Dose (mg) | Physiological conditions | Number of subjects | Body weight of participants (kg) (mean ± SD) | Ages of participants (year) (mean ± SD) |
|---|---|---|---|---|---|
| C2105 | 200 | Fasted and fed | 20 | 76.5 ± 9.4 | 55.2 ± 8.6 |
| C2118 | 300 | Fasted | 58 | 76.8 ± 13.2 | 44.1 ± 7.4 |
| A2106 | 400 | Fasted and fed | 44 | 79.4 ± 12.6 | 37.0 ± 9.0 |
| A2108 | 600 | Fasted | 18 | 73.1 ± 8.6 | 39.9 ± 11.0 |
Data Analysis of Clinical Studies
The PK profiles of cpd X were determined by collecting series of blood samples at pre-dose, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, and 72 h post-dose. Plasma concentrations of the cpd X were measured using a validated liquid chromatography/tandem mass spectrometry (LC-MS/MS) assay. The plasma concentration versus time data were analyzed with non-compartmental analysis (NCA) methods to calculate PK parameters (e.g., AUC, Cmax, tmax, CL/F, etc.) using WinNonlin Version 5.0.1 (Pharsight Corporation, Mountain View, CA). In addition, arithmetic mean plasma concentrations at each sampling time were calculated, and the mean plasma concentration versus time profiles at 200, 300, 400, and 600 mg were individually transferred to the GastroPlus™ software database for simulation.
Modeling and Simulations
Disposition Pharmacokinetics of Compound X in Humans
The PK of cpd X following intravenous administration has never been investigated in humans. The key disposition parameters in humans used in this study (e.g., clearance and volume of distribution) were collected from literature based on a verified model, with minor modifications according to the CL/F values obtained following NCA (Table II).
Table II.
Physicochemical Parameters, Default Physiological Values, and Pharmacokinetic Parameter Used in the Simulation at Various Doses
| Parameters | Value(s) |
|---|---|
| Compound parameters | |
| M w: g/mol | >475 |
| cLogP: | >4 |
| pK a (base): | 3.2, 6.2 |
| Dosage: | IR capsule |
| Solubility (mg/mL ): | 1.8 (pH 1), 0.3 (pH 2), 0.001 (pH 6.8) |
| Biorelevant solubility (mg/mL) : | 0.023 (fasted); 0.190 (fed) |
| Mean precipitation time (s) : | 450 s (fasted); 2,000 s (fed) |
| Effective permeability (cm/s): | 1.48 × 10−4 |
| Particle radius of API (μm): | 19 |
| Physiological parameters | |
| Stomach pH | 1.2 (Fasted); 1.2–4.9 (Fed) |
| Duodenum/jejunum pH | 6.0–6.4 (Fasted); 5.4–6.0 (Fed) |
| Ileum pH | 6.6–7.4 (Fasted); 6.6–7.4 (Fed) |
| Cecum–colon pH | 6.4–6.8 |
| Stomach transit time (h) | 2.0 (Fasted); 5.4 (Fed) |
| Small intestine transit time (h) | 3.3 |
| Cecum transit time (h) | 4.2 |
| Ascending colon transit time (h) | 12.6 |
| Pharmacokinetics | |
| First pass extraction (%): | 9.0 |
| Blood/plasma ratio: | 0.68 |
| Plasma unbound (%): | 1.6 |
| Clearance (L/h/kg) | 0.070 |
| V c (L/kg) | 0.4 |
| k 12 (1/h) | 0.64 |
| k 21 (1/h) | 0.17 |
| V t (L/kg) | 1.5 |
Prediction of Human Plasma Concentration–Time Profiles and Estimation of Food Effect
The GastroPlus™ (Version 8, Simulation Plus, Inc., CA) was used to simulate oral pharmacokinetic profiles of cpd X following a single dose administration under both fasted and high fat meal conditions. The default human physiology model (Opt LogD model SA/V 6.1) was selected, and fraction of absorption was estimated following simulation. Experimentally measured physicochemical properties of the cpd X, including pKa, LogP, pH-dependent aqueous solubility, and permeability are shown in Table II. The apparent permeability measured across Caco-2 cell layers (Papp, apical to basolateral at 30 μM, 2.7 × 10−6 cm/s) was converted to a human effective permeability (Peff, 1.5 × 10−4 cm/s) using a reported correlative equation (10).
Estimation of In Vivo Solubility and Dissolution Profiles
The cpd X is a weak base with two pKa values (pKa1 = 3.0 and pKa2 = 6.2) displaying high solubility under acidic conditions (1.81 mg/ml at pH 1). The solubility is sharply reduced with increasing pH (0.3 mg/mL at pH 2 and 0.001 mg/mL at pH 6.8) (Table II). Therefore, the in vivo dissolution kinetics of cpd X is expected to be altered accordingly with physiological pH changes in the GI tract following a meal intake. To estimate the in vivo solubility of the cpd X, the solubility in biorelevant media including a simulated gastric fluid (SGF), a fasted simulated small intestinal fluid (FaSSIF), and a fed simulated small intestinal fluid (FeSSIF) was measured. These solubility values, together with other biopharmaceutic properties of cpd X, such as diffusion coefficient and particle size were utilized to generate in vivo dissolution profiles under fasted and fed conditions in the model using a modified Noyes–Whitney equation (11). The in vivo precipitation time used in this in silico model was adjusted to best fit the in vivo human pharmacokinetic profiles, because no feasible method was currently available to quantitatively determine the precipitation time. An in vitro two-step dissolution test in the SGF/FaSSIF and SGF/FeSSIF was investigated and referenced to provide useful insights of drug precipitation.
Estimation of pH Values in Stomach
The pH value in stomach was reported to be time dependent within subjects and variables during the digestion process following food intake (12). This value, however, was constant in the current ACAT model (pH = 4.9) and failed to reflect a dynamic change of the gastric acidity. In this study, human stomach pH was switched to a set of sequential values following meal intake, similar to the values reported by Gardner et al. and Dressman et al. (12,13) (Table III).
Table III.
Stomach pH and ACAT Physiology Models under High Fat Fed Conditions
| Time after dose (h) | Stomach pH | ACAT physiology model |
|---|---|---|
| 0–3 | 4.9 | Human physiological fed |
| 4–5 | 3.5 | Human physiological fed |
| 4–4.5 | 2.0 | Human physiological fed |
| >4.5 | 1.2 | Human physiological fasted |
IMPACT OF DRUG PARTICLE SIZE ON DRUG ABSORPTION AND PARAMETER SENSITIVITY ANALYSIS
For the solid active pharmaceutical ingredients (API), the dissolution rate is proportional to drug solubility and the surface area of drug particles according to a modified Noyes–Whitney equation and therefore, inversely proportional to the particle size. In addition, drug solubility is inversely proportional to the particle size according to Frudlich–Ostwald equation (Eq. 1) (14).
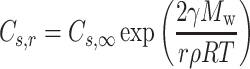 |
1 |
where Cs,r is the solubility of drug particle of radius r, Cs,∞ is the solubility of the same drug with no curvature at temperature T, r is the particle radius, R is the gas constant (8.314 J/K/mol), ρ is the density, Mw is the molecular weight, and γ is the interfacial tension. Drug particles less than 1 μm are easily trapped in apical microvilli of the intestinal wall, leading to an abnormally higher concentration gradient across intestinal wall and higher permeation rate. When the particle size is within nanoscale range, the Noyes–Whitney dissolution equation is modified by taking consideration of the nanoparticle factor effect (NFE), which serves as a solubility scaling factor of Cs,r to describe the increased dissolution rate of nanoparticles (Eq. 2),
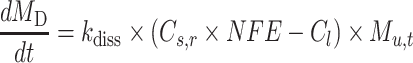 |
2 |
where dMD/dt is the dissolution rate, kdiss is the dissolution rate constant, Cl is the total drug concentration in the lumen of the compartment, MD is the amount of dissolved drug, and Mu,t is the undissolved amount at time of t.
To investigate the correlation of the particle size with fraction of absorption at various doses, a parameter sensitivity analysis (PSA) was conducted. A three-dimension surface response plot was generated to elucidate the interactive effect of particle sizes and doses on the extent of cpd X absorption under both fasted and fed condition. In addition, the positive food effects, represented by AUC0-inf ratio of fed versus fasted, was estimated based on the predicted AUC values in the PSA.
RESULTS
Observed Pharmacokinetics of Compound X
The cpd X was rapidly absorbed with measurable plasma concentrations at 0.5 h post-dose, and tmax occurred at approximately 4 h post-dose under fasted conditions. The AUC and Cmax of the cpd X were increased with increasing doses in an under dose-proportional manner (Table IV). The Cpd X displayed significant and dose-dependent food effects. After dosing with high-fat food, the tmax was prolonged to 5 h, and AUCinf was increased by 49.6% and 74.3% at 200 and 400 mg dose levels, respectively (Fig. 1 and Table IV).
Table IV.
Calculated and Predicted Pharmacokinetic Parameters of Cpd X at Various Dose Levels under Both Fasted and Fed Conditions
| Dose (mg) | Meal condition | C max(ng/ml) | AUCinf (ng × h/ml) | t max (h) | F a (%) | F (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| Observed | Predicted | Observed | Predicted | Observed | Predicted | Predicted | Predicted | ||
| 200 | Fasted | 320 | 302 | 8,595 | 11,170 | 4.0 | 3.1 | 29.7 | 27.4 |
| 200 | Fed | 601 | 547 | 12,857 | 15,555 | 5.0 | 5.5 | 41.8 | 38.0 |
| 300 | Fasted | 451 | 387 | 11,486 | 13,080 | 4.0 | 3.0 | 24.0 | 22.1 |
| 400 | Fasted | 508 | 449 | 14,656 | 15,920 | 4.0 | 3.1 | 20.3 | 18.7 |
| 400 | Fed | 1068 | 892 | 25,542 | 25,010 | 5.0 | 6.1 | 33.2 | 30.6 |
| 600 | Fasted | 453 | 529 | 14,576 | 18,840 | 4.0 | 3.6 | 15.6 | 14.4 |
Fig. 1.
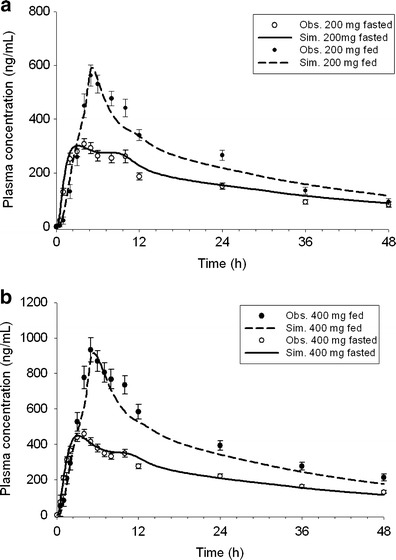
Mean clinically observed (solid circles with standard error) and model-simulated plasma concentration versus time profiles of cpd X after a single oral dose of a 200 mg or b 400 mg cpd X under fasted and fed condition
Modeling and Simulations
Prediction of Concentration–Time Profiles and Pharmacokinetic Parameters
Simulation results of cpd X at 200 and 400 mg under both fasted and fed conditions are shown in Fig. 1. The modeling was evaluated by comparing predicted pharmacokinetic parameters including Cmax, AUCinf, and tmax to those mean values calculated from observed clinical data. The predicted pharmacokinetic values were observed to be within ±25% of the observed mean values at various doses under both meal conditions (Table IV).
Gastrointestinal Dissolution and Absorption of Compound X under Fasted and Fed Conditions
Absorption of the cpd X in different GI regions was estimated by the calculated fraction absorbed (Fa) (Fig. 2). The cpd X was primarily absorbed in the upper GI tract including duodenum and jejunum. The absorption in ileum was minimal, and less than 5% absorption occurred in colon over the dose range from 200 to 600 mg under both meal conditions, likely due to the limited solubility at elevated pH in ileum and colonic regions. The cpd X displayed dose-dependent absorption, and Fa decreased with increasing doses. For example, Fa decreased from 29.7% to 15.6% when the dose was increased from 200 to 600 mg under fasted conditions and decreased from 41.8% to 33.2% when the dose was increased from 200 to 400 mg under fed conditions (Table IV). The in vivo dissolution profiles of cpd X were derived following simulations (Fig. 3). Under fasted conditions, cpd X achieved rapid dissolution in the stomach within the first hour post-dose and then, proceeded with rapid precipitation when accessing intestinal tract with relatively high pH values. The fraction of amount of drug dissolved (as% of dose), dropped from 30% to 20% when the dose was increased from 200 to 400 mg under fasted conditions. In contrast, slow but gradually enhanced dissolution was observed for the cpd X under fed conditions and faster dissolution of the cpd X occurred 3 h after meal when gastric pH decreased from 4.9 to 3.5. The simulated triphasic dissolution profile indicated that drug dissolution rate was correlated to the time-dependent gastric pH variation after a high fat meal. In addition, drug precipitation appeared to be delayed with minimal impact on drug absorption.
Fig. 2.
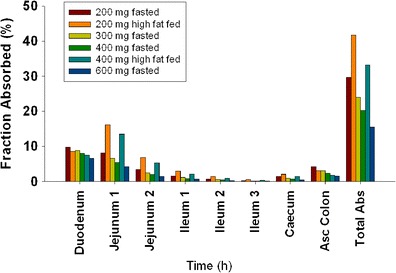
Fraction of regional absorption under fasted and fed conditions in healthy volunteers after a single dose of cpd X
Fig. 3.
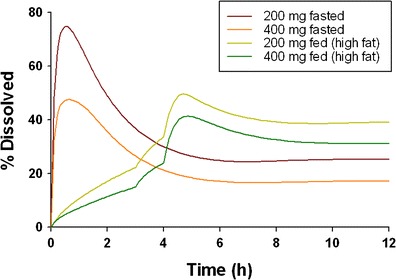
Percentage of dissolved cpd X in healthy volunteers after single 200 and 400 mg cpd X doses with and without receiving a high fat meal 30 min prior to the dose
Parameter Sensitivity Analysis
The PSA was conducted using three-dimensional surface response plots to investigate the sensitivity of Fa to mean drug particle radius in the range from 0.01 to 1 μm at doses ranging from 200 to 800 mg under fasted and fed conditions. Under fasted condition (Fig. 4a), a steep drop in Fa was observed over the therapeutic dose range from 200 to 400 mg when the particle radius was increased above 0.05 μm. In addition, the plot suggested that 60–80% Fa can be achieved by reducing the particle radius to less than 0.1 μm for the dose ranging from 200 to 400 mg (therapeutic dose range) under fasted conditions. Under fed condition (Fig. 4b), drug absorption was nearly complete (>85%) if the particle radius is less than 0.1 μm, regardless of doses in the selected range (200–800 mg). The Fa was reduced to 35–80% if the particle radius was in the range of 0.1–1 μm and the reduction of Fa increased when the dose increased from 200 to 800 mg. Comparing the surface response plots under fasted and fed condition, the Fa between fasted and fed state was still large if the particle radius was greater than 0.1 μm, whereas such difference was significantly reduced when the particle radius was in nanoparticle range (0.1–1 μm).
Fig. 4.
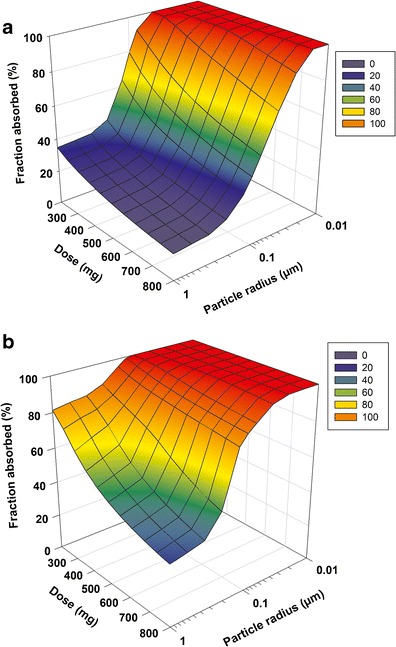
Surface response plot describing interactive effect of particle size and dose on fraction absorbed (F a) of cpd X under fasted (a) and fed (b) conditions
DISCUSSION
Food may significantly impact drug absorption and distribution by various mechanisms including changing the physiological conditions or direct drug–food interactions. Positive food effects have been commonly observed for BCS class II/IV compounds displaying low solubility within the GI tract. These compounds are primarily lipophilic and weak bases with pH-dependent solubility profiles (15), exhibiting decreased solubility and are thus susceptible to precipitation with increased pH in the intestine. The precipitated drug will exists as aggregates of fine particles and will only partially redissolve in the GI tract due to limited solubility under the conditions with elevated pH, leading to incomplete absorption under fasted conditions. Following food intake, these compounds are retained in the stomach with prolonged dissolution resulting in an improved absorption. In addition, the intestinal solubility of the cpd X increases remarkably in the presence of bile salts that are secreted following the food intake, leading to the additional enhancement in absorption of the cpd X.
A rapid and significant precipitation was observed for the cpd X under fasted conditions. In comparison, the precipitation was significantly delayed under fed conditions (Fig. 3), suggesting that precipitation was prevented in the presence of food. In addition, tmax of the cpd X under fed conditions was prolonged from 4 to 5 h at 400 mg, suggesting a continuous absorption in the downstream GI tract as a result of a sufficient concentration of the cpd X in the dissolved form. This is consistent with a previous report demonstrating that in vivo precipitation was delayed in the presence of a high fat meal due to a high degree of supersaturation in the secreted bile following a food stimulation (16). The true in vivo precipitation time, which is the mean time for particles to precipitate from a supersaturated state when the local concentration exceeds the drug solubility, is technically difficult to measure. In this study, the precipitation time was estimated via data-fitting of multiple pharmacokinetic datasets obtained from the clinical studies at various dose levels. The precipitation time of 2,000 and 450 s used in this model may reflect the realistic precipitation rates of the cpd X in vivo under fed and fasted conditions, respectively.
The reduced precipitation under fed conditions might be attributable to improved wettability of a drug substance with food content such as lipids, which reduces the interfacial surface energy between the cpd X drug particles and the GI fluids, thus enhancing the solubilization of the cpd X along the GI tract. Bile salt has been reported to increase drug absorption by enhancing drug solubility in the GI tract, which is a function of bile salt concentration expressed by the equation proposed by Mithani et al. (17). Solubility measurement in biorelevant media containing bile salts and lecithin provided a valuable information to estimate in vivo solubility under physiological conditions, enabling accurate estimation of absorption and bioavailability. The formulae of FaSSIF and FeSSIF were designed based on the most recent research evidence in humans. Nicolaides et al. suggested adding pepsin or lipase to improve correlations between in vitro solubility and in vivo absorption (18). In the case of the cpd X, the measured solubility in FaSSIF and FeSSIF, as expected, assisted to better predict the plasma levels.
A commonly accepted strategy to reduce positive food effects is to enhance drug absorption under fasted conditions, therefore reducing the gap in absorption between fasted and fed conditions. Wu et al. and Jinno et al. demonstrated suppression of food effect by improving drug absorption using drug particle size reduction strategies for two BCS II compounds (19,20). The cpd X exhibits similar pharmaceutical properties to those BCS II compounds suggesting that food effect may be mitigated using the same strategies. In general, if a compound has sufficient absorption (Fa > 70%) under fasted state, positive food effect tends to be clinically insignificant because the extent of absorption increased by a meal will be limited. From the PSA, greater than 70% of Fa under fasted state can be achieved for a particle radius less than 0.1 μm at a dose up to 400 mg, and a particle size less than 0.05 μm is required to reach similar absorption at higher doses. Therefore, API particle size reduction to nanoparticle range appears to be an effective strategy to mitigate the food effect of cpd X.
Physiologically based absorption modeling has been recognized as a useful tool to understand the effects of API and formulation properties on bioavailability and explore specific mechanisms for drug absorption. The predictability of drug absorption using a dog absorption model to explore the correlation between particle size and bioavailability has been evaluated by Kesisoglou et al. (21). This model predicted the dependency of drug absorption extent on the particle radius for the particle size greater than 1.8 μm. However, for the particle size smaller than 500 nm, the predicted exposure deviated significantly from the observed results. These results suggest that NFE may play an essential role in the enhancement of solubility and should be considered in the model for nano-sized API. In this model, the NFE was incorporated in PBPK model and the PSA results clearly suggested a strong influence of nanoparticle effects on bioavailability.
CONCLUSION
A PBPK modeling was developed to elucidate the mechanism of food effect of cpd X, a weak base compound. The observed low bioavailability was primarily due to a significant precipitation under fasted conditions leading to incomplete absorption. The positive food effect of the cpd X was resulted from a prolonged precipitation time and increased in vivo solubility under fed conditions. The PBPK based model indicates that the oral absorption is sensitive to the particle size of API. The reduction of particle size may potentially improve absorption under fasted conditions and minimize differences in drug exposures observed under fasted and fed conditions.
Abbreviations
- ACAT
advanced compartmental absorption and transit
- AUC
area under the plasma concentration–time curve
- BCS
Biopharmaceutics Classification System
- CAT
compartmental absorption and transit
- Cmax
maximum plasma concentration
- ECG
electrocardiogram
- FaSSIF
fasted-state simulating intestinal fluid
- FeSSIF
fed-state simulating intestinal fluid
- GI
gastrointestinal
- IR
immediate release
- PBPK
physiologically based pharmacokinetics
- PSA
parameter sensitivity analysis
- PK
pharmacokinetics
- SGF
simulated gastric fluid
- tmax
time to reach maximum concentration
- Fa
fraction of absorption
- API
active pharmaceutical ingredients
- NFE
Nanoparticle factor effect
Footnotes
Binfeng Xia and Hefei Zhang contributed equally to this work.
REFERENCES
- 1.Yu LX, Lipka E, Crison JR, Amidon GL. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19(3):359–376. doi: 10.1016/0169-409X(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 2.Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(Suppl. 1):S41–S67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 3.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–420. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 4.Fleisher D, Li C, Zhou Y, Pao L-H, Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clinical implications. Clin Pharmacokinet. 1999;36(3):233–254. doi: 10.2165/00003088-199936030-00004. [DOI] [PubMed] [Google Scholar]
- 5.Parrott N, Lukacova V, Fraczkiewicz G, Bolger MB. Predicting pharmacokinetics of drugs using physiologically based modeling—application to food effects. AAPS J. 2009;11(1):45–53. doi: 10.1208/s12248-008-9079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu C-H, Li H, Levons J, Lentz K, Gandhi RB, Raghavan K, et al. Predicting effect of food on extent of drug absorption based on physicochemical properties (Pharmaceutical Research (2007) DOI: 10.1007/s11095-007-9236-1) Pharm Res. 2008;25(4):979. doi: 10.1007/s11095-007-9337-x. [DOI] [PubMed] [Google Scholar]
- 7.Heimbach T, Xia BF, Lin TH, He HD. Case studies for practical food effect assessments across BCS/BDDCS class compounds using in silico, in vitro, and preclinical in vivo data. AAPS J. 2013;15(1):143–158. doi: 10.1208/s12248-012-9419-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner C, Jantratid E, Kesisoglou F, Vertzoni M, Reppas C, Dressman JB. Predicting the oral absorption of a poorly soluble, poorly permeable weak base using biorelevant dissolution and transfer model tests coupled with a physiologically based pharmacokinetic model. Eur J Pharm Biopharm. 2012;82(1):127–138. doi: 10.1016/j.ejpb.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 9.Xia BF, Heimbach T, Lin TH, Li SF, Zhang HF, Sheng J, et al. Utility of physiologically based modeling and preclinical in vitro/in vivo data to mitigate positive food effect in a BCS class 2 compound. AAPS Pharmscitech. 2013;14(3):1255–1266. doi: 10.1208/s12249-013-0018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Usansky HH, Sinko PJ. Estimating human drug oral absorption kinetics from Caco-2 permeability using an absorption-disposition model: model development and evaluation and derivation of analytical solutions for k(a) and F(a) J Pharmacol Exp Ther. 2005;314(1):391–399. doi: 10.1124/jpet.104.076182. [DOI] [PubMed] [Google Scholar]
- 11.Lu AT, Frisella ME, Johnson KC. Dissolution modeling: factors affecting the dissolution rates of polydisperse powders. Pharm Res. 1993;10(9):1308–1314. doi: 10.1023/A:1018917729477. [DOI] [PubMed] [Google Scholar]
- 12.Gardner JD, Ciociola AA, Robinson M. Measurement of meal-stimulated gastric acid secretion by in vivo gastric autotitration. J Appl Physiol. 2002;92(2):427–434. doi: 10.1152/japplphysiol.00956.2001. [DOI] [PubMed] [Google Scholar]
- 13.Dressman JB, Berardi RR, Dermentzoglou LC, Russell TL, Schmaltz SP, Barnett JL, et al. Upper gastrointestinal (GI) Ph in young, healthy men and women. Pharm Res. 1990;7(7):756–761. doi: 10.1023/A:1015827908309. [DOI] [PubMed] [Google Scholar]
- 14.Muller RH, Peters K. Nanosuspensions for the formulation of poorly soluble drugs: I. Preparation by a size-reduction technique. Int J Pharm. 1998;160(2):229–237. doi: 10.1016/S0378-5173(97)00311-6. [DOI] [Google Scholar]
- 15.Benet LZ, Broccatelli F, Oprea TI. BDDCS applied to over 900 drugs. AAPS J. 2011;13(4):519–547. doi: 10.1208/s12248-011-9290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shono Y, Jantratid E, Dressman JB. Precipitation in the small intestine may play a more important role in the in vivo performance of poorly soluble weak bases in the fasted state: case example nelfinavir. Eur J Pharm Biopharm. 2011;79(2):349–356. doi: 10.1016/j.ejpb.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Mithani SD, Bakatselou V, TenHoor CN, Dressman JB. Estimation of the increase in solubility of drugs as a function of bile salt concentration. Pharm Res. 1996;13(1):163–167. doi: 10.1023/A:1016062224568. [DOI] [PubMed] [Google Scholar]
- 18.Nicolaides E, Galia E, Efthymiopoulos C, Dressman JB, Reppas C. Forecasting the in vivo performance of four low solubility drugs from their in vitro dissolution data. Pharm Res. 1999;16(12):1876–1882. doi: 10.1023/A:1018959511323. [DOI] [PubMed] [Google Scholar]
- 19.Jinno J, Kamada N, Miyake M, Yamada K, Mukai T, Odomi M, et al. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J Control Release. 2006;111(1–2):56–64. doi: 10.1016/j.jconrel.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, Loper A, Landis E, Hettrick L, Novak L, Lynn K, et al. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int J Pharm. 2004;285(1–2):135–146. doi: 10.1016/j.ijpharm.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 21.Kesisoglou F, Wu YH. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008;10(4):516–525. doi: 10.1208/s12248-008-9061-4. [DOI] [PMC free article] [PubMed] [Google Scholar]