

Abstract
Pressurized metered dose inhalers (MDIs) are a long-standing method to treat diseases of the lung, such as asthma and chronic obstructive pulmonary disease. MDIs rely on the driving force of the propellant, which comprises the bulk of the MDI formulation, to atomize droplets containing drug and excipients, which ideally should deposit in the lungs. During the phase out of chlorofluorocarbon propellants and the introduction of more environmentally friendly hydrofluoroalkane propellants, many improvements were made to the methods of formulating for MDI drug delivery along with a greater understanding of formulation variables on product performance. This review presents a survey of challenges associated with formulating MDIs as solution or suspension products with one or more drugs, while considering the physicochemical properties of various excipients and how the addition of these excipients may impact overall product performance of the MDI. Propellants, volatile and nonvolatile cosolvents, surfactants, polymers, suspension stabilizers, and bulking agents are among the variety of excipients discussed in this review article. Furthermore, other formulation approaches, such as engineered excipient and drug-excipient particles, to deliver multiple drugs from a single MDI are also evaluated.
KEY WORDS: ethanol, formulation, metered dose inhaler (MDI), propellant, solubilization aids, suspending agents
INTRODUCTION TO MDI FORMULATION TECHNOLOGY
In 1956, Riker Laboratories (later acquired by 3M Pharmaceuticals) introduced the first pressurized metered dose inhaler (MDI), Medihaler Epi™, for the management of asthma and chronic obstructive pulmonary disease (COPD) (1). Upon introduction of the MDI, medical treatment of lung diseases changed significantly. Since that time, MDIs have become the most widely used treatment for controlling symptoms of asthma and COPD. More recently, formulation and device modifications were merited when chlorofluorocarbon (CFC) propellants were linked to the depletion of the ozone layer (2). With the successful transition to new propellant systems, MDIs are still well accepted and highly utilized by patients across the globe, with the annual production of over a half billion units and nearly one trillion MDI doses inhaled by patients to date (3,4). Looking forward, the effectiveness, ease of use, and relatively low cost of these aerosol preparations in combination with modifications in delivery technology and formulation sciences, will likely result in MDI use expanding to include the treatment of diseases previously untreated via the respiratory tract.
In developing MDI systems, there are two major areas that need to be considered: the device hardware and the formulation. The hardware consists of the vial (i.e., aluminum can or plasticized glass vial), metering valve, actuator, and for newer MDIs usually a dose counter. The formulation comprises primarily of the propellant, drug, and often other excipients. In many respects, modern hydrofluoroalkane (HFA) MDIs appear very similar to patients as their CFC predecessors. However, beneath the apparently unchanged surface of the MDI device, significant technological changes have occurred and new hardware components and formulation approaches are in development for the next generation of MDIs.
Although only the current state and future prospects of MDI formulations are in this review, it is important to note that, in reality, they function together with the device hardware to determine the eventual performance characteristics of the MDI system (5). Key performance attributes of an MDI include the delivered dose content uniformity, aerodynamic particle size distribution (APSD) of the delivered aerosol, chemical and physical stability of the drug over the product shelf life, and extent of leachables from device components, among other attributes.
The fine particle mass of drug (mass of aerosol particles with aerodynamic diameters that are approximately less than 5 μm) and the residual APSD are critical performance metrics that are intuitively linked to the efficacy of the product. The fine particle mass, is frequently represented by the fine particle fraction (FPF; the fraction of total mass of aerosol particles delivered from the device with aerodynamic diameters that are approximately less than 5 μm), and is a characteristic in vitro metric that represents the amount of drug that is considered respirable. The residual APSD is characterized by in vitro performance attributions, such as mass median aerodynamic diameter (MMAD; aerodynamic diameter at which 50% of the aerosolized mass lies below the stated value) and geometric standard deviation (GSD). Typically, aerosolized particles with aerodynamic diameters between 0.5 and 5 μm are delivered to the lungs, and smaller particles are more likely to deposit in the deep lung compared to larger particles (6).
This review seeks to present current state-of-the-art and future prospects for various formulation components for MDI drug delivery systems. The article is organized to review formulation strategies based on whether the drug is in solution or suspension in the propellant system, with additional excipients. Thus, topics such as cosolvents and suspension stabilizers are described as they pertain to solution or suspension formulations.
PROPELLANTS
Propellants comprise the bulk of any MDI formulation and are thus required to be toxicologically safe, nonflammable, and chemically inert with appropriate boiling points and densities. They are liquefied compressed gas, which function as a driving force and energy source for atomization of the formulation upon actuation. Propellant within the canister exists in two phases (liquid and saturated vapor) and ideally provides the same vapor pressure regardless of whether the MDI canister is full or nearly empty. For example, carbon dioxide is not suitable for MDI formulations even though it is a compressed gas, because the vapor pressure steadily declines as the canister empties, which leads to changing performance characteristics over the lifetime of the inhaler (4).
MDIs were initially formulated with CFCs as the propellant. However, the signing of the Montreal Protocol on Substances that Deplete the Ozone Layer (more commonly referred to as “the Montreal Protocol”) in 1989 led to the reformulation of MDIs with environmentally acceptable alternative propellants. HFAs were found to not deplete stratospheric ozone and were demonstrated to be safe as pharmaceutical excipients. Thus, they were developed to replace CFC propellants. However, HFAs could not directly substitute for CFC propellants, as previously used excipients and hardware components were not compatible with HFA formulations. As a result, significant effort was required to develop new device hardware and formulation approaches.
The Transition from CFCs to HFAs
Historically, MDIs utilized CFC propellants because of their limited toxicity, inertness, and suitable vapor pressures (7). The CFC propellants in marketed MDIs contained trichlorofluoromethane (CFC 11), dichlorodifluoromethane (CFC 12), dichlorotetrafluoroethane (CFC 114), or blends of these propellants (see Table I). CFC 12 has a lower boiling point and is more volatile than CFCs 11 and 114, thus it was widely used to provide a formulation vapor pressure sufficient to achieve suitable atomization of the CFC MDI formulations. CFCs 11 and 114 mainly functioned to modify the vapor pressure of CFC 12 and to facilitate manufacturing when used in formulations with propellant blends (10). CFCs were not only readily used in MDIs but were also highly utilized in household aerosol sprays, air conditioners (as refrigerants), fire extinguishers, industrial manufacturing of foams and insulations, as well as many other industrial applications.
Table I.
Physicochemical and Environmental Properties of CFC and HFA Propellants
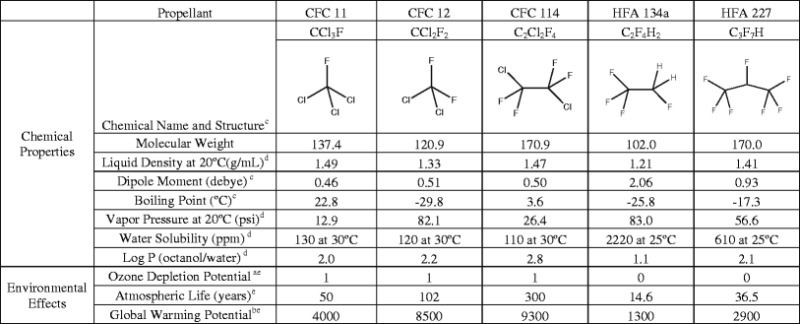
A factor that led to their widespread use was the extremely low reactivity of CFC propellants. However, CFCs were implicated in the depletion of stratospheric ozone (2). The extensive destruction of the ozone by CFCs is due to two factors: (1) the chemical stability under ambient environmental conditions and low aqueous solubility of CFCs result in long lifespans of these chemicals that permit ample time for CFC molecules to diffuse into the upper atmosphere (11) and (2) once in the stratosphere, CFCs break down under exposure to ultraviolet light and form chlorine radicals (2). The chlorine radicals formed from a single CFC propellant molecule can destroy 100,000 molecules of the ozone (12).
Considering the environmental ramifications of CFC use, the Montreal Protocol was devised, and then ratified in 1989, initiating the phase out of CFC propellants, including those used in MDIs. As of February 2013, the Montreal Protocol has been ratified by 197 countries (13). However, as pharmaceutical inhalers are considered life saving for many asthmatic and COPD patients, they were exempted from the protocol pending availability of suitable alternatives (14).
The Montreal Protocol provided motivation to the pharmaceutical industry to develop non-CFC-containing inhaler products. As a result, there were significant studies and investments in dry powder inhaler and liquid nebulizer technologies, in addition to the identification of suitable propellants to replace CFCs for use in MDIs. Two candidates for CFC replacement were identified, 1,1,1,2-tetrafluoroethane (HFA 134a) and 1,1,1,2,3,3,3-heptafluoropropane (HFA 227). These HFAs, first mentioned in patents as suitable propellants for MDIs in 1987 (15,16), lack the ozone-depleting characteristics of their predecessors; however, they still contribute to the greenhouse effect, albeit to a lesser degree than their CFC counterparts, as displayed in Table I (17). Additionally, the half-life of these HFA propellants in the atmosphere is a fraction of that of the CFCs they replaced (3). Other propellants have been explored as replacements for CFCs, namely, 1,1-difluoroethane (HFA 152a), propane, n-butane, isobutane, n-pentane, isopentane, neopentane, dimethylether, and hydrofluoro-olefins (HFO) (18–24). Many of these propellants have not been extensively studied and toxicological risks have not been assessed because they are flammable and thus pose an inherent safety risk.
Both HFAs 134a and 227 have broadly similar thermodynamic properties (i.e., boiling point and vapor pressure) as CFC 12 but are chemically different. Presumably, this is due to the lack of polarizability of the fluorinated hydrocarbons as compared with the partially chloro-substituted CFCs (7). This decrease in polarizability relative to CFC propellants could explain some solubility differences of solutes in HFA-based systems, despite their increased polarity over CFCs. Another difference between the propellants is the hydrogen(s) on the HFAs, resulting in an increased dipole moment relative to CFC propellants which are completely chloro- and fluoro-substituted. As a result of this dipole, the highly electropositive hydrogen(s) appear to make the environment much less amiable to nonpolar solutes while potentially enabling a degree of hydrogen bonding. The propellant polarity affects the solubility of drugs and excipients in the liquefied propellant. The reformulation from CFC to HFA propellants is further complicated by the fact that no comparable HFA equivalent for CFCs 11 and 114 is available. These considerations prevent simple substitution of HFA propellants for CFCs and contribute to the challenge of transitioning to HFA MDI products.
Characteristics of HFAs 134a and 227
Although the above characteristics may begin to explain the difference in observed propellant–excipient or propellant–drug interactions, it is arguably academic, as CFC propellants are not options for future therapeutics. Thus, when formulating MDIs, there are only two propellants currently available, HFAs 134a and 227.
HFAs 134a and 227 share many similar characteristics. Both propellants show a very low degree of impurity, with both being more than 99.9% pure (25). Compared with CFC propellants, both HFAs have relatively low boiling points (as seen in Table I) which afford sufficient vapor pressure, even at reduced temperatures, to enable efficient drug delivery (26–29). Additionally, they are completely miscible in one another and vapor pressure upon mixing behaves ideally, thus they may be blended in different proportions to obtain a specific vapor pressure or density (30). Both propellants have excellent safety profiles (31) and are chemically stable under normal storage conditions (25).
Although, seemingly subtle, some differences in the physical and chemical properties of HFAs may be significant for formulating a given drug in the HFA formulation. HFA 227 has a logP of 2.05 versus 1.06 for HFA 134a (7), and as such, water has nearly 4-fold increased solubility in HFA 134a versus 227 (2,200 and 610 ppm, respectively) (7). Of note, both HFAs 134a and 227 have significantly greater water uptake as compared to the aforementioned CFC propellants (all approximately 120 ppm), likely due to the relatively increased polarity (25). Thus, when formulating a suspension MDI of a compound, physical stability as a function of water is a consideration. Likewise, for compounds that are susceptible to degradation pathways in which water is involved, the amount of water in the formulation could be important. While the absolute solubility of water in HFAs 134a and 227 may be different, it is important to note that water levels start relatively low and increase slowly over time. The migration rate of water will depend not only on the propellant and additional excipients (e.g., ethanol) but also on the valve components and storage conditions (32). Williams and Hu (33) showed that the emitted particle size and FPF can change depending on the drug and extent of moisture ingress. Interestingly, water scavengers (such as hydroxypropyl methylcellulose coated silica gel, aluminum desiccant, and molecular sieve beads) have shown some promise as desiccants in prototypical HFA formulations (33). An additional factor to consider for suspension formulations is the difference in density of the two propellants (see Table I), which can affect particle settling or creaming behavior. It may be advantageous in some cases to match the density of the formulation to the density of the suspended drug particles.
Novel Propellants
The political dynamics of the global warming debate has the potential to influence the future of MDIs as HFA propellants are greenhouse gases that may contribute to global warming, albeit to a lesser degree than CFCs. As a result, there is the potential for future restriction of their use in MDI formulations. In reality, the contribution to global warming of medicinal HFA MDIs is minimal. HFA propellants contributed approximately 3% of total emissions of CO2 equivalents in 2007 (34); of those emissions, less than 2% is due to MDIs, resulting in a minimal contribution of HFA MDIs to global warming (35). Nevertheless, given the global warming potential of HFA propellants, new propellants have been evaluated.
Recently, isobutane has been investigated as an alternative to HFA. Laboratorio Pablo Cassara, in Argentina, began exploring the use of isobutane, a commonly used flammable refrigerant, as a propellant for MDIs. Cassara supplies 60–70% of the market's albuterol CFC MDIs and has made plans to phase out these inhalers and replace them with isobutane, as a propellant (24,36). Thus far, Ding and Zhang (37) have begun expanding upon the current knowledge of the toxicology (38) and application of tracheal instillation of albuterol sulfate MDI driven by isobutane in guinea pigs; clinical studies have yet to establish safety in humans. In comparison to HFAs, isobutane has a significantly lower global warming potential (3.3 for 100-year global warming potential relative to CO2). Isobutane has a boiling point of −11.7°C, liquid density of 0.563 g/mL (at 21°C), vapor pressure of 31.1 psi (at 21°C) with a water solubility of 80 ppm (39).
Additionally, HFA 152a has also received attention as an alternative propellant in MDIs (40,41). HFA 152a has a boiling point of −24.7°C, liquid density of 2.70 g/mL, vapor pressure of 88 psi (at 25°C), dipole moment of 2.30 debye and a water solubility of 2.671 g/L (at 25°C) (42). It has a 100-year global warming potential relative to CO2 of 140. Abuse of HFA 152a, found in canned air, has been linked to transient central nervous system symptoms including euphoria, confusion, and tremor. Furthermore, it is also linked to pulmonary irritation, asphyxia, cardiac arrhythmias, and death (43). However, short-term inhalation of 200 and 1,000 ppm HFA 152a for 2 h with light exercise did not prove to have significant central nervous system symptoms or pulmonary irritation in human subjects (44). Further investigation of HFA 152a will determine if formulating MDIs, rescue inhalers or control medications, is feasible from the safety standpoint.
HFO propellants have been developed by Honeywell Special Chemicals and they include trans-1,3,3,3,-tetrafluoropro-1-ene (HFO 1234ze) and 2,3,3,3,-tetrafluoroprop-1-ene (HFO 1234yf) (20,21). These propellants are not flammable. HFO 1234ze and HFO 1234yf have significantly lower global warming potential (6 and 4, respectively for 100-year global warming potential relative to CO2) than HFAs. HFO 1234ze, known as Honeywell's Solstice™, has a boiling point of −19°C, liquid density of 1.12 g/mL, vapor pressure of 46.4 psi (at 21°C), dipole moment of 1.443 debye, and a water solubility of 225 ppm (20,21,45). HFO 1234yf, known as Dupont's Opteon™, is an air conditioning refrigerant commonly used in motor vehicles; it has a boiling point of −29°C, liquid density of 1.09 g/mL, vapor pressure of 98.2 psi (at 25°C), dipole moment of 2.543 debye, and a water solubility of 260 ppm (20,21,46). These characteristics closely mimic those of HFA propellants, suggesting that they may be suitable alternative for MDI formulations. Furthermore, HFOs appear to be as compatible as HFAs 134a and 227 with standard MDI valves designed by Aptar Pharma (20). Results from toxicology studies for HFOs still remain to be published.
While lower global warming potential propellants for MDI are being explored, no serious discussion of banning HFA MDIs has been made to date. Indeed, despite the strong scientific justification for eliminating CFC propellants, CFC MDIs were not phased out until two decades after the signing of the Montreal Protocol. There is a far weaker scientific rationale to eliminate HFA use and it is unlikely that current MDIs will be forced off the market in the near future (35).
SOLUTION FORMULATIONS
MDIs can be formulated with the drug completely dissolved in the formulation, rendering a solution formulation, or with the drug practically insoluble in the formulation, rendering a suspension formulation. Compared with suspension formulations, solution MDIs offer the benefits of homogenous formulation (i.e., patients do not need to shake the vial immediately prior to use and there is no concern related to sampling homogeneity), a finer residual aerosol (47) and potentially larger fine particle doses (i.e., fine particle mass per actuation) (4,48). When formulating solution MDIs, the total amount of fine particle drug delivered cannot simply be increased by increasing the drug concentration in a formulation. Many drugs are not readily soluble in HFA propellants, which frequently limits the amount of drug that can be dosed using MDIs. Previously, surfactants or complexation aids were used in MDIs to increase drug solubility in CFC systems (7,8). However, many of the conventional excipients used in CFC formulations and approved for human use, are insoluble in HFA systems (49). Thus, to create a solution MDI and use previously approved excipients (see Table II), cosolvents are often added to the formulation to help increase the solubility of the drug or other excipients. These excipients may also alter the dissolution of residual particles from the aerosol spray in the lungs, which results in modulating the pharmacological effect (52,53).
Table II.
Excipients Used in Inhalable Drug Products
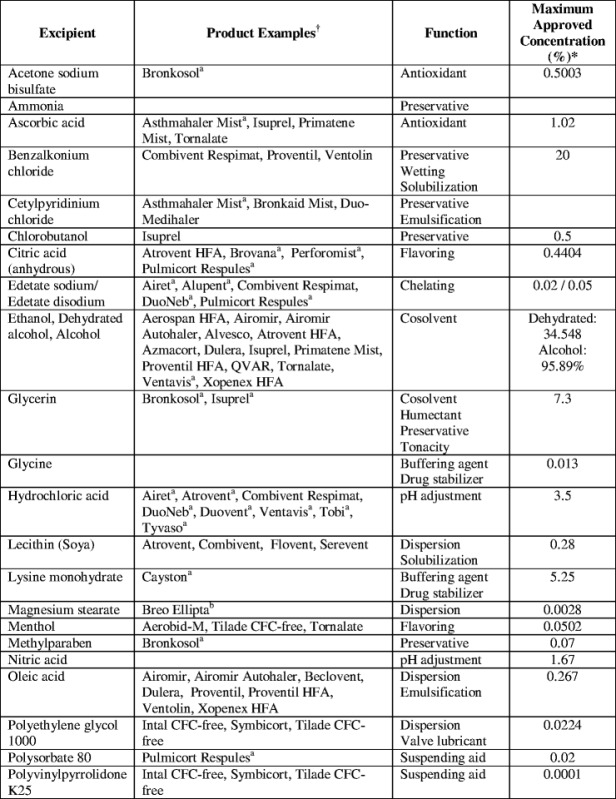
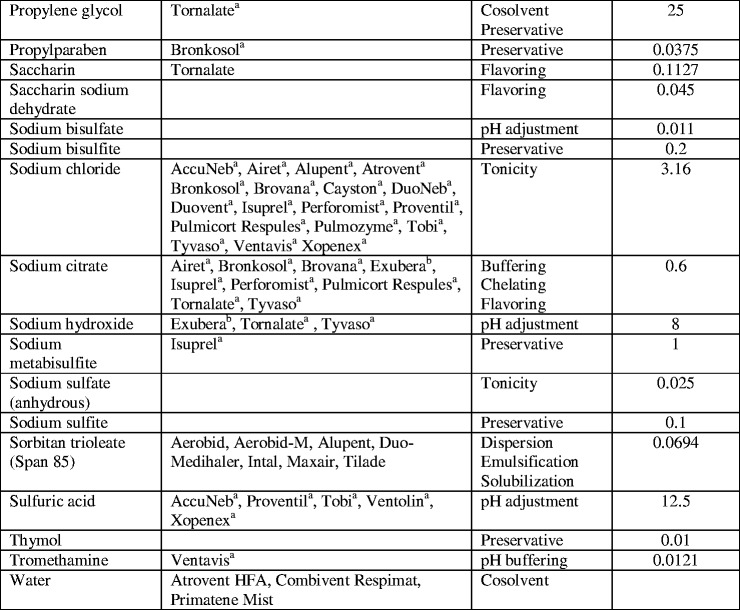
aProduct examples are subscripted based on formulation type. All other products are MDIs
bNebulizer products
cDry powder inhalers
dThe data provided in this table are from the FDA Inactive Ingredient Search for Approved Drug Products (last updated 29 March 2013) and Drugs@FDA databases (50,51), along with the product information for each medication
Effect of Ethanol on Solubility and Performance
The primary cosolvent utilized in MDI formulations is ethanol. Typically, it is utilized in an HFA formulation to increase drug or excipient solubility or to enhance valve function. However, the effect of ethanol on solubility varies significantly based on solute structure. Hoye et al. has investigated the effect of ethanol on the solubility of 21 different compounds, having logP's of −0.17 to 9.85 (54). It was found that the addition of 20% ethanol in HFA 134a could increase the solubility of these compounds by as little as 1.3 times to as much as 99.4 times, relative to the solubility of these compounds in pure HFA 134a. Interestingly, solubility increased for all compounds; however, a direct correlation with logP was not observed. Representative ethanol cosolvent solubility profiles for a variety of compounds are given in Fig. 1.
Fig. 1.
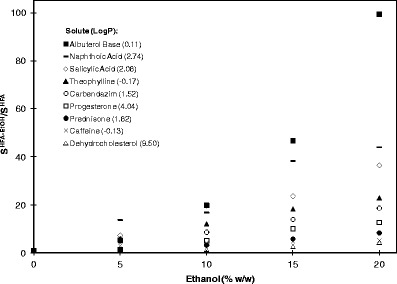
Solubility of a variety of solutes in ethanol cosolvent systems in HFA 134a (S HFA-EtOH) relative to their solubility in a pure HFA 134a system (S HFA) as a function of ethanol concentration. LogP for each solute is represented in parentheses in the legend of the graph. Adapted from Hoye and Myrdal (54)
The addition of semivolatile ethanol has multiple effects on the delivery process. Ethanol concentration can influence the delivery characteristics of MDIs in three ways: (1) by changing the formulation density and thus changing the total mass of formulation atomized during actuation of the device, (2) by changing atomization of the formulation and the size of the atomized droplets, and (3) by changing the evaporation rate of these droplets towards their residual particle sizes (55). As the concentration of ethanol increases, the vapor pressure of the formulation decreases, this in turn affects the atomization process. The decreased atomization force leads to an increase in the initial droplet size distribution (see Fig. 2). This results in larger residual particles being present after evaporation of the droplets in the aerosol spray. Additionally, the larger droplets cause increased deposition in the mouth and throat. Thus, increased ethanol concentration leads to a decrease in FPF and fine particle mass, thereby decreasing the overall dosing efficiency (55).
Fig. 2.
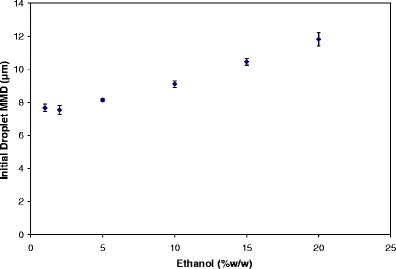
The effect of ethanol concentration on the resulting initial droplet MMD for HFA 134a solution MDI with 1% (w/w) drug. Andersen cascade impaction measurements were made using a large volume chamber as the inlet. From Stein and Myrdal (55)
Several investigators have illustrated the effect of ethanol concentration on product performance for solution MDIs (56–58). As presented in Fig. 3, Gupta et al. showed that when ethanol concentration increased from 0% to 20% (w/w), the solubility of beclomethasone dipropionate (BDP) increased linearly in HFA 134a (56). While the drug concentration in the formulation increased linearly with ethanol, the corresponding FPF decreases. As a result, the net gain in fine particle mass delivered (or “effective solubility”) diminishes as the ethanol concentration increases to 20% (w/w).
Fig. 3.
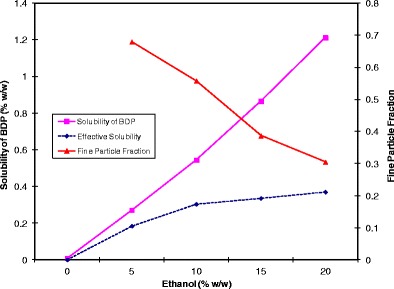
The effect of ethanol concentration on the solubility of beclomethasone dipropionate (BDP) and the resulting FPF in HFA 134a. The dashed line represents the effective solubility of BDP, which is the net gain in the delivered fine particle mass. Adapted from Gupta et al. (56)
Myrdal et al. (57) found similar effects of ethanol on product performance for HFA 227 using the drug cyclosporine, as shown in Fig. 4. For example, a cyclosporine solution MDI in HFA 227 could achieve a fine particle mass of approximately 750 μg/actuation with no ethanol, at a drug concentration of 1.5% (w/w). However, the fine particle mass decreases to approximately 350 μg/actuation when 10% (w/w) ethanol is added for the same drug concentration. This corresponds to a reduction in FPF from approximately 66% to 38% when ethanol is increased from 0% to 10% (w/w) in HFA 227. These trends were found to be true for dissolved cyclosporine drug concentrations ranging from 0.1% to 1.5% (w/w). Similar results were also found with a model drug (fluorescein sodium) and HFA 227 (59). Thus, when the concentration of ethanol is increased, the overall delivery efficiency of the formulation decreases, thereby limiting effectiveness of using ethanol as a solubilizing aid.
Fig. 4.
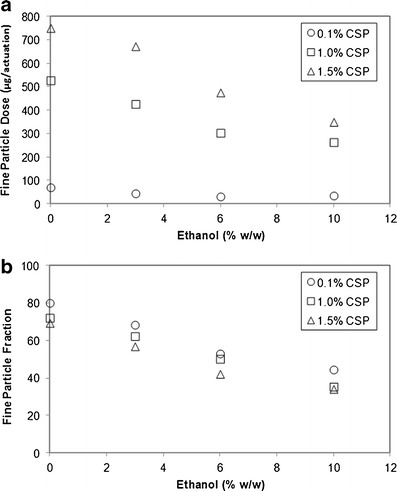
a The effect of ethanol on the fine particle dose and b the effect of ethanol on FPF for 0.1%, 1%, and 1.5% (w/w) cyclosporine (CSP) formulations in HFA 227. Adapted from Myrdal et al. (57)
There are two primary mechanisms that lead to reduced delivery efficiency for formulations with higher ethanol concentration. First, the size of the atomized droplets is larger with increased ethanol in a formulation. Second, the atomized droplets that contain a greater proportion of ethanol evaporate more slowly than droplets containing less ethanol or only the propellant (55). The net result of both of these mechanisms is that the droplets from a formulation with high ethanol concentration remain for a longer duration at sizes that are likely to deposit in the turbulent region of the airways (i.e., oropharynx and upper airways) (60). Figure 2 presents the effect of ethanol concentration on initial droplet mass median diameter (MMD) for HFA 134a formulations; the initial droplet MMDs were calculated based on experimental measurements of the residual MMAD using Eq. 1 (61). Modest increases in ethanol concentrations resulted in a notable increase in initial droplet MMD (62). Stein and Myrdal (55) theoretically and experimentally evaluated several semivolatile cosolvents (such as butyl acetate, ethyl acetate, acetonitrile, methanol, and methyl acetate) and demonstrated that FPF increases as the rate of evaporation increases. It was hypothesized that the droplet size of the atomized spray decreases more rapidly for formulations containing cosolvents that evaporate more rapidly. This results in decreased turbulent deposition in the mouth–throat region (55). Furthermore, it was theoretically determined that for the same concentration of drug, modulating the initial droplet MMD had a greater impact on solution formulations compared to suspension formulations (62).
| 1 |
where MMADR is the MMAD of the residual particles, MMDI is the MMD of the initial droplets, ρI is the density of the initial droplet (which is assumed to be the same as the formulation density), ρR is the density of the nonvolatile residual particles, and CNV is the weight fraction of the nonvolatiles in the formulation.
Although the addition of ethanol to the formulation decreases the rate of evaporation of the atomized droplets, the droplets still evaporate rapidly (typically in less than approximately 10 ms) (55). Thus, while depending on ethanol level, the atomized droplets could reach a “dry” residual particle size prior to depositing in the lung. Other cosolvents, such as water or propylene glycol, are significantly less volatile and may not evaporate prior to deposition in the lung. In some cases, the use of low volatility cosolvents can be used to increase the residual aerodynamic particle size to a target range.
Effect of Nonvolatile Concentration on Performance
The primary determinants of residual particle size from solution MDIs are the size of the initial droplets and the composition of the formulation. For a solution formulation, this relationship is summarized by Eq. 1 (61). In the simplest case, where the drug is not volatile and no other volatile excipients are in the formulation, the nonvolatile concentration, CNV, is simply the concentration of the drug and the residual particle density, ρR, is the density of the drug. As presented in Fig. 5, the residual MMAD increases as a cube-root function of the concentration of the drug (for formulations without other nonvolatile excipients). It is important to recognize that if the drug density value utilized in Eq. 1 is obtained from crystalline material, this may be an overestimation of the of the true density since the residual particle generally contains amorphous drug that typically has a lower density than the crystalline form.
Fig. 5.
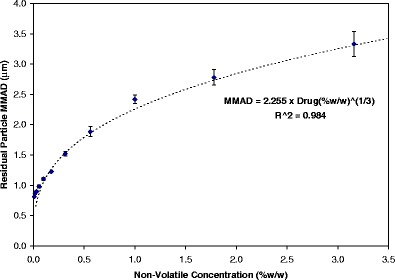
Depiction of the influence of nonvolatile drug concentration (oligolatic acid was used as a drug surrogate) on the residual particle MMAD for a series of HFA 134a solution MDIs. Each data point represents an average of four tests, and the error bar represents the standard deviation. Measurements were made using the USP inlet. From Stein and Myrdal (61)
In formulations where nonvolatile (or low volatility) excipients are present in addition to the drug, the CNV in Eq. 1 is the sum of the weight fractions of the drug and the nonvolatile excipients and the ρR is the density of the residual particle. The addition of nonvolatile excipients is expected to increase the residual MMAD of the formulation and potentially decrease the FPF. For instance, with the addition of 1.22% (w/w) Pluronic L81 to a formulation with 0.04% (w/w) dissolved drug in HFA 227 with ethanol, the residual MMAD increased from 1.56 to 3.70 μm while the FPF did not change significantly (59). However, further increase in Pluronic L81 concentration (up to 5.45%), resulted in a significant increase in the MMAD and decrease in the FPF compared to formulations with 0% and 1.22% Pluronic L81.
Novel Solubilization Aids
Using traditional cosolvents, the fine particle dose that can be achieved is limited based on the drug solubility in the cosolvent–propellant solution and the decrease in delivery efficiency at high cosolvent levels. Novel solubilization aids have been investigated which avoid, to an extent, the decrease in delivery efficiency associated with the use of ethanol. Below, several approaches to improve the solubility of drugs in HFA systems are provided. This list is not exhaustive; however, it does present the variety of compounds that are currently explored to solubilize drugs to render solution MDI formulations.
Micellar solubilization was used to enhance the solubility of albuterol and triamcinolone actonide in CFC–solution formulations using an isotropic solution of soya phosphatidylcholine (63). The solubility of the drugs increased proportionally with the addition of the surfactant but decreased with the addition of increased water relative to the surfactant.
More recently, Stein et al., Scherrer et al., and Stefely et al. (64–66) studied the effect of solubilizing two new chemical entities (NCE) using carboxylic acid functionalized methyl polyethylene glycol (f-mPEG) and/or oligolactic acid (OLA) in combination with ethanol. These excipients were shown to be synergistic with ethanol in increasing drug solubility in the formulation (67,68) such that acceptable dissolved drug concentrations can be obtained at reduced ethanol concentrations. Stein et al. (64) found that with 20% ethanol in HFA 134a, 0.82% NCE #1 rendered a maximum fine particle dose of 69 μg/actuation, which was significantly improved to 245 μg/actuation with 0.82% drug, 2.1% f-mPEG, and 5.3% ethanol, by weight. Similar results were seen with NCE #2, whose conventional formulation (25% ethanol and 0.3% NCE #2 by weight) had poor delivery efficiency but changing the formulation by adding 1.6% OLA or 1.1% f-mPEG (with 1% or 2.1% ethanol, respectively) improved drug delivery.
In addition, Stefely et al. (67) explored the use of two classes of HFA-compatible excipients: hydrophobic and hydrophilic counterions. Hydrophobic counterions, such as lauric acid or mono-functionalized lauric acid with an amide or ester (e.g., lauroyl sarcosine and lauroyl lactylate), may be used along with ethanol to synergistically increase the solubility of drugs that contain an amine functional group. Hydrophilic counterions, such as functionalized polyethers (i.e., carboxylic acid functionalized PEG) (68) could also be used to increase drug solubility. Increasing the amount of the excipient in a formulation or decreasing the length of the PEG chain resulted in increased drug solubility of a drug with amine functionality. Furthermore, the excipient was found to be synergistic with ethanol in solubilizing the drug.
Rogueda found that partially and fully acetylated cyclodextrins (CD), while commonly studied as suspension stabilizers, may also solubilize drug (69). Peractylated β-CD has a solubility of 0.1% (w/w) in HFA 227 but is significantly more soluble in HFA 134a (>1%, w/w). It was found that for a fixed 1:1 molar ratio of budesonide/CD, co-spray drying the two agents yielded a solution, whereas a physical mixture of budesonide and CD or simply budesonide in HFA 227 formed a suspension in HFA.
Solution Formulation Strategies
As companies began developing HFA MDI products to replace marketed CFC products, the fact that HFA propellants differ from CFC propellants in both chemical and physical properties caused numerous challenges. For example, Beclovent™ MDIs were CFC suspension formulations of BDP. However, HFA suspension formulations of BDP proved to be problematic due to the increased solubility of BDP in the HFA propellant systems. As a result, HFA BDP MDIs have been developed as solution formulations. Two distinct formulation strategies have been utilized to create HFA solution formulations of BDP: (1) take advantage of the extrafine aerosol production of HFA solutions and produce an MDI with increased efficacy and decreased deposition in the central airways, or (2) try to match the dose and particle size to the respective CFC formulation so that patients can continue with the same dose that they were used to. An example of the first approach is the development of QVAR® (70); an example of the latter approach is BDP Modulite® MDI (71).
The Modulite® approach provides a rational and empirical methodology that allows for the modulation of several dependent variables to anticipate the performance of an HFA-based MDI solution formulation. These variables include the quantity of the cosolvent, actuator orifice geometry, nonvolatile concentration, metering valve size and the vapor pressure of the propellant. The Modulite® approach has been utilized to formulate HFA formoterol fumerate in 6 and 12 μg/actuation strengths with 12% (w/w) ethanol, 0.024% or 0.038% 0.1 M aqueous hydrochloric acid and 50 or 63 μL valves, respectively for the 6 and 12 μg doses (72). To match the residual particle size distribution from CFC formoterol suspension formulation, the actuator nozzle orifice diameter (OD) was selected to be 0.3 mm, which provided an HFA formulation that would replace the CFC formulation without a change in FPF and the efficacy of the medication. This approach has also been utilized to develop an HFA formulation of budesonide to match CFC formulations, Pulmicort® and Desonac® DA (73). In this case, the addition of a nonvolatile component and the actuator OD were varied such that the aerosol cloud would have similar characteristics as the CFC suspension formulations and the residual particle APSD would also be equivalent. Since the CFC formulations were suspensions, adding a nonvolatile component (water and glycerol), in addition to the drug, increased the residual particle APSD of the Modulite® solution MDIs. Furthermore, actuator ODs were decreased from those used with the marketed CFC formulations, in order to reduce the velocity of the MDI plume. Similar approaches have been taken in transitioning from the CFC formulation of BDP (suspension, Beclazone®) to the BDP-HFA formulation (solution, Clenil Modulite®) (74). Beclazone® and Clenil Modulite® have a similar residual particle size distribution (3.1 μm with a GSD of 3.26 versus 2.8 μm with a GSD of 2.71 for 50 μg/actuation dose, respectively), similar FPF (34.2% versus 31.6%, respectively) and fine particle dose (14.4 versus 16.6 μg/actuation, respectively). Chaplin and Head (75) propose that these factors permit switching patients from the CFC formulation directly to the Modulite® formulation, without changing the drug dosage.
By contrast, an alternate solution formulation strategy leverages the extrafine aerosol formulation for HFA solution MDIs to improve the fine particle dose of the HFA MDI compared to the CFC-formulation (70). This strategy enables similar efficacy to be obtained using a decreased total dosage of the drug compared to the CFC or Modulite® formulation approaches (75,76). Alternatively, if the dose of the extrafine formulation is comparable to that of the CFC or Modulite® formulations, the extrafine formulation will have an increase fine particle dose and a “leftward shift” in the dose–response curve with potentially an increase in the maximum response (77). QVAR® 80 μg/actuation, an HFA formulation of dissolved BDP, has a residual MMAD of 1.1 μm (70). With a small residual particle size, it is expected that a greater extent of the drug deposits in the peripheral airways (airway diameters, ≤2 mm), compared with a CFC BDP formulation with a residual size of approximately 3 μm (74), which primarily deposited in the central airways (78). The extrafine aerosol formulation approach permits QVAR® to have a lower formulation drug concentration, increased inhalation technique tolerance and increased ratio of therapeutic efficacy to adverse effects because of the deposition of the drug in the peripheral airways compared to the CFC formulation (79,80). Treating the peripheral airways is important especially for a large proportion of asthmatic patients who experience persistent small airway dysfunction (77) and has been shown to improve the probability of patients achieving asthma control over a period of 1 year (81). Other marketed extrafine HFA MDI formulations include ciclesonide (Alvesco® HFA), flunisolide hemihydrate (Aerospan® HFA), and formoterol fumarate (Atimos®). In addition, a solution combination product with BDP and formoterol (Fostair®) is available in Europe. A solution formulation of salmeterol xinafoate is being investigated, which utilizes up to 2% (w/w) water to solubilize the drug (82).
Chiesi Farmaceutici has claimed that certain solution formulations can be stabilized using small amounts of strong mineral acids, such as hydrochloric, nitric or phosphoric acids (83). For instance, the marketed CFC formoterol formulation, Foradil®, had a shelf life of 12 months in the refrigerator and only three months at room temperature. It is speculated that phenylakylamino β2-agonists may be inherently unstable due to their susceptibility to oxidation and the presence of a highly polar vehicle may accelerate their degradation (83). It has been disclosed that the chemical stability of dissolved formoterol in HFA can be substantially improved by the selection of appropriate vials (i.e., canisters) and tight control of the pH of the formulation (83). The formulation is much more stable at apparent pH values below 5.6; the inventors selected an apparent pH range of 3.0 to 3.5 for this formulation. At an apparent pH of 7.4, 67.2% of the initial formoterol content was still present after 20 days, while at an apparent pH of 3.3, 89.9% of the initial drug content was still present after 20 days. In addition, the use of inert canisters (stainless steel, anodized aluminum, or organic coated) that do not leach metal ions or alkali as a consequence to the addition of acid to the formulation appear to inhibit catalysis of radical oxidative reactions.
PARTICLE PREPARATION FOR MDI FORMULATIONS
The solid form of a drug can affect solubility, dissolution and stability in a formulation. For instance, different salts of drugs can have different solubility, dissolution and stability properties in propellant systems, which significantly impact the MDIs product performance (e.g., albuterol base versus albuterol sulfate) (84). The primary objectives for inhalation drug particle engineering are to produce drug particles with an appropriate particle size distribution and desired dispersibility. For instance, surface modifications of drug particles with magnesium stearate or glycerol monostearate can be done to improve the aerosolization and deaglomeration of micronized drug particles (85). In addition, particle engineering can be utilized for optimizing bioavailability, targeting receptors, evading clearance mechanisms and affording controlled drug release. While particle engineering for inhalation drug products is briefly discussed below, please refer to Shoyele and Cawthorne's (86) and Chow et al.'s (87) articles for an extensive review of the topic.
Typically, prior to formulating a drug, the size of the crystalline material needs to be reduced to obtain suspension MDI formulations with particles of a suitable size for inhalation. This can be achieved by milling, spray drying, or using supercritical fluids (88,89). Ball mills and fluid-energy mills (such as jet mills) are the primary modes of milling powders to achieve particles with diameters of 1 to 5 μm (90). Ball mills utilize balls that grind the drug as the balls tumble inside the mill. This method is relatively slow and is difficult to scale up (91). Jet milling, which is the primary method of micronizing drugs, reduces particle size of coarse powders by high velocity particle-particle collisions. The mechanical process of milling can affect the crystallinity of the material and amorphous regions can be produced at the newly formed surfaces of the micronized material (92). In addition, milling typically yields nonspherical particles, with flat surfaces that may increase adhesion between the micronized particles (90). Alternatively, spray drying may be used to manufacture drug particles for MDI formulations. Spray drying converts a solution or liquid dispersion (also known as “feed”) to dried particulates by the process of atomizing a spray of the liquid containing the drug followed by quickly drying the droplets, which yields solid particles. Factors such as the feed composition, drug concentration, liquid feed rate, drying rate, temperature and relative humidity can be varied, allowing one to optimize the size distribution, shape, morphology and density of the particles. Compared to milling, spray drying often produces relatively spherical, amorphous particles. Finally, supercritical fluids may also be utilized to manufacture particles for inhalation. A supercritical fluid is any substance at a temperature and pressure above its critical point. Supercritical fluids can be used in multiple ways to micronize drug particles. They may be used to micronize drug material through rapid expansion of supercritical solutions, using supercritical fluid as an antisolvent and precipitation of particles from gas saturated solutions (90). All three of these methods rely on dissolving the drug in the supercritical fluid, at high pressure and temperature, followed by decrease in pressure and/or temperature which yields a reduction in the density of the solution, thereby decreasing the solvation power of the supercritical fluid, leading to precipitation of the drug.
The method for preparing drug particles for MDI formulations needs to be selected based on the chemical stability of the drug. Proteins, for instance, require additional care when micronizing, due to being heat-labile and need to preserve any three-dimensional conformation. Frequently, spray-drying with another agent (i.e., sodium carboxymethylcellulose (93,94), trehalose with polyvinyl alcohol (95), and/or polyvinylpyrrolidone (PVP) (96)) is utilized for protein drugs due to the need to preserve the three-dimensional conformation and biological activity of the protein. Proteins and nucleic acids have been lyophilized providing a morphology that reduces van der Waals interactions, which may serve to protect their integrity and also decrease suspension settling rate relative to milling (97). In addition, well-dispersed nanoparticles containing proteins have been produced by freeze-drying with the intention to be used with HFA systems by Tan et al. (98). The process involves dissolving the protein in a tert-butyl alcohol–water system with lecithin (as a surfactant) and lactose (as a cryoprotectant) followed by freeze-drying and purifying.
More complicated preparation of the drug matrix may be required if it is desired to modify drug release. For example, drug loaded into swellable hydrogel microparticles (99) has been shown to modify drug release. The swelling of these particles are governed by the hygroscopic growth of the particles (excipients and/or drug) in the respiratory tract. Hygroscopic growth depends on the hygroscopicity of the excipient and the drug, the properties of the engineered particle, and the respiratory parameters (100). Namely, the particles developed by Selvam et al. (99) are composed of drug-loaded polylactic-co-glycolic acid nanoparticles which are encapsulated in PEG-chitosan copolymer microparticles. These microparticles swell upon deposition in the deep lung, thus evading alveolar macrophages and permitting modified drug release. Some other hygroscopic excipients, in decreasing order of hygroscopic potential, include: sodium chloride, citric acid, propylene glycol, and mannitol (100). Alternatively, chitosan microspheres have been investigated to protect and afford sustained release of proteins and plasmid nucleic acids via MDI drug delivery (101); the addition of glycerol to solution BDP MDI formulations has shown to affect the extent of drug metabolism across a cell layer, suggesting increased residence time of the drug particles in the lungs (102).
SUSPENSION FORMULATIONS
Many MDI applications are formulated as suspensions in which the drug particles are suspended in the HFA system, creating a heterogenous formulation. A primary concern for formulating suspension MDIs is instability due to nonideal dispersion of the drug. This can occur because of phase separation, flocculation, agglomeration, drug particle interaction with other drug particles or device material, or moisture ingress (103). Depending on the relative density of the suspended drug to that of the continuous phase, the drug will either cream or settle in the formulation. The drug content of each subsequent dose can increase or decrease over time if the drug is not adequately dispersed by shaking the vial. Considering this nature, suspensions inherently present the concern of dose uniformity, in general, as well as over the life of the MDI (8). As drug particles associate to form large flocculates that cream or settle, a nonuniform suspension gradient is created, leading to variability in metered dose (104,105). Even more detrimental to suspension formulations is the irreversible agglomeration or caking of particles.
A principal consideration for a suspension formulation is that the drug must be practically insoluble in the formulation. The inherent drug properties or the addition of ethanol will afford different levels of drug solubility in the formulation. Thermodynamic solubility in combination with kinetics, over time, can lead to an increased particle size distribution, a phenomenon known as Ostwald ripening (7). Surface molecules on relatively smaller particles have a higher free energy than molecules inside of the particle or molecules on the surface of larger particles. Thus, thermodynamics favor the dissolution of the smaller particles and a corresponding growth of the larger particles, which results in the reduction of the overall free energy of the system (8).
Interestingly, the formulation and drug form may affect particle growth. For instance, BDP grows rapidly when exposed to CFC propellants. For instance, 6 h post-exposure to CFC 11, the mean particle size of micronized BDP grew from 1.6 to 22.2 μm. However, the ethyl acetate solvate of BDP experiences minimal growth when exposed to CFC 11 (103). Aside from the growth of micronized material affecting the residual APSD, it may also affect the propensity of the particles to settle or cream within the MDI vial.
Solid drug particles in a suspension formulation often cream or settle. The sedimentation velocity (or creaming velocity) of suspended drug particles can be determined by Stokes law (Eq. 2),
| 2 |
where ν is the sedimentation velocity (such that ν > 0 is in the direction of gravity), g is the gravitational acceleration constant, dp is the diameter of the suspended particle, ρp is the density of the suspended particle, ρform is the density of the formulation, and η is the viscosity of the formulation (7). Thus, suspensions are inherently susceptible to gravitational sedimentation or creaming. Larger suspended particles will cream or settle faster than smaller particles. Furthermore, particles with diameters of less than 0.5 μm will be affected by Brownian motion, which may oppose settling or creaming, assuming that the particle density is not starkly different from the formulation density (103). Brownian motion and particle diffusivity increases with lower viscosity of the formulation and smaller particle size. Therefore, at ambient temperatures, increasing particle diffusivity can lead to increased particle–particle or particle–device interactions, leading to increased coagulation rates or larger flocculate sizes. In addition, most crystal drug densities vary between 1.15 and 1.40 g/cm3. Gravitational stability can be enhanced by matching the density of the system to the density of the drug by adding excipients or blending propellants to increase or decrease the overall density of the HFA formulation (106). For formulations with only blended HFAs 134 and 227, the density of the formulation can range between 1.21 and 1.41 g/cm3; however, the addition of ethanol (ρ = 0.789 g/cm3) can lower the overall formulation density. However, the benefit of this density-matching approach is limited by the fact that the density of propellant formulations varies significantly with temperature changes. Alternatively, the drug particles can be engineered to match the density of the formulation (107), which is the approach utilized by the PulmoSpheres® platform, described below. Secondly, excipients can be utilized to ensure decreased agglomeration of suspended drug particles, which will be discussed in much detail in the proceeding sections.
Effect of Nonvolatile Content
Unlike solution formulations, the nonvolatile concentration does not impact the residual particle size of suspension MDIs in a direct, predictable manner. In fact, the drug concentration along with the properties of the micronized drug (i.e., raw drug MMAD, GSD, and density) impact the residual particle size distribution. For formulations with dilute suspended drug content, the MMAD of the residual particles is very close to that of the micronized drug (see Fig. 6). This occurs as most of the drug-laden atomized droplets contain only a single suspended drug particle. However, as the drug concentration increases, more of the atomized droplets contain multiple drug particles which lead to an increase in the residual particle MMAD. Consequently, the residual MMAD increases more rapidly with change in drug concentration for suspension formulations containing smaller micronized drug than that for larger micronized drug (108,109). Major predictors for the residual particle MMAD of suspension MDIs are the size of the micronized drug, the number of drug particles per unit volume in a given formulation (which is a factor of the concentration, size distribution, and density of the micronized drug and the density of the formulation), and the initial droplet size distribution. As droplet size and particle concentration increase, droplets have increased propensity to contain multiple drug particles (62), resulting in larger residual particle size distributions, which may further decrease the FPF (110). Thus, suspension MDI formulations with very fine micronized drug at a low concentration may result in a relatively high FPF (111).
Fig. 6.
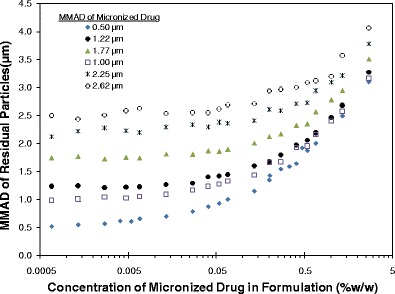
The theoretical effect of concentration and MMAD of micronized drug on the residual APSD (MMAD of residual particles) derived from simulations with 50 μL metering chambers, 0.3 mm orifice diameters (OD), 8.5% (w/w) ethanol in HFA 134a. From Stein et al. (108)
Sometimes nonvolatile impurities can leach into the formulation from device components or other sources and impact suspension formulations. For example, silicone is often added to valve components during valve manufacturing. Silicone can leach into the formulation over time, particularly when the MDIs are stored at high temperatures. It appears that the silicone in the formulation can lead to an appreciable particle size coarsening of the formulation potentially through the aggregation of particles into clusters (112,113). Furthermore, depending on the material utilized in the valve, there may be an increased attraction of drug suspended in propellants to the component, resulting in poor dose uniformity (114).
Stabilizing and Suspending Agents
Surfactants, a primary class of suspension stabilizers, required extensive reevaluation with the transition from CFC to HFA propellants. Surfactants, such as soya lecithin, sorbitan trioleate and oleic acid, which are readily soluble in CFC propellants (especially CFC 11) have very low solubility in HFA propellants (see Table III). Surfactant polarity, indicated by their respective hydrophilic–lipophilic balance (HLB), correlates with the incompatibility of the aforementioned surfactants in the more polar HFA environment (8,115). A high HLB value indicates that the surfactant is highly hydrophilic and a low HLB value indicated that the surfactant is highly lipophilic.
Table III.
Solubility of Select Surfactants in HFAs
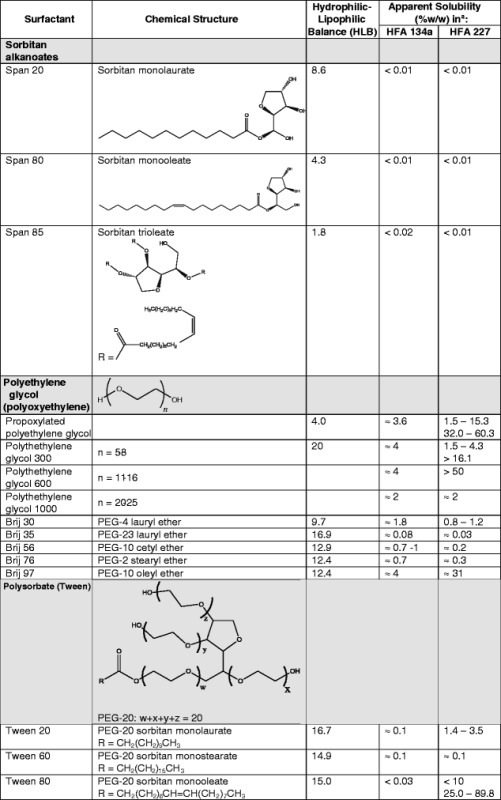
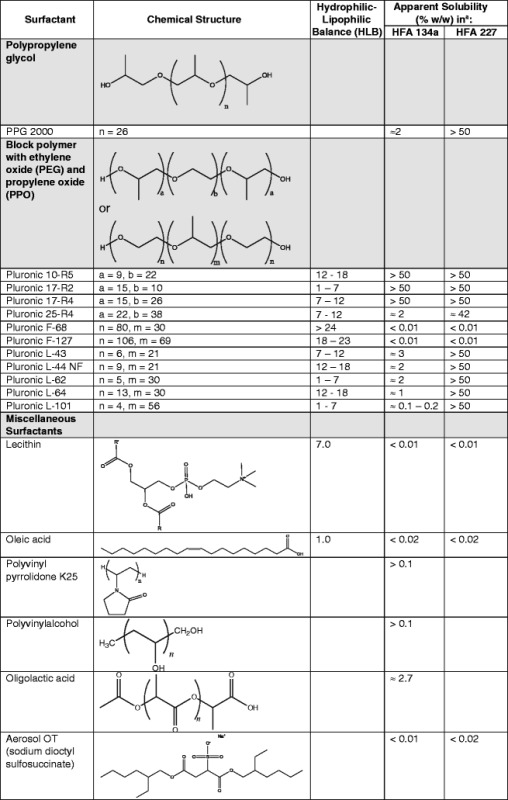
Data in this table are compiled from BASF Corp. Product Information, Griffin, Alexandridis and Hatton, Vervaet and Byron, da Rocha et al., and Ridder et al. (8, 115–119)
aIf a range of values are presented for solubility, the HFA-surfactant system appeared to produce a single phase at the stated concentrations of the surfactant. All solubility values presented were carried out between 19.5°C and 25°C at saturated pressure
Surfactants are frequently utilized in MDI formulations for a myriad of reasons. In solution formulations, surfactants serve to increase drug solubility, moderate temperature-dependent drug solubility and overcome valve sticking issues (8). However for suspensions, surfactants are primarily used to prevent irreversible particle agglomeration, prevent drug particle adhesion to the container walls and valve components, decrease the rate of separation between the drug and the propellant system, and prevent valve sticking problems. Surfactants stabilize the dispersion by decreasing the electrostatic forces between the micronized drug particles thus decreasing crystal growth or particle agglomeration during storage conditions (7). Surfactants typically must be adequately soluble and stable in HFA systems to be used for suspension formulations. For instance, oleic acid has substantially lower solubility in HFA propellants than in CFCs. In order to utilize oleic acid to stabilize albuterol sulfate suspension in the Proventil® HFA formulation, sufficient ethanol is used to solubilize oleic acid. Similarly, although lecithin is effectively insoluble in HFA (<0.01% (w/w) in HFAs 134a and 227 (8)), it is soluble in dimethyl ether and propane propellant systems, and can be utilized to form water-in-oil (inverse) microemulsions that further stabilize suspension formulations (22,120).
Several novel surfactant excipients that function as dispersion aids for HFA suspension MDI formulations have also been explored. For instance, OLA, presented above as a method to increase drug solubility, is similar to polylactic acid except that it is generally shorter than most polylactic acid chains (OLA's generally consists of only 5 to 20 repeating units) and the terminal alcohol can be modified by acetylation (121). In addition to its application as an excipient in solution MDI formulations, it has been shown to function as a suspension aid. The head group of OLA interacts with the drug and the tail interacts with HFA, thus permitting a surfactant-like effect in stabilizing the suspension formulation (66). Furthermore, it has been shown to enable improved through life medication delivery compared with conventional suspension MDI formulations. Interestingly, OLA's have been presented to modify drug release by forming in situ microspheres for a variety of drugs including steroids, 5-lipoxygenase inhibitors, and immune response modifiers (122).
More common excipients have also been investigated as suspending aids in HFA propellants. PEG at 0.05% to 0.5% (w/w) with 0.001% (w/w) PVP has been shown to decrease interparticulate cohesive forces between albuterol sulfate drug particles suspended in a model propellant (123). This effect was inversely dependent on the molecular weight of PEG and increased with increasing concentration of PEG. Hydrophilic counterions, such as functionalized polyethers, can be used to stabilize suspension formulations depending on the physicochemical properties of the drug and the amount of the hydrophilic counterion used (67). Examples of such include carboxylic acid functionalized PEGs and glycine functionalized PEGs (68). For instance, it was found that the addition of glycine functionalized PEG to a pirbuterol acetate formulation in HFA 134a with ethanol increased the time of flocculation compared with a formulation without PEG. Wu and da Rocha (124) explored the use of polylactic acid–PEG–polylactic acid (PLA-PEG-PLA) to disperse albuterol base in HFA 227 and showed that the concentration, molecular weight, length of the surfactant tail and the ratio between number of PLA and PEG have a large impact on the drug's cohesive forces. PLA-PEG-PLA microspheres have also been shown to modify drug release. Rogueda showed that 1H,1H,2H,2H-perfluorooctan-1-ol and methyl-PEG-1,2-distearoyl-phosphatidylethanolamine conjugate decrease adhesion of drug particles to the headspace and also retard phase separation time (125). Glaxo Group Ltd. has also shown that other novel surfactants can also be utilized in HFA formulations for the aforementioned purposes (126,127) (see Fig. 7). Propylene glycol diesters (Miglyol 840) and triglyceride esters (Miglyol 812) of medium-chain fatty acids may also be utilized as surfactants and have been shown to decrease discharge pressure upon actuation, which may positively influence oropharyngeal drug deposition from MDIs (128). Furthermore, volatile mono- and sesquiterpene hydrocarbon oils, such as citral, menthol, eucalyptus oil, cinnamaldehyde, and cineole (129) are being investigated as stabilizers for suspension MDI formulations (130). Interestingly, cineole along with n-heptane (129) has been shown to improve suspension quality of a peptide nanoparticle, engineered by the mechanism developed by Tan et al. (98), as described under the subheading “Particle Preparation for MDI Formulations” within this article. Other excipients have also been explored, which includes diethylene glycol monoethyl ether, polyoxyethylene 20 sorbitan monolaurate, polyoxytheylene 20 sorbitan mono-oleate, propoxylated PEG, and polyoxyethylene lauryl ether (131,132). These excipients have been found to have favorable solubility in HFAs 134a and 227. It is postulated that surfactants with elevated HLB can be functional in HFA 227 as suspension stabilizers, while all others studied by Byron and Blondino (131,132) are primarily functional in HFA 134a. It should be noted that many of these aforementioned excipients have yet to be used clinically and none of these excipients are in current marketed products.
Fig. 7.
Structures of novel surfactants from Glaxo Group Ltd. Adapted from Looker et al. (126,127)
As an alternative to surfactants, surface-modified nanoparticle excipients (133) can be utilized to modulate interparticulate interactions of drug particles in suspension MDI formulations. These surface-modified nanoparticles are spherical with 5- to 10-nm diameters and function to eliminate flocculation of drug particles, leading to improved dosing reproducibility. The core of the nanoparticle can be composed of amorphous silica or iron oxide and the functionalized surface is designed to be hydrophilic to enable compatibility with HFA. Since these particles are extremely small, a large number of nonaggregating nanoparticles can be used in the formulation. They provide a steric barrier, preventing drug particles from interacting with each other, thereby eliminating flocculation. Some examples of surface modification investigated for MDI formulations include magnesium stearate (85), glycerol monostearate (85), and crosslinked chitosan-PEG 1,000 (134). Furthermore, the gravitational settling or creaming rate of the drug is decreased as the drug is not permitted to flocculate. Similarly, the principle to provide a steric barrier to prevent drug particles from flocculating is also used in the following approaches: (1) formulating an in situ precipitation of the drug creating a microsuspension or nanosuspension complexes of the drug with hydroxypropyl-β-CD (135,136) or acylated-β-CD (69), PEG and/or ethanol in HFA, or (2) trapping HFA-philic (i.e., PEG) moieties (137) at the surface of polar drug particles using a modified emulsification–diffusion method.
Two innovative approaches have been taken to formulate polar drugs in dispersion by creating microenvironments that are aqueous with the drug enclosed in a “shell” that is HFA-philic. One approach is to use suspended core–shell particles in HFA, where the particles are made by emulsification diffusion (138). The shell consists of HFA-philic oligolactide grafts attached to short chitosan backbone, while the active drug moiety is found within the particle core and is protected by the shell. This approach has been used for albuterol sulfate and bovine serum albumin in HFA 227. The experiments revealed improved dispersion stability and aerosol characteristics (i.e., FPF and residual MMAD) compared to conventional formulations. A similar approach is to formulate drugs in water-in-oil (reverse) microemulsions, where the emulsions create an aqueous microenvironment within the propellant system that enables the delivery of water-soluble compounds. For instance, Selvam et al. and Chokshi et al. utilized various ethylene oxide–propylene oxide–ethylene oxide (EOn−PO~30−EOn) polymers to form reverse aqueous microemulsions in HFAs 134a and 227 (139,140). It was found that PO-based amphiphiles can reduce the tension of the propellant-water interface, thus permitting the formation of reverse aggregates, which can be utilized to deliver polar solutes via MDIs. Others have formed reverse emulsions using fluorine moieties, such as perfluorooctyl bromide with perfluoroalkylated dimorpholinophosphate (a fluorinated surfactant) (141) or polar fluorinated nonionic oxyethylene glycol with ethanol, propanol or pentanol as a cosolvent (142). It was found in the latter case that in order to successfully form microemulsions in HFA 134a, the surfactant had to possess a short fluorocarbon tail and/or a relatively long ethylene oxide head group. However, the amount of surfactant required to get reasonable water incorporation into the formulation adversely affects the drug delivery since it leads to decreased volatility which results in a coarser aerosol and increased oropharyngeal deposition (142). Thus, while reverse microemulsions in HFA appear to be an appealing mode for delivering polar drugs, their utility can be limited due to poor delivery efficiency and detrimental interaction between the device and formulation.
Another consideration for the stability of suspensions is the effect of low levels of water in the formulation. For instance, an accelerated stability study of isoproterenol sulfate and atropine methylbromide HFA MDIs at 40°C/75% relative humidity compared to 40°C/ambient humidity for 3 months reveals that at higher levels of humidity, the emitted doses were not uniform and the FPF was significantly reduced; however, moisture ingress can be limited with selected sealing gasket materials and controlled manufacturing conditions (143,144). However, the inclusion of water does not always have a detrimental effect on the suspension HFA formulation. In fact, in certain situations, adding a minute amount (<0.18% (w/w) of water (145)), in excess to the amount of water that would nominally be in the formulation (i.e., water ingress by process or storage of the MDI) has been found to improve the re-dispersibility of the formulation (146). For instance, 0.015% (w/w) water was added to a BDP monohydrate formulation resulting in a formulation that formed a weakly flocculated suspension, which was readily redispersed upon shaking (147).
PulmoSpheres®
The PulmoSpheres® technology relies on creating suspensions of lipid porous microspheres, which allows the propellant to permeate within the particles creating particles with an effective density that is virtually identical to the propellant regardless of the formulation temperature (148). By decreasing the density differential between the suspended drug particles and the continuous phase of MDIs, particle settling or creaming is greatly reduced resulting in improved formulation stability. Furthermore, the larger geometric size of PulmoSpheres®, results in an overall decrease in surface area of particles compared to conventional formulations (103). This reduces the subsequent contact area for particle–particle interactions, thereby reducing the probability of agglomeration. It is believed that this is due to a decreased van der Waals potential between particles, which depends on the difference in polarizability of the particles and the propellant. Since PulmoSpheres® are hollow porous particles, the incorporation of the propellant into the particles generates particles with similar polarizability as the propellant, which reduces the van der Waals potential between particles.
PulmoSpheres® for inhalation are manufactured in one of three ways: (1) the drug can be dissolved along with the lipid in the feed stock (148); (2) the drug can be suspended as crystals in the aqueous phase of the spray-drying feed stock (149); or (3) creating excipient-only lipid particles (105). In the case of the suspended drug particles, the drug particles can in fact be partially dissolved, yielding a mixed phase feed stock (105,149). While the PulmoSphere® technology affords benefits over conventional formulations of suspension MDIs, the spray drying process may not readily be utilized for compounds with low glass transition temperatures (e.g., glycopyrrolate), measureable propellant solubility (e.g., mometosone furoate), or detectable chemical lability (i.e., proteins and peptides). The excipient-only case overcomes challenges associated with drug stability during the spray drying process. In this case, the lipid microsphere excipient particles are combined with crystalline drug during preparation of the MDI formulation (105). This approach greatly simplifies the process required to manufacture the PulmoSphere® particles.
Cromolyn sodium, albuterol sulfate, and fomoterol fumerate PulmoSpheres® have been effectively made with spray drying. While micronization leads to a broad range of particle size distributions and little control over morphology and density, engineering the drug into PulmoSpheres® provides the opportunity to control these factors. PulmoSpheres® can be engineered to enable delivery of a broad range of drug concentrations (10 μg of fomoterol fumerate to 1 mg of cromolyn sodium) with decreased rate of phase separation and particle aggregation over a period of hours compared to commercial Intal® CFC (commercial cromolyn sodium formulation) and Proventil® HFA (commercial albuterol sulfate formulation) formulations (148). Furthermore, albuterol sulfate was formulated in 99mTc-radiolabeled PulmoSphere® particles in HFA 134a and was compared to Ventolin® HFA (commercial albuterol sulfate formulation) in nine subjects using gamma scintigraphy. It was found that the lung deposition was doubled for the PulmoSphere® formulation compared with the commercial product and oropharyngeal deposition was significantly reduced. In addition, distearoylphosphatidylcholine (DSPC)-coated budesonide microcrystals dispersed in HFA 134a were found to have residual MMADs of 3.2 to 3.4 μm and did not present “loss of prime” concerns or variability in dose delivery over the course of the life of the vial (150). These proof-of-concept studies demonstrate potential advantages of PulmoSphere® formulations over conventional suspension MDI formulations. While, no PulmoSphere® MDI formulations have been approved, Tobi® Podhaler®, a dry powder inhaler PulmoSphere® tobramycin formulation, was approved in Europe in 2010 (151).
Excipients Used as Bulking Agents
Inherently, suspension MDI formulations exhibit some degree of variable dosing behavior. Variability in dose delivery for suspension formulations can be caused by, among other things, (1) variable drug concentration because of drug deposition on the canister or valve surfaces, and (2) variable sampling of the formulation by the valve because of flocculation and creaming/settling (152). This behavior is especially apparent for low-dose formulations. One method to overcome dosing variability is to use bulking agents along with ethanol in the formulation (64, 152). These bulking agents can be made of saccharides (i.e,. lactose and maltose), amino acids (i.e., glycine and leucine) and salts (i.e., sodium chloride); however, most research has been conducted with α-lactose monohydrate (153,154). Whereas most suspension formulation approaches seek to reduce particle flocculation, this approach actually improves dosing reproducibility by greatly enhancing flocculation. Submicron bulking excipients are especially useful for forming a stable drug-excipient coflocculated matrix, which improves dosing reproducibility by minimizing the ability of the drug to migrate into and out of the valve metering volume (64). The underlying mechanism of how submicron lactose can be utilized as a bulking agent relies on the low sedimentation and high tendency of the submicron lactose to flocculate (133). Thus, the resulting effect is a suspension with a loosely flocculated matrix that houses the micronized drug and decreases the mobility of the drug in the formulation and which can easily be redispersed upon shaking the vial. By minimizing the mobility of the drug particles, the bulking excipients minimize segregation of the drug in the formulation and thus ensure that the drug is, on the macroscopic level, uniformly distributed in the canister. This allows for consistent sampling of the formulation by the valve. While submicron-sized lactose has received the most attention as a bulking agent, larger sized lactose (greater than 1 μm in diameter) can also decrease drug adherence to the canister walls and valves while also increasing resistance to moisture ingress (155).
Submicron-sized lactose has been used to stabilize a micronized formoterol fumarate HFA suspension leading to long-term stability at room temperature and decreased dosing variability over the life of the MDI while preventing loss of prime issues (64). l-leucine particles (38 to 125 μm) have been used to stabilize albuterol sulfate suspensions and fluticasone propionate in HFA formulations (156,157). HFA formulations of albuterol sulfate and fluticasone propionate both had fewer irreversible drug agglomerates and increased FPFs when l-leucine was incorporated into formulation. A preferred process for making suspended bulking excipients is high pressure homogenization of a slurry of the micronized bulking agent in ethanol, until the desired size of the bulking agent is achieved (152). The slurry is then mixed with other components of the formulation to achieve the desired concentration of the drug and bulking agent. Formulations containing bulking excipient with particles with sizes between 100 and 200 nm and excipient/drug ratios between 0.1:1 and 25:1 have been shown to result in stable MDI suspensions (153). Lactose was found to have stronger cohesive effects with a model drug, sibenadet hydrochloride, compared with mannitol, resulting in more consistent FPF over the life of the inhaler; this suggests that the particulate excipient's aggregation behavior plays a key role in its utility (158).
COMBINATION DRUG MDI PRODUCTS
Frequently, there are therapeutic advantages with simultaneously administering two or three drugs in the same dose (i.e., combination drug product) because of synergistic effects of the different drugs (159). Combination therapies facilitate improved medication therapy adherence among patients. Chronic lung disease guidelines recommend treating severe COPD with a combination of inhaled medications, including long-acting β-agonists (LABA) and corticosteroids (CS). Furthermore, some evidence-based medicine suggests that combining these with a long-acting muscarinic antagonist (LAMA) will afford improved quality of life for patients due to complementary pharmacologic activities of these drugs (105,159).
There are several approaches used to formulate combination MDI products. One such method currently being investigated involves tailored particle engineering by cocrystallizing two or more drugs (160). In this approach, a solution containing the drugs dissolved at the desired ratio is atomized and the droplets are collected in a crystallization vessel containing an antisolvent, while ultrasonic waves are utilized to induce nucleation and crystal growth. The solvent is then evaporated and micrometer-sized crystals are collected and delivered as suspension MDIs. For this method, coformulating two physicochemically dissimilar drugs is especially challenging since the solubility of LABAs, LAMAs, and CSs can differ significantly relative to each other for a given solvent, thus making it difficult to find an appropriate solvent system and antisolvent for the crystallization technique. Alternatively, two or more drugs can be spray dried, creating microparticles that contain both (or more) drugs. In this approach, the resulting particle may consist of crystalline, partially crystalline, and/or amorphous solids. Finally, two or three of the drugs can be formulated in an HFA system as a solution formulation. To formulate ipratropium, formoterol, and budesonide in HFA, the drugs were dissolved in the system using ethanol (161). The resulting formulation delivered 5 μg ipratropium bromide, 2.25 μg fomoterol fumarate, and 80 μg budesonide. The residual particle MMAD and GSD and FPF were identical for all three drugs. In addition, it is expected that the residual MMAD increases and the FPF decreases with the increase in nonvolatile concentration.
The most common approach being used in combination MDIs is to develop suspension formulations in which the two drugs are both in the form of micronized, suspended drug particles. Combination suspension formulation approaches that have been used in marketed combination MDI products include: (1) excipient-free suspensions (e.g., Advair® HFA, fluticasone propionate, and salmeterol as a xinafoate salt); (2) using HFA-soluble polymers, such as PEG and PVP as suspension stabilizers (e.g., Symbicort®, budesonide, and fomoterol fumarate dihydrate); or (3) using cosolvents, such as ethanol, to dissolve sufficient oleic acid to formulate a stable suspension (e.g., Dulara®, formoterol fumarate, and mometosone furoate) (105).
In the aforementioned marketed approaches, the coformulation effect must be considered. When formulating a combination product of two drugs, the residual APSDs of the two drugs in the formulation are frequently different from each other. Moreover, this effect is generally dose dependent on the individual components of the MDI formulation, resulting in differing FPFs for the drugs in the combination product (105). For instance, Advair® HFA (available in 44/21, 110/21, and 220/21 μg/actuation of fluticasone propionate/salmeterol in the form of a xinafoate salt) has a FPF of 48–52% for fluticasone propionate for the three strengths of fluticasone (44 to 220 μg/actuation) and 63–75% for a fixed dose of salmeterol xinafoate. Whereas, fluticasone propionate, as monotherepy from Flovent®, at a dose range of 44 to 220 μg/actuation yields a FPF of 41–50%. Thus, dose proportionality may not be achieved as a ratio of lung dose to the labeled dose as the individual strengths of the drugs in the combination MDI increases. Considerations of chemical interactions need to also be made when formulating a combination formulation. For instance, the interparticulate interactions for the two drugs found in Advair® and Symbicort® were found to be quite different, based on atomic force microscopy and Raman spectroscopy (162). It was found that budesonide and formoterol appeared as discrete particulates, whereas salmeterol and fluticasone appeared agglomerated once aerosolized and deposited on a cascade impactor plate. It was proposed that the drugs in Symbicort® interact with each other through weak van der Waals forces, while the drugs in Advair® interact with each other on a chemical level. The flocculation seen with the drugs in Advair® can potentially lead to a decrease in FPF, as the flocculates inherently have a larger aerodynamic diameter than that found for the individual drugs.
Pearl Therapeutics has enabled consistent aerosol performance of inhaled medication, regardless of if the drug(s) is/are emitted from a single-, double-, or triple-therapy product, thereby eluding the traditional coformulation effect (105,163). This technology relies on the incorporation of drug-free microparticles into the suspension MDI formulation. The phospholipid microparticles are made by spray drying from an aqueous emulsion containing perfluorooctyl bromide as the organic phase and DSPC and calcium chloride dispersed in the aqueous phase. Water and perfluorooctyl bromide are removed during the spray-drying process resulting in microparticles of DSPC with calcium chloride. The microparticles are optimized for aerodynamic properties while designed to be ideal cosuspending agents for conventional drug crystals within suspension MDIs. They are designed to have relatively large geometric particle size which maximizes the contact surface area for micronized drug particles, and the microparticles are porous thereby reducing the particle density, hence reducing the aerodynamic particle diameter. This technology involves cosuspending micronized drug crystals with the previously mentioned microparticles in HFA propellants, where the drug crystals are irreversibly associated with the porous DSPC and calcium chloride particles (163). Overall, this technology potentially offers the ability to match in vitro performance regardless of if the drug is delivered alone or in combination with other drugs.
The Pearl technology platform has been utilized for a variety of agents (105). It has been successfully utilized to formulate combinations of up to three drugs in a formulation, including a triple therapy of CS, LABA, and LAMA. For the triple therapy suspension MDI, the FPF and the MMAD of the drug components assessed in the combination and as single components were comparable despite the 10-fold difference in dose between the drugs. The approach was found to be useful for MDI suspension drug strengths ranging from 1 to 100 μg/actuation. The technology has shown a linear relationship between labeled dose and fine particle mass for glycopyrrolate suspension MDI with doses ranging from 0.3 to 18 μg/actuation (164). Within this range, the formulation was found to be stable in a 6-week temperature cycling study (between −5°C and 40°C) and in a 3-month accelerated stability study (at 40°C with 75% relative humidity).
CONCLUSIONS
Drug delivery via pressurized MDIs has been a longstanding therapeutic modality for the treatment of respiratory diseases, such as asthma and COPD. Over the past two decades, MDI formulation approaches have been reexamined primarily as a result of the transition from CFC to HFA propellants. The development efforts have led to not only new methods for formulating solution and suspension formulations, but they have also led to an increased understanding of critical formulation (and device) variables that influence the consistency and efficiency of the drug delivery. For example, while drug or excipient solubility in HFA systems may be enhanced by increasing ethanol concentration, this has a detrimental effect on atomization and droplet evaporation and thus has limited potential to increase the fine particle dose that can be delivered. It has also been found that the stability and dosing uniformity of suspension formulations have been improved through the use of a variety of surfactants, bulking agents and phospholipid microparticles. Other formulation approaches, such as engineered drug-excipient particles or engineered excipient particles, are likely to expand the range of drugs that can be delivered from MDIs and may enable sustained drug delivery. Expansion into new therapeutic areas, combination products, increased access to medication in developing markets and increasing cost pressures in developed markets will ensure that MDIs will remain a mainstay in the treatment of pulmonary diseases for many years to come.
Acknowledgments
Conflict of Interest
Stephen W. Stein is currently employed by 3M Drug Delivery Systems. Poonam Sheth and Paul B. Myrdal declared that no conflict of interest exists.
Contributor Information
Paul B. Myrdal, Email: myrdal@pharmacy.arizona.edu
Poonam Sheth, Phone: +1-520-6263847, FAX: +1-520-6267355, sheth@email.arizona.edu.
Stephen W. Stein, Email: swstein@mmm.com
References
- 1.Thiel CG. From Susie's question to CFC free: an inventor's perspective on forty years of MDI development and regulation. Respir Drug Deliv. 1996;1:115–24. [Google Scholar]
- 2.Evans M, Telfer A, Smith R, Smith B, Lang G, Chen J, et al. Stratospheric sink for chlorofluoromethanes: chlorine atomc-atalysed destruction of ozone. Nature. 1974;249:811. [Google Scholar]
- 3.McDonald KJ, Martin GP. Transition to CFC-free metered dose inhalers—into the new millennium. Int J Pharm. 2000;201(1):89–107. doi: 10.1016/s0378-5173(00)00401-4. [DOI] [PubMed] [Google Scholar]
- 4.Newman SP, Peart J. Pressurized metered dose inhalers. In: Newman SP, editor. Respiratory drug delivery: essential theory and practice. Richmond: Respiratory Drug Delivery Online; 2009. pp. 117–216. [Google Scholar]
- 5.Stein SW, Sheth P, Hodson PD, Myrdal PB. Advances in metered dose inhaler technology: Hardware development. AAPS PharmSciTech. doi:10.1208/s12249-013-0062-y [DOI] [PMC free article] [PubMed]
- 6.Carvalho TC, Peters JI, Williams RO., III Influence of particle size on regional lung deposition—what evidence is there? Int J Pharm. 2011;406(1):1–10. doi: 10.1016/j.ijpharm.2010.12.040. [DOI] [PubMed] [Google Scholar]
- 7.Smyth HDC. The influence of formulation variables on the performance of alternative propellant-driven metered dose inhalers. Adv Drug Deliv Rev. 2003;55(7):807–28. doi: 10.1016/s0169-409x(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 8.Vervaet C, Byron PR. Drug–surfactant–propellant interactions in HFA-formulations. Int J Pharm. 1999;186(1):13–30. doi: 10.1016/s0378-5173(99)00134-9. [DOI] [PubMed] [Google Scholar]
- 9.McCulloch A. CFC and halon replacements in the environment. J Fluor Chem. 1999;100(1):163–73. [Google Scholar]
- 10.Newman SP. Principles of metered-dose inhaler design. Respir Care. 2005;50(9):1177–90. [PubMed] [Google Scholar]
- 11.Wallington TJ, Schneider WF, Worsnop DR, Nielsen OJ, Sehested J, Debruyn WJ, et al. The environmental impact of CFC replacements HFCs and HCFCs. Environ Sci Technol. 1994;28(7):320A–6. doi: 10.1021/es00056a714. [DOI] [PubMed] [Google Scholar]
- 12.US Environmental Protection Agency. The process of ozone depletion. Available at: http://www.epa.gov/ozone/science/process.html. Accessed July 17, 2012.
- 13.United Nations Environment Programme Website. Available at: http://www.unep.org. Accessed June 5, 2013.
- 14.Federal Registrar. Food and Drug Administration 21 Code of Federal Regulations (2), Use of ozone-depleting substances, Essential-use determination. 2002.
- 15.Rogueda P, Lallement A, Traini D, Iliev I, Young PM. Twenty years of HFA pMDI patents: facts and perspectives. J Pharm Pharmacol. 2012;64(9):1209–16. doi: 10.1111/j.2042-7158.2011.01387.x. [DOI] [PubMed] [Google Scholar]
- 16.Gupte AJ, Bogardus RE, inventors; Richardson Vicks Inc., assignee. Dry aerosol foam. European patent EP 247,608. 1987 Dec 2.
- 17.Smith IJ. The challenge of reformulation. J Aerosol Med. 1995;8(Suppl 1):S19–27. doi: 10.1089/jam.1995.8.suppl_1.s-19. [DOI] [PubMed] [Google Scholar]
- 18.Dalby RN. Prediction and assessment of flammability hazards associated with metered-dose inhalers containing flammable propellants. Pharm Res. 1992;9(5):636–42. doi: 10.1023/a:1015850026577. [DOI] [PubMed] [Google Scholar]
- 19.Dalby RN, Byron PR, inventors; Virginia Commonwealth University, assignee. Formulations for delivery of beclomethasone diproprionate by metered dose inhalers containing no chlorofluorocarbon propellants. US patent US 5,202,110. 13 Apr 1993.
- 20.Decaire B, Ghelani K, Conviser S, Sarrailh S, Le Corre B, Baron C. Materials compatibility testing of new low global warming potential propellants. Respir Drug Deliv Eur. 2011;2:281–4. [Google Scholar]
- 21.Knopeck G, Decaire B, Ghelani K. A new generation of aerosol propellants for metered dose inhalers. Respir Drug Deliv. 2010;2:591–4. [Google Scholar]
- 22.Sommerville ML, Johnson CS, Jr, Cain JB, Rypacek F, Hickey AJ. Lecithin microemulsions in dimethyl ether and propane for the generation of pharmaceutical aerosols containing polar solutes. Pharm Dev Technol. 2002;7(3):273–88. doi: 10.1081/pdt-120005724. [DOI] [PubMed] [Google Scholar]
- 23.Sommerville ML, Hickey AJ. Aerosol generation by metered-dose inhalers containing dimethyl ether/propane inverse microemulsions. AAPS PharmSciTech. 2003;4(4):455–61. doi: 10.1208/pt040458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vega JC, Toneguzzo F, inventors; Laboratorio Pablo Cassera, S.R.L., assignee. Non-ozone depleting medicinal formulations with low greenhouse effect. US patent US 207,685. 16 Aug 2012.
- 25.Solvay Fluoro GmbH. Solkane 227 pharma and Solkane 134a pharma Product Information. Available at: http://www.solvay-fluor.com. Accessed June 21, 2012.
- 26.Hoye WL, Mogalian EM, Myrdal PB. Effects of extreme temperatures on drug delivery of albuterol sulfate hydrofluoroalkane inhalation aerosols. Am J Health-Syst Ph. 2005;62(21):2271–7. doi: 10.2146/ajhp050067. [DOI] [PubMed] [Google Scholar]
- 27.Ross DL, Gabrio BJ. Advances in metered dose inhaler technology with the development of a chlorofluorocarbon-free drug delivery system. J Aerosol Med. 1999;12(3):151–60. doi: 10.1089/jam.1999.12.151. [DOI] [PubMed] [Google Scholar]
- 28.Stein S, Stefely J. Reinventing metered dose inhalers: from poorly efficient CFC MDIs to highly efficient HFA MDIs. Drug Deliv Technol. 2003;3(1):46–51. [Google Scholar]
- 29.Stein SW, Cocks PM. Size distribution measurements from metered dose inhalers at low temperatures. Respiratory Drug Delivery Europe. 2013;2:203–8. [Google Scholar]
- 30.Williams R, III, Liu J. Formulation of a protein with propellant HFA 134a for aerosol delivery. Eur J Pharm Sci. 1999;7(2):137–44. doi: 10.1016/s0928-0987(98)00015-3. [DOI] [PubMed] [Google Scholar]
- 31.Leach CL. The CFC, to HFA transition and its impact on pulmonary drug development. Respir Care. 2005;50(9):1201–8. [PubMed] [Google Scholar]
- 32.Reynolds JM, McNamara DP. Model for moisture transport into inhalation aerosols. Pharm Res. 1996;13(5):809–11. doi: 10.1023/a:1016028423118. [DOI] [PubMed] [Google Scholar]
- 33.Williams R, Hu C. Investigation of moisture scavengers in pressurized metered-dose inhalers. STP Pharm Sci. 2000;10(3):243–50. [Google Scholar]
- 34.Velders GJ, Fahey DW, Daniel JS, McFarland M, Andersen SO. The large contribution of projected HFC emissions to future climate forcing. Proc Natl Acad Sci. 2009;106(27):10949–54. doi: 10.1073/pnas.0902817106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stein SW, Fradley G. What is the future of MDIs? Respir Drug Deliv. 2010;2:373–6. [Google Scholar]
- 36.UNEP Technology and Economic Assessment Panel. Montreal protocol on substances that deplete the ozone layer: TEAP 2010 progress report. Available at: http://www.unep.org. Accessed July 1, 2012.
- 37.Ding L, Zhang J. Isobutane driven salbutamol sulfate metered dose inhaler: formulation selection and respiratory tract absorption in guinea pigs. Yao Xue Xue Bao. 2009;44(9):1040–5. [PubMed] [Google Scholar]
- 38.Moore A. Final report of the safety assessment of isobutane, isopentane, n-butane, and propane. J Am Coll Toxicol. 1982;1:127–42. [Google Scholar]
- 39.Aeropres Corporation. MSDS. Available at: http://www.aeropres.com/msds. Accessed June 21, 2013.
- 40.Corr S, Noakes TJ, inventors; Mexichem Amanco Holding S.A DE C.V, assignee. Compositions comprising salbutamol sulfate. World Intellectual Property Organization patent WO 2013/054135. 2013 Apr 18.
- 41.Corr S, Noakes TJ, inventors; Mexichem Amanco Holding S.A DE C.V, assignee. Pharmaceutical compositions. World Intellectual Property Organization patent WO 2012/156711. 2012 Nov 22.
- 42.International Programme on Chemical Safety. 1,1-difluoroethane (HFC-152a) (Screening Information Data Set – SIDs). 2008; Available at: http://inchem.org/. Accessed May 23, 2013.
- 43.Vance C, Swalwell C, McIntyre IM. Deaths involving 1, 1-difluoroethane at the San Diego County Medical Examiner's Office. J Anal Toxicol. 2012;36(9):626–33. doi: 10.1093/jat/bks074. [DOI] [PubMed] [Google Scholar]
- 44.Ernstgård L, Sjögren B, Dekant W, Schmidt T, Johanson G. Uptake and disposition of 1,1-difluoroethane (HFC-152a) in humans. Toxicol Lett. 2012;209(1):21–9. doi: 10.1016/j.toxlet.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 45.Honeywell International Inc. Solstice propellant technical brochure. Available at: http://honeywell-solstice-propellants.com. Accessed June 17, 2012.
- 46.Dupont. Opteon YF FAQ. Available at: http://www2.dupont.com. Accessed June 17, 2012.
- 47.Leach CL, Kuehl PJ, Chand R, Ketai L, Norenberg JP, McDonald JD. Characterization of respiratory deposition of fluticasone-salmeterol hydrofluoroalkane-134a and hydrofluoroalkane-134a beclomethasone in asthmatic patients. Ann Allergy Asthma Immunol. 2012;108(3):195–200. doi: 10.1016/j.anai.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 48.Dalby RN, Byron PR. Comparison of output particle size distributions from pressurized aerosols formulated as solutions or suspensions. Pharm Res. 1988;5(1):36–9. doi: 10.1023/a:1015859311228. [DOI] [PubMed] [Google Scholar]
- 49.Noakes T. Medical aerosol propellants. J Fluor Chem. 2002;118(1):35–45. [Google Scholar]
- 50.U.S. Food and Drug Administration. Inactive Ingredient Search for Approved Drug Products. 2013; Available at: http://www.accessdata.fda.gov/scripts/cder/iig/. Accessed June 21, 2013.
- 51.U.S. Food and Drug Administration. Drugs@FDA: FDA Approved Drug Products. Available at: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm. Accessed June 21, 2013.
- 52.Grainger C, Saunders M, Buttini F, Telford R, Merolla L, Martin G, et al. Critical characteristics for corticosteroid solution metered dose inhaler bioequivalence. Mol Pharm. 2012;9(3):563–9. doi: 10.1021/mp200415g. [DOI] [PubMed] [Google Scholar]
- 53.Riley T, Christopher D, Arp J, Casazza A, Colombani A, Cooper A, et al. Challenges with developing in vitro dissolution tests for orally inhaled products (OIPs) AAPS PharmSciTech. 2012;13(3):978–89. doi: 10.1208/s12249-012-9822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoye JA, Myrdal PB. Measurement and correlation of solute solubility in HFA-134a/ethanol systems. Int J Pharm. 2008;362(1):184–8. doi: 10.1016/j.ijpharm.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 55.Stein SW, Myrdal PB. The relative influence of atomization and evaporation on metered dose inhaler drug delivery efficiency. Aerosol Sci Tech. 2006;40(5):335–47. [Google Scholar]
- 56.Gupta A, Stein SW, Myrdal PB. Balancing ethanol cosolvent concentration with product performance in 134a-based pressurized metered dose inhalers. J Aerosol Med. 2003;16(2):167–74. doi: 10.1089/089426803321919924. [DOI] [PubMed] [Google Scholar]
- 57.Myrdal PB, Karlage KL, Stein SW, Brown BA, Haynes A. Optimized dose delivery of the peptide cyclosporine using hydrofluoroalkane-based metered dose inhalers. J Pharm Sci. 2004;93(4):1054–61. doi: 10.1002/jps.20025. [DOI] [PubMed] [Google Scholar]
- 58.Mogalian E, Myrdal PB. Application of USP inlet extensions to the TSI impactor system 3306/3320 using HFA 227 based solution metered dose inhalers. Drug Dev Ind Pharm. 2005;31(10):977–85. doi: 10.1080/03639040500306211. [DOI] [PubMed] [Google Scholar]
- 59.Saleem IY, Smyth HD. Tuning aerosol particle size distribution of metered dose inhalers using cosolvents and surfactants. BioMed Res Int. 2013 doi: 10.1155/2013/574310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stein SW, Gabrio BJ. Understanding throat deposition during cascade impactor testing. Respir Drug Deliv. 2000;2:287–90. [Google Scholar]
- 61.Stein SW, Myrdal PB. A theoretical and experimental analysis of formulation and device parameters affecting solution MDI size distributions. J Pharm Sci. 2004;93(8):2158–75. doi: 10.1002/jps.20116. [DOI] [PubMed] [Google Scholar]
- 62.Sheth P, Stein SW, Myrdal PB. The influence of initial atomized droplet size on residual particle size from pressurized metered dose inhalers. Int J Pharm. 2013;455(1–2):57–65. doi: 10.1016/j.ijpharm.2013.07.061. [DOI] [PubMed] [Google Scholar]
- 63.Warren SJ, Farr SJ. Formulation of solution metered dose inhalers and comparison with aerosols emitted from conventional suspension systems. Int J Pharm. 1995;124(2):195–203. [Google Scholar]
- 64.Stein SW, Forsyth BR, Stefely JS, Christensen JD, Alband TD, Jinks PA. Expanding the dosing range of metered dose inhalers through formulation and hardware optimization. Respir Drug Deliv. 2004;1:125–34. [Google Scholar]
- 65.Scherrer RA, Stefely JS, Stein SW, inventors; 3M Innovative Properties Company, assignee. Medicinal aerosol formulations comprising ion pair complexes. World Intellectual Property Organization patent WO 2003/059316. 24 Jul 2003.
- 66.Stefely JS, Duan DC, Myrdal PB, Ross DL, Schultz DW, Leach CL. Design and utility of a novel class of biocompatible excipients for HFA based MDIs. Respir Drug Deliv. 2000;1:83–90. [Google Scholar]
- 67.Stefely JS, Brown BA, Hammerbeck DM, Stein SW. Equipping the MDI for the 21st century by expanding its formulation options. Respir Drug Deliv. 2002;1:207–14. [Google Scholar]
- 68.Stefely JS, Duan DC, inventors; 3M Innovative Properties Company, assignee. Medicinal aerosol compositions with a functionalized polyethyleneglycol excipient. US patent US 7,718,162. 18 May 2010.
- 69.Rogueda P, inventor; AstraZeneca, assignee. Pharmaceutical spray formulation comprising a hypro fluor alkane amd an acylated cyclodextrin. World Intellectual Property Organization patent WO 2005/053637. 16 Jun 2005.
- 70.Tashkin DP. Extra-fine corticosteroid aerosols from hydrofluoroalkane-134a metered-dose inhalers: potential advantages and disadvantages. Chest. 1999;115(2):316–8. doi: 10.1378/chest.115.2.316. [DOI] [PubMed] [Google Scholar]
- 71.Magnussen H. Budesonide Modulite®: improving the changeover to CFC-free treatments. Respir Med. 2003;97(Suppl D):S1–3. doi: 10.1016/j.rmed.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 72.Lewis D, Ganderton D, Meakin B, Brambilla G. Modulite®: a simple solution to a difficult problem. Respiration. 2005;72(Suppl. 1):3–5. doi: 10.1159/000083686. [DOI] [PubMed] [Google Scholar]
- 73.Ganderton D, Lewis D, Davies R, Meakin B, Church T. The formulation and evaluation of a CFC-free budesonide pressurised metered dose inhaler. Respir Med. 2003;97:S4–9. doi: 10.1016/j.rmed.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 74.Majury C, Tran CH, Taylor G. Assessing the influence of ethanol on the aerosol properties of beclomethasone pMDIs using the next generation impactor. Respir Drug Deliv. 2012;2:433–6. [Google Scholar]
- 75.Chaplin S, Head S. Clenil Modulite, a CFC-free MDI with no adjustment on switching. Prescriber. 2007;18(13):43–6. [Google Scholar]
- 76.Busse WW, Brazinsky S, Jacobson K, Stricker W, Schmitt K, Vanden Burgt J, et al. Efficacy response of inhaled beclomethasone dipropionate in asthma is proportional to dose and is improved by formulation with a new propellant. J Allergy Clin Immunol. 1999;104(6):1215–22. doi: 10.1016/s0091-6749(99)70016-3. [DOI] [PubMed] [Google Scholar]
- 77.Lipworth B. Targeting the small airways asthma phenotype: If we can reach it, should we treat it? Ann Allergy Asthma Immonol. 2013;110(4):233–9. doi: 10.1016/j.anai.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 78.Leach C, Davidson P, Boudreau R. Improved airway targeting with the CFC-free HFA-beclomethasone metered-dose inhaler compared with CFC-beclomethasone. Eur Respir J. 1998;12(6):1346–53. doi: 10.1183/09031936.98.12061346. [DOI] [PubMed] [Google Scholar]
- 79.Price D, Thomas M, Haughney J, Lewis RA, Burden A, von Ziegenweidt J, et al. Real-life comparison of beclometasone dipropionate as an extrafine-or larger-particle formulation for asthma. Respir Med. 2013 doi: 10.1016/j.rmed.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 80.Robinson CA, Tsourounis C. Inhaled corticosteroid metered-dose inhalers: how do variations in technique for solutions versus suspensions affect drug distribution? Ann Pharmacother. 2013;47(3):416–20. doi: 10.1345/aph.1R480. [DOI] [PubMed] [Google Scholar]
- 81.Barnes N, Price D, Colice G, Chisholm A, Dorinsky P, Hillyer E, et al. Asthma control with extrafine-particle hydrofluoroalkane–beclometasone vs. large-particle chlorofluorocarbon–beclometasone: a real-world observational study. Clin Exp Allergy. 2011;41(11):1521–32. doi: 10.1111/j.1365-2222.2011.03820.x. [DOI] [PubMed] [Google Scholar]
- 82.Church TK, Lewis DA, Ganderton D, Meakin BJ, Brambilla G, inventors; Chiesi Farmaceutici S.p.A, assignee. Salmeterol superfine formulation. US patent US 8,088,362. 3 Jan 2012.
- 83.Lewis D, Ganderton D, Meakin B, Brambilla G, Ferraris A, inventors; Cheiesi Farmaceutici S.p.A., assignee. Stable pharmaceutical solution formulations for pressurized metered dose inhalers. US patent US 7,018,618. 28 Mar 2006.
- 84.Tzou T, Pachuta RR, Coy RB, Schultz RK. Drug form selection in albuterol-containing metered-dose inhaler formulations and its impact on chemical and physical stability. J Pharm Sci. 1997;86(12):1352–7. doi: 10.1021/js970225g. [DOI] [PubMed] [Google Scholar]
- 85.Stank K, Steckel H. Physico-chemical characterisation of surface modified particles for inhalation. Int J Pharm. 2013;114:9–18. doi: 10.1016/j.ijpharm.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 86.Shoyele SA, Cawthorne S. Particle engineering techniques for inhaled biopharmaceuticals. Adv Drug Deliv Rev. 2006;58(9):1009–29. doi: 10.1016/j.addr.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 87.Chow AH, Tong HH, Chattopadhyay P, Shekunov BY. Particle engineering for pulmonary drug delivery. Pharm Res. 2007;24(3):411–37. doi: 10.1007/s11095-006-9174-3. [DOI] [PubMed] [Google Scholar]
- 88.Murnane D, Martin GP, Marriott C. Investigations into the formulation of metered dose inhalers of salmeterol xinafoate and fluticasone propionate microcrystals. Pharm Res. 2008;25(10):2283–91. doi: 10.1007/s11095-008-9622-3. [DOI] [PubMed] [Google Scholar]
- 89.Jones SA, Martin GP, Brown MB. Manipulation of beclomethasone–hydrofluoroalkane interactions using biocompatible macromolecules. J Pharm Sci. 2006;95(5):1060–74. doi: 10.1002/jps.20608. [DOI] [PubMed] [Google Scholar]
- 90.Pilcer G, Amighi K. Formulation strategy and use of excipients in pulmonary drug delivery. Int J Pharm. 2010;392(1):1–19. doi: 10.1016/j.ijpharm.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 91.Telko MJ, Hickey AJ. Dry powder inhaler formulation. Respir Care. 2005;50(9):1209–27. [PubMed] [Google Scholar]
- 92.Malcolmson RJ, Embleton JK. Dry powder formulations for pulmonary delivery. Pharm Sci Technol Today. 1998;1(9):394–8. [Google Scholar]
- 93.Li H, Seville PC. Novel pMDI formulations for pulmonary delivery of proteins. Int J Pharm. 2010;385(1):73–8. doi: 10.1016/j.ijpharm.2009.10.032. [DOI] [PubMed] [Google Scholar]
- 94.Li H, Song X, Seville PC. The use of sodium carboxymethylcellulose in the preparation of spray-dried proteins for pulmonary drug delivery. Eur J Pharm Sci. 2010;40(1):56–61. doi: 10.1016/j.ejps.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 95.Liao Y, Brown MB, Jones SA, Nazir T, Martin GP. The effects of polyvinyl alcohol on the in vitro stability and delivery of spray-dried protein particles from surfactant-free HFA 134a-based pressurised metered dose inhalers. Int J Pharm. 2005;304(1):29–39. doi: 10.1016/j.ijpharm.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 96.Jones SA, Martin GP, Brown MB. Stabilisation of deoxyribonuclease in hydrofluoroalkanes using miscible vinyl polymers. J Control Release. 2006;115(1):1–8. doi: 10.1016/j.jconrel.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 97.Tam JM, Engstrom JD, Williams RO, III, Johnston KP. Suspensions of protein and poorly water soluble drug particles for high dosages with pressurized metered dose inhalers. Respir Drug Deliv. 2008;3:931–6. [Google Scholar]
- 98.Tan Y, Yang Z, Peng X, Xin F, Xu Y, Feng M, et al. A novel bottom-up process to produce nanoparticles containing protein and peptide for suspension in hydrofluoroalkane propellants. Int J Pharm. 2011;413(1):167–73. doi: 10.1016/j.ijpharm.2011.03.069. [DOI] [PubMed] [Google Scholar]
- 99.Selvam P, El-Sherbiny IM, Smyth HD. Swellable hydrogel particles for controlled release pulmonary administration using propellant-driven metered dose inhalers. J Aerosol Med. 2011;24(1):25–34. doi: 10.1089/jamp.2010.0830. [DOI] [PubMed] [Google Scholar]
- 100.Longest PW, Hindle M. Numerical model to characterize the size increase of combination drug and hygroscopic excipient nanoparticle aerosols. Aerosol Sci Tech. 2011;45(7):884–99. doi: 10.1080/02786826.2011.566592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Williams RO, III, Barron MK, Alonso MJ, Remuñán-López C. Investigation of a pMDI system containing chitosan microspheres and P134a. Int J Pharm. 1998;174(1):209–22. [Google Scholar]
- 102.Haghi M, Bebawy M, Colombo P, Forbes B, Lewis D, Salama R, et al. Towards the bioequivalence of pressurised metered dose inhalers 2. Aerodynamically equivalent particles (with and without glycerol) exhibit different biopharmaceutical profiles in vitro. Eur J Pharm Biopharm. 2013 doi: 10.1016/j.ejpb.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 103.O'Donnell KP, Williams RO., III Pulmonary dispersion formulations: The impact of dispersed powder properties on pressurized metered dose inhaler stability. Drug Dev Ind Pharm. 2013;39(3):413–24. doi: 10.3109/03639045.2012.664145. [DOI] [PubMed] [Google Scholar]
- 104.Rogueda PG, Buckin V, Kudryashov E. Size and concentration monitoring of HFA suspensions. Respir Drug Deliv. 2006;2:453–6. [Google Scholar]
- 105.Lechuga-Ballesteros D, Noga B, Vehring R, Cummings RH, Dwivedi SK. Novel cosuspension metered-dose inhalers for the combination therapy of chronic obstructive pulmonary disease and asthma. Futur Med Chem. 2011;3(13):1703–18. doi: 10.4155/fmc.11.133. [DOI] [PubMed] [Google Scholar]
- 106.Sukasamea N, Boonmea P, Srichanaa T. Development of budesonide suspensions for use in an HFA pressurized metered dose inhaler. ScienceAsia. 2011;37(1):31–7. [Google Scholar]
- 107.Rogueda P. Novel hydrofluoroalkane suspension formulations for respiratory drug delivery. Expert Opin Drug Deliv. 2005;2(4):625–38. doi: 10.1517/17425247.2.4.625. [DOI] [PubMed] [Google Scholar]
- 108.Stein SW, Sheth P, Karayiannis C, Chiou H, Myrdal PB. Modeling MDI delivery: A priori predictions, empirical models and experiments. Respir Drug Deliv. 2010;1:353–64. [Google Scholar]
- 109.Stein SW, Sheth P, Myrdal PB. A model for predicting size distributions delivered from pMDIs with suspended drug. Int J Pharm. 2012;422(1):101–15. doi: 10.1016/j.ijpharm.2011.10.035. [DOI] [PubMed] [Google Scholar]
- 110.Berry J, Kline LC, Sherwood JK, Chaudhry S, Obenauer-Kutner L, Hart JL, et al. Influence of the size of micronized active pharmaceutical ingredient on the aerodynamic particle size and stability of a metered dose inhaler. Drug Dev Ind Pharm. 2004;30(7):705–14. doi: 10.1081/ddc-120039213. [DOI] [PubMed] [Google Scholar]
- 111.Sharpe SA, Sequeira JA, inventors; Schering Corporation, assignee. Metered dose inhaler containing an aerosol suspension formulation. European patent EP 1,785,156. 2012 Jun 27.
- 112.Sherwood JK, Alex S, Salama G, Obenauer-Kutner L, Huyck S, Berry J, et al. Particle size coarsening induced by valve silicone in a metered dose inhaler. Drug Dev Ind Pharm. 2007;33(2):155–62. doi: 10.1080/03639040600814650. [DOI] [PubMed] [Google Scholar]
- 113.Berry J, Kline L, Naini V, Chaudhry S, Hart J, Sequeira J. Influence of the valve lubricant on the aerodynamic particle size of a metered dose inhaler. Drug Dev Ind Pharm. 2004;30(3):267–75. doi: 10.1081/ddc-120030420. [DOI] [PubMed] [Google Scholar]
- 114.James J, Davies M, Toon R, Jinks P, Roberts CJ. Particulate drug interactions with polymeric and elastomeric valve components in suspension formulations for metered dose inhalers. Int J Pharm. 2009;366(1):124–32. doi: 10.1016/j.ijpharm.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 115.Ridder KB, Davies-Cutting CJ, Kellaway IW. Surfactant solubility and aggregate orientation in hydrofluoroalkanes. Int J Pharm. 2005;295(1):57–65. doi: 10.1016/j.ijpharm.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 116.da Rocha SR, Bharatwaj B, Saiprasad S. Science and technology of pressurized metered-dose inhalers. In: Smyth HDC, Hickey AJ, editors. Controlled pulmonary drug delivery. New York: Springer; 2011. pp. 165–201. [Google Scholar]
- 117.BASF: The Chemical Company. Pluronic®: product Information. Available at: http://worldaccount.basf.com/wa/NAFTA~en_US/Catalog/ChemicalsNAFTA/pi/BASF/Brand/pluronic. Accessed July 7, 2013.
- 118.Alexandridis P, Alan HT. Poly (ethylene oxide)-poly (propylene oxide)-poly (ethylene oxide) block copolymer surfactants in aqueous solutions and at interfaces: thermodynamics, structure, dynamics, and modeling. Colloids Surf A Physicochem Eng Asp. 1995;96(1):1–46. [Google Scholar]
- 119.Griffin WC. Calculation of HLB values of non-ionic surfactants. Am Perfumer Essent Oil Rev. 1955;65:26–9. [Google Scholar]
- 120.Sommerville ML, Cain JB, Johnson CS, Jr, Hickey AJ. Lecithin inverse microemulsions for the pulmonary delivery of polar compounds utilizing dimethylether and propane as propellants. Pharm Dev Technol. 2000;5(2):219–30. doi: 10.1081/pdt-100100537. [DOI] [PubMed] [Google Scholar]
- 121.Sheth P, Myrdal PB. Polymers for pulmonary drug delivery. In: Smyth HDC, Hickey AJ, editors. Controlled pulmonary drug delivery. New York: Springer; 2011. pp. 265–82. [Google Scholar]
- 122.Leach CL, Hameister WM, Tomaie MA, Hammerbeck DM, Stefely JS. Oligolactic acid (OLA) biomatrices for sustained release of asthma therapeutics. Respir Drug Deliv. 2000;1:75–82. [Google Scholar]
- 123.Traini D, Young P, Rogueda P, Price R. Investigation into the influence of polymeric stabilizing excipients on inter-particulate forces in pressurised metered dose inhalers. Int J Pharm. 2006;320(1):58–63. doi: 10.1016/j.ijpharm.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 124.Wu L, da Rocha SR. Biocompatible and biodegradable copolymer stabilizers for hydrofluoroalkane dispersions: a colloidal probe microscopy investigation. Langmuir. 2007;23(24):12104–10. doi: 10.1021/la702108x. [DOI] [PubMed] [Google Scholar]
- 125.Rogueda PG. Pushing the boundaries: searching for novel HFA suspension formulations. Respir Drug Deliv. 2004;1:117–24. [Google Scholar]
- 126.Looker BE, Lunniss CJ, Redgrave AJ, inventors; Glaxo Group Limited, assignee. Compounds for use as surfactants. World Intellectual Property Organization patent WO 2003/035237. 1 May 2003.
- 127.Looker BE, Lunniss CJ, Redgrave A, inventors; Glaxo Group Limited, assignee. Carboxylic acid compounds for use as surfactants. World Intellectual Property Organization patent WO 2003/068722. 21 Aug 2003.
- 128.Berry J, Chaudry IA, Sequeira JA, Kopcha M, inventors; Schering Corporation, assignee. Non-chlorofluorocarbon aerosol formulations. European patent EP 0656206. 4 June 1995.
- 129.Tan Y, Yang Z, Pan X, Chen M, Feng M, Wang L, et al. Stability and aerosolization of pressurized metered dose inhalers containing thymopentin nanoparticles produced using a bottom-up process. Int J Pharm. 2012;427(2):385–92. doi: 10.1016/j.ijpharm.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 130.Kellaway IW, Taylor K, Nyambura BK, inventors; School of Pharmacy, University of London, assignee. Formulations for delivery via pressurised metered dose inhalers comprising an essential oil as suspension stabiliser. European patent EP 2,089,008. 20 Jul 2011.
- 131.Byron PR, Blondino FE. Metered dose inhaler fomulations which include the ozone-friendly propellant HFC 134a and a pharmaceutically acceptable suspending, solubilizing, wetting, emulsifying or lubricating agent 1996.
- 132.Byron P, Blondino F inventors; The Center for Innovative Technology, Virginia Commonwealth University, assignees. Pharmaceutically acceptable agents for solubilizing, wetting, emulsifying, or lubricating in metered dose inhaler formulations which use HFC-227 propellant. US patent US 5,492,688. 20 Feb 1996.
- 133.Cantor AS, Stefely JS, Jinks PA, Baran JR, Ganser JM, Mueting MW, et al. Modifying interparticulate interactions using surface-modified excipient nanoparticles. Respir Drug Deliv. 2008;1:309–18. [Google Scholar]
- 134.Sharma K, Somavarapu S, Colombani A, Govind N, Taylor KM. Crosslinked chitosan nanoparticle formulations for delivery from pressurized metered dose inhalers. Eur J Pharm Biopharm. 2012;81(1):74–81. doi: 10.1016/j.ejpb.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 135.Steckel H, Wehle S. A novel formulation technique for metered dose inhaler (MDI) suspensions. Int J Pharm. 2004;284(1):75–82. doi: 10.1016/j.ijpharm.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 136.Sawatdee S, Phetmung H, Srichana T. Sildenafil citrate monohydrate–cyclodextrin nanosuspension complexes for use in metered-dose inhalers. Int J Pharm. 2013 doi: 10.1016/j.ijpharm.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 137.Wu L, Al-Haydari M, da Rocha SR. Novel propellant-driven inhalation formulations: engineering polar drug particles with surface-trapped hydrofluoroalkane-philes. Eur J Pharm Sci. 2008;33(2):146–58. doi: 10.1016/j.ejps.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 138.Wu L, Bharatwaj B, Panyam J, da Rocha SR. Core–shell particles for the dispersion of small polar drugs and biomolecules in hydrofluoroalkane propellants. Pharm Res. 2008;25(2):289–301. doi: 10.1007/s11095-007-9466-2. [DOI] [PubMed] [Google Scholar]
- 139.Chokshi U, Selvam P, Porcar L, da Rocha SR. Reverse aqueous emulsions and microemulsions in HFA227 propellant stabilized by non-ionic ethoxylated amphiphiles. Int J Pharm. 2009;369(1):176–84. doi: 10.1016/j.ijpharm.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 140.Selvam P, Bharatwaj B, Porcar L, da Rocha SR. Reverse aqueous microemulsions in hydrofluoroalkane propellants and their aerosol characteristics. Int J Pharm. 2012;422(1):428–35. doi: 10.1016/j.ijpharm.2011.10.038. [DOI] [PubMed] [Google Scholar]
- 141.Butz N, Porte C, Courrier H, Krafft M, Vandamme TF. Reverse water-in-fluorocarbon emulsions for use in pressurized metered-dose inhalers containing hydrofluoroalkane propellants. Int J Pharm. 2002;238(1):257–69. doi: 10.1016/s0378-5173(02)00086-8. [DOI] [PubMed] [Google Scholar]
- 142.Patel N, Marlow M, Lawrence MJ. Formation of fluorinated nonionic surfactant microemulsions in hydrofluorocarbon 134a (HFC 134a) J Colloid Interface Sci. 2003;258(2):345–53. doi: 10.1016/s0021-9797(02)00072-3. [DOI] [PubMed] [Google Scholar]
- 143.Murata S, Ito H, Izumi T, Chikushi A. Effect of the moisture content in aerosol on the spray performance of Stmerin® D HFA preparations. Chem Pharm Bull. 2006;54(9):1276–80. doi: 10.1248/cpb.54.1276. [DOI] [PubMed] [Google Scholar]
- 144.Murata S, Izumi T, Ito H. Effect of the moisture content in aerosol on the spray performance of Stmerin® D hydrofluoroalkane preparations (2) Chem Pharm Bull. 2012;60(5):593–7. doi: 10.1248/cpb.60.593. [DOI] [PubMed] [Google Scholar]
- 145.Adjei A, Cutie AJ, inventors. Medicinal aerosol formulation. US patent US 6,261,539. 17 Jul 2001.
- 146.DeStefano G, Kelash-Cannova LJ, inventors; Boehringer Ingelheim Pharmaceutics, Inc., assignee. Formulation for metered dose inhaler using hydro-fluoro-alkanes as propellants. US patent US 7,914,770. 29 Mar 2011.
- 147.Neale PJ, Taylor AJ, inventors; Glaxo Group Limited, assignee. Medicaments for treating respiratory disorders. United States patent US 5,688,782. 1997 Nov 18.
- 148.Dellamary LA, Tarara TE, Smith DJ, Woelk CH, Adractas A, Costello ML, et al. Hollow porous particles in metered dose inhalers. Pharm Res. 2000;17(2):168–74. doi: 10.1023/a:1007513213292. [DOI] [PubMed] [Google Scholar]
- 149.Weers JG, Tarara TE, Malcolmson RJ, Leung D. Embedded crystals in low density particles: formulation, manufacture, and properties. Respir Drug Deliv. 2006;1:297–306. [Google Scholar]
- 150.Tarara TE, Hartman MS, Gill H, Kennedy AA, Weers JG. Characterization of suspension-based metered dose inhaler formulations composed of spray-dried budesonide microcrystals dispersed in HFA-134a. Pharm Res. 2004;21(9):1607–14. doi: 10.1023/b:pham.0000041455.13980.f1. [DOI] [PubMed] [Google Scholar]
- 151.Geller DE, Weers J, Heuerding S. Development of an inhaled dry-powder formulation of tobramycin using PulmoSphere™ technology. J Aerosol Med. 2011;24(4):175–82. doi: 10.1089/jamp.2010.0855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jinks PA. Preparation and utility of sub-micron lactose, a novel excipient for HFA MDI suspension formulations [abstract]. Drug Del to the Lungs, 14. 2003.
- 153.James J, Crean B, Davies M, Toon R, Jinks P, Roberts CJ. The surface characterisation and comparison of two potential sub-micron, sugar bulking excipients for use in low-dose, suspension formulations in metered dose inhalers. Int J Pharm. 2008;361(1):209–21. doi: 10.1016/j.ijpharm.2008.05.032. [DOI] [PubMed] [Google Scholar]
- 154.Adjei A, Cutie AJ, inventors; Aeropharm Technology Incorporated, assignee. Medicinal aerosol formulation. European patent EP 1,731,140. 13 Apr 2011.
- 155.Toneguzzo F, Vega JC, inventors; Laboratorio Pablo Cassara S.R.L., assignee. Stabilized metered dose inhaler. US patent US 104,488. 2 My 2013.
- 156.Jones R, Evans RM, Warren SJ, Taylor G. Development of a fluticasone propionate suspension pMDI formulation using a second particulate system. Respir Drug Deliv. 2004;2:401–4. [Google Scholar]
- 157.Jones R, Evans RM, Warren SJ, Taylor G. Development of a novel suspension MDI formulation using a low energy dispersion system. Respir Drug Deliv. 2002;2:799–802. [Google Scholar]
- 158.Young PM, Adi H, Patel T, Law K, Rogueda P, Traini D. The influence of micronised particulates on the aerosolisation properties of pressurised metered dose inhalers. J Aerosol Sci. 2009;40(4):324–37. [Google Scholar]
- 159.Johnson M. Inhaled corticosteroid—long-acting ß2-agonist synergism: therapeutic implications in human lung disease. Respir Drug Deliv. 2004;1:99–108. [Google Scholar]
- 160.Kaerger JS, Price R. Processing of spherical crystalline particles via a novel solution atomization and crystallization by sonication (SAXS) technique. Pharm Res. 2004;21(2):372–81. doi: 10.1023/b:pham.0000016252.97296.f1. [DOI] [PubMed] [Google Scholar]
- 161.Adi H, Young PM, Traini D. Co-deposition of a triple therapy drug formulation for the treatment of chronic obstructive pulmonary disease using solution-based pressurised metered dose inhalers. J Pharm Pharmacol. 2012;64(9):1245–53. doi: 10.1111/j.2042-7158.2011.01370.x. [DOI] [PubMed] [Google Scholar]
- 162.Rogueda PG, Price R, Smith T, Young PM, Traini D. Particle synergy and aerosol performance in non-aqueous liquid of two combinations metered dose inhalation formulations: an AFM and Raman investigation. J Colloid Interface Sci. 2011;361(2):649–55. doi: 10.1016/j.jcis.2011.05.073. [DOI] [PubMed] [Google Scholar]
- 163.Vehring R, Lechuga-Ballesteros D, Joshi V, Noga B, Dwivedi SK. Cosuspensions of microcrystals and engineered microparticles for uniform and efficient delivery of respiratory therapeutics from pressurized metered dose inhalers. Langmuir. 2012;28(42):15015–23. doi: 10.1021/la302281n. [DOI] [PubMed] [Google Scholar]
- 164.Noga B, Cummings H, Joshi V, Lechuga-Ballesteros D, Schultz RD, Speck JH, et al. Product performance, stability and dose proportionality of glycopyrrolate metered dose inhaler with sub-microgram doses using Pearl cosuspension technology. Respir Drug Deliv. 2012;2:645–8. [Google Scholar]