

Abstract
Subunit/split influenza vaccines are less reactogenic compared with the whole virus vaccines. However, their immunogenicity is relatively low and thus required proper adjuvant and/or delivery vehicle for immunogenicity enhancement. Influenza vaccines administered intramuscularly induce minimum, if any, mucosal immunity at the respiratory mucosa which is the prime site of the infection. In this study, chitosan (CS) nanoparticles were prepared by ionic cross-linking of the CS with sodium tripolyphosphate (TPP) at the CS/TPP ratio of 1:0.6 using 2 h mixing time. The CS/TPP nanoparticles were used as delivery vehicle of an intranasal influenza vaccine made of hemagglutinin (HA)-split influenza virus product. Innocuousness, immunogenicity, and protective efficacy of the CS/TPP-HA vaccine were tested in influenza mouse model in comparison with the antigen alone vaccine. The CS/TPP-HA nanoparticles had required characteristics including nano-sizes, positive charges, and high antigen encapsulation efficiency. Mice that received two doses of the CS/TPP-HA vaccine intranasally showed no adverse symptoms indicating the vaccine innocuousness. The animals developed higher systemic and mucosal antibody responses than vaccine made of the HA-split influenza virus alone. The CS/TPP-HA vaccine could induce also a cell-mediated immune response shown as high numbers of IFN-γ-secreting cells in spleens while the HA vaccine alone could not. Besides, the CS nanoparticle encapsulated HA-split vaccine reduced markedly the influenza morbidity and also conferred 100% protective rate to the vaccinated mice against lethal influenza virus challenge. Overall results indicated that the CS nanoparticles invented in this study is an effective and safe delivery vehicle/adjuvant for the influenza vaccine.
KEY WORDS: chitosan nanoparticles, delivery systems, influenza virus, intranasal, split influenza vaccine
INTRODUCTION
Influenza is a highly contagious devastating respiratory disease which causes public health and socio-economic problems worldwide (1). Currently available influenza vaccines are made of either inactivated whole viruses or the virus subunit/split products (2). The former vaccine is highly immunogenic but often causes adverse reactions particularly in infants and children (3,4). Therefore many countries prefer the subunit/split vaccines for their people (5,6). However, the less reactogenic vaccines confer limited immunogenicity and require either high antigenic dose or, in the case of naive subjects, a booster dose (7,8). Co-administration of an immunological adjuvant with the vaccine components improved immunogenicity leading to antigen sparing and adequate protective immune response after a single dose (9,10). Immunogenicity of the vaccine was enhanced better by emulsion adjuvants, i.e., AS03 (oil-in-water preparation) or MF59 (a squalene-based oil-in-water preparation), than alum due probably to recruitment of more inflammatory cells, lymphocytes, and antigen-presenting cells (dendritic cells and macrophages) (11). Nevertheless, the emulsion adjuvants also increase adverse reactions, both local and systemic (12,13).
Influenza vaccines are administered mostly by intramuscular route which has some limitations. Systemic immune response is induced but low, if there would be any, the respiratory mucosal response. Usually muscles have little antigen-presenting cells including dendritic cells and macrophages. The skeletal muscles do not express MHC molecules under normal conditions. Besides, human muscle cells express B7-H3, a B7 homolog, which functions as a co-inhibitory molecule of T cells for local immune regulatory processes (14). Intranasal route for influenza vaccine administration, on the contrary, offers several advantages. Besides this route that is needle- and painless, evidences have indicated also that the antigen activated immune cells at the nasal-associated lymphoid tissue commonly home at the respiratory immune effector sites including larynx- and bronchus-associated lymphoid tissues (15) and exert their defensive activities at the prime sites of influenza virus infection. Moreover, the vaccine components applied intranasally may reach the macrophages which roam about in the trachea and lungs. These cells may engulf the vaccine, become mature during their drainage to local lymph nodes (acquire ability to express co-signaling and MHC molecules and secrete cytokines), and present antigen to the T cells for induction of local immune response (16). The vaccine antigen applied intranasally may also reach the blood circulation and the systemic immune response is stimulated in spleen and peripheral lymph nodes (17). Thus, both mucosal and systemic immune responses can be expected after the intranasal immunization of appropriate vaccine formulation that could overcome the normal mucosal tolerance and breach the nasal epithelium tight junction (18).
In recent decades, chitosan (CS), which is a nontoxic, bio-adhesive, -degradable, and -compatible material, has been used widely as a carrier for peptide, protein, and DNA-based vaccines (19–21). CS is an attractive intranasal vaccine delivery vehicle. Its mucoadhesive property could overcome the mucociliary clearance, thus increasing the resident time of the vaccine component in the nasal passage. CS promotes paracellular transportation of the cargo antigen through the nasal mucosa (22). It served as an immunopotentiating agent to augment vaccine immunogenicity and effectiveness (23). Besides, annotated data have demonstrated that the CS nanoparticles induced both mucosal and systemic immune responses to the entrapped antigen after intranasal administration (24,25). In this study, a split influenza vaccine containing hemagglutinin of H1N1 virus formulated with CS nanoparticles was administered intranasally. The vaccine innocuousness, immunogenicity, and protective efficacy in a mouse model of influenza are reported herein.
MATERIALS AND METHODS
Materials
Low viscous CS and sodium tripolyphosphate (TPP) were from Sigma-Aldrich, MO, USA. Sodium acetate trihydrate and acetic acid were from Nacalai Tesque, Kyoto, Japan. All other chemicals were reagent grade. Purified rat monoclonal anti-mouse IFN-γ, rat anti-mouse IFN-γ-biotin conjugate, streptavidin-horseradish peroxidase (HRP) conjugate, and BD OptEIATM assay diluent were from Becton-Dickinson (BD) Biosciences, CA, USA. HRP-labeled goat anti-mouse IgG and IgA were from Southern Biotech, AL, USA. TMB-E and TMB-H substrates were from Moss, MD, USA.
Animals, Hemagglutinin-Split Influenza Vaccine, and Virus
All animal experiments were approved by the Ethical Committee of the National Institute of Biomedical Innovation (NIBIO), Osaka, Japan. Animal manipulation was performed according to the NIBIO Guidelines for Care and Use of Laboratory Animals. Female BALB/c mice, 5–7 weeks old, were from Japan SLC, Hamamatsu, Japan. They were maintained under specific pathogen-free conditions in an animal room at the NIBIO.
Influenza vaccine [split product of inactivated virions of A/Brisbane/59/2007(H1N1) strain] which contained mainly hemagglutinin (HA) was purified, inactivated, and disrupted by Kanonji Institute, Research Foundation for Microbial Diseases of Osaka University, Japan (26).
Mouse-adapted A/Brisbane/59/2007(H1N1) (26) was used for animal challenge when testing the vaccine protective efficacy. The virus was propagated in Mardin–Darby canine kidney cells grown in complete Eagle’s Minimum Essential Medium containing heat-inactivated 10% fetal bovine serum (FBS) (Nichirei Biosciences, Tokyo, Japan), 2 mM l-glutamine, 1.5 mg/ml sodium bicarbonate, and 2 μg/ml gentamycin. The viruses harvested from the culture supernatant were inactivated by ultraviolet irradiation in biosafety level-3 (BSL-3) facility at the NIBIO.
Preparation of CS/TPP Nanoparticles
The CS/TPP nanoparticles were prepared as described previously (27) with modifications. A 0.5% (w/v) CS stock solution was prepared by dissolving the CS powder in 1% v/v aqueous acetic acid under magnetic stirring with gentle heating until a transparent solution was obtained. The preparation was adjusted to pH 5.4 and filtered through a 0.45-μm membrane. Working CS solutions of different concentrations, i.e., 0.05, 0.1, 0.15, 0.2, 0.25, and 0.3%, were prepared by diluting the stock solution with 25 mM sodium acetate buffer, pH 5.4. TPP working solutions (0.05, 0.1, 0.2, and 0.3%) were prepared in ultrapure distilled water. Plain CS/TPP nanoparticles were obtained by ionic cross-linking of positively charged nitrogen groups in CS and negatively charged phosphate groups in TPP. Preliminary screenings for the optimal concentrations of CS and TPP in forming colloidal solution(s) were done by mixing various working concentrations of CS (5 ml) and solutions of TPP (0.5, 1, 1.5, 2, 2.5, and 3 ml). Thereafter, a selected CS concentration was mixed with varying amounts of TPP to yield different CS/TPP mass ratios, i.e., 1:0.2 to 1:5.0 (w/w), for selection of the CS/TPP ratio that formed the desired CS/TPP nanoparticles. The mixing time was varied from 1 to 5 h to investigate the effect on the nanoparticle sizes.
Encapsulation of HA-Split Influenza Virus Product into CS/TPP Nanoparticles
Encapsulation of the HA-split influenza product into CS/TPP nanoparticles was performed by mixing 0.7 ml containing 1 μg of HA-split virus component in acetate buffer, pH 5.4, with 3.3 ml of 0.05% w/v CS solution; then 1 ml of 0.1% TPP solution was admixed. The preparation was kept stirring at 25°C for 2 h. The CS/TPP-HA nanoparticles were harvested by centrifugation at 14,000×g for 20 min; the pellet was re-suspended in 0.5 ml sterile PBS.
Characterization of CS-Encapsulated HA-Split Virus Nanoparticles
Morphology of CS-encapsulated HA-split virus nanoparticles (CS/TPP-HA) was examined using transmission electron microscopy (TEM) (HT7700; Hitachi, Tokyo, Japan). The sizes and surface charges were determined by using the automated measurement program of Zetasizer-nano series instrument (Malvern, Worcestershire, UK). In order to estimate percent antigen encapsulation efficiency (% EE), the CS/TPP-HA preparation was centrifuged at 14,000×g for 20 min. The % EE was calculated: [(total amount of HA-split virus added − amount of HA-split virus in the supernatant)/amount of HA-split virus in supernatant] × 100. Measurement was performed in triplicate.
Vaccine Immunogenicity
The CS/TPP-HA were centrifuged and the pellet was resuspended in 500 μl of PBS (1 μl contained 0.05 μg of HA-split virus product). Mice were divided into four groups of five mice each. Group 1 mice were immunized intranasally (i.n.) twice at a 3-week interval with 20 μl of CS/TPP-HA (contained 1 μg of the antigen). Mice of groups 2–4 (controls) received individually two doses at a 3-week interval of 20 μl of HA-split virus alone (1 μg), plain CS/TPP nanoparticles, and PBS, respectively. Two weeks after the booster, all mice were bled and antigen-specific IgG antibodies in their sera were determined by indirect enzyme-linked immunosorbent assay (ELISA). After bleeding, bronchoalveolar lavage fluid (BALF) was harvested from each mouse by flushing 1 ml of PBS containing gentamycin via a Surflo® Teflon I.V. catheter (Terumo, Tokyo, Japan) inserted through a hole made in proximal trachea into the lower respiratory tract; then the fluid was drawn back through the catheter. Another catheter was used to flush 1 ml of PBS upward from the tracheal hole through nasal passage in order to collect the nasal wash fluid (NW). The BALF and NW were centrifuged to remove tissue or cell debris. The supernatants were collected separately and concentrated 4× by using 30K membrane Amicon® Ultrafiltration before use in antigen-specific IgA antibody determination by indirect ELISA. Spleen was aseptically excised from the mouse and single cells were prepared in RPMI-1640 medium supplemented with 5% FBS for enumeration of the antigen-specific IFN-γ-secreting cells by ELISPOT assay.
Indirect ELISA
Indirect ELISA (28) was used for determining antigen-specific serum IgG and IgA in mouse sera and BALF and NW, respectively. Briefly, MicrotestTM 96-well ELISA plates (BD Biosciences) were coated with 50 μl of 1 μg/ml HA-split vaccine and kept overnight at 4°C. After washing with 0.1% PBS-T, all wells were blocked with BD OptEIATM assay diluent. Serial twofold dilutions of sera (started from 1:128), BALF, and NW (started from 1:2) from all mouse groups were added appropriately to the antigen-coated wells, and the plates were kept at 25°C for 2 h. For serum IgG detection, goat anti-mouse IgG-HRP conjugate (diluted 1:4,000) were added to the wells. Goat anti-mouse IgA-HRP conjugate (diluted 1:8,000) was used for specific IgA detection in BALF and NW. The plates were incubated at 25°C for 1 h, washed, and TMB-E substrate was used for color development. The enzymatic reaction was stopped by adding 25 μl of 1 N HCl. OD450nm of the content in each well was determined against blank (wells to which PBS was added instead of the mouse sample). The specific antibody titer was the highest dilution of the sample that the OD450nm was ≥0.05.
ELISPOT Assay
The number of IFN-γ-secreting splenocytes was determined by using IFN-γ ELISPOT procedure (29). Briefly, Multiscreen® HTS HA 96-well filtration plates (Millipore, Ireland) were coated with 100 μl of 5 μg/ml purified rat monoclonal anti-mouse IFN-γ antibody and incubated at 4°C overnight. The plates were washed and each well was blocked with 300 μl of 5% FBS in RPMI-1640 medium at 37°C for 3 h. Single spleen cells of the vaccinated/control mice were added to appropriate wells (106 cells/well) followed by UV-inactivated A/Brisbane/59/2007(H1N1) (106 pfu/well; optimal amount from titration) in 5% complete RPMI-1640 medium, and the plates were incubated at 37°C in 5% CO2 incubator for 48 h. The content in each well was discarded and the wells were washed before adding 1 μg/ml of rat anti-mouse IFN-γ-biotin conjugate (100 μl/well) and incubated at 25°C for 2 h. Streptavidin–HRP conjugate (100 μl of 1:800 dilution) and TMB-H substrate were used for spot revelation. The enzymatic reaction was stopped by rinsing the wells with distilled water. Automatic KS-ELISPOT reader (Carl Zeiss, Oberkochen, Germany) was used for spot enumeration. Data were expressed as the number of spots per 106 splenocytes. Three independent experiments were performed.
Vaccine Protective Efficacy
Two sets of four mouse groups (five mice per group) were prepared. Groups 1 of both sets were immunized intranasally with the CS/TPP-HA for the vaccine immunogenicity study while groups 2–4 of each set served as respective controls. Two weeks after the booster dose, mice of set 1 were challenged intranasally with 5 LD50 of the mouse-adapted A/Brisbane/59/2007(H1N1) and set 2 animals received individually 20 LD50 of the virus intranasally. The animals were observed daily for morbidity (body weight loss) and mortality. Experiments were terminated at day 14 post-challenge.
Statistical Analysis
Data were analyzed using GraphPad Prism 5 (San Diego, CA, USA). Differences among mouse groups were compared by unpaired t test. A Mann–Whitney t test was used when the normality was not obtained. The p value of <0.05 was statistically different.
RESULTS
Plain CS/TPP Nanoparticles
Optimum proportion of CS and TPP in forming ionic cross-linked nanoparticles was screened randomly from a total of 144 CS/TPP formulations including six CS working solutions (0.05–0.3%) and 6 vol (0.5–3 ml) of four concentrations of TPP working solutions (0.05–0.3%). The CS/TPP colloidal solutions (with turbidity) were obtained when 0.05% w/v CS was used (final concentration 0.333 mg/ml) to mix with variable volumes of various TPP solutions (data not shown). When the CS final concentration was deviated from 0.333 mg/ml, either a clear solution, suspension, or precipitation was obtained.
The formulations containing CS at 0.333 mg/ml that yielded colloidal solutions were investigated further to find out the optimal TPP concentration for CS/TPP nanoparticle formation which was performed by setting up CS/TPP ratios (w/w) from 1:0.2 to 1:5.0. For this experiment, 3.3 ml of 0.05% w/v CS (final concentration at 0.333 mg/ml) were mixed with 1 ml of various TPP concentrations (0.007–0.167% w/v equivalent to final concentrations at 0.067–1.665 mg/ml, respectively). The final volume of the preparations was adjusted with acetate buffer, pH 5.4, to 5 ml. It was found that only CS/TPP ratio of 1:0.6 (Table I, formulation C) provided colloidal nanoparticles while other CS/TPP ratios formed microparticles (formulations D–H), aggregates (formulations I–M) or clear solution (no particles; formulations A and B) (Table I). The CS/TPP ratio 1:0.6 colloid was then prepared by varying the CS/TPP mixing times for 2, 3, 4, and 5 h. The results indicated that the particle sizes increased in a time-dependent manner, i.e., the longer the mixing period, the larger the particle sizes (Table II). Taken together, the optimal condition for producing the CS/TPP nanoparticles was 0.333 mg/ml CS solution, 0.200 mg/ml TPP solution (CS/TPP mass ratio was 1:0.6), and 2 h mixing time. The C1 formulation (Table II) which produced the nanoparticles of 302.88 ± 6.20 nm was used as the HA-split influenza vaccine delivery vehicle.
Table I.
Systematic Patterns, Particle Sizes, and Polydispersity Index of Plain CS/TPP Nanoparticles Prepared by Using Different CS/TPP Ratios
| Formulation | CS/TPP ratioa | Systematic patternb | Size in nm (mean ± SD) | Polydispersity index (mean ± SD) |
|---|---|---|---|---|
| A | 1:0.2 | Clear solution | n/d | n/d |
| B | 1:0.4 | Clear solution | n/d | n/d |
| C | 1:0.6 | Colloidal solution | 151.67 ± 2.89 | 0.20 ± 0.003 |
| D | 1:0.8 | Colloidal solution | 1793.33 ± 136.14 | 0.33 ± 0.07 |
| E | 1:1.0 | Suspension | 1.62E + 04 ± n/d | 0.72 ± n/d |
| F | 1:1.5 | Suspension | 5.57E + 04 ± n/d | 1.000 ± n/d |
| G | 1:2.0 | Suspension | 3.85E + 04 ± n/d | 0.168 ± n/d |
| H | 1:2.5 | Suspension | 2.31E + 04 ± n/d | 0.103 ± n/d |
| I | 1:3.0 | Precipitate | n/d | n/d |
| J | 1:3.5 | Precipitate | n/d | n/d |
| K | 1:4.0 | Precipitate | n/d | n/d |
| L | 1:4.5 | Precipitate | n/d | n/d |
| M | 1:5.0 | Precipitate | n/d | n/d |
n/d not determined
aChitosan concentration was fixed at 0.333 mg/ml while TPP concentration was varied from 0.067 to 1.665 mg/ml
bMixing time was 1 h
Table II.
Effects of Mixing Time on Particle Sizes and Polydispersity Index
| Formulation | Mixing time (h) | Size in nm (mean ± SD) | Polydispersity index (mean ± SD) |
|---|---|---|---|
| C1 | 2 | 302.88 ± 6.20 | 0.20 ± 0.02 |
| C2 | 3 | 824.67 ± 24.58 | 0.47 ± 0.08 |
| C3 | 4 | 1066.67 ± 11.55 | 0.52 ± 0.05 |
| C4 | 5 | 1490.00 ± 91.65 | 0.44 ± 0.02 |
Nanoparticles were prepared using formulation C (ratio of CS/TPP was 1:0.6)
Characteristics of CS/TPP-Encapsulated HA-Split Vaccine Nanoparticles
The CS/TPP-HA nanoparticles revealed a spherical shape by TEM (Fig. 1). The physicochemical characteristics of the CS/TPP-HA vaccine are shown in Table III. The zeta potential of the CS-formulated HA-split vaccine nanoparticles was cathodic (ranged from 21.93 to 21.97 mV). The sizes of the vaccine particles (351.00 ± 2.00 nm in the prime preparation and 358.67 ± 5.13 nm in the booster preparation) were larger than those of the respective plain CS/TPP preparations (315.67 ± 5.13 and 300.50 ± 8.50 nm, respectively) (p < 0.05). Both CS/TPP-HA and plain CS/TPP revealed rather uniformed nano-sizes. The CS/TPP-HA had 78.13–78.54% encapsulation efficiencies (% EE).
Fig. 1.
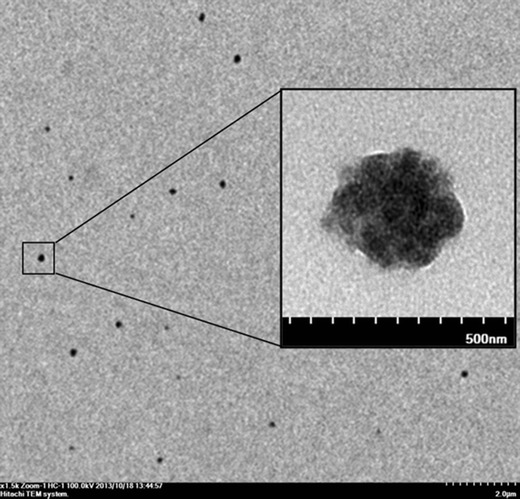
Transmission electron micrograph of CS/TPP-HA nanoparticles produced at CS/TPP ratio of 1:0.6
Table III.
The Physicochemical Characteristics of CS/TPP-HA Vaccine
| Physicochemical characteristics | Preparations for immunization | |||
|---|---|---|---|---|
| CS/TPP-HAa | CS/TPPb | |||
| Prime dose | Boost dose | Prime dose | Boost dose | |
| Size in nm (mean ± SD) | 351.00 ± 2.00 | 358.67 ± 5.13 | 315.67 ± 5.13 | 300.50 ± 8.50 |
| Polydispersity index (mean ± SD) | 0.28 ± 0.04 | 0.28 ± 0.02 | 0.24 ± 0.02 | 0.23 ± 0.02 |
| Zeta potential in mV (mean ± SD) | +21.97 ± 0.45 | +21.93 ± 0.49 | +22.97 ± 1.19 | +22.05 ± 0.85 |
| Encapsulation efficiency ± SD (%) | 78.13 ± 9.39 | 78.54 ± 3.83 | n/d | n/d |
a CS/TPP-HA chitosan/TPP-encapsulated split-HA vaccine
b CS/TPP chitosan/TPP without split-HA vaccine used as mock control; n/d encapsulation efficiency was not determined because no HA vaccine was incorporated in nanoparticles
Immune Responses Induced by CS/TPP-HA Vaccine
Mice immunized intranasally with two doses of the CS-encapsulated HA-split virus vaccine and vaccine controls did not show any sign and symptoms of adverse effects. The CS-encapsulated HA-split virus vaccinated mice had both systemic and mucosal immune responses. The levels of HA-specific serum IgG and secretory IgA in BALF and NW of mice vaccinated with CS/TPP-HA vaccine, HA alone, plain CS/TPP, and PBS are shown in Fig 2. The HA-specific IgG antibody titers in sera of mice immunized with CS/TPP-HA vaccine were significantly higher than those stimulated with HA-split vaccine alone, plain CS/TPP, and PBS (Fig. 2a) (p < 0.05). The HA-specific IgA levels in BALF (Fig. 2b) as well as NW (Fig. 2c) of mice vaccinated with CS/TPP-HA were also higher than the control groups.
Fig. 2.
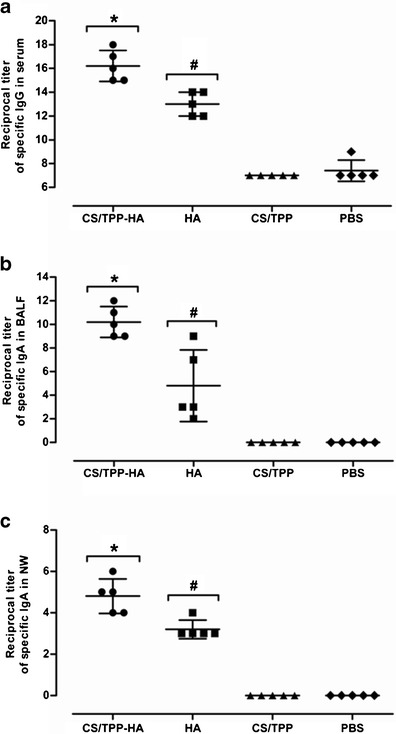
Indirect ELISA antibody titers in sera, BALF, and NW of vaccinated and control mice. Reciprocal titers of HA-specific serum IgG (a), bronchoalveolar lavage fluids (BALF) specific IgA (b), and nasal wash (NW) specific IgA (c). Mean and SD of the reciprocal titers of each treatment group are indicated. *Different significantly from HA, CS/TPP, and PBS at p <0.05; #different significantly from CS/TPP and PBS at p <0.05
The intranasally administered CS/TPP-HA-split influenza vaccine could induce also specific cell-mediated immune response. Significant numbers of IFN-γ-secreting cells were found in spleens of CS/TPP-HA vaccinated mice and were absent in the control mice including HA-split virus alone, CS/TPP, and PBS stimulated groups (Fig. 3).
Fig. 3.
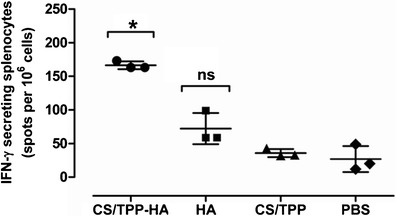
Numbers of influenza-specific IFN-γ-secreting splenocytes measured by ELISPOT assay. Data are the average number of spots in well containing 106 influenza virus stimulated spleen cells from three independent experiments. Bars indicate mean and SD of the numbers of IFN-γ secreting cells. *Different significantly from HA, CS/TPP, and PBS at p < 0.01, p < 0.0001, and p < 0.0001, respectively. ns no significant difference from CS/TPP and PBS
Protective Efficacy of the Vaccines
After being challenged intranasally with 5 LD50 of the mouse adapted A/Brisbane/59/2007(H1N1) virus, mice that received CS/TPP-HA, HA-split vaccine alone, CS/TPP, and PBS had 100, 60, 0, and 0% survival, respectively (Fig. 4a). When the challenge dose was increased to 20 LD50, all mice vaccinated with the CS/TPP-HA survived until the end of the experiments (day 14) while mice of the other groups died within day 9 post-infection (Fig. 4c). Figure 4b shows body weights of all mouse groups after receiving the 5 LD50 virus challenge. All mice of the group that received CS/TPP-HA-split vaccine and 60% of the HA-split vaccine group lose their body weights during the first 4 and 7 days after challenge, respectively; their body weights were regained thereafter. Mice of the CS/TPP and PBS groups had steady body weight loss until death within day 6 post-infection. Morbidity of the mice that received the 20 LD50 challenge is shown in Fig. 4d. CS/TPP-HA vaccinated mice had their weight loss during the first 4 days and then regained while all mice of the other groups continued to loss their weights, became moribund, and died.
Fig. 4.
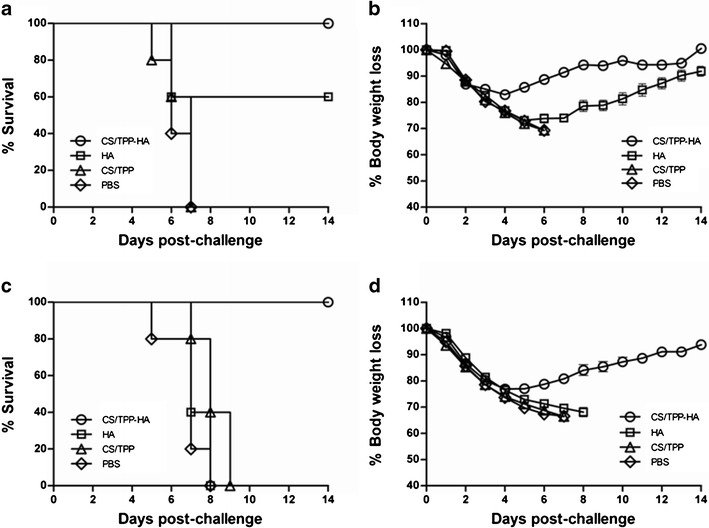
Protective efficacies of the vaccines shown as percent survival and body weight loss of vaccinated and control mice after infecting intranasally with 5 LD50 (a and b, respectively) or 20 LD50 (c and d, respectively) of mouse adapted A/Brisbane/58/2007(H1N1)
DISCUSSION
Vaccination is an effective measure for prevention of influenza (30). The first prerequisite step of the influenza virus infection occurs at the respiratory epithelium. Thus, immunological factor that operates at the infection prime site is highly important for prevention of the virus replication in the epithelial cells and further systemic spread. Intramuscular vaccines rarely induce the local immune response. Also, the antibody induced systemically by the parenteral vaccine usually do not reach the mucosal surface except by passive transudation which would occur only when the circulating IgG level is high and sustained or in the situation of pathotopic potentiation. Vaccine administration by intranasal route, on the other hand, stimulates both systemic and mucosal immune responses, particularly specific secretory IgA which are mucophilic and protease resistant, and therefore functions well as the first-line defense at the mucosa (31,32). However, immunogenicity acquired from the mucosal vaccine per se may be inadequate and thus effective adjuvant and/or vaccine delivery vehicle is required to improve the magnitude of the immune response (28,33,34). Among various developing adjuvant/delivery systems, CS is favorable for intranasal vaccine due to its mucoadhesive property and ability to overcome the formidable nasal epithelial barrier (23,35). Several studies have demonstrated success in using CS nanoparticles as delivery systems for intranasal protein and DNA vaccines (36–38).
In this study, CS nanoparticles were used to encapsulate HA-split influenza virus vaccine. The ionic cross-linking technique is a shear force-, organic solvent-, and heat-free procedure; thus, it is suitable for vulnerable molecules such as the vaccine antigen of this study (39). There are many factors to be considered in formulating suitable CS nanoparticles for the vaccine. These include concentrations of the CS and the TPP, the CS/TPP mass ratio, and the reagent mixing time. Therefore, the preliminary experiments were performed to screen and select the best CS and TPP combination in formation of the CS/TPP colloid. The ionic cross-linking between the CS cationic amine groups and the polyanion of TPP phosphate groups was used for preparing the CS/TPP nanoparticles. Data in the literature have demonstrated that spontaneous particle formation in the ionic cross-linkage occurred only at suitable concentrations of CS and TPP (40). In this study, the nanoparticulate system of CS/TPP with good physicochemical properties, i.e., nanometer range in size, positive charge, and relatively high encapsulation efficiency, was achieved at CS/TPP ratio of 1:0.6 by using 2 h mixing time However, physicochemical properties of CS nanoparticles product formed by ionic-cross-linking are affected by chemical properties of CS (molecular weight, degree of deacetylation, and chemical modification) and condition of preparation (pH, volume, and mixing time) (41–43). Therefore, the characteristic of the CS/TPP nanoparticles produced in one study may not be similar to that of another (38,44).
Incorporation of the HA-split influenza virus into CS/TPP nanoparticles was done at pH 5.4. At this acidic pH, the amine groups of CS were positively charged because each deacetylated amine group contained in the CS had a pKa value of about 6.5 (45). In addition, HA-split virus product had pI 4.8; thus, at pH 5.4 it was negatively charged. Therefore, the antigen could be entrapped into the CS/TPP nanoparticles with relatively high percentage of encapsulation efficiency. According to the TEM image, the HA-split virus component might be condensed by the CS polymer chains along with the TPP to form the encapsulated nanoparticles. The CS/TPP-HA might be linked together through two different bonds. One possible association mechanism of the vaccine component with the nanoparticles besides the entrapment of the HA-split influenza virus product in the CS/TPP nanoparticles was the electrostatic interaction between the cathodic CS and the anodic vaccine component (46).
The bioadhesive cationic polymeric CS was used to prolong residence time in the nasal passage and promote absorption of CS vaccine via nasal mucosa (47,48). The established CS/TPP-HA nanoparticles in this study possessed positive surface charge (21.93–21.97 mV) indicating a suitable property for intranasal vaccine delivery (49). The positively charged CS/TPP-HA should bind with the negatively charged mucus glycoproteins (50) and also dendrites of some dendritic cells (DCs) that protruded through the paracellular junction of the epithelial cells (51). The intimate contact between the vaccine formulation and the mucus/mucosa should increase retention time of CS/TPP-HA vaccine and increased absorption opportunity through the nasal mucosa (35). The binding of the nanoparticles to dendritic cells should allow antigen delivery into the cytoplasm for antigen processing via the cytosolic pathway (MHC class I) and followed by Th1/cell-mediated immune response stimulation. The phagocytosed component can be processed and the peptides are presented via the MHC class II pathways which the humoral immune response should be induced (52). Moreover, cross-antigen presentation could be expected also (53).
Sizes are known to influence the mucosal uptake of the particulate delivery system (54). However, the optimal size for nasal vaccine delivery vehicles is still controversial. Many studies revealed that stronger and more robust immune responses were induced by submicron sizes of CS nanoparticles because the particles of this size range are readily phagocytosed by the antigen-presenting cells (55–57). Data from mucus adsorption studies revealed that mucoadhesive particles, which are fine nanoparticles (230–320 nm), increased in penetration through the mucus layer. On the other hand, larger particles (2 μm) were adsorbed on the surface of mucosa (Langmuir-type adsorption) (58). CS ionic cross-linking-HA nanoparticles of this study had appropriate sizes of 351.00–358.67 nm as measured by using the Zetasizer. The particles when applied intranasally should be engulfed by the microfold (M) cell overlying the nasal associated lymphoid tissue and the intact antigen should be delivered appropriately to the follicular dendritic cells for local immune response stimulation. The antigen may be carried by the DCs to the local draining lymph nodes or distant lymphoid tissues where immune responses can be incited therein (59,60). The ability of the CS/TPP-HA in inducing systemic and mucosal immune responses has been demonstrated in this study by the high levels of HA-specific serum IgG antibodies and IFN-γ-secreting cells in spleen and specific IgA antibodies in BALF and NW, respectively.
Not only the immunogenicity of the CS/TPP-HA split vaccine over the HA-split vaccine alone was demonstrated but also the former vaccine formulation could provide higher protective efficacies than the latter against both low and high lethal challenges (5 and 20 LD50, respectively) with highly virulent influenza virus. All mice vaccinated intranasally with the CS nanoparticle-entrapped HA-split vaccine survived the lethal infections while only partial (60%) and no protection (0%) were observed for mice that received HA-split vaccine alone against the low and the high doses, respectively. Overall results indicated that CS nanoparticle preparation produced in this study is a suitable delivery vehicle/adjuvant that not only enhanced the influenza vaccine immunogenicity but also increased the encapsulated vaccine protective efficacy. Unfortunately, experiments comparing the adjuvanticity of the CS nanoparticles and other approved human vaccine adjuvants such as alum and water-in-oil/oil-in-water/water-in-oil-in water emulsions have not been done for this HA-split influenza vaccine.
CONCLUSIONS
A suitable condition for production of CS/TPP nanoparticles by means of ionic cross-linking was studied. The nanoparticles were used as a successful delivery vehicle of HA-split influenza virus vaccine for intranasal immunization. Mice that received the CS/TPP nanoparticle-encapsulated HA-split influenza vaccine intranasally developed higher systemic and mucosal antibody responses than the vaccine made of the HA-split influenza virus alone. The nanoparticle-encapsulated vaccine could induce also a cell-mediated immune response shown as high numbers of IFN-γ-secreting cells in spleens while the HA vaccine alone could not do so. Besides, the CS nanoparticle-encapsulated HA-split vaccine reduced markedly the influenza morbidity and conferred 100% protective rate to the vaccinated mice against lethal influenza virus challenges. Taken together, CS nanoparticles invented in this study is an effective and safe delivery vehicle/adjuvant for the monovalent HA-split influenza vaccine tested. They should be investigated further for multivalent HA-split vaccine or universal influenza vaccine made of other influenza virus conserved proteins such as matrix protein-1 (M1), ion channel protein (M2), nucleoprotein (NP), and multifunctional non-structural protein-1 (NS1).
Acknowledgments
This work was supported by a grant from the Thailand Research Fund (TRF) through the Royal Golden Jubilee (RGJ) Ph.D. Program (PHD/0004/2550). We are extremely grateful to the staff of the NIBIO, especially Pranee Somboonthum, Sumiko Matsuoka, Ahmed M. Haredy, Megumi Ota, and Eiko Moriishi, for their technical assistance and advice. We thank Urai Chaisri for help with TEM. We also thank Prof. Wanpen Chaicumpa of the Department of Parasitology, Faculty of Medicine Siriraj Hospital, Mahidol University for proofreading and correcting this manuscript.
Declaration of Interest
The authors report no declaration of interest.
References
- 1.Gasparini R, Amicizia D, Lai PL, Panatto D. Clinical and socioeconomic impact of seasonal and pandemic influenza in adults and the elderly. Hum Vaccin Immunother. 2012;8:21–28. doi: 10.4161/hv.8.1.17622. [DOI] [PubMed] [Google Scholar]
- 2.Wong SS, Webby RJ. Traditional and new influenza vaccine. Clin Microbiol Rev. 2013;26:476–492. doi: 10.1128/CMR.00097-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gross PA, Ennis FA, Gaerlan PF, Denson LJ, Denning CR, Schiffman D. A controlled double-blind comparison of reactogenicity, immunogenicity, and protective efficacy of whole-virus and split-product influenza vaccines in children. J Infect Dis. 1977;136:623–632. doi: 10.1093/infdis/136.5.623. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein DI, Zahradnik JM, DeAngelis CJ, Cherry JD. Clinical reactions and serologic responses after vaccination with whole-virus or split-virus influenza vaccines in children aged 6 to 36 months. Pediatrics. 1982;69:404–408. [PubMed] [Google Scholar]
- 5.Kim YK, Eun BW, Kim NH, Kang EK, Lee BS, Kim DH, et al. Comparison of immunogenicity and reactogenicity of split versus subunit influenza vaccine in Korean children aged 6–35 months. Scand J Infect Dis. 2013;45:460–468. doi: 10.3109/00365548.2012.755267. [DOI] [PubMed] [Google Scholar]
- 6.Wright PF. The use of inactivated influenza vaccine in children. Semin Pediatr Infect Dis. 2006;17:200–205. doi: 10.1053/j.spid.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention (CDC) Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP)—United States, 2012–13 influenza season. Morb Mortal Wkly Rep. 2012;61:613–618. [PubMed] [Google Scholar]
- 8.Neuzil KM, Jackson LA, Nelson J, Kilmov A, Cox N, Bridges CB, et al. Immunogenicity and reactogenicity of 1 versus 2 doses of trivalent inactivated influenza vaccine in vaccine-naive 5–8-year-old children. J Infect Dis. 2006;194:1032–1039. doi: 10.1086/507309. [DOI] [PubMed] [Google Scholar]
- 9.Nicholson KG, Colegate AE, Podda A, Stephenson I, Wood J, Ypma E, et al. Safety and antigenicity of non-adjuvanted and MF59-adjuvanted influenza A/duck/Singapore/97 (H5N3) vaccine: a randomized trial of two potential vaccines against H5N1 influenza. Lancet. 2001;357:1937–1943. doi: 10.1016/S0140-6736(00)05066-2. [DOI] [PubMed] [Google Scholar]
- 10.Vesikari T, Pellegrini M, Karvonen A, Groth N, Borkowski A, O′Hagan DT, et al. Enhanced immunogenicity of seasonal influenza vaccines in young children using MF59 adjuvant. Pediatr Infect Dis J. 2009;28:563–571. doi: 10.1097/INF.0b013e31819d6394. [DOI] [PubMed] [Google Scholar]
- 11.Calabro S, Tortoli M, Baudner BC, Pacitto A, Cortese M, O′Hagan DT, et al. Vaccine adjuvants alum and MF59 induce rapid recruitment of neutrophils and monocytes that participate in antigen transport to draining lymph nodes. Vaccine. 2011;29:1812–1823. doi: 10.1016/j.vaccine.2010.12.090. [DOI] [PubMed] [Google Scholar]
- 12.Lippi G, Targher G, Franchinni M. Vaccination, squalene and anti-squalene antibodies: facts or fiction? Eur J Intern Med. 2010;21:70–73. doi: 10.1016/j.ejim.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Whitehouse M. Oily adjuvants and autoimmunity: now time for reconsideration? Lupus. 2012;21:217–222. doi: 10.1177/0961203311429818. [DOI] [PubMed] [Google Scholar]
- 14.Waschbisch A, Wintterle S, Lochmüller H, Walter MC, Wischhusen J, Kieseier BC. Human muscle cells express the costimulatory molecule B7-H3, which modulated muscle–immune interactions. Arthritis Rheum. 2008;58:3600–3608. doi: 10.1002/art.23997. [DOI] [PubMed] [Google Scholar]
- 15.Davis SS. Nasal vaccines. Adv Drug Deliv Rev. 2001;51:21–42. doi: 10.1016/S0169-409X(01)00162-4. [DOI] [PubMed] [Google Scholar]
- 16.Kiyono H, Fukuyama S. NALT- versus Peyer's-patch-mediated mucosal immunity. Nat Rev Immunol. 2004;4:699–710. doi: 10.1038/nri1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- 18.Svindland SC, Jul-Larsen Å, Pathirana R, Anderson S, Madhun A, Montomoli E, et al. The mucosal and systemic immune responses elicited by a chitosan-adjuvanted intranasal influenza H5N1 vaccine. Influenza Other Respi Viruses. 2011;6:90–100. doi: 10.1111/j.1750-2659.2011.00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amidi M, Romeijn SG, Verhoef JC, Junginger HE, Bungener L, Huckriede A, et al. N-Trimethyl chitosan (TMC) nanoparticles loaded with influenza subunit antigen for intranasal vaccination: biological properties and immunogenicity in a mouse model. Vaccine. 2007;25:144–153. doi: 10.1016/j.vaccine.2006.06.086. [DOI] [PubMed] [Google Scholar]
- 20.Chua BY, Al Kobaisi M, Zeng W, Mainwaring D, Jackson DC. Chitosan microparticles and nanoparticles as biocompatible delivery vehicles for peptide and protein-based immunocontraceptive vaccines. Mol Pharm. 2012;9:81–90. doi: 10.1021/mp200264m. [DOI] [PubMed] [Google Scholar]
- 21.Yang X, Yuan X, Cai D, Wang S, Zong L. Low molecular weight chitosan in DNA vaccine delivery via mucosa. Int J Pharm. 2009;375:123–132. doi: 10.1016/j.ijpharm.2009.03.032. [DOI] [PubMed] [Google Scholar]
- 22.van der Lubber IM, Verhoef JC, Borchard G, Junginger HE. Chitosan and its derivatives in mucosal drug and vaccine delivery. Eur J Pharm Sci. 2001;14:201–207. doi: 10.1016/S0928-0987(01)00172-5. [DOI] [PubMed] [Google Scholar]
- 23.Alpar HO, Somavarapu S, Atuah KN, Bramwell VW. Biodegradable mucoadhesive particulates for nasal and pulmonary antigen and DNA delivery. Adv Drug Deliv Rev. 2005;57:411–430. doi: 10.1016/j.addr.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 24.Sui Z, Chen Q, Fang F, Zheng M, Chen Z. Cross-protection against influenza virus infection by intranasal administration of M1-based vaccine with chitosan as an adjuvant. Vaccine. 2010;28:7690–7698. doi: 10.1016/j.vaccine.2010.09.019. [DOI] [PubMed] [Google Scholar]
- 25.Svindland SC, Pedersen GK, Pathirana RD, Bredholt G, Nøstbakken JK, Jul-Larsen A, et al. A study of chitosan and c-di-GMP as mucosal adjuvants for intranasal influenza H5N1 vaccine. Influenza Other Respi Viruses. 2012 doi: 10.1111/irv.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okamoto S, Matsuoka S, Takenaka N, Haredy AM, Tanimoto T, Gomi Y, et al. Intranasal immunization with a formalin-inactivated human influenza A virus whole-virion vaccine alone and intranasal immunization with split-virion vaccine with mucosal adjuvants show similar levels of cross-protection. Clin Vaccine Immunol. 2012;19:979–990. doi: 10.1128/CVI.00016-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calvo P, Remuñán-López C, Vila-Jato JL, Alonso MJ. Novel hydrophilic chitosan–polyethylene oxide nanoparticles as protein carriers. J Appl Polym Sci. 1997;63:125–132. doi: 10.1002/(SICI)1097-4628(19970103)63:1<125::AID-APP13>3.0.CO;2-4. [DOI] [Google Scholar]
- 28.Okamoto S, Matsuura M, Akagi T, Akashi M, Tanimoto T, Ishikawa T, et al. Poly(γ-glutamic acid) nano-particles combined with mucosal influenza virus hemagglutinin vaccine protects against influenza virus infection in mice. Vaccine. 2009;27:5896–5905. doi: 10.1016/j.vaccine.2009.07.037. [DOI] [PubMed] [Google Scholar]
- 29.Okamoto S, Yoshii H, Akagi T, Akashi M, Ishikawa T, Okuno Y, et al. Influenza hemagglutinin vaccine with poly(γ-glutamic acid) nanoparticles enhances the protection against influenza virus infection through both humoral and cell-mediated immunity. Vaccine. 2007;25:8270–8278. doi: 10.1016/j.vaccine.2007.09.051. [DOI] [PubMed] [Google Scholar]
- 30.Nichol KL. The efficacy, effectiveness and cost-effectiveness of inactivated influenza virus vaccine. Vaccine. 2003;21:1769–1775. doi: 10.1016/S0264-410X(03)00070-7. [DOI] [PubMed] [Google Scholar]
- 31.Horton RE, Vidarsson G. Antibodies and their receptors: different potential roles in mucosal defense. Front Immunol. 2013;4:200. doi: 10.3389/fimmu.2013.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodríquez A, Tjärnlund A, Ivanji J, Singh M, García I, Williams A, et al. Role of IgA in the defense against respiratory infections IgA deficient mice exhibited increased susceptibility to intranasal infection with Mycobacterium bovis BCG. Vaccine. 2005;23:2565–2572. doi: 10.1016/j.vaccine.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 33.Hagenaars N, Mastrobattista E, Verheul RJ, Mooren I, Glansbeek HL, Heldens JG, et al. Physicochemical and immunological characterization of N, N, N-trimethyl chitosan-coated whole inactivated influenza virus vaccine for intranasal administration. Pharm Res. 2009;26:1353–1364. doi: 10.1007/s11095-009-9845-y. [DOI] [PubMed] [Google Scholar]
- 34.Liu H, Patil HP, de Vries-Idema J, Wilschut J, Huckriede A. Enhancement of the immunogenicity and protective efficacy of a mucosal influenza subunit vaccine by the saponin adjuvant GPI-0100. PLoS One. 2012;7:e52135. doi: 10.1371/journal.pone.0052135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma S, Mukkur TK, Benson HA, Chen Y. Pharmaceutical aspects of intranasal delivery of vaccines using particulate systems. J Pharm Sci. 2009;98:812–843. doi: 10.1002/jps.21493. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Zhang W, Liu F, Zheng M, Zheng D, Zhang T, et al. Intranasal immunization with live attenuated influenza vaccine plus chitosan as an adjuvant protects mice against homologous and heterologous virus challenge. Arch Virol. 2012;157:1451–1461. doi: 10.1007/s00705-012-1318-7. [DOI] [PubMed] [Google Scholar]
- 37.Raghuwanshi D, Mishra V, Das D, Kaur K, Suresh MR. Dendritic cell targeted chitosan nanoparticles for nasal DNA immunization against SARS CoV nucleocapsid protein. Mol Pharm. 2012;9:946–956. doi: 10.1021/mp200553x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Figueiredo L, Cadete A, Gonçalves LM, Corvo ML, Almeida AJ. Intranasal immunization of mice against Streptococcus equi using positively charged nanoparticulate carrier systems. Vaccine. 2012;30:6551–6558. doi: 10.1016/j.vaccine.2012.08.050. [DOI] [PubMed] [Google Scholar]
- 39.Amidi M, Mastrobattista E, Jiskoot W, Hennick WE. Chitosan-based delivery systems for protein therapeutics and antigens. Adv Drug Deliv Rev. 2010;62:59–82. doi: 10.1016/j.addr.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Csaba N, Köping-Höggård M, Alonso MJ. Ionically cross-linked chitosan/tripolyphosphate nanoparticles for oligonucleotide and plasmid DNA delivery. Int J Pharm. 2009;382:205–214. doi: 10.1016/j.ijpharm.2009.07.028. [DOI] [PubMed] [Google Scholar]
- 41.Xu Y, Du Y. Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles. Int J Pharm. 2003;250:215–226. doi: 10.1016/S0378-5173(02)00548-3. [DOI] [PubMed] [Google Scholar]
- 42.Gan Q, Wang T, Cochrane C, McCarron P. Modulation of surface charge, particle size and morphological properties of chitosan–TPP nanoparticles intended for gene delivery. Colloids Surf B Biointerfaces. 2005;44:65–73. doi: 10.1016/j.colsurfb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Akbuğa J, Bergisadi N. Effect of formulation variables on cis-platin loaded chitosan microsphere properties. J Microencapsul. 1999;16:697–703. doi: 10.1080/026520499288645. [DOI] [PubMed] [Google Scholar]
- 44.Garcia-Fuentes M, Alonso MJ. Chitosan-based drug nanocarriers: where do we stand? J Control Release. 2012;161:496–504. doi: 10.1016/j.jconrel.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 45.Mao S, Sun W, Kissel T. Chitosan-based formulations for delivery of DNA and siRNA. Adv Drug Deliv Rev. 2010;62:12–27. doi: 10.1016/j.addr.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 46.Prego C, García M, Torres D, Alonso MJ. Transmucosal macromolecule drug delivery. J Control Release. 2005;101:151–162. doi: 10.1016/j.jconrel.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 47.Vila A, Sánchez A, Janes K, Behrens I, Kissel T, Vila Jato JL, et al. Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur J Pharm Biopharm. 2004;57:123–131. doi: 10.1016/j.ejpb.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Illum L. Nasal drug delivery—possibilities, problems and solutions. J Control Release. 2003;87:187–198. doi: 10.1016/S0168-3659(02)00363-2. [DOI] [PubMed] [Google Scholar]
- 49.Forged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298:315–322. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 50.Smart JD. The basics and underlying mechanisms of mucoadhesion. Adv Drug Deliv Rev. 2005;57:1556–1568. doi: 10.1016/j.addr.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Davis SS. The use of soluble polymers and polymer microparticles to provide improved vaccine responses after parenteral and mucosal delivery. Vaccine. 2006;24(2):S2-7–10. doi: 10.1016/j.vaccine.2005.01.102. [DOI] [PubMed] [Google Scholar]
- 52.De Temmerman ML, Rejman J, Demeester J, Irvine DJ, Gander B, De Smedt SC. Particulate vaccines: on the quest for optimal delivery and immune response. Drug Discov Today. 2011;16:569–582. doi: 10.1016/j.drudis.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 53.Bevan MJ. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J Exp Med. 1976;143:1283–1288. doi: 10.1084/jem.143.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Correia-Pinto JF, Csaba N, Alonso MJ. Vaccine delivery carriers: insights and future perspectives. Int J Pharm. 2013;440:27–38. doi: 10.1016/j.ijpharm.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 55.He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 56.Nagamoto T, Hattori Y, Takayama K, Maitani Y. Novel chitosan particles and chitosan-coated emulsions inducing immune response via intranasal vaccine delivery. Pharm Res. 2004;21:671–674. doi: 10.1023/B:PHAM.0000022414.17183.58. [DOI] [PubMed] [Google Scholar]
- 57.Durrey C, Irache JM, Puisieux F, Duchêne D, Ponchel G. Mucoadhesion of latexes. II. Adsorption isotherms and desorption studies. Pharm Res. 1994;11:680–683. doi: 10.1023/A:1018920128007. [DOI] [PubMed] [Google Scholar]
- 58.Sailaja AK, Amareshwar P, Chakravarty P. Chitosan nanoparticles as a drug delivery system. Res J Pharm Biol Chem Sci. 2010;1:474–484. [Google Scholar]
- 59.Källenius G, Pawlowski A, Brandtzaeg P, Svenson S. Should a new tuberculosis vaccine be administered intranasally? Tuberculosis. 2007;87:257–266. doi: 10.1016/j.tube.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 60.Köping-Höggård M, Sánchez A, Alonso MJ. Nanoparticles as carriers for nasal vaccine delivery. Expert Rev Vaccines. 2005;4:185–196. doi: 10.1586/14760584.4.2.185. [DOI] [PubMed] [Google Scholar]