

Abstract
This study presents a formulation approach that was shown to mitigate the dramatic food effect observed for a BCS Class II drug. In vitro (dissolution), in vivo (dog), and in silico (GastroPlus®) models were developed to understand the food effect and design strategies to mitigate it. The results showed that such models can be used successfully to mimic the clinically observed food effect. GastroPlus® modeling showed that food effect was primarily due to the extensive solubilization of the drug into the dietary lipid content of the meal. Several formulations were screened for dissolution rate using the biorelevant dissolution tests. Surfactant type and binder amount were found to play a significant role in the dissolution rate of the tablet prototypes that were manufactured using a high-shear wet granulation process. The performance of the lead prototypes (exhibiting best in vitro dissolution performance) was tested in dogs and human subjects. A new formulation approach, where vitamin E TPGS was included in the tablet formulation, was found to mitigate the food effect in humans.
KEY WORDS: bioavailability, food effect, in vivo in vitro models, vitamin E TPGS, wet granulation
INTRODUCTION
Food is known to affect the pharmacokinetics of orally administered drugs and can do so in a negative, positive or neutral manner (1–3). It is for this reason that the Food and Drug Administration (FDA) stipulates the food-label for all prescription products, codified in the Product Labeling (Code of Federal Regulations, Title 21, Vol. 4, Part 201, Sections 201.5, 201.57) and provides guidance for the industry entitled ‘Food-effect bioavailability and fed bioequivalence studies’ (4). It is estimated that out of 106 approved NMEs (new molecule entities) between 2001 and 2010, about 28% showed positive food effect and 31% showed negative food effect. Food can influence the drug absorption through physicochemical (drug-meal interactions mediated by altering dissolution, degradation, and diffusion mechanisms) and/or physiological mechanisms (interactions mediated by altering the residence times, volumes, and content of gastric and intestinal secretions, as well as membrane transport mechanisms) (2,5). Frameworks establishing qualitative prediction of drug-meal interactions have been published (6,7). An understanding of the drug substance and formulation properties can often be useful in predicting such food-drug interactions.
Biopharmaceutical Classification System (BCS) developed by Amidon et al. (8) helps categorize drugs based on the drug aqueous solubility and GI permeability. Biopharmaceutics Drug Disposition Classification System (BDDCS) further categorized the drugs based on their solubility and metabolism rates (1,9,10). Gu et al. (3) developed a statistical model to predict the food effect on drug absorption based on drug solubility, permeability, and dose of 92 drug candidates. While these qualitative methods are useful and, in many cases, guide the clinical/pharmaceutical development efforts (including seeking in vivo biowaivers, where applicable), they do not provide quantification with respect to food effect. There has been considerable effort in developing quantitative prediction tools based on in vitro dissolution/diffusion tests, in vivo animal models, and in silico models that would enable estimation of the magnitude of observed food effect in humans. Such tools are critical in developing food effect mitigation strategies and potentially reducing the number of clinical trials in humans.
The commonly used in vitro tests to study food effect involves using artificial dissolution media to mimic the in vivo (fed or fasted state) conditions, including biorelevant fluids (6,11–15), and other discriminatory dissolution media (water, simulated gastric fluid—SGF, simulated intestinal fluid—SIF) (13,16,17). In addition to drug dissolution, permeability component was also included by some researchers in their in vitro system, which proved to be useful to study food effect (12,18,19). Use of animal models, has been the common practice for estimation of the potential food effect in humans (6,15,20). While in certain instances, inconsistencies between the dog and human PK have been reported based on the physiological differences, the use of dog model continues to be prevalent in estimating the food effect (6,21).
Another upcoming methodology to study food effect is in silico modeling using commercial software packages such as GastroPlus®, PK-Sim®, SimCyp Simulator®, Stella® etc. (22). These models, referred to as physiologically based pharmacokinetic (PBPK) models, are able to incorporate physiological parameters together with available in vitro data for the drug to simulate plasma drug-concentration profiles. Case studies where such models were successfully used to predict the PK behavior in fed and fasted humans have been published (7,23,24).
While several generalities have been established on estimating the food effect in humans, it is clear that the model that works well will be drug and dosage form and formulation-specific. It would therefore be necessary to understand the physicochemical and physiological mechanisms affecting the specific drug molecule and establish prediction models based on the approaches discussed here, recognizing that certain iterations to them may be required to have the best prediction and utility.
Once it is recognized that a certain drug molecule will exhibit food effect, it is desirable to devise strategies to mitigate it, in order to minimize meal-dependent PK/PD effects, as well as to increase patient compliance of the product. Formulation strategies to enhance drug solubility/dissolution rates and in turn bioavailability for poorly water-soluble drugs include: (a) modification of the drug molecule (formation of salts, pro-drugs, and complexes), (b) crystal modification (amorphization, co-crystal formation), (c) particle modification (micronization, nanosizing), (d) microenvironmental control (pH modification, surfactants), (e) solid dispersions/solutions (dispersions in hydrophilic carriers via hot-melt extrusion, spray-drying), (f) liquid dispersions/solutions (solubilization in lipid/surfactant/polymer vehicles), and (g) others (modified-release, including gastro-retentive, delayed formulations). These strategies work well to mitigate food effect as well, if the underlying cause of it is the limited drug solubility.
The present work describes and rationalizes the formulation development strategy utilized in overcoming the food effect on oral pharmacokinetics of a BCS Class II drug. This is a retrospective analysis to understand the primary cause for the large food effect so that a mitigation strategy could be devised to minimize the impact. Reconciliation between the predicted and the observed food effect is done as part of this assessment, while also establishing the clinical relevance and the business case for food effect mitigation. The first part of this strategy included assessment of the magnitude of the food effect, when presented as simple, immediate-release solid dosage forms, while also delineating the effects of dissolution-rates and solubilization. In vitro, in vivo, and in silico models were developed in order to study this food effect and use this knowledge to devise a new tablet formulation strategy. Such models efficiently guide the subsequent formulation strategy (second part of the study), reducing the efforts (on a mechanistic basis) to only a selective few that are appropriate for the drug candidate/properties under consideration. Several formulation prototypes were screened using dissolution and in silico models, where the strategy was to improve the wetting and solubilization of the drug with surfactants. In silico physiologically based pharmacokinetic modeling (PBPK) was used to confirm the hypothesis on the role of solubilization on the observed food effect. The lead formulation prototypes were tested in dogs and humans. Where applicable, this clinical study can (a) inform the dosing/administration for Phase-III/pivotal trials, (b) serve as a definitive food-effect study, and (c) be used to define the labeling instructions for dosage and administration.
MATERIAL AND METHODS
Drug Molecule Physicochemical Properties
The API used in this study is a lipophilic, weak acid and meets the USP definition of practically insoluble (<100 μg/mL) at pH values less than 7. The aqueous solubility is almost eight times higher at neutral pH than at pH in the range of 1.2–6. The pKa and logD values are 4.85 and 5.09, respectively (25). The Peff value is 5,000 cm/s. The molecule has poor wetting characteristics in water with a high contact angle of 160°. The API has high solubility in oils and lipids. The API is classified as BCS Class II displaying poor aqueous solubility across the physiological pH range and high permeability based on clinical data (25).
In Vitro Dissolution
For dissolution testing, reagent-grade chemicals were used unless otherwise indicated. Boric acid, 10 N sodium hydroxide (J.T. Baker, Phillipsburg, NJ), potassium chloride, sodium phosphate monobasic (EM Science, Gibbstown, NJ) were used to prepare the dissolution media. Sodium dodecyl sulfate (SDS, EM Science) and Brij 35 (J.T. Baker, Phillipsburg, NJ) and were used as surfactant additives to the medium. Barnstead purified water (Barnstead International, Dubuque, IA) was used to prepare both dissolution media and HPLC mobile phases. Trifluoroacetic acid and HPLC-grade acetonitrile (J.T. Baker) were used for the preparation of the mobile phases. The borate buffers (pH 8.5 and 9.0, 0.05 M) were prepared according to USP. Sodium phosphate buffer (pH 6.8) was prepared by dissolving 6.9 g of sodium phosphate monobasic monohydrate per liter of water. The pH was adjusted to the desired value with 10 N sodium hydroxide. The media containing surfactant was prepared by dissolving the surfactants in the buffer.
Dissolution testing was performed in compliance with USP. The final method conditions were: Apparatus 2 at a paddle speed of 50 rpm; 1-L glass vessels. The dissolution medium was 1,000 mL of sodium phosphate buffer (pH 7.5; 0.05 M) with 0.5% Brij (v/v), maintained at 37 ± 0.5°C. Tests were conducted on a Distek Premiere 5100 system (Distek, North Brunswick, NJ) with a Vankel VK 8000 autosampling system. A sample volume of 1.5 mL was filtered and collected directly into a 2-mL HPLC vial using a 0.45 μm PVDF membrane filter for sample collection (Millipore Corp., Billerica, MA). The samples were collected at time points of 10, 20, 30, 45, and 60 min and an additional 30 min at a paddle speed of 200 rpm. During the dissolution method development process, the sample collection time points were typically set as 15, 30, 45, 60, and 90 min at the desired paddle speed with an additional 30 min at a paddle speed of 200 rpm for tests using USP apparatus 2.
The HPLC system used was a Waters Alliance® (Waters Corporation, Milford, MA). Chromatographic data were recorded and processed using Waters Empower. The final reversed-phase HPLC method utilized a Phenomenex Gemni NX C18 column (5 μm, 5 cm × 3.0 mm i.d.; Phenomenex Inc., Torrance, CA) maintained at 30°C with a mobile phase of water-acetonitrile-trifluoroacetic acid (20:80:0.1 v/v/v), a flow rate of 0.4 mL/min, UV detection at 306 nm, an injection volume of 10 μL, and a run time of 2 min.
In Vitro Biorelevant Dissolution
The assessment of various tablet formulations using biorelevant dissolution media, namely FaSSIF and FeSSIF, was conducted using USP apparatus 4, a Sotex flow-through dissolution system (Sotax AG, Basel) with the closed-loop configuration. FaSSIF and FeSSIF media were prepared as formulations proposed by Galia et al. (16). During the testing, the dissolution media at 37 ± 0.5°C was pumped through each flow-through cell by a piston pump at the flow rate of 8 mL/min. The media volume was 250 mL for each channel. In each flow-through cell, about 6.4 g of glass beads (1 mm in diameter) were used to fill up the conical bottom part of the cell, with a ruby bead (5 mm in diameter) at the cone apex. Tablets were horizontally positioned on top of the glass beads in the flow-through cells. Millipore filters (0.45 μm pore size) were used in the filter-head in each experiment. A fraction collector was used collect the samples for offline HPLC analysis.
In Silico Modeling
A physiologically based pharmacokinetic (PBPK) model was developed using commercial software (GastroPlus®, v7.0, Simulations Plus, Inc., Lancaster, CA) to simulate plasma concentrations for this drug. The clearance and distribution input used in the model were determined by fitting a three-compartment model to iv data. The absorption profile of the drug in the small and large intestine was determined from the plasma concentration versus time profiles over a range of doses. The logD, pH-solubility profile, and solubility in dietary lipids were determined experimentally, and the aqueous diffusivity was estimated based on molecular weight. To simulate the fed-state condition, the peak kinetic solubility in FeSSIF with added 10% microlipid (surrogate for high-fat meal media) media was used as the model input (25).
In Vivo Dog Study
The dog studies discussed in the section ‘Food Effect Evaluation’ under sub-heading “Dog Study” (solution and tablet formulation) and under ‘Formulation Evaluation’ section under sub-heading “Dog Study” (prototype food effect mitigating formulations) were crossover studies in four male dogs, with 1 week washout period between doses. The fasted dogs were pretreated with pentagastrin. The dose was flushed with 50 ml water for the fasted state, and with 50 ml high-fat meal supplement for the fed state. The high-fat meal supplement used in this case comprised of a similar fat, protein, and carbohydrate make-up as a high-fat American breakfast that was weight-adjusted for a dog (4). Each dog received about 100 cal meal comprising a calorie breakdown of roughly 15% protein, 25% carbohydrate and 60% fat. Sampling was done at time points 0, 1, 2, 3, 4, 8, and 24 h. A solution formulation was also included as part of this study for comparison purposes. A crash-resistant solution formulation (containing drug dissolved at 20 mg/mL in 50% PEG 400/35% ethanol/15% polysorbate 80) was used as the reference for bioavailability calculations.
Clinical Studies
The first trial was conducted (open-label, four-period, four-treatment, crossover study, 600 mg dose) in 18 healthy subjects randomized to receive either of the three formulations (section ‘Food Effect Evaluation’ under sub-heading “Clinical Study”) in the fasted state in Periods 1–3 with a minimum 5-day washout between each dose according to one of six randomly assigned treatment sequences. All subjects received formulation #2 with a high-fat meal (treatment D) in Period 4. Blood samples were collected at selected time points for pharmacokinetic analysis up to 72 h post-dosing.
The second study (discussed in the section ‘Formulation Evaluation’ under sub-heading “Clinical Study”) was an open-label, randomized, crossover study in healthy subjects (n = 35). The subjects were randomized to receive five single doses (200 mg dose) of the tablets (formulation #12, section “Formulation Screening”) according to a randomization scheduled with a 5-day washout period in between each dose. Healthy male or female subjects (18–49 years of age) as determined by medical history, physical examination, 12-lead ECG, and clinical laboratory evaluations were eligible to participate in this study. Blood samples were collected at several time points for pharmacokinetic analysis up to 72 h post-dosing.
Materials and Process for Formulation Screening
Batches with nine different formulations were manufactured using high-shear wet granulation process in order to maximize the rate of drug dissolution, which was postulated to enhance the in vivo solubility. These formulations had varying levels and types of disintegrants, binders and surfactants, all of which have a direct impact on drug dissolution.
Microcrystalline cellulose and croscarmellose sodium were purchased from FMC Co. (Philadelphia, PA), hydroxypropyl cellulose (HPC) from Ashland Specialty Ingredients (Wilmington, DE), lactose monohydrate from Kerry BioSciences (Beloit, WI), polyvinylpyrrolidone (PVP K-12) from Ashland Specialty Ingredients (Covington, KY) and magnesium stearate from Mallinckrodt Chemicals (Saint Louis, MO). Vitamin E TPGS (d-alpha-tocopheryl polyethylene glycol 1000 succinate) was purchased from Eastman Chemical Company (Kingsport, TN), and Poloxamer (Pluronic 188) and SLS (sodium lauryl sulphate) from BASF (Florham Park NJ).
The batches were manufactured using Diosna® high-shear granulator fitted with 1 or 2 L bowls for 200- and 500-g batch sizes, respectively. Intragranular excipients along with the drug were added to the granulator bowl and mixed for 2 min. A 20% w/w aqueous solution of vitamin E TPGS was used for granulation for formulations containing TPGS. In order to reach the desired granulation end-point, additional water was added separately as required. To prepare the vitamin E TPGS solution, it was first completely melted at 60°C, and then stirred with water to form a homogenous solution. Vitamin E TPGS or the binder solution (when binder was added wet) was added in the granulator over a 3-min period. After the solution addition, the granules were wet-massed for 30 s and then passed through an eight-mesh sieve. The wet mass was dried in a hot-air convection oven set at 40°C to a desired LOD (within 1% of the initial LOD). The dried material was passed through a Comil (Quadro, Ontario) running at 2,600 rpm using a 045R (1.1 mm opening) screen. The extra-granular components were added to the milled granules and mixed in a bin-blender for 10 min. Magnesium stearate (prescreened) was then added and blended for additional 5 min.
RESULTS AND DISCUSSION
Food Effect Evaluation
Clinical Study
Three formulations were evaluated initially in a clinical study to establish the impact of food on drug bioavailability. The formulations included a hard gelatin capsule (formulation #1, treatment A), a film-coated tablet (formulation #2, treatment B), and formulation 2 using micronized API (formulation #3, treatment C). All of these formulations were prepared using a dry granulation process. Another difference between formulations #1 and #2 was the drug to SLS ratio, where SLS was added to improve drug wetting. Formulation #1 had a SLS/drug ratio of 1:30 whereas the ratio was 1:50 for formulation #2.
The results from this study are shown in Fig. 1. A comparison of formulation #2 between the fasted and the fed state clearly demonstrates a dramatic positive food effect, with much higher BA observed when the tablet is administered with food. No significant BA differences were observed between formulations #2 and #3 indicating that the particle size of the drug substance had no role to play in the observed BA. This clearly indicates that particle size reduction techniques are unlikely to work as a food-effect mitigation strategy for this drug. The reason for this observation could be the insignificant effect of particle size on the drug dissolution. The relative comparison statistics between the four treatments are shown in Table I. The administration of the tablet with a high-fat meal (Treatment D), resulted in an approximately 29- and 11-fold increase in Cmax, and AUC(INF) values respectively, compared to the tablet administered while fasted (Treatment B). An approximately 30% and 50% reduction in Cmax and AUC values, respectively was observed when using a tablet formulation #2 compared to the hard gelatin capsule formulation #1. It may be speculated that the favorable SLS:drug ratio in the capsule formulation may be the underlying reason for this difference.
Fig. 1.
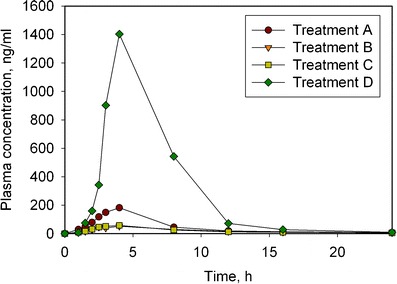
Plasma concentration profiles of the 4 treatments showing the dramatic food effect observed in human subjects. Treatments A, B, and C correspond to a hard gelatin capsule (formulation #1), a film-coated tablet (formulation #2), and formulation #2 manufactured using micronized API (formulation #3), respectively, all under fasted state. Treatment D corresponds to formulation #2 under fed state
Table I.
Treatment Comparisons for Selected Pharmacokinetic Parameters Showing a Dramatic Food Effect Observed for Dry Granulated Tablet (D vs B)
| Treatment comparison | C max GMR (90% CI) | AUC(INF) GMR (90% CI) | AUC(0–T) GMR (90% CI) |
|---|---|---|---|
| B vs A (Formulation 2 vs 1) | 0.31 (0.22, 0.45) | 0.48 (0.37, 0.61) | 0.47 (0.37, 0.61) |
| D vs B (Formulation 2: fed vs fasted) | 29.62 (17.56, 49.95) | 11.46 (8.32, 15.79) | 11.79 (8.50, 16.36) |
| C vs B (Formulation 3 vs 2) | 1.01 (0.74, 1.38) | 1.03 (0.84, 1.27) | 1.03 (0.83, 1.28) |
AUC(0-T) area under the plasma concentration-time curve from zero to the time of the last quantifiable concentration, AUC(INF) area under the plasma concentration-time curve from time zero extrapolated to infinite time, C max observed maximum plasma concentration, CI confidence interval, GMR geometric mean ratio
Such a strong positive food effect can be attributed to several factors: physicochemically based (solubilization of the drug, sensitivity to GI pH), altered permeability (bile and lipid-influenced influx or efflux), reduced GI emptying and transit, or other physiological changes such as increased spanchnic blood flow or altered gut metabolism (25). In order to devise a strategy to mitigate this food effect, it was necessary to investigate the key rate-limiting factors. To breakdown this understanding in vitro dissolution (USP and biorelevant dissolution), in vivo dog food-effect model and in silico GastroPlus modeling was used. In vitro tests were also used to screen formulation candidates and approaches before testing them in dog studies and human subjects.
In order to study the food effect further, it was desirable to qualify a dog model and/or establish ways of using in vitro tests to be able to screen formulation candidates and approaches before testing them in human subjects.
Dog Study
Formulation #2 (dry granulated tablet) was tested in dogs in both fed and fasted state. The study design is discussed in the section “In Vivo Dog Study”. The dog data showed that dry granulated tablet has a significant positive food effect (Table II), similar to that observed in humans. The food effect (fed/fasted % BA ratio) was found to be 3.64-fold in dogs. The crash-resistant solution formulation (containing drug dissolved at 20 mg/mL in 50% PEG 400/35% ethanol/15% polysorbate 80) showed no food effect and was used as the reference for BA calculation. The relative BA of the tablet was only 16% of the solution formulation in fasted state and increased to about 60% with a high-fat meal. This suggests that the food effect is mostly related to API dissolution/solubility in lipid environment and the slower GI motility (longer Tmax, 6 h for fed state versus 4 h for fasted). The study also demonstrates that this dog food-effect model can be useful indicator of the clinical food effect for this compound.
Table II.
Summary of Pharmacokinetic Parameters Demonstrating Food Effect Observed in Dogs
| Treatment | C max mean (SD) ng/ml, 104 | AUC(0-24) mean (SD) ng-h/ml, × 104 | Relative BA mean (SD) % |
|---|---|---|---|
| Solution, fasted | 4.3 (1.4) | 50.4 (26.6) | Reference |
| Solution, fed | 4.9 (1.2) | 51.4 (30.4) | – |
| Dry granulated tablet, fasted | 1.4 (0.8) | 8.3 (5.9) | 16 (11) |
| Dry granulated tablet, fed | 3.0 (0.6) | 30.4 (9.7) | 59 (19) |
Biorelevant Media Dissolution
Biorelevant dissolution (in FaSSIF and FeSSIF media) was conducted on formulations #1 (hard gelatin capsule) and #2 (dry granulated tablet). Both formulations showed a significant difference in dissolution profiles between the FaSSIF and FeSSIF media, with higher rates and extent of dissolution observed with the FeSSIF media (Fig. 2). Additionally, capsules showed higher dissolution than tablets in FaSSIF media, however no difference was observed in FeSSIF media. These results were consistent with that observed in human subjects. It can be concluded that in vitro dissolution testing using FaSSIF and FeSSIF media can be a good tool to estimate food effect for this drug, as observed for the tablet formulation.
Fig. 2.
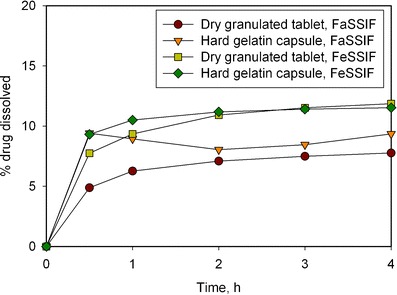
Drug dissolution profiles in biorelevant media showing food effect for dry granulated and hard gelatin capsule formulations (using USP 4 closed loop, 250 mL of media, 37°C, 8 mL/min)
In Silico Modeling Using GastroPlus® Software
In silico PBPK modeling using GastroPlus® was used to simulate the fed and fasted state in order to further understand the underlying cause of food effect. As reported in the section “In Silico Modeling”, fasted and fed conditions were simulated using biorelevant media. The model simulations were then compared to a clinical study where subjects were dosed with the drug in the morning under fasted conditions and evening, 12 h later, with food (Fig. 3). When kinetic biorelevant media solubility, i.e., FaSSIF and FeSSIF, was included in the model, the model predicted the fasted-state clinical data but not the fed-state data. Solubility experiments showed that the drug was very soluble in digestible lipids and subsequent experiments, where additional lipid was included in the FeSSIF, indicated a greater than tenfold increase in drug solubility from 109 μg/ml in FaSSIF media to 1,340 μg/ml in FeSSIF + Microlipid media (all adjusted to pH 6.3) (25). The model was run using FeSSIF + Microlipid media, assuming a 1 h gastric emptying T50 (as is typically observed with a high-fat meal). As can be seen from Fig. 3, the model was then able to simulate accurately both the fed and fasted state drug plasma concentration profiles. Based on these simulations, the clinically observed food effect appeared to be caused primarily due to the extensive solubilization of the drug into the dietary lipid content of the meal.
Fig. 3.
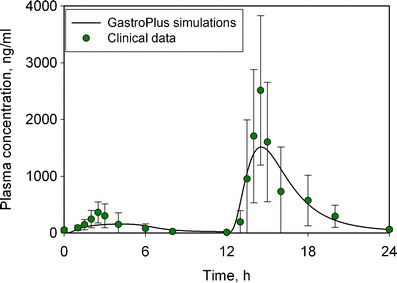
In silico modeling of observed food effect using GastroPlus software. The solid circles corresponds to clinical data, whereas the solid line represents GastroPlus simulations predictions
In order to mitigate food effect, a formulation change strategy was used, as is discussed in subsequent sub-sections.
Formulation Selection
In Vitro Dissolution Method Development
An in vitro dissolution method was developed that would allow for quick screening of several formulation prototypes and identify lead prototypes that can then be tested for their in vivo performance. It was desirable that the developed test is representative of in vivo performance (BA) so that the prototypes selected based on this test have a higher chance of in vivo success. The dissolution method conditions were selected based on obtaining adequate dissolution rate of drug and its ability to discriminate two particular dosage forms (formulations #1—hard gelatin capsule and #2—dry granulated tablet) that only differed slightly in the formulation composition (drug/SLS ratio) but had shown significant bioavailability differences as discussed before.
The dissolution media was evaluated for the dissolution behaviors of the two formulations using USP Apparatus II (paddle) with paddle speed of 50 rpm. Other dissolution parameters such as apparatus type and paddle speed were evaluated after appropriate media was chosen (data not shown). Based on results of method screening, the final dissolution conditions chosen were 50 mM potassium phosphate buffer at pH 7.5 containing 0.5% Brij 35 (at 50 rpm paddle speed). The dissolution profiles of formulations #1 and #2 based on this dissolution method are shown in Fig. 4. The differences between the dissolution profiles, especially during the early time points, demonstrate the discriminatory capability of the method. This was further confirmed to be the case when various prototype formulations were screened, as discussed in the next section.
Fig. 4.
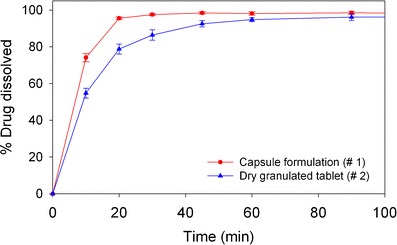
Dissolution profiles in 1,000 mL dissolution media of pH 7.5 phosphate buffer containing 0.5% Brij 35; using USP apparatus 2 at 37°C and paddle speed of 50 rpm
Formulation Screening
Given that the food effect for this drug is governed by its solubility, it is likely that formulation strategies involving the use of wetting agents or lipids, etc. can help in the food effect mitigation. This paper focuses on a wet granulated dosage form, which was one of many formulation options pursued in an effort to overcome the dramatic food effect observed for this drug. A high-shear wet granulation process was chosen to enable intimate contact of the drug with surfactant(s). A series of formulations were screened using the dissolution method described in the section “In Vivo Dissolution Method Development”. The aim was to maximize the dissolution rate of the formulation and then test the lead candidates in dogs and eventually in a human study for food effect mitigation.
The formulations variables screened included:
Surfactant concentration (1–4%), type (Poloxamer, SLS, Vitamin E TPGS) and mode of addition (wet vs dry)
Binder concentration (0–4%) and type (PVP, HPC)
The details of the formulation compositions are provided in Table III The drug loading for these formulations was fixed at 50% w/w. The filler was split equally (50:50) between lactose monohydrate and microcrystalline cellulose. The details of the high-shear wet granulation manufacturing process are described in the section “Materials and Process for Formulation Screening”.
Table III.
Key Composition Differences Among Various Formulations Screened
| Formulation # | Concentration w/w, % | |||||
|---|---|---|---|---|---|---|
| Surfactant | Binder | Disintegrant | ||||
| SLS | Poloxamer | Vitamin E TPGS | PVP | HPC | Croscarmellose sodium | |
| 4 | 4.0 (D) | – | – | – | 4.0 (W) | 5.0 |
| 5 | – | 4.0 (D) | – | – | 4.0 (W) | 5.0 |
| 6 | 4.0 (W) | – | – | – | – | 11.5 |
| 7 | 4.0 (D) | – | – | 4.0 (W) | – | 11.5 |
| 8 | – | 4.0 (D) | – | 2.0 (W) | – | – |
| 9 | 4.0 (D) | – | 4.7 (W) | – | 2.0 (D) | 11.5 |
| 10 | 4.0 (D) | – | 4.7 (W) | – | – | 11.5 |
| 11 | 4.0 (D) | – | 8.0 (W) | 2.0 (D) | – | 11.5 |
| 12 | – | 4.0 (D) | 4.7 (W) | – | – | 11.5 |
W added wet, D added dry
Dissolution testing was conducted on tablet formulations presented in Table III, and compared to the dissolution of dry granulated tablet (formulation #2) and hard gelatin capsule (formulation #1) as references. Formulations #4 and #5 vary in their surfactant type and were the first tested wet-granulated prototypes. The results show that formulations #4 and #5 have a slower dissolution profile than both the reference formulations, even when the surfactant/drug ratio was fourfold higher than the dry granulated tablet (Fig. 5). Also, poloxamer formulation showed significantly faster dissolution than SLS. The overall slower dissolution for these cases compared to the references could be due to the concentrations of the binder and disintegrant, which were not optimized at this stage. Therefore, formulation #6 was designed with no binder and higher disintegrant level (11.5% w/w). The removal of binder and the higher disintegrant level did result in a significant increase in the dissolution rates. Even with no binder, the dissolution for formulation #6 was only slightly higher than the dry granulated tablet, and not as high as the capsule formulation (Fig. 6). When a different binder (PVP) was added to formulation #6 (formulation #7), a significant reduction in dissolution rate was observed. Therefore, it was clear that binder and disintegrant concentration, and surfactant type were playing a significant role, but not the binder type. Keeping this in mind, formulation #8 was manufactured with 4% poloxamer and a reduced binder level (2% binder). Formulation #8 showed a significantly higher dissolution rate than the dry granulated tablet and compared well to the capsule formulation (Fig. 6). Formulation #8 was therefore selected for further testing.
Fig. 5.
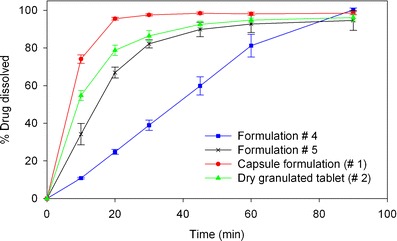
Dissolution profiles of formulations #4 and 5 compared to the reference dry granulated and hard gelatin capsule formulations using in vitro dissolution test
Fig. 6.
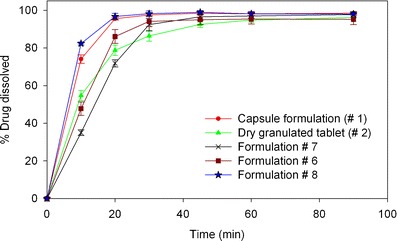
Dissolution profiles of formulations #6, #7, and #8 compared to the reference dry granulated and hard gelatin capsule formulations using in vitro dissolution test
Due to the drug’s high solubility in vitamin E TPGS (>25% w/w at 65°C), it was selected for further evaluation. The concept of using vitamin E TPGS (low melting waxy solid) in a solid dosage form is relatively new (26,27). Vitamin E TPGS is known to enhance the bioavailability of poorly water-soluble drugs by various mechanisms such as: (a) stabilization of amorphous drug form, (b) enhancing drug solubilization due to its surfactant properties, and (c) enhancing drug permeability by P-glycoprotein efflux inhibition (27). Pandey et al. (27) showed that vitamin E TPGS also has binder-like properties and therefore a formulation consisting of vitamin E TPGS may not require a conventional binder. Formulations #9 and #10 were designed using vitamin E TPGS as the surfactant and a lower amount of binder (2% and 0% w/w, respectively). A significant improvement in dissolution rate was observed for formulation #10 compared to the dry granulation tablet, as shown in Fig. 7. However, formulation #9 did not show the same magnitude of increase, possibly due to the presence of binder in the formulation. A higher concentration (8% w/w) of vitamin E TPGS was also tested as prototype formulation #11, which also included 2% w/w binder. No change in dissolution rate was observed compared to the case with 4.5% w/w (formulation #9) due to an increase in Vitamin E TPGS concentration, as shown in Fig. 7. Both formulations #9 and #11 did not offer much improvement in dissolution over the dry granulated tablet possibly due to the presence of binder at the same time as vitamin E TPGS (also has binder-like properties). Formulation #12 was designed with 4.7% Vitamin E TPGS with poloxamer at 4% w/w without the use of any conventional binder. This formulation showed the best dissolution profile, with significantly higher dissolution than the capsule formulation.
Fig. 7.
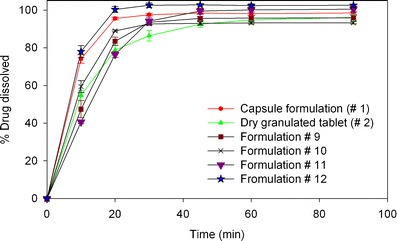
Dissolution profiles of formulations #9–12 compared to the reference dry granulated and hard gelatin capsule formulations using in vitro dissolution test
Based on these observations, formulations #8 (4% w/w poloxamer + 2% w/w PVP) and #12 (4% w/w poloxamer + 4.7% w/w vitamin E TPGS) were chosen as the two lead candidates for further evaluation. Both these formulations showed slightly higher dissolution rates than the capsule formulation.
Formulation Evaluation
Biorelevant Media Dissolution
The two lead wet-granulated tablet formulations (#8 and #12) based on results from dissolution testing (using phosphate buffer media) were evaluated using biorelevant media for food effect. As can be seen from Fig. 8, in FaSSIF state, both formulations #8 (poloxamer formulation) and #12 (vitamin E TPGS + Poloxamer), showed significantly faster dissolution rate than the dry granulated tablet (#2), with #12 showing superior performance. One of the primary reasons for the dissolution enhancement for formulations #8 and #12 would be the presence of surfactants in the formulation. In FeSSIF media, formulations #8 and #12 gave almost similar performance, with only a slightly higher dissolution for #12. Interesting to note is that the difference between FaSSIF and FeSSIF media (commonly considered an indication of food effect) was the least for formulation #12, but only after the 6-h time point.
Fig. 8.
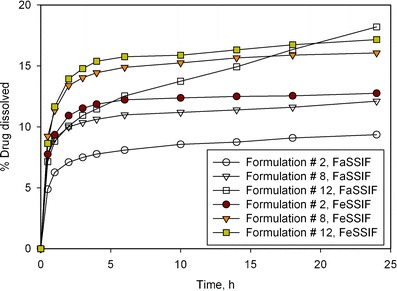
Dissolution profiles of lead prototype formulations (#8 and #12) compared to the reference dry granulated tablet (#2) using in vitro dissolution test using FaSSIF and FeSSIF biorelevant media
Based on these data, it was confirmed that both the wet-granulated tablet formulations gave superior performance in biorelevant media, both in FaSSIF and FeSSIF media, and there were indications, especially in the FaSSIF media, that formulation #12 (with Vitamin E TPGS) may give a better in vivo performance in terms of the food effect.
Dog Study
The lead formulation candidates (formulations #8 and #12) based on in vitro testing results were tested in dogs. The study set-up is described in the section “In Vivo Dog Study”. All formulations for this study were tested in fasted state and the results are shown in Fig. 9. The quantitative summary of the results is shown in Table IV. Relative BA calculations were done based on a solution formulation reference that was also used in this study. Formulation #8 (poloxamer) did not show a significant increase in relative BA (9.1%) when compared to the dry granulated tablet (8%). Interestingly, formulation #12 (poloxamer + Vitamin E TPGS) shows significantly higher relative BA of 20.7% compared to the 8% observed for the dry granulated tablet (Table IV). This is consistent with what was observed in the biorelevant dissolution media, however, the effect is more pronounced in dogs. Also interesting to note is that this functionality of vitamin E TPGS was not captured by the dissolution test (using phosphate buffer media). Based on these data, it was decided to evaluate formulation #12 as the lead candidate in a human clinical study for food-effect mitigation.
Fig. 9.
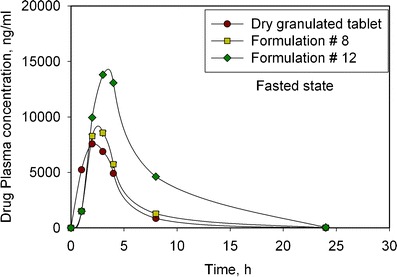
Plasma concentration profiles of the lead formulation prototypes (#8 and #12) in a dog study (fasted state) demonstrating a significant increase in exposure for Vitamin E TPGS-based formulation (#12)
Table IV.
Summary of Pharmacokinetic Parameters Demonstrating Enhanced Bioavailability with the Use of Vitamin E TPGS-based Formulation
| Fasted state | C max (ng/mL) | T max (h) | AUC 0-24 h (ng · h/mL) | Relative BA (%) | |||
|---|---|---|---|---|---|---|---|
| Formulation | Mean | SD | Median | Mean | SD | Mean | SD |
| Solution | 43126 | 13761 | 3 | 504019 | 265726 | Reference | – |
| Dry granulated tablet | 8694 | 586 | 2.5 | 40519 | 8796 | 8.0 | 1.8 |
| Formulation #8 | 8944 | 6744 | 3 | 45714 | 38369 | 9.1 | 7.6 |
| Formulation #12 | 14382 | 3776 | 3 | 104507 | 28685 | 20.7 | 5.7 |
Clinical Study
The vitamin E TPGS-based wet granulation tablet formulation (#12) that showed the best in vitro dissolution data and in vivo dog results was used in a clinical study to establish if this formulation can be confirmed to mitigate food effect in humans. The main objective of the clinical study was to estimate the oral bioavailability of the test formulations when administered fasted or with a standard meal relative to that of a reference tablet (that showed dramatic food effect as discussed in the section ‘Food Effect Evaluation’ under sub-heading “Clinical Study”) administered with a standard meal. A statistical treatment comparison summary of selected pharmacokinetic parameters is shown in Table V. There are no significant differences between the PK parameters shown in Table V when vitamin E TPGS-based formulation was dosed in fed versus fasted state. Therefore, this study confirmed that the vitamin E TPGS-based wet granulation tablet (formulation #12) was able to successfully mitigate the dramatic food effect observed before (Table I). This is in agreement to what was observed in the dog study and in vitro data (using biorelevant media) and confirms that improved wetting and solubilization of the drug is one of the prevailing mechanisms to mitigate the food effect.
Table V.
Estimated Ratios of Adjusted Geometric Means (90% CI) for Selected Pharmacokinetic Parameters
| Treatment comparison | AUC(INF) GMR (90% CI) | AUC(0-T) GMR (90% CI) | C max GMR (90% CI) |
|---|---|---|---|
| TPGS-based wet-granulated tablet—fed vs fasted | 1.280 (1.024, 1.598) | 1.470 (1.162, 1.860) | 1.405 (1.004, 1.966) |
| TPGS-based wet-granulated tablet—fed vs dry granulated fed | 0.869 (0.722, 1.045) | 0.861 (0.702, 1.055) | 0.967 (0.723, 1.293) |
AUC(0-T) area under the plasma concentration-time curve from zero to the time of the last quantifiable concentration, AUC(INF) area under the plasma concentration-time curve from time zero extrapolated to infinite time, C max observed maximum plasma concentration, CI confidence interval, GMR geometric mean ratio
Discussion and Conclusions
This paper presents a case study of a BCS Class II compound that was observed to have a dramatic positive food effect. In vitro, in vivo, and in silico modeling techniques were used to dissect the primary mechanism behind the observed food effect and devise a formulation strategy to mitigate the food effect. The solubility of the drug was observed to increase ten-fold in the presence of lipid indicating a very strong solubilization capacity from dietary lipids in a high-fat meal. In vitro biorelevant dissolution and Gastroplus modeling confirmed drug solubilization to be one of the key drivers for the positive food effect. However, in silico modeling also suggested that solubilization alone may not fully explain the observed food effect (25). Other factors such as gastric pH environment in the fed-state, efflux, and influx transporters, and/or first-pass metabolism can also play a role. Further studies are needed to investigate their impact to the overall food effect.
This paper also presents a unique formulation approach to mitigate the observed food effect. The formulation strategy was to maximize the in vitro dissolution of the drug by manipulating formulation components such as surfactant and binder. The incorporation of surfactants such as poloxamer and vitamin E TPGS in a solid dosage form by a high-shear wet granulation process was found to enhance significantly the dissolution of the tablet formulation. This was observed to be the case both in phosphate buffer media as well as biorelevant media. The dog study data showed that the presence of vitamin E TPGS was essential in enhancing BA in the fasted state and therefore can potentially reduce the food effect. This was confirmed in human subjects, where the vitamin E TPGS-based wet-granulated tablet was successful in mitigating the food effect.
Acknowledgments
The authors would like to thank Elena Zour, Robert Jerzewski, Robert Francis, Joan Ruan, Anne Kelly, and Bing He from Bristol-Myers Squibb for their support and help with this work.
References
- 1.Custodio JM, Wu C-Y, Benet LZ. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60(6):717–33. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fleisher D, Li C, Zhou Y, Pao LH, Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clinical implications. Clin Pharmacokinet. 1999;36(3):233–54. doi: 10.2165/00003088-199936030-00004. [DOI] [PubMed] [Google Scholar]
- 3.Gu CH, Li H, Levons J, Lentz K, Gandhi RB, Raghavan K, et al. Predicting effect of food on extent of drug absorption based on physicochemical properties. Pharm Res. 2007;24(6):1118–30. doi: 10.1007/s11095-007-9236-1. [DOI] [PubMed] [Google Scholar]
- 4.FDA. Guidance for Industry: Food-effect bioavailability and fed bioequivalence studies. Food and Drug Administration. 2002;Rockville, MD.
- 5.Charman WN, Porter CJ, Mithani S, Dressman JB. Physiochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J Pharm Sci. 1997;86(3):269–82. doi: 10.1021/js960085v. [DOI] [PubMed] [Google Scholar]
- 6.Lentz KA. Current methods for predicting human food effect. AAPS J. 2008;10(2):282–8. doi: 10.1208/s12248-008-9025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parrott N, Lukacova V, Fraczkiewicz G, Bolger MB. Predicting pharmacokinetics of drugs using physiologically based modeling—application to food effects. The AAPS Journal. 2009;11(1):45–53. doi: 10.1208/s12248-008-9079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 9.Dahan A, Miller JM, Amidon GL. Prediction of solubility and permeability class membership: provisional BCS classification of the world’s top oral drugs. AAPS J. 2009;11(4):740–6. doi: 10.1208/s12248-009-9144-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 11.Dressman JB, Reppas C. In vitro–in vivo correlations for lipophilic, poorly water-soluble drugs. European Journal of Pharmaceutical Sciences. 2000;11(2):S73–80. doi: 10.1016/S0928-0987(00)00181-0. [DOI] [PubMed] [Google Scholar]
- 12.Kataoka M, Masaoka Y, Sakuma S, Yamashita S. Effect of food intake on the oral absorption of poorly water-soluble drugs: in vitro assessment of drug dissolution and permeation assay system. J Pharm Sci. 2006;95(9):2051–61. doi: 10.1002/jps.20691. [DOI] [PubMed] [Google Scholar]
- 13.Persson E, Gustafsson A-S, Carlsson A, Nilsson R, Knutson L, Forsell P, et al. The effects of food on the dissolution of poorly soluble drugs in human and in model small intestinal fluids. Pharm Res. 2005;22(12):2141–51. doi: 10.1007/s11095-005-8192-x. [DOI] [PubMed] [Google Scholar]
- 14.Shono Y, Jantratid E, Kesisoglou F, Reppas C, Dressman JB. Forecasting in vivo oral absorption and food effect of micronized and nanosized aprepitant formulations in humans. Eur J Pharm Biopharm. 2010;76(1):95–104. doi: 10.1016/j.ejpb.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y, Loper A, Landis E, Hettrick L, Novak L, Lynn K, et al. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int J Pharm. 2004;285(1–2):135–46. doi: 10.1016/j.ijpharm.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Galia E, Nicolaides E, Horter D, Lobenberg R, Reppas C, Dressman JB. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm Res. 1998;15(5):698–705. doi: 10.1023/A:1011910801212. [DOI] [PubMed] [Google Scholar]
- 17.Klein S. The use of biorelevant dissolution media to forecast the in vivo performance of a drug. The AAPS Journal. 2010;12(3):397–406. doi: 10.1208/s12248-010-9203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ginski MJ, Taneja R, Polli JE. Prediction of dissolution-absorption relationships from a continuous dissolution/Caco-2 system. AAPS PharmSci. 1999;1(2):E3. doi: 10.1208/ps010203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Motz SA, Schaefer UF, Balbach S, Eichinger T, Lehr CM. Permeability assessment for solid oral drug formulations based on Caco-2 monolayer in combination with a flow through dissolution cell. Eur J Pharm Biopharm. 2007;66(2):286–95. doi: 10.1016/j.ejpb.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 20.Zhou R, Moench P, Heran C, Lu X, Mathias N, Faria TN, et al. pH-dependent dissolution in vitro and absorption in vivo of weakly basic drugs: development of a canine model. Pharm Res. 2005;22(2):188–92. doi: 10.1007/s11095-004-1185-3. [DOI] [PubMed] [Google Scholar]
- 21.Fotaki N, Symillides M, Reppas C. Canine versus in vitro data for predicting input profiles of l-sulpiride after oral administration. Eur J Pharm Sci. 2005;26(3–4):324–33. doi: 10.1016/j.ejps.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Rowland M, Peck C, Tucker G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45–73. doi: 10.1146/annurev-pharmtox-010510-100540. [DOI] [PubMed] [Google Scholar]
- 23.Jones HM, Parrott N, Ohlenbusch G, Lave T. Predicting pharmacokinetic food effects using biorelevant solubility media and physiologically based modelling. Clin Pharmacokinet. 2006;45(12):1213–26. doi: 10.2165/00003088-200645120-00006. [DOI] [PubMed] [Google Scholar]
- 24.Nicolaides E, Galia E, Efthymiopoulos C, Dressman JB, Reppas C. Forecasting the in vivo performance of four low solubility drugs from their in vitro dissolution data. Pharm Res. 1999;16(12):1876–82. doi: 10.1023/A:1018959511323. [DOI] [PubMed] [Google Scholar]
- 25.Mathias NR, Crison J. The use of modeling tools to drive efficient oral product design. AAPS J. 2012;14(3):591–600. doi: 10.1208/s12248-012-9372-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin F, Tatavarti A. Tabletability assessment of conventional formulations containing vitamin E tocopheryl polyethylene glycol succinate. Int J Pharm. 2010;389(1–2):58–65. doi: 10.1016/j.ijpharm.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 27.Pandey P, Sinko PD, Bindra DS, Hamey R, Gour S, Vema-Varapu C. Processing challenges with solid dosage formulations containing vitamin E TPGS. Pharm Dev Technol. 2013;18(1):296–304. doi: 10.3109/10837450.2012.737807. [DOI] [PubMed] [Google Scholar]