

Abstract
Normal cellular function is dependent on a number of highly regulated homeostatic mechanisms, which act in concert to maintain conditions suitable for life. During periods of nutritional deficit, cells initiate a number of recycling programs which break down complex intracellular structures, thus allowing them to utilize the energy stored within. These recycling systems, broadly named “autophagy”, enable the cell to maintain the flow of nutritional substrates until they can be replenished from external sources. Recent research has shown that a number of regulatory components of the autophagy program are controlled by lysine acetylation. Lysine acetylation is a reversible post-translational modification that can alter the activity of enzymes in a number of cellular compartments. Strikingly, the main substrate for this modification is a product of cellular energy metabolism: acetyl-CoA. This suggests a direct and intricate link between fuel metabolites and the systems which regulate nutritional homeostasis. In this review, we examine how acetylation regulates the systems that control cellular autophagy, and how global protein acetylation status may act as a trigger for recycling of cellular components in a nutrient-dependent fashion. In particular, we focus on how acetylation may control the degradation and turnover of mitochondria, the major source of fuel-derived acetyl-CoA.
Keywords: Acetylation, Sirt3, GCN5L1, Mitophagy, Autophagy
1. Introduction
Extreme starvation induces the intracellular recycling of nutrients in order to sustain energy resources and life. This nutrient deprived state results in the progressive shrinkage of organs [1], which is mediated in part by cellular self (auto) eating (phagy), a biological program termed macroautophagy or autophagy. The intracellular autophagy program was initially recognized and described histologically approximately 50 years ago [2, 3]. The molecular characterization and consequences of the induction of this program are actively being explored. Although excessive starvation itself is destructive [1], the induction of autophagy is required for recycling of intracellular contents to maintain energetic homeostasis under restricted nutrient conditions. The absolute requirement of this intracellular recycling for energetic homeostasis is evident in neonates during the postpartum period where the initial maternal production of milk is insufficient for the infant’s energy needs [4]. Similarly, in association with additional ameliorative benefits of exercise, acute exercise evokes autophagy in the heart and skeletal muscle of mice [5]. At the opposite side of the spectrum, the disruption of autophagy is associated with pathologies such as malignancies and neurodegenerative diseases [6]. Although this concept is more complex in that autophagy itself can sustain nutrient recycling for tumor growth and metastasis [7] and its inhibition can promote cancer cell death [8]. Together, these findings suggest that autophagy is central to the maintenance of cellular homeostatic control. While extreme nutrient depletion induces autophagy, less extreme forms of starvation, i.e. caloric restriction, also confer beneficial lifespan and health effects. Therefore, it is not surprising that the programs at the molecular level that have been identified to trigger starvation-induced autophagy are similarly modulated in response to chronic caloric-restriction [9, 10] and to alternate day fasting [11].
The quality control of individual organelles within cells is also regulated during macroautophagy and/or can be selectively regulated at the single organelle level. This is epitomized by mitochondria, which can undergo selective mitophagy during development or in response to direct mitochondrial stressors [12]. The molecular programs controlling selective organelle ‘phagy’ are less well defined and whether the nutrient-restriction stressors inducing macroautophagy are operational in distinct organelles has not been as well established.
A mechanism whereby starvation and caloric restriction exert biological effects includes the modulation of levels of metabolic intermediates. An example being the acetyl group from acetyl-CoA, an intermediate of glucose, fat and protein metabolism, which when bound to protein lysine residues, modifies that protein’s properties in a myriad of ways [13]. Lysine residue acetylation status has been found to modify: allosteric DNA:protein and protein:protein interactions; protein stability or subcellular localization and enzymatic activities [14]. Another metabolic intermediate that is modulated with nutrition status is the NAD/NADH ratio. Interestingly, one family of deacetylases linked to caloric-restriction mediated longevity, the sirtuins or Sirts, employ NAD as a cofactor to mediate lysine residue deacetylation. The nutrient-sensing enzymes that regulate protein acetylation/deacetylation have begun to be characterized [15, 16], and of note, the mitochondrial enriched deacetylase Sirt3 is itself regulated in response to changing nutrient levels [17-19]. Despite the accumulating evidence supporting the role of this pathway in the modulation of mitochondrial function, this biology requires additional study as epitomized by the lack of metabolic phenotype in specific genetic depletion of Sirt3 in tissues such as the liver and skeletal muscle [20].
Due to the increasing metabolic disease burden in response to macronutrient excess [21], and the associated disruption in mitochondrial function in response to caloric overload [19, 22], our understanding as to how metabolic intermediates may regulate innate quality control programs (such as autophagy and mitophagy) should give us insight into disease pathophysiology. Given that nutrient restriction can modulate both autophagy and acetylation status, it is not surprising that emerging evidence suggests that these two cellular processes may be intricately linked. Furthermore, understanding the links between metabolic intermediates, acetylation and autophagy may facilitate the design of therapeutic strategies to counter macronutrient-overload diseases through improved cellular housekeeping and homeostasis. To explore these concepts, the objective of this review will focus on nutrient load, the regulation and role of acetylation and its control of autophagy and mitophagy.
2. The modulation of acetylation in response to nutrient levels
Acetyl-CoA is a metabolic intermediate that resides within mitochondrial, cytosolic and nuclear compartments [23]. In mitochondria, acetyl-CoA is generated during the oxidation of pyruvate and fatty acids, and its major function is to convey carbon atoms to the citric acid cycle for energy production. Therefore, the availability of acetyl-CoA, via its catabolism in the citric acid cycle with the generation of NADH, may also affect the NAD/NADH ratio, and potentially sirtuin deactylation activity. These effects may also place the mitochondria at the hub of cellular acetylation status. Furthermore, the citric acid cycle intermediate citrate can be exported from the mitochondria into the cytosol and nucleus respectively. In both these compartments ATP citrate lyase (ACL) converts citrate to acetyl-CoA. In the cytosol, acetyl-CoA serves as a substrate for lipogenesis and for acetyltransferase enzymes. In the nucleus, the acetyl-CoA, at least derived from growth factor stimulation, excess glucose oxidation and ACL activity in the cytosol, functions as a substrate for histone acetyltransferase reactions. Therefore nutrient status directly alters acetyl-CoA levels in the mitochondria and cytosol leading to coordinating histone acetylation and gene expression [24].
Acute restriction in caloric intake provokes adipose tissue lipolysis, resulting in elevated levels of circulating non-esterified fatty acids [25]. When levels of circulating fatty acids are high, the production of acetyl-CoA from fat breakdown exceeds the cellular energy requirements. In an organ restricted fashion, the excess acetyl-CoA can be employed in lipid, cholesterol and ketone body synthesis, and as a substrate for protein acetylation. Nutrient overload, by increasing fatty acid and/or glucose levels, can similarly increase acetyl-CoA levels resulting in similar, albeit possibly more chronic, effects. Although longer-term caloric restriction and/or starvation results in a reduction in circulating free fatty acid and triglyceride levels once fat stores are catabolized [26, 27], these nutrient depleted states increase hepatic and renal ketogenesis, with concurrent ketone catabolism as an acetyl-CoA source for high-energy consuming organs such as the brain and heart [28].
In parallel, within mitochondria there is evidence that with changes in acetyl-CoA levels there is concurrent modulation of acetylation levels on a multitude of proteins in response to diverse conditions including fasting, caloric restriction and overload, and following ethanol intoxication [19, 29-32]. As mentioned and discussed below nutritional status also affects the NAD/NADH ratio and sirtuin activity leading to enzymatic deacetylation. The molecular regulatory control of protein acetylation and the functional consequences of this post-translational modification have been actively pursued over the last decade. These programs and effects are reviewed in the following section.
Additionally, the recognition of non-enzymatic acetylation of proteins in the presence of acetyl-CoA was described many decades ago [33] and denatured mitochondrial proteins undergo acetylation in the presence of acetyl-CoA [16]. In fact, it has been suggested that the high acetyl-CoA concentration and pH of mitochondria promotes non-enzymatic protein acetylation [34]. However, as mentioned below, GCN5L1 may counter Sirt3 mediated deacetylation in the mitochondria. The concept of non-enzymatic protein acetylation may be operational in diabetes where metabolic inflexibility, which is defined as the inability to switch from fatty acid to glucose oxidation during the transition from the fasted to fed state, results in part from the allosteric inhibition of pyruvate dehydrogenase by increased mitochondrial acetyl-CoA levels [35, 36]. The role of non-enzymatic protein acetylation has not been extensively investigated, although its’ potentially important regulatory role has been recently been reviewed [37].
3. Nutrient-sensing regulatory control of protein acetylation
There are three major acetyltransferase (also known as histone acetyltransferases [HATs] or, more accurately, lysine acetyltransferases [KATs]) families, and member proteins from each group have been implicated in the control of cellular homeostasis (reviewed [15]). Like KATs, deacetylase proteins (also known as histone deacetylases [HDACs] or lysine deacetylases [KDACs]) are grouped into families. Class I, II and Class IV are zinc-dependent and are either localized in the nucleus or can shuttle between the nucleus and the cytoplasm [38]. These enzymes are predominantly regulated independent of nutrient-status. In contrast, the Class III KDACs are predominantly NAD+-dependent deacetylases (Sirtuins), and as mentioned, function as sensors of the energetic status of the cell in response to the subcellular compartment levels of NAD+ and nicotinamide and/or to the ratio of NAD+:NADH [39-43]. Mammals have 7 sirtuin enzymes designated Sirt1 through Sirt7, which have distinct tissue distributions and subcellular localizations that, in part, contribute to their distinct biological functions [44-46]. The mammalian sirtuins are further phylogenetically divided into five subclasses based on the homology of their 250 amino acid core domain [47]. Sirt1, 2 and 3 constitute subclass I and predominantly function in the nuclear (Sirt1), cytoplasmic (Sirt1 and 2) and mitochondrial (Sirt3) compartments. These three enzymes show closest homology to the yeast longevity protein Sir2, exhibit the most robust deacetylase activity and will be reviewed here, in concert with their counter-regulatory KATs, in the control of the autophagy/mitophagy regulatory programs.
Sirt1 is the most extensively explored sirtuin, and it deacetylates multiple targets in the nucleus and cytoplasm (see reviews [45, 48]). The essential role for Sirt1-mediated deacetylation of nuclear regulatory proteins and metabolic pathway enzymes is underscored in that the genetic depletion of Sirt1 results in embryonic lethality. Interestingly, Sirt1 is activated by both starvation and caloric restriction which align with its nutrient-sensing role and possible role in longetivy. Sirt1’s deacetylation targets include LKB1, NF-κB, PGC1-α and Foxo3a therefore linking Sirt1 activity to AMPK, nutrient sensing enzymes, inflammation, mitochondrial biogenesis and oxidant defense systems, respectively [45, 46, 49]. Sirt1 also regulates numerous aspects of cell metabolism through deacetylation of PPARγ (β-oxidation) and FOXO1 (gluconeogenesis). In addition, Sirt1 plays a role in autophagy through deacetylation of numerous autophagy factors (see below). Sirt2 also has nuclear and cytosolic targets, and in contrast to Sirt1, the depletion of Sirt2 has ameliorative effects against multiple stressors [50-52]). Its regulatory role in autophagy is less well established, but is discussed further in the section 4.3 of this review. Sirt3 is the third major deacetylase and it functions predominantly in mitochondria [53]. The depletion of Sirt3 has a subtle phenotype [54] which is unmasked in response to prolonged fasting [18], or following chronic perturbations in caloric intake [19, 55, 56]. Sirt3 mediated deacetylation has been shown to regulate numerous aspects of mitochondrial function including ROS generation, ATP production and apoptosis by regulating MPTP opening [17, 57-59]. In addition, Sirt3 regulates β-oxidation, amino acid metabolism, the electron transport chain, ATP production and the urea cycle [60, 61].
The counter-regulatory acetyltransferase enzyme system is less well characterized, although in the nucleus both Gcn5 and p300 have been shown to counter the actions of Sirt1 [48]. It is interesting to note that the levels of Gcn5 are reduced when Sirt1 is genetically depleted as a putative adaptive process to ‘compensate’ for the reduction in the deacetylase suggesting that mechanisms exist within the cell to regulate protein acetylation status by regulating enzyme expression [62]. These acetyltransferase proteins have not been extensively studied in autophagy, although the role of p300 will be discussed below. The process of protein acetylation in the mitochondria is even less well understood, although GCN5L1 has been identified as a critical component of this program and was recently shown to regulate mitophagy [63, 64].
4. Autophagy as a nutrient-sensing housekeeping program
The first 30 years of research after the description of autophagy focused on its role in protein and intracellular content catabolism in response to nutrient deprivation [65]. Our knowledge of the diverse spectrum of intracellular functions linked to autophagy has expanded significantly as a result of the phenotypes uncovered by genetic manipulation of canonical molecules regulating autophagy [3, 66-68]. The broad array of functions attributed to autophagy has recently been reviewed [3, 69, 70], and will not be extensively discussed here. Nevertheless, characterization of these regulatory programs has also enhanced our understanding of cellular energetic sensing in the regulation of autophagy, cellular refueling and metabolism.
4.1 Canonical Mediators of Autophagy
The autophagy program begins, from a structural perspective, with the formation of an isolation membrane which is termed the phagophore. This structure develops from membrane components gleaned from the endoplasmic reticulum (ER), the plasma membrane and/or the mitochondria [71]. Numerous membranes can contribute to phagophore formation as evidenced by this structure being formed at ER-mitochondria contact sites [72]. The process is followed by elongation of the phagophore and closure to form the autophagosome, and then by fusion of the autophagosome to the lysosome. During this process, portions of the cytoplasm (microautophagy), protein aggregates (chaperone-mediated autophagy), or complete organelles, for example mitochondria (mitophagy), are engulfed by the double membraned autophagosome for subsequent fusion with lysosomes. The degradation of proteins and organelles by the lysosomal hydrolytic enzymes provides the cell with the nutrients that are required to adapt to unfavorable conditions such as starvation. Other important functions of autophagy include: a role in cell differentiation, as shown by the involvement of mitophagy in mitochondrial depletion during erythrogenesis (Mortensen et al., 2010); the elimination of aggregate-prone proteins that would otherwise accumulate in the cytoplasm and jeopardize the integrity of the cell [73]; the homeostatic quality control of damaged organelles [12]; and the concomitant regulation of the inflammasome [74, 75].
Molecular and genetic studies in yeast and mammalian cells have begun to define the protein complexes required to initiate autophagosome assembly and to facilitate the progression through the autophagosome-lysosomal pathway. A simplified schematic illustrating the regulatory and structural proteins involved is shown in Figure 1. Autophagosome assembly includes the sequential activation of the Ulk1 multi-protein complex; the Beclin1/class III phosphatidylinotisol 3-kinase (PI3K) complex and two ubiquitin-like protein complexes. These apical events in the initiation of autophagy and in the autophagosome nucleation have been described previously [76] [77, 78].
Fig 1.
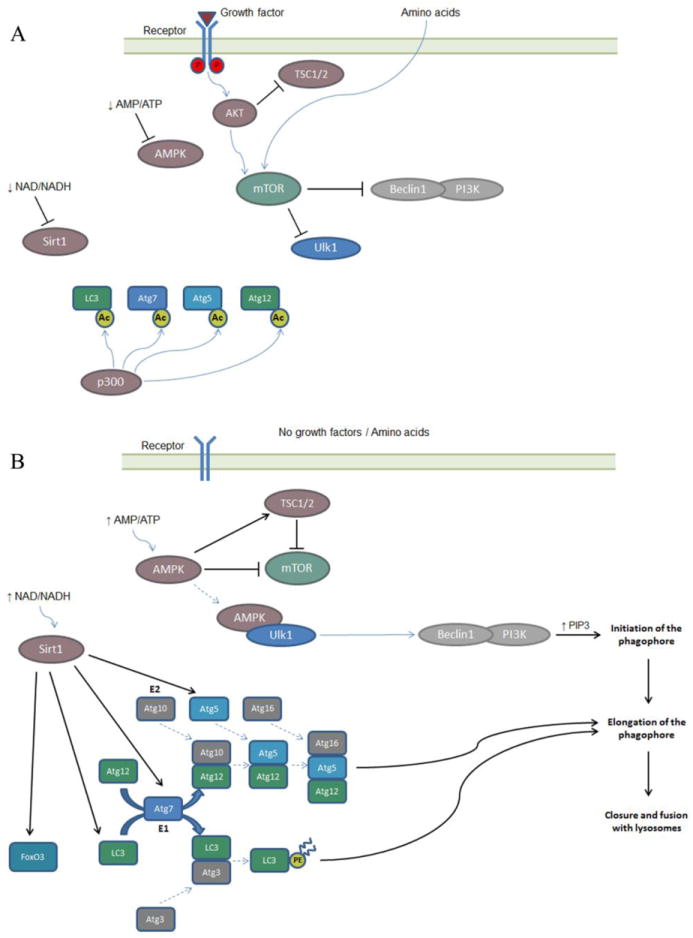
Nutrient depletion pathways in the activation of autophagy. A. In the presence of nutrients, growth factor and amino acids, Sirt1 and AMPK are inactive, whereas AKT is activated by the growth factor receptors. AKT activates mTOR directly, and indirectly by blocking its inhibitor TSC1/2. The mTOR complex sequesters and phosphorylates the Ulk1 complex, and phosphorylates and inhibits the Beclin/class III PI3K complex. P300 acetylates and inhibits LC3, Atg7, Atg5 and ATg12, members of the ubiquitin-like systems involved in the elongation of the phagophore. Autophagy is inhibited. B. In the absence of nutrients, growth factors and amino acids AKT is inactive, whereas AMPK is activated by the low AMP/ATP ratio. AMPK inhibits mTOR directly and indirectly, by activating its inhibitor TSC1/2. AMPK also bind to the Ulk1 complex, phosphorylating Ulk1. The Ulk1 complex phosphorylates the Beclin/class III PI3K complex, which generates PIP3 and this leads to the initiation of the phagophore. Caloric restriction also activates Sirt1, which deacetylates LC3, Atg7, Atg5 and FoXO3. This leads to the activation of the ubiquitin-like systems (Atg5, 12, 16 and LC3-II) involved in the elongation of the phagophore and also to the activation of transcription by FoxO3. The combination of these programs activates cellular autophagy.
The two ubiquitin-like systems involved in autophagy generate the Atg5/12/16 complex and promotes the lipidation of microtubule-associated protein 1 light-chain 3 (LC3). Atg12 is covalently attached to Atg5 by the action of Atg7 and Atg10, E1-like and E2-like enzymes respectively. The Atg5-Atg12 conjugate interacts with Atg16 non-covalently. LC3 (Atg8) is first cleaved by Atg4 and then covalently conjugated to phosphatidylethanolamine (PE) by the action of Atg7 and Atg3, a E2-like enzyme, to form LC3-II. LC3-II is a component of the phagophore membrane and thus is the classical marker for autophagosome assembly. These conjugation systems are proposed to promote elongation of the phagophore and are regulated by nutrient availability, in that some components (Atg5, Atg7, Atg8 and Atg12) are modified by acetylation [79, 80](see below).
The absolute requirement of autophagy in the transition from placental nutrition to lactation is evident in that the complete genetic depletion of numerous canonical autophagy proteins in mice results in death within the first day of life (for review, [81]). These deficiencies can be bypassed by the conditional knockout of some of these proteins in a tissue restricted manner, as shown with the conditional depletion of Atg5 or Atg7. These mice do survive but show severe defects including the accumulation of swollen and deformed mitochondria – a phenotype highlighting the role of autophagy as a intrinsic organelle quality control program (for review, [81]).
4.2 Regulation of autophagy by nutrient availability
The nutrient-sensing control of autophagy is predominantly regulated by two interconnected signaling pathways, i.e. the mammalian target of rapamycin1 (mTOR1) complex and adenosine monophosphate-activated protein kinase (AMPK).
The mTOR1 complex consists on mammalian target of Rapamycin (mTOR), Raptor and additional regulatory proteins. This complex is controlled by the availability of growth factors and of amino acids. When growth factors are present, they trigger signaling through the insulin-like growth factor receptor (IGF1R), with the subsequent activation of the Akt kinase. The activation of Akt results in a plethora of phosphorylation-dependent events that coordinately activate and/or inactivate counter-regulatory programs. Although, this has been extensively reviewed elsewhere [3, 82, 83], examples of this coordinate regulation include the activation of Raptor for mTOR1 activation and the inhibition of the TSC1-TSC2 complex, which is an inhibitor of mTOR. Active mTOR1 inhibits autophagy via the phosphorylation of numerous canonical autophagy mediators including the inhibition of Ulk1 complex via direct and indirect effects [77, 84]. On the other hand, under starvation conditions, active TSC1-TSC2 inhibits mTOR1 leading to the activation of the Ulk1 complex and downstream regulatory complexes. This ultimately leads to the initiation of autophagy. The mechanisms whereby amino acids levels modulate mTOR1 activity are being defined [85].
AMPK is a nutrient sensing kinase that is activated by the elevated ratio of the AMP:ATP nucleotides in the cell. AMPK activation triggers a multitude of energy production and sparing effects (see recent review [86]), which includes the activation of autophagy [87]. Here, the activation of AMPK directly interacts and activates Ulk1 via the phosphorylation of distinct serine residues [84]. The activation of Ulk1 complex therefore appears to function as a common node in both AMPK activation and mTOR1 inhibition nutrient-sensing induction of autophagy.
It has recently been recognized that a mitochondrial associated BH3 domain protein Beclin 1 also functions in a nutrient dependent manner in the modulation of autophagy in part via its interaction with other BH3 domain proteins including Bcl-2 or Bcl-xl. When bound to these antiapoptotic proteins, Beclin 1 is precluded from interacting with its cognate partners in the Beclin/class III PI3K complex. However, under starvation conditions Beclin 1 dissociates from Bcl-2 (reviewed [88]).
4.3. Acetylation as a post-translational modifier of autophagy
Numerous post-translational modifications (PTMs) are utilized in the regulation of autophagy including phosphorylation, lipidation and ubiquitination. Emerging evidence suggests that acetylation is another major PTM orchestrating the regulation of autophagy (and mitophagy) in a nutrient-dependent manner. The nutrient-sensing deacetylase Sirt1 is induced by starvation and modifies numerous proteins in the nucleus and cytosol. Evidence suggests that it plays a major role in autophagy induction during nutrient depletion through deacetylation of canonical autophagy mediators. Interestingly, the transient overexpression of Sirt1 is sufficient to induce autophagy in the absence of nutrient deprivation and Sirt1 KO MEFs display attenuated starvation-activated autophagy [79]. In parallel with this phenotype, Sirt1 directly interacts with and deacetylates canonical autophagy mediators, including Atg5, 7 and 8 [79]. In a concomitant pattern, the knockdown of the acetyltransferase protein p300 diminished acetylation of Atg5, 7, 8 and 12, thereby enabling autophagy [80]. Additionally the overexpression of p300 impairs starvation-induced autophagy [80]. Taken together it appears that acetylation status of canonical autophagy mediators regulates autophagy induction in a nutrient dependent manner by the longevity protein Sirt1 and counter-regulated by the acetyltransferase p300. In addition, Sirt1 has been found to drive autophagy via its deacetylation and activation of FoxO3, a transcription factor that regulates the expression of the GTP-binding protein Rab7 and mediates late autophagosome-lysosome fusion [89].
Interestingly, and via a completely distinct mechanism, the cytosolic microtubule-associated histone deacetylase HDAC6 also functions to stimulate autophagosome-lysosome fusion [90]. The mechanisms underpinning this are discussed in the context of mitophagy in the following section. Recently, another histone deacetylase in the same family as HDAC6, HDAC10, was shown to influence autophagy. Loss of HDAC10 disrupts autophagic flux leading to the accumulation of autophagic structures in neuroblastoma cells suggesting that acetylation status regulates autophagy in cancer. It is thought that tumor cells use autophagy as a defense mechanism when exposed to chemotherapy agents. HDAC10 has been shown to deacetylate and activate heat shock 70 protein family members, which plays a role in lysosomal protein degradation and membrane integrity during the final stage of autophagy. Therefore HDAC10 may function in tumor cells to augment lysosomal function and the autophagic process through its actions on Hsp70 [91].
Overall, emerging evidence suggests that numerous deacetylases regulate global autophagy at a variety of stages of the autophagic process through modulation of the ‘acetylome.’ This concept of acetylation-mediated macroautophagy is also evident using pharmacologic compounds through the induction of autophagy by resveratrol, which promotes sirtuin activity and deacetylation, and by spermidine, which functions as an inhibitor of acetylation [92]. Administering spermidine to yeast leads to deacetylation of histone H3 with subsequent upregulation of various autophagy transcripts including ATG7, 11 and 15. This augmented autophagy was essential for attenuated oxidative stress and necrosis ultimately promoting longevity in yeast [93]. Furthermore, global changes in the acetylome using the acetyltransferase inhibitor spermidine and the sirtuin activator resveratrol trigger autophagy in independent but converging mechanisms. Resveratrol mediated autophagy induction requires Sirt1, while spermidine acts independently of Sirt1 suggesting that these compounds use different mechanisms to trigger autophagy. Despite these potential different mechanisms it appears that both compounds trigger similar changes in the global acetylome of the cell [92]. Also of note, spermidine and resveratrol alter the acetylation status of over 100 proteins that are part of the human autophagy system network [94]. It should be noted that although resveratrol and spermidine are thought to promote protein deacetylation, treatment with these compounds promotes both deacetylation and acetylation changes in the global acetylome. Therefore autophagy induction through global deacetylation or acetylation changes appear not to be unidirectional. This is highlighted by the fact that alterations in the acetylome of autophagy mediators is not directly concordant with the modulation of autophagy, as the histone acetyltransferase Esa1 directly acetylates Atg3 to increase autophagy in yeast [95]. Additionally, in cancer cells the disassociation of the cytosolic deacetylase Sirt2, from the transcription factor FoxO1, promotes autophagy [96]. Here, the acetylation of FoxO1 facilitates its interaction, in a transcription independent manner, with Atg7 to augment autophagy [96].
Taken together, these data make a strong case for Sirt1 and HDAC6 dependent deacetylation in starvation-induced autophagy. However, the findings discussed above also suggest a more complex role for acetylation PTMs in the overall control of these nutrient-dependent and independent autophagic housekeeping programs. Furthermore, in metazoans it has recently been shown that the acetylation of Ulk1, by a nuclear acetyltransferase (TIP60), in response to growth factor deprivation, can activate autophagy [97]. Taking all these findings into account suggest a more complex system that is not completely characterized. Nevertheless, what is becoming apparent is that acetylation plays a major role in the regulation of macroautophagy.
5. Acetylation in the control of selective mitophagy
5.1 Mitophagy as a regulated and selective housekeeping program
Mitophagy, a mitochondrial specific form of autophagy, is vital for homeostatic control of this organelle. Mitophagy is essential for: depletion of mitochondria during the maturation of erythrocytes [98]; the post-fertilization removal of mitochondria in embryos [99]; maintenance of mitochondrial genomic integrity [100] and control of mitochondrial homeostasis in response to cellular stressors [12, 101]. Mitochondrial density and function are tissue specific [102] and mature red blood cells lack mitochondria to ensuring efficient systemic oxygen delivery and fertilized eggs degrade sperm-derived mitochondria to retain maternal mitochondrial genome inheritance, respectively. These two cell types epitomize the biological extreme of mitochondrial clearance through the mitophagy program as a mechanism to optimize their respective unique functions. Selective mitochondrial removal is also evident in response to mitochondrial damage, either via chemical mitochondrial uncoupling by carbonyl cyanide m-chlorophenyl hydrazone (CCCP) [103], or through ischemia induced injury that triggers mitophagy to eliminate damaged ROS generating mitochondria [101, 104]. Numerous protein mediators have been identified that link to the mitochondria thereby targeting this organelle for sequestration by the autophagolysome machinery [12, 105]. The elimination of defective mitochondria through mitophagy has potentially interesting implications in aging [106] and age-related diseases such as neurodegeneration [107]. Although it has not been comprehensively demonstrated in mammalia, the mitophagy program has been shown to be central to maintenance of mitochondrial content and integrity in yeast [100]. Here, wildtype yeast maintains mitochondrial content and genomic integrity in response to nitrogen deprivation stress. In contrast, yeast devoid of the mitophagy receptor Atg32 display augmented mitochondrial mass with increased levels of damaged mitochondria due to an inability to properly regulate mitophagy. This creates a vicious cycle of accumulated damaged mitochondria generating oxidative stress that further leads to a loss of mitochondrial genome integrity following nitrogen deprivation. To date, a mammalian equivalent of ATG32 has not been described.
5.2 Regulatory proteins operational in selective mitophagy
In addition to the (de)acetylation of lysine residues of autophagy proteins by mediators such as Sirt1 as described above other PTMs appear to be crucial for mitophagy regulation. The ubiquitination of lysine residues on mitochondrial proteins has been identified as an important ‘signature’ in the initiation of mitophagy. This was initially uncovered when the E3-ubiquitin ligase Parkin was found to target damaged mitochondrial outer membrane proteins for ubiquitination as a mitophagy initiating event [103]. The molecular program orchestrating this has been more extensively characterized and involves multiple proteins. An important outer mitochondrial membrane (OMM) protein involved in this regulation is the serine-threonine kinase Pink1. Under basal conditions, Pink1 is degraded by mitochondrial proteases such as mitochondrial processing peptidase (MPP) and presenilin-associated rhomboid-like protease (PARL) [108]. However, in response to extensive mitochondrial injury, Pink1 levels accumulate on the OMM enabling it to attract and phosphorylate the cytosolic protein Parkin. The enrichment of this E3-ligase on the OMM directly ubiquitinates constitutive OMM proteins including: VDAC1; mitofusin-1 and -2 (Mfn1 & 2) and Miro. It is interesting to note that the Mitofusins are central to mitochondrial fusion and ubiquitination promotes their degradation, which is mediated by the proteosome and p97. Inhibition of fusion would yield smaller fragmented or “bite sized” mitochondria that would be physically ideal for degradation in autophagosomes. Additional evidence of the importance of fragmented mitochondria for mitophagy is the fact that Drp1 (a fission mediator) knockout MEFs are impaired in fasting and uncoupling induced mitophagy [109-111]. A fundamental role for mitochondrial dynamics during mitophagy is also supported by the fact that acute fasting promotes hyper-fusion of mitochondria via protein kinase A mediated inhibition of Drp1. This hyperfused state of mitochondria is thought to prevent mitochondrial degradation by mitophagy during short periods of nutrient limitations although mitochondria are eliminated following chronic starvation conditions [112]. The role of the modulation of mitochondrial dynamics in mitophagy is probably more complex than initially proposed, as Parkin has also been shown to target and promote the degradation of Drp1 [113]. Also, the Parkin target Miro facilitates mitochondrial mobility along microtubules, and its ubiquitination inhibits mitochondrial motility [114]. This is proposed to contribute to damaged mitochondrial immobility, a physical property that probably is conducive to efficient mitophagy.
Parkin mediated ubiquitination of OMM proteins targets mitochondria to autophagosomes. The decoration of the OMM with ubiquitinated lysine residues attracts the adaptor protein p62/Sequestosome1 [115] from the cytosol to the mitochondria. p62 also binds to LC3-II (Atg8) on the autophagosomes [116]. Although p62 has been found to be an important mediator in the initiation of mitophagy, p62 chaperone independent programs are also operational [105]. Also, the integrated roles of mitophagy and the proteosome in mitochondrial turnover is elegantly illustrated where the Pink1/Parkin/Ubiquitin/p62/LC3-II cascade occur in parallel with the degradation of the OMM components by the proteosome [117]. Here, mitochondrial depolarization initiates OMM protein degradation via the p97 AAA+-ATPase protease family member, in a Parkin dependent manner [117]. As an E3-ligase, Parkin has numerous other substrates that are probably not directly linked to mitophagy and those pathways are not discussed further here [118-120]. However, Parkin can also link mitophagy to the macroautophagy machinery by directly binding to a protein within the the Beclin1/class III phosphatidylinotisol 3-kinase (PI3K) complex [121] to promote autophagosome nucleation.
In light of the large number of E3-ubiquitin ligases, it is not surprising that additional family members can also initiate this program. To date it has been found that the overexpression of the mitochondrial localized RNF185 E3-ligase induces BNIP1 ubiquitination on the OMM, with subsequent p62 co-localization and targeting of mitochondria to autophagosomes [122]. Also the E3-ligase Smurf1, has been shown to function in concert with Parkin in CCCP-induced mitophagy [123].
In contrast to ubiquitin and p62 dependent mitophagy, alternate mitochondrial linked ‘receptors’ have been identified that can directly interact with LC3-II to sequester mitochondria to the autophagosome. Examples of such OMM ‘receptors’ include Nix/Bnip3L and Bnip3, which as BH3 domain proteins can directly interact with LC3-II [124, 125]. This mammalian mechanism mirrors a program operational in yeast where Atg32 on the OMM directly interacts with the autophagosome, although ATG32 and Nix do not show homology [126]. Interestingly, ‘nutrient’ sensing transcription factors such as HIF-1, and Foxo3, regulate expression of Nix and BNIP3. This is also linked to acetylation, in that Sirt3 mediated deacetylation of Foxo3 in response to redox stressors has also been shown to drive mitophagy [127]. Additionally, another OMM mitophagy ‘receptor’ protein that has recently identified is called FUNDC1. Under an oxygen-replete milieu FUNDC1 is inhibited in response to Src kinase phosphorylation, while under hypoxia this inhibition is relieved and FUNDC1 directly binds to LC3-II to promulgate mitophagy [128]. Various proposed mechanisms of autophagy are depicted in Figure 2.
Fig 2.
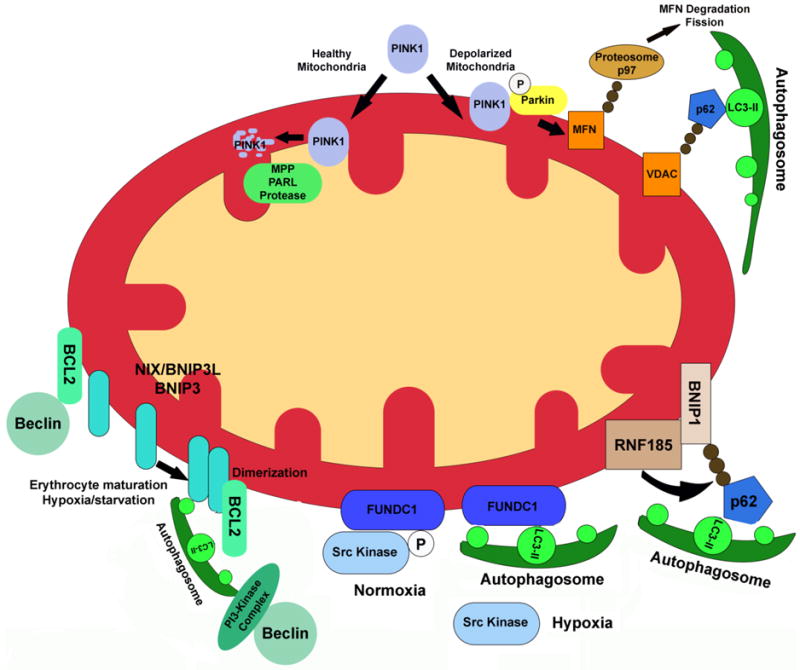
Multiple mechanisms are employed in the degradation of mitochondria through mitophagy. Pink1 protein levels are stabilized in depolarized mitochondria, following a reduction in its proteolytic processing by PARL, which triggers the activation of the E3 ubiquitin ligase, Parkin. Parkin ubiquitinylates mitochondrial membrane proteins, which concurrently blocks mitochondria fusion (via the degradation of mitofusin proteins) and directs depolarized organelles to autophagosomes, mediated by p62 and LC3-II. Both hypoxia and cellular differentiation induce mitophagy via NIX and BNIP3, which homo-dimerize and directly bind to LC3-II during autophagosome formation. This dimerization is also thought to free Beclin1 from its interaction with anti-apoptotic BCL-2 family proteins. Hypoxia has also been shown to block FUNDC1 phosphorylation, which results in FUNDC1 binding to LC3-II. Finally, the mitochondria-localized E3 ligase RNF185 ubiquitinylates BNIP3, leading to the targeting of mitochondria to autophagosomes via p62 and LC3-II. (B-cell lymphoma 2, BCL-2; BCL2/adenovirus E1B 19 kDa protein-interacting protein 1/3, BNIP1/3; ring finger protein 185, RNF185; nucleoporin 62, p62; PTEN-induced putative kinase 1, Pink1; voltage dependent anion channel, VDAC; mitofusin, MFN; Presenilins-associated rhomboid-like protein, PARL).
5.3 Acetylation as a nutrient-sensing PTM in mitophagy
The emerging understanding of mitophagy programs has uncovered the role of ubiquitination and phosphorylation PTMs in regulating organelle degradation. As acetylation is both a nutrient sensing PTM and important in the modulation of the activity of canonical cytosolic autophagy regulators [79, 80], it is not surprising that this PTM is also emerging as an important sensor in the mitophagy induction program. In a fashion somewhat reminiscent of the role of Miro, the deacetylases Sirt2 and HDAC6 negatively regulate mitochondrial movement on microtubules in response to their deacetylation of α-tubulin. Although the role of Sirt2 in mitophagy does not appear to have been directly explored, HDAC6 has been found to also play a role in Parkin mediated mitophagy [129]. In response to CCCP administration, HDAC6 interacts with the OMM, where its effect on microtubule function is proposed to augment mitophagy by stalling mitochondrial motility, as a putative promoter of the aggregome [129]. In addition to negatively regulating mitochondrial motility, HDAC6 promotes autophagosome-lysosomal fusion by deacetylating Cortactin and promoting F-actin remodeling [90]. Therefore, an emerging concept is that Parkin enables mitophagy by both promoting the immobilization of damaged mitochondria and by catalyzing their subsequent ubiquitination for damaged mitochondrial targeting to autophagosomes [12, 90, 129].
Similarly to the role of Sirt1 in modifying macroautophagy [79, 80], the mitochondrial deacetylase Sirt3 is activated, and a component of the mitochondrial acetyltransferase machinery, GCN5L1, are downregulated by nutrient deprivation [18, 63]. By extension, the question arises as to whether the mitochondrial acetylation profile can modulate mitophagy. We find that the isolated genetic depletion of Sirt3 does not appreciably modulate mitophagy. However, in stark contrast, GCN5L1 knockdown MEFs show robust accumulation of LC3-II and p62 on mitochondria. Using various cell lines and knockdown MEFs where GCN5L1 was depleted by either lentiviral shRNA or genetically, we have identified that the augmentation of mitophagy in response to GCN5L1 depletion, and concurrent restricted mitochondrial deacetylation, is associated with: the robust increase in mitochondrial protein ubiquitination; is dependent on Atg5 and p62; and does not require Parkin [63]. Interestingly, mitophagy induction through GCN5L1 depletion improved mitochondrial homeostasis as measured by reduced ROS generation and MPTP opening [63]. Major outstanding issues to be resolved in this mitochondrial deacetylation initiated mitophagy are: does the deacetylation of lysine residues of mitochondrial proteins enable or facilitate ubiquitylation in an allosteric manner (as shown for extra-mitochondrial proteins [130]); what are the targets of mitochondrial protein ubiquitination and, if ubiquitination of OMM is driving this mitophagy induction, how does the depletion of GCN5L1 promote this PTM? Interestingly, the siRNA screen utilized by Orvedahl et al. that found SMURF1 regulates mitophagy also found that GCN5L1 may regulate mitophagy [123].
Despite no effect under basal conditions, Sirt3, in response to oxidative stress, initiated mitophagy via deacetylation of FoxO3 with the subsequent upregulation of Nix and Bnip3 [127] suggesting that Sirt3 and GCN5L1 are working opposite each other to regulate mitophagy function through global changes in the mitochondrial acetylome. Further work will be required to investigate whether this exogenous stress-induced loss of mitochondrial proteins shares features with the mitophagy program induced by discrete mitochondrial protein deacetylation in response to GCN5L1 depletion, i.e. is there overlap in targets of Sirt3 and GCN5L1 mediated (de)acetylation during mitophagy induction. Although not explored to date, a potential mechanism whereby changes in mitochondrial acetylation may activate mitophagy could result from metabolic change mediated reactive oxygen signaling. This mechanism has recently been found to drive mitophagy in response to the modest blunting of electron transfer chain activity in drosophila [131]. Additionally, via as yet undefined retrograde signaling, the genetic depletion of GCN5L1 also activates a master regulator of lysosomal biogenesis [64], which in itself may potentiate autophagy. These and other potential pathways whereby GCN5L1 and/or Sirt3 may regulate these programs are illustrated in Figure 3. Additionally, when combined with the actions of cytosolic (de)acetylases, it appears that the acetylation plays a significant role in mitochondrial autophagy, dynamics, turnover and homeostasis [129, 132, 133].
Fig 3.
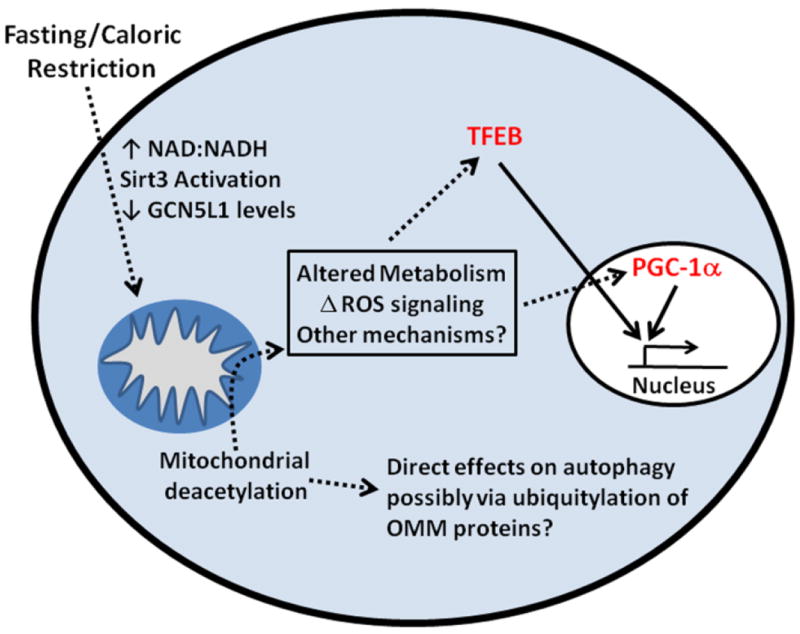
A schematic model of the role of mitochondrial deacetylation in the modulation of selective mitophagy. Fasting or caloric restriction result in the deacetylation of mitochondrial proteins via activation of Sirt3 and the downregulation of GCN5L1. Through an unknown signaling mechanism, perhaps by the direct ubiquitinylation of mitochondrial proteins or via retrograde signaling, mitophagy is initiated. In turn, lysosomal biogenesis in upregulated via the lysosome biogenesis transcription factor EB (TFEB), which is countered be reciprocal upregulation of the mitochondrial biogenesis master regulator peroxisome-proliferator activated receptor gamma, coactivator 1 alpha (PGC-1α). Together these pathways drive mitophagy, mitochondrial turnover and mitochondrial replacement via the mitochondrial biogenesis program.
6. Conclusions/Future Directions
Our understanding of how autophagy and mitophagy play a pivotal role in maintaining cellular homeostasis is advancing in parallel with the recognition that acetylation, as a major nutrient sensing PTM, is an integral regulatory component of these programs. This review highlights the integration of these two concepts and we propose that the increased understanding of these programs may identify approaches to modify the autophagy/mitophagy program in pathology amelioration. The potential to modify these pathways to ameliorate disease is already being suggested in that the inhibition of Sirt2 and HDAC6 has been shown to have beneficial effects in models of Huntington, Parkinson and Alzheimer disease [50, 134, 135], suggesting that the control of mitochondrial motility plays a role in the pathogenesis of neurodegeneration. Although, whether this is directly linked to autophagy or mitophagy in neuronal cell quality control, remains to be addressed. More direct evidence where the disruption of autophagy and mitophagy exacerbate pathology has been demonstrated in fatty liver disease, in pressure-overload and ischemia mediated heart disease and in the exacerbation in immune responses [74, 104, 136, 137]. Taken together these early studies suggest that as we advance our understanding of the nutrient regulatory role in augmenting the autophagy/mitophagy housekeeping programs we should gain insight and potential targets to study to prevent or reverse nutrient excess associated diseases.
Highlights.
Nutrient levels modulate the protein acetylation
The acetylation of proteins modifies protein function
The autophagy and mitophagy quality control programs are regulated by acetylation
Mitochondrial acetylation is integral in mitochondrial quality control
Acknowledgments
This study was supported by the Division of Intramural Research of the National Heart Lung and Blood Institute of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Altun G, Akansu B, Altun BU, Azmak D, Yilmaz A. Deaths due to hunger strike: post-mortem findings. Forensic Sci Int. 2004;146:35–38. doi: 10.1016/j.forsciint.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 2.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 3.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 4.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 5.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, Levine B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Claerhout S, Dutta B, Bossuyt W, Zhang F, Nguyen-Charles C, Dennison JB, Yu Q, Yu S, Balazsi G, Lu Y, Mills GB. Abortive autophagy induces endoplasmic reticulum stress and cell death in cancer cells. PLoS One. 2012;7:e39400. doi: 10.1371/journal.pone.0039400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- 10.Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I, Michaud M, Madeo F, Tavernarakis N, Kroemer G. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madorsky I, Opalach K, Waber A, Verrier JD, Solmo C, Foster T, Dunn WA, Jr, Notterpek L. Intermittent fasting alleviates the neuropathic phenotype in a mouse model of Charcot-Marie-Tooth disease. Neurobiol Dis. 2009;34:146–154. doi: 10.1016/j.nbd.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xiong Y, Guan KL. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J Cell Biol. 2012;198:155–164. doi: 10.1083/jcb.201202056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31:449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Z, Scott I, Webster BR, Sack MN. The emerging characterization of lysine residue deacetylation on the modulation of mitochondrial function and cardiovascular biology. Circ Res. 2009;105:830–841. doi: 10.1161/CIRCRESAHA.109.204974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyl transferase program; a novel role for GCN5L1. Biochem J. 2012;443:627–634. doi: 10.1042/BJ20120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, Sack MN. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic Biol Med. 2010;49:1230–1237. doi: 10.1016/j.freeradbiomed.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, Bao J, Gius D, Sack MN, Jing E, Kahn CR, Friedman JE, Jonscher KR. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433:505–514. doi: 10.1042/BJ20100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez-Marcos PJ, Jeninga EH, Canto C, Harach T, de Boer VC, Andreux P, Moullan N, Pirinen E, Yamamoto H, Houten SM, Schoonjans K, Auwerx J. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Sci Rep. 2012;2:425. doi: 10.1038/srep00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hossain P, Kawar B, El NM. Obesity and diabetes in the developing world--a growing challenge. N Engl J Med. 2007;356:213–215. doi: 10.1056/NEJMp068177. [DOI] [PubMed] [Google Scholar]
- 22.Pagel-Langenickel I, Bao J, Pang L, Sack MN. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev. 2010;31:25–51. doi: 10.1210/er.2009-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rathmell JC, Newgard CB. Biochemistry. A glucose-to-gene link. Science. 2009;324:1021–1022. doi: 10.1126/science.1174665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldrick RB, Hirsch J. Serial studies on the metabolism of human adipose tissue. II. Effects of caloric restriction and refeeding on lipogenesis, and the uptake and release of free fatty acids in obese and nonobese individuals. J Clin Invest. 1964;43:1793–1804. doi: 10.1172/JCI105053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci U S A. 2004;101:6659–6663. doi: 10.1073/pnas.0308291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammer S, Snel M, Lamb HJ, Jazet IM, van der Meer RW, Pijl H, Meinders EA, Romijn JA, de RA, Smit JW. Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases myocardial triglyceride content and improves myocardial function. J Am Coll Cardiol. 2008;52:1006–1012. doi: 10.1016/j.jacc.2008.04.068. [DOI] [PubMed] [Google Scholar]
- 28.Owen OE, Felig P, Morgan AP, Wahren J, Cahill GF., Jr Liver and kidney metabolism during prolonged starvation. J Clin Invest. 1969;48:574–583. doi: 10.1172/JCI106016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, Zhao Y. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 30.Schwer B, Eckersdorff M, Li Y, Silva JC, Fermin D, Kurtev MV, Giallourakis C, Comb MJ, Alt FW, Lombard DB. Calorie restriction alters mitochondrial protein acetylation. Aging Cell. 2009;8:604–606. doi: 10.1111/j.1474-9726.2009.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Picklo MJ., Sr Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochem Biophys Res Commun. 2008;376:615–619. doi: 10.1016/j.bbrc.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 33.Paik WK, Pearson D, Lee HW, Kim S. Nonenzymatic acetylation of histones with acetyl-CoA. Biochim Biophys Acta. 1970;213:513–522. doi: 10.1016/0005-2787(70)90058-4. [DOI] [PubMed] [Google Scholar]
- 34.Wagner GR, Payne RM. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J Biol Chem. 2013;288:29036–29045. doi: 10.1074/jbc.M113.486753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, Covington JD, Bajpeyi S, Ravussin E, Kraus W, Koves TR, Mynatt RL. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012;15:764–777. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schrenk DF, Bisswanger H. Measurements of electron spin resonance with the pyruvate dehydrogenase complex from Escherichia coli. Studies on the allosteric binding site of acetyl-coenzyme A. Eur J Biochem. 1984;143:561–566. doi: 10.1111/j.1432-1033.1984.tb08406.x. [DOI] [PubMed] [Google Scholar]
- 37.Ghanta S, Grossman R, Brenner C. Mitochondrial protein acetylation as a cell-intrinsic, evolutionary driver of fat storage: chemical and metabolic logic of acetyl-lysine modifications. Critical Reviews in Biochemistry & Molecular Biology. 2013 doi: 10.3109/10409238.2013.838204. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riccio A. New endogenous regulators of class I histone deacetylases. Sci Signal. 2010;3:pe1. doi: 10.1126/scisignal.3103pe1. [DOI] [PubMed] [Google Scholar]
- 39.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 40.Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- 41.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 42.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwer B, Verdin E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008;7:104–112. doi: 10.1016/j.cmet.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 45.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Webster BR, Lu Z, Sack MN, Scott I. The role of sirtuins in modulating redox stressors. Free Radic Biol Med. 2012;52:281–290. doi: 10.1016/j.freeradbiomed.2011.10.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 48.Sack MN, Finkel T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a013102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verdin E, Hirschey MD, Finley LW, Haigis MC. Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci. 2010;35:669–675. doi: 10.1016/j.tibs.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 51.Lynn EG, McLeod CJ, Gordon JP, Bao J, Sack MN. SIRT2 is a negative regulator of anoxia-reoxygenation tolerance via regulation of 14-3-3 zeta and BAD in H9c2 cells. FEBS Lett. 2008;582:2857–2862. doi: 10.1016/j.febslet.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Narayan N, Lee IH, Borenstein R, Sun J, Wong R, Tong G, Fergusson MM, Liu J, Rovira II, Cheng HL, Wang G, Gucek M, Lombard D, Alt FW, Sack MN, Murphy E, Cao L, Finkel T. The NAD-dependent deacetylase SIRT2 is required for programmed necrosis. Nature. 2012;492:199–204. doi: 10.1038/nature11700. [DOI] [PubMed] [Google Scholar]
- 53.Bao J, Lu Z, Joseph JJ, Carabenciov D, Dimond CC, Pang L, Samsel L, McCoy JP, Jr, Leclerc J, Nguyen P, Gius D, Sack MN. Characterization of the murine SIRT3 mitochondrial localization sequence and comparison of mitochondrial enrichment and deacetylase activity of long and short SIRT3 isoforms. J Cell Biochem. 2010;110:238–247. doi: 10.1002/jcb.22531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Jr, Weissman S, Verdin E, Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol. 2007;27:8807–8814. doi: 10.1128/MCB.01636-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV, Jr, Kahn CR, Verdin E. SIRT3 Deficiency and Mitochondrial Protein Hyperacetylation Accelerate the Development of the Metabolic Syndrome. Mol Cell. 2011 doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hallows WC, Yu W, Smith BC, Devires MK, Ellinger JJ, Someya S, Shortreed MR, Prolla T, Markley JL, Smith LM, Zhao S, Guan KL, Denu JM. Sirt3 Promotes the Urea Cycle and Fatty Acid Oxidation during Dietary Restriction. Mol Cell. 2011;41:139–149. doi: 10.1016/j.molcel.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, Westphall MS, Pagliarini DJ, Prolla TA, Assadi-Porter F, Roy S, Denu JM, Coon JJ. Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol Cell. 2013;49:186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC, Baar K, Schenk S. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation following endurance exercise. J Biol Chem. 2011;286:30561–30570. doi: 10.1074/jbc.M111.261685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Webster BR, Scott I, Han K, Li JH, Lu Z, Stevens MV, Malide D, Chen Y, Samsel L, Connelly PS, Daniels MP, McCoy JP, Jr, Combs CA, Gucek M, Sack MN. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J Cell Sci. 2013;126:4843–4849. doi: 10.1242/jcs.131300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scott I, Webster BR, Chan CK, Okonkwo JU, Han K, Sack MN. GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J Biol Chem. 2013 doi: 10.1074/jbc.M113.521641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mortimore GE, Poso AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;7:539–564. doi: 10.1146/annurev.nu.07.070187.002543. [DOI] [PubMed] [Google Scholar]
- 66.Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 69.Hubbard VM, Valdor R, Macian F, Cuervo AM. Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology. 2012;13:21–35. doi: 10.1007/s10522-011-9331-x. [DOI] [PubMed] [Google Scholar]
- 70.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 73.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 74.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 76.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, Cecconi F. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol. 2013;15:406–416. doi: 10.1038/ncb2708. [DOI] [PubMed] [Google Scholar]
- 78.Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, Pagliarini V, Matteoni S, Fuoco C, Giunta L, D’Amelio M, Nardacci R, Romagnoli A, Piacentini M, Cecconi F, Fimia GM. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155–168. doi: 10.1083/jcb.201002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem. 2009;284:6322–6328. doi: 10.1074/jbc.M807135200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ichimura Y, Komatsu M. Pathophysiological role of autophagy: lesson from autophagy-deficient mouse models. Exp Anim. 2011;60:329–345. doi: 10.1538/expanim.60.329. [DOI] [PubMed] [Google Scholar]
- 82.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 83.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 84.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martina JA, Puertollano R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 2013;200:475–491. doi: 10.1083/jcb.201209135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wong PM, Puente C, Ganley IG, Jiang X. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013;9:124–137. doi: 10.4161/auto.23323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao TP. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oehme I, Linke JP, Bock BC, Milde T, Lodrini M, Hartenstein B, Wiegand I, Eckert C, Roth W, Kool M, Kaden S, Grone HJ, Schulte JH, Lindner S, Hamacher-Brady A, Brady NR, Deubzer HE, Witt O. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc Natl Acad Sci U S A. 2013;110:E2592–2601. doi: 10.1073/pnas.1300113110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Morselli E, Marino G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, Cabrera S, Benit P, Rustin P, Criollo A, Kepp O, Galluzzi L, Shen S, Malik SA, Maiuri MC, Horio Y, Lopez-Otin C, Andersen JS, Tavernarakis N, Madeo F, Kroemer G. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol. 2011;192:615–629. doi: 10.1083/jcb.201008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, Ma C, Sun Y, Zhang S, Feng W, Zhu L, Le Y, Gong X, Yan X, Hong B, Jiang FJ, Xie Z, Miao D, Deng H, Yu L. Function and molecular mechanism of acetylation in autophagy regulation. Science. 2012;336:474–477. doi: 10.1126/science.1216990. [DOI] [PubMed] [Google Scholar]
- 96.Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12:665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 97.Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Ruan K, Wang Z, Zhang CS, Chien KY, Wu J, Li Q, Han J, Lin SC. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science. 2012;336:477–481. doi: 10.1126/science.1217032. [DOI] [PubMed] [Google Scholar]
- 98.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- 100.Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, Kang D. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem. 2012;287:3265–3272. doi: 10.1074/jbc.M111.280156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yogalingam G, Hwang S, Ferreira JC, Mochly-Rosen D. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase C delta (deltaPKC) inhibits mitochondrial elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J Biol Chem. 2013 doi: 10.1074/jbc.M113.466870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Johnson DT, Harris RA, French S, Blair PV, You J, Bemis KG, Wang M, Balaban RS. Tissue heterogeneity of the mammalian mitochondrial proteome. Am J Physiol Cell Physiol. 2007;292:C689–697. doi: 10.1152/ajpcell.00108.2006. [DOI] [PubMed] [Google Scholar]
- 103.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012;111:1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Takeda K, Yoshida T, Kikuchi S, Nagao K, Kokubu A, Pluskal T, Villar-Briones A, Nakamura T, Yanagida M. Synergistic roles of the proteasome and autophagy for mitochondrial maintenance and chronological lifespan in fission yeast. Proc Natl Acad Sci U S A. 2010;107:3540–3545. doi: 10.1073/pnas.0911055107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, Waddington SN, Schapira AH, Duchen MR. Mitochondria and quality control defects in a mouse model of Gaucher disease-links to Parkinson’s disease. Cell Metab. 2013;17:941–953. doi: 10.1016/j.cmet.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;301:H1924–1931. doi: 10.1152/ajpheart.00368.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kristensen AR, Schandorff S, Hoyer-Hansen M, Nielsen MO, Jaattela M, Dengjel J, Andersen JS. Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics. 2008;7:2419–2428. doi: 10.1074/mcp.M800184-MCP200. [DOI] [PubMed] [Google Scholar]
- 113.Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem. 2011;286:11649–11658. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Seibenhener ML, Du Y, Diaz-Meco MT, Moscat J, Wooten MC, Wooten MW. A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim Biophys Acta. 2013;1833:452–459. doi: 10.1016/j.bbamcr.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim KY, Stevens MV, Akter MH, Rusk SE, Huang RJ, Cohen A, Noguchi A, Springer D, Bocharov AV, Eggerman TL, Suen DF, Youle RJ, Amar M, Remaley AT, Sack MN. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J Clin Invest. 2011;121:3701–3712. doi: 10.1172/JCI44736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim KY, Sack MN. Parkin in the regulation of fat uptake and mitochondrial biology: emerging links in the pathophysiology of Parkinson’s disease. Curr Opin Lipidol. 2012;23:201–205. doi: 10.1097/MOL.0b013e328352dc5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Muller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, Lichtenthaler SF, Motori E, Hrelia S, Wurst W, Trumbach D, Langer T, Krappmann D, Dittmar G, Tatzelt J, Winklhofer KF. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. 2013;49:908–921. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- 121.Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, Vandenberghe W. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci. 2011;31:10249–10261. doi: 10.1523/JNEUROSCI.1917-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tang F, Wang B, Li N, Wu Y, Jia J, Suo T, Chen Q, Liu YJ, Tang J. RNF185, a novel mitochondrial ubiquitin E3 ligase, regulates autophagy through interaction with BNIP1. PLoS One. 2011;6:e24367. doi: 10.1371/journal.pone.0024367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Orvedahl A, Sumpter R, Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, Forst CV, Wrana JL, Zhang YE, Luby-Phelps K, Xavier RJ, Xie Y, Levine B. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, Gustafsson AB. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011;18:721–731. doi: 10.1038/cdd.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kondo-Okamoto N, Noda NN, Suzuki SW, Nakatogawa H, Takahashi I, Matsunami M, Hashimoto A, Inagaki F, Ohsumi Y, Okamoto K. Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J Biol Chem. 2012;287:10631–10638. doi: 10.1074/jbc.M111.299917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tseng AH, Shieh SS, Ling Wang D. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med. 2013 doi: 10.1016/j.freeradbiomed.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 128.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 129.Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bosch-Presegue L, Raurell-Vila H, Marazuela-Duque A, Kane-Goldsmith N, Valle A, Oliver J, Serrano L, Vaquero A. Stabilization of Suv39H1 by SirT1 is part of oxidative stress response and ensures genome protection. Mol Cell. 2011;42:210–223. doi: 10.1016/j.molcel.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 131.Owusu-Ansah E, Song W, Perrimon N. Muscle Mitohormesis Promotes Longevity via Systemic Repression of Insulin Signaling. Cell. 2013;155:699–712. doi: 10.1016/j.cell.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kim C, Choi H, Jung ES, Lee W, Oh S, Jeon NL, Mook-Jung I. HDAC6 inhibitor blocks amyloid beta-induced impairment of mitochondrial transport in hippocampal neurons. PLoS One. 2012;7:e42983. doi: 10.1371/journal.pone.0042983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shieh JM, Wei TT, Tang YA, Huang SM, Wen WL, Chen MY, Cheng HC, Salunke SB, Chen CS, Lin P, Chen CT, Wang YC. Mitochondrial apoptosis and FAK signaling disruption by a novel histone deacetylase inhibitor, HTPB, in antitumor and antimetastatic mouse models. PLoS One. 2012;7:e30240. doi: 10.1371/journal.pone.0030240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, Marsh JL. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Govindarajan N, Rao P, Burkhardt S, Sananbenesi F, Schluter OM, Bradke F, Lu J, Fischer A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol Med. 2013;5:52–63. doi: 10.1002/emmm.201201923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J, Dorn GW, 2nd, Brady MJ, Macleod KF. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol. 2012;32:2570–2584. doi: 10.1128/MCB.00167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]