

Abstract
Histone side chains are post-translationally modified at multiple sites, including at Lys36 on histone H3 (H3K36). Several enzymes from yeast and humans, including the methyltransferases SET domain-containing 2 (Set2) and nuclear receptor SET domain- containing 1 (NSD1), respectively, alter the methylation status of H3K36, and significant progress has been made in understanding how they affect chromatin structure and function. Although H3K36 methylation is most commonly associated with the transcription of active euchromatin, it has also been implicated in diverse processes, including alternative splicing, dosage compensation and transcriptional repression, as well as DNA repair and recombination. Disrupted placement of methylated H3K36 within the chromatin landscape can lead to a range of human diseases, underscoring the importance of this modification.
The packaging of DNA with basic histones and a vast array of additional factors mediates the formation of chromatin and thereby determines the outcome of virtually all of the DNA processes in eukaryotes. Acetylation and methylation of histones were originally identified in radiolabelling studies using cell extracts1,2. Today, enzymes catalysing numerous histone post-translational modifications or ‘marks’ have been identified; these include phosphorylation, various modes of methylation at Arg and Lys side chains, acetylation and ubiquitylation. These marks are thought to exist in dynamic combinations to generate a ‘code’, or ‘language’, that can enforce the regulatory features of chromatin during nearly all of the aspects of cellular metabolism: a given modification or permutation of modifications dictates a distinct biological output. Given the critical biological roles of chromatin and the numerous pathologies related to its misregulation, understanding the role of histone modifications is a paramount, yet complicated task. In this Review, we focus on our current understanding of a key modification, the methylation of Lys36 at histone H3 (H3K36). Widely described to be associated with active chromatin, H3K36 methylation has also been implicated in transcriptional repression, alternative splicing, dosage compensation, DNA replication and repair, DNA methylation and the transmission of the memory of gene expression from parents to offspring during development. We highlight the specificity, regulation and functional role of the enzymes that methylate H3K36, but we refer readers to informative reviews for a detailed discussion of the counteracting demethylases3–5.
Regulating the methylation of H3K36
Histone methyltransferase (HMTase) enzymes use S-adenosylmethionine to add methyl groups to specific histone Lys or Arg residues. To date, at least eight distinct mammalian enzymes have been described that methylate H3K36 in vitro and/or in vivo (FIG. 1). All of the H3K36-specific methyltransferases identified thus far have the catalytic SET domain in common, but they have varying preferences for Lys36 residues in different methylation states. In yeast, SET domain-containing 2 (Set2) performs all three methylation events at H3K36 (REF. 6), but in higher eukaryotes there is accumulating evidence that these events require a division of labour between the mono- and dimethylases and the SET2-type trimethylases (FIG. 1). Several of these enzymes also possess multiple chromatin-interacting domains, including those known to interact with methylated H3K36 itself (such as the PWWP domain) and with additional methylated histone residues (such as plant homeodomain fingers (PHD fingers))7–9 (FIG. 1). Notably, SET2 proteins from multiple species have a carboxy-terminal domain that interacts with the large subunit of RNA polymerase II (RNAPII), which is known as RNAPII subunit B1 (RPB1)10.
Figure 1. Domain structures of enzymes that methylate H3K36.

a | A schematic of the enzymes that have been shown to promote the formation of methylated Lys36 on histone H3 (H3K36). With the exception of fly maternal-effect sterile 4 (MES-4), only human enzymes are shown. The SET domain is shown with its pre (AWS) and post domains; C5HCH is a zinc-finger (ZNF) domain; WW domains are known to interact with Pro-rich peptides; the PWWP domain is known to interact with trimethylated H3K36; and the AT hook is a DNA-binding domain. All domain assignments were derived from Ensembl. b | Depiction of the transitions between the multiple H3K36 methylation states, highlighting the enzymes that have been shown to function in changing a given methylation state. ASH1L, ASH1-like; BAH, bromo- associated homology; BROM, bromodomain; HMG, high mobility group; MYND, myeloid, Nervy and DEAF-1 ZNF; NIDs, nuclear receptor interaction domains; NSD, nuclear receptor SET domain-containing; PHD, plant homeodomain; RuBisCo, ribulose-1,5-bisphosphate carboxylase oxygenase; SETD, SET domain-containing; SETMAR, SET domain and mariner transposase fusion gene-containing; SMYD2, SET and MYND domain-containing 2.
Determining HMTase specificity
The most parsimonious scenario would predict that each H3K36 methyltransferase enzyme catalyses a transition between two distinct methylation states (for example, between unmethylated H3K36 and monomethylated H3K36 (H3K36me1)). However, this is not necessarily the case. There are many discrepancies as to the level of methylation (mono-, di- and tri-) imparted, as well as the residue (or residues) that is targeted, by the various H3K36-specific methyltransferases; these discrepancies may arise from multiple factors, including the nature of the substrate tested (for example, peptides and histones versus nucleosomes), the source of enzyme (for example, full length versus SET domain only) and the assay conditions themselves (for example, antibody specificity versus mass spectrometry). Physiologically relevant substrates, characterized using nucleosomes and/or loss-of-function experiments, have been determined for some H3K36 methyltransferases (TABLE 1); but for enzymes such as SETD3, the enzymatic activities reported so far are based on analyses of peptides and core histones11,12.
Table 1.
Reported substrate specificities for enzymes that methylate H3K36
| Enzyme* | Assay conditions | Histone substrates identified | Refs |
|---|---|---|---|
| NSD1 (KMT3B) | Nucleosomes | H3, H3K36, H3K36me1, H3K36me2, H4K20 | 14, 16–18 |
| Cell based and/or in vivo | H3K36me1, H3K36me2, H4K20me3 | 17–19, 78 | |
| NSD2 (WHSC1, MMSET) | Nucleosomes | H3, H3K36, H3K36me1, H3K36me2 | 16,17, 19,30 |
| Cell based and/or in vivo | H3K27me2‡, H3K36me2 | 17,28, 36 | |
| NSD3 (WHSC1L1) | Nucleosomes | H3, H3K36 | 16 |
| Cell based and/or in vivo | H3K36me2 | 40 | |
| ASH1; ASH1L | Nucleosomes | H3, H3K36, H3K36me2, H3K36me3§ | 41,48, 49 |
| Cell based and/or in vivo | H3K4, H3K4me3 | 49 | |
| SMYD2 | Core histones (octamers) | H3, H3K4, H3K36me2 | 42,43 |
| SETMAR (METNASE) | Core histones (octamers) | H3K4, H3K36me2 | 12 |
| Cell based and/or in vivo | H3K36me2 | 96 | |
| SETD2 (KMT3A, HYPB) | Nucleosomes | H3, H3K36me1, H3K36me2, H3K36me3 | 34,64 |
| Cell based and/or in vivo | H3K36me3 | 25,33,34 | |
| SETD3 | Core histones (octamers) | H3K4, H3K36 | 11 |
Table entries for the knockdown (cell based) or knockout (in vivo) analysis indicate a reported decrease in a given methylation state. ASH1, Absent, small and homeotic discs 1; ASH1L, ASH1-like; H, histone; NSD, nuclear receptor SET domain-containing; me1, monomethylated; me2, dimethylated; me3, trimethylated; SETD, SET domain-containing; SETMAR, SET domain and mariner transposase fusion gene-containing; SMYD2, SET and MYND domain-containing 2.
Alternative names are provided in brackets.
RE-IIBP isoform of NSD2.
Gln2265Ala mutant of ASH1L.
NSD1 as a mono- and dimethylase for H3K36
Although it was originally shown to bind steroid nuclear receptors13, nuclear receptor SET domain-containing 1 (NSD1; also known as KMT3B) was subsequently reported to use its SET domain to methylate H3K36 as well as H4K20 (REF. 14). However, NSD1 also methylates non-histone substrates, such as the p65 subunit of nuclear factor-κB (NF-κB)15. Indeed, several HMTases have multiple substrates, including non-histones, and it is important to consider this when interpreting phenotypes derived from loss-of-function experiments.
NSD1 has specific mono- and dimethylase activity for H3K36 (REFS 16–18), generating H3K36me1 and dimethylated H3K36 (H3K36me2), and there are also mixed reports as to whether it has specificity for H4K20 (REFS 14,19,20). Enzymatic assays using recombinant nucleosomes containing unmethylated H3K36 or a mimic of H3K36me1 (REF. 21) were shown to serve as specific substrates for NSD1 (REF. 16). However, when histone octamers were used as substrates, NSD1 was found to additionally methylate histone H4 as well as histones H2A and H2B, suggesting that nucleosomes contribute to H3K36 specificity. Structural data have also confirmed the specificity of NSD1 as a mono- and dimethylase that targets H3K36 (REF. 18). Intriguingly, this study showed that the substrate-binding channel of NSD1 is blocked by an autoinhibitory loop that resides between the SET and post-SET domains. As Lys36 resides near the core of the nucleosome, interactions between the enzyme and DNA may allosterically relieve inhibition by this loop, a result that is consistent with the preference of NSD enzymes for nucleosome substrates, as opposed to octamers. This furtherunderscores the necessity for using physiologically relevant substrates in order to properly characterize enzyme specificity towards H3K36.
NSD1 controls the levels of methylated H3K36 within and surrounding the body of the bone morphogenic protein 4 (BMP4) gene in human HCT116 colorectal cancer cells17. In the absence of NSD1, BMP4 expression is significantly impaired and the levels of all three forms of methylated H3K36 are reduced within the body of the gene. These data could support a model in which NSD1 catalyses H3K36 trimethylation (H3K36me3), but a more likely scenario is that the loss of mono- and dimethylation at H3K36 also inhibits trimethylation by failing to provide another HMTase, SETD2 (also known as KMT3A and HYPB), with its substrate. This is an important distinction to make when considering which HMTases regulate the formation of a trimethylated residue.
The role of NSD1 as a mono- and dimethylase also seems to be conserved in other metazoans. Worms and flies each have one NSD1 orthologue called maternal-effect sterile 4 (MES-4)22,23. In Caenorhabditis elegans, MES-4 has been reported to be a dimethylase that is specific for H3K36 (REF. 22). Similarly to NSD1, Drosophila melanogaster MES-4 catalyses global mono- and dimethylation of H3K36 in vivo, but SET2, the fly orthologue of human SETD2, regulates all trimethylation at this site; again, MES-4 is probably required to provide the proper dimethylated H3K36 substrate for the SET2 enzyme.
In worms, mutation of the SETD2 orthologue histone methyltransferase-like 1 (met-1) significantly reduced global H3K36me3 levels24. This supports the notion that the worm MES-4 and MET-1 proteins coordinate dimethylation and trimethylation, respectively24. These experiments are generally consistent with studies of mammalian NSD proteins and SETD2 (REFS 16,25). By contrast, separate studies showed by immunostaining that worm embryos individually deficient in mes-4 or met-1 retain significant levels of H3K36me3, but embryos doubly deficient in mes-4 and met-1 are completely devoid of H3K36me3 (REFS 26,27). This suggests that MES-4 collaborates with MET-1 to perform trimethylation. As each of these studies has used different antibodies, it will be important to validate these experiments using additional, antibody-independent techniques, such as mass spectrometry. Collectively, the combination of in vitro and in vivo data suggests that NSD1 and its orthologues probably catalyse the addition of either the monomethyl or dimethyl groups onto H3K36 and so indirectly regulate the levels of trimethylation by altering the availability of monomethyl and dimethyl substrates for the trimethylating enzymes.
NSD2 as a mono- and dimethylase for H3K36
Similarly to NSD1, NSD2, the product of Wolf–Hirschhorn syndrome candidate gene 1 (WHSC1; also known as MMSET), has been reported to target H3K36, as well as H4K20, H3K4 and H3K27 (REFS 16,28–32). Here again, these discrepancies may lie, at least in part, with the nature of the substrates used in the assays. For example, NSD2 acts as a dimethylase towards H3K36 when presented with nucleosomes, but it preferentially dimethylates H4K44 when presented with octamers16. Interestingly, when short single-stranded and double-stranded DNA molecules that are notably smaller than the length that is necessary to generate a nucleosome are added to octamers, NSD2 preferentially dimethylates H3K36, a result that has been attributed to the ability of DNA to act as an allosteric effector of NSD2 (REF. 16). This is consistent with another study, in which mass spectrometry and immunoblotting against native and recombinant nucleosomes showed that NSD2, as well as its major splicing variant, RE-IIBP, acts as a mono- and dimethylase specific for H3K36 (REF. 30). By contrast, there have been reports that the NSD2 isoform RE-IIBP is an H3K27-specific HMTase28 and that H3K36 trimethylation is lost in embryonic stem (ES) cells derived from NSD2-defective mice32. But the idea that NSD2 might act as a trimethylase would not be consistent with a previous study that identified SETD2 as the sole trimethylase in mammalian cells25. Furthermore, H3K36me3 levels are reduced when SETD2 is depleted, despite normal levels of H3K36me2 (REFS 33,34). Regardless, these data are all consistent with a role for NSD2 in H3K36 methylation. This is also supported by the observation that, in ~15% of patients with multiple myeloma (a haematological malignancy that accounts for 1% of all cancers35), NSD2 is overexpressed as a result of a translocation with the immunoglobulin locus (t(4;14)+) and the global levels of H3K36me2 are increased36.
Possible specificity of NSD2 for H4K20
Despite strong evidence linking NSD2 to H3K36me, it may also act as an H4K20-specific methyltransferase during the cellular response to DNA double-strand breaks (DSBs)37,38. NSD2 was isolated in an RNA interference screen for genes involved in resistance to hydroxyurea, a DNA-replication inhibitor38. Consistently, NSD2-defective cells are sensitive to DNA damage and NSD2 localizes to sites of DNA damage. Moreover, NSD2 is phosphorylated at Ser102 by the kinase ataxia telangiectasia mutated (ATM) upon damage37, and this mediates the recruitment of NSD1 to sites of DNA damage, where it has been suggested to dimethylate H4K20, which is an important event in recruiting the DNA damage response regulator p53-binding protein 1 (53BP1)39. Although one could envision that NSD2 phosphorylation and damaged DNA may both alter the catalytic specificity of NSD2 through allosterism, it should be noted that recombinant NSD2 methylates H4 in in vitro assays with H4 peptides37. Thus, it is possible that NSD2 does have some specificity for H4 in addition to H3 even in the absence of DNA damage.
The Set2 trimethylase
In yeast, Set2 is non-essential, performs all three methylation reactions at H3K36, and couples H3K36me3 with transcriptional elongation through an interaction with Rpb1, the large subunit of RNAPII10. The human orthologue of Set2, SETD2, also interacts with RNAPII during elongation, but it is an essential protein and has been identified as a huntingtin-interacting protein. The role of SETD2 in Huntington’s disease is obscure. SETD2 is a trimethylase25,33,34, and some reports have indicated that, in vitro, recombinant human SETD2 can add all three methyl groups to H3K36 (REFS 32,34). By contrast, in vivo knockdown experiments targeting SETD2 revealed reduced levels of H3K36me3 only34. This underscores the importance of using multiple approaches to determine enzyme specificity and also raises an important point about enzyme processivity. Does a trimethylase obligatorily require a dimethyl substrate or can it processively place all three groups on a Lys substrate? SETD2 associates with heterogeneous nuclear ribonucleoprotein L (hnRNPL), and hnRNPL knockdown analyses show decreased levels of H3K36me3 but not H3K36me1 or H3K36me2 (REF. 34). Thus, the ability to act as a processive enzyme could depend on the presence and regulation of distinct cofactors, as well as on the presence of a previously methylated substrate.
Defining further HMTase specificities for H3K36
Like other NSD family members, the HMTase NSD3 (also known as WHSC1L1) appears to be specific for H3K36 (REFS 16,40). So far, relatively little is known about the substrate specificities of the Trithorax protein ASH1-like (ASH1L) (an orthologue of the fly protein Absent, small and homeotic discs 1 (ASH1)), SET domain and mariner transposase fusion gene-containing (SETMAR; also known as METNASE), SETD3 and SET and MYND domain-containing 2 (SMYD2). Each has been reported to methylate H3K36, particularly with respect to dimethylation, but other substrate specificities have also been described for these enzymes12,41–43. However, the assays for SETD3 specificity were performed with peptides11, and it will be important to confirm this enzyme specificity using nucleosome-based assays16,44.
In addition to targeting H3K36, SMYD2 methylates H3K4 and non-histone substrates, such as p53 and retinoblastoma (RB)45,46. The in vitro methylase activity of SMYD2 towards H3K4, but not towards H3K36, depends on its ability to bind heat shock protein 90α (HSP90α)42. Although the expression of SMYD2 appears to be largely confined to the brain and heart, its conditional deletion from mouse cardiomyocytes showed that it is dispensable for heart development and, surprisingly, its deletion had no effect on the global levels of methylated H3K36 (REF. 47). This may suggest that the physiological target for SMYD2 is a non-histone substrate, providing an additional example of how the interpretation of knockout phenotypes for HMTases must be carefully considered.
Rigorous experiments using recombinant nucleosomes have demonstrated that ASH1L is a dimethylase that is specific for H3K36 (REFS 41,48). However, ASH1L has also been reported to target H3K4 and to localize within the transcribed regions of active target genes49; and in vivo levels of H3K4me3 were reduced in the absence of ASH1L. Additional studies have shown that ASH1L also targets H3K36 and H4K20 (REFS 44,46). Thus, it remains to be seen what the full substrate repertoire of ASH1L is. Notably, similarly to NSD1, ASH1L has an autoinhibitory loop that resides between the SET and post-SET domains, which is a feature that has been proposed as a hallmark feature of H3K36-specific enzymes41,48.
Additional factors that influence H3K36 methylation
Several mechanisms have been described for how H3K36 methylation levels are regulated at a given locus (REFS 50–52). In addition to cofactors such as HSP90α and hnRNPL, Pro isomerization of histone H3 and additional histone modifications on both H3 and H4 have been shown to regulate the total levels of H3K36 methylation by Set2, although it is not clear how this occurs. Additional studies in yeast have shown that cyclins (encoded by bypass UAS requirement 1 (BUR1) and BUR2) are required for H3K36me3 (REF. 53). Moreover, the yeast histone chaperone anti-silencing function 1 (Asf1), the RNAPII kinase C-terminal domain (CTD) kinase 1 (Ctk1) and the elongation factor Spt6 (a subunit of the FACT (facilitates chromatin transcription) complex) all regulate the levels of H3K36 trimethylation but not dimethylation54,55. Other elongation factors, such as polymerase-associated factor 1 (Paf1), have also been shown to alter the levels of H3K36 methylation56. Large cells 1 (Lge1), a factor associated with the ubiquitin pathway, was also identified in a genome-wide screen for candidate genes that specifically regulate H3K36 methylation57. This study also confirmed the role of Paf1 in regulating H3K36me3: mutations in Paf1 disrupted the levels of H3K36me3, but not those of H3K36me2 (REF. 56). Collectively, these studies suggest an emerging theme: that HMTase cofactors may alter substrate specificity on a gene-to-gene basis, providing an additional layer of regulation for their activity at distinct targets.
Roles of H3K36 methylation in gene expression
Methylation of H3K36 has been observed at multiple stages of RNA biosynthesis and is not always associated with gene activation. Rather, its ultimate output and where it fits into gene expression pathways are functions of multiple variables: where in the gene body the methylated H3K36 mark is placed, when H3K36 is methylated and what reader protein binds to this modification. Further complicating these variables is the fact that the degree of methylation can result in different biological outcomes.
H3K36 methylation and transcriptional activation
Numerous studies in multiple systems support a role for H3K36 methylation in transcriptional activation. It has been observed that, in general, there is a progressive shift from monomethylation to trimethylation of H3K36 between the promoters and the 3′ ends of genes58. However, in zebrafish, somatic cells also show a bias for H3K36me3 in the 3′ end of actively transcribed genes, but this mark is curiously present in the 5′ promoter regions of quiescent genes that are developmentally regulated during spermatogenesis59. The functional significance of this is unknown, and it will be important to determine whether promoter-proximal H3K36me3-containing nucleosomes can recruit repressive histone deacetylases to promote gene silencing.
Multiple lines of evidence in budding yeast have showed that Set2, a non-essential enzyme that is responsible for all three forms of methylated H3K36, is coupled to transcriptional elongation. Deletions in yeast Set2 cause sensitivity to the elongation inhibitor 6-azauracil and phenocopy mutations in other known elongation factors10. However, it remains possible that this phenotype may arise from indirect effects on nucleotide metabolism. Nevertheless, Set2 associates with the hyperphosphorylated form of RNAPII and deposits the trimethyl group onto H3K36 during elongation10,60–62 in various systems, including yeast and humans. This interaction is regulated by phosphorylated residues in the CTD of Rbp1, the large subunit of RNAPII: the Ser2-phosphorylated form of the CTD is indicative of elongation and the Ser5-phosphorylated form is characteristic of a paused polymerase at a promoter63. Indeed, human SET2 proteins also appear to bind RNAPII and to target H3K36 (REFS 34,64).
One well-established function of Set2 in yeast is the prevention of aberrant transcriptional initiation within coding sequences (FIG. 2). Set2 catalyses H3K36 methylation co-transcriptionally and recruits the reduced potassium dependency 3 small (Rpd3S) deacetylase complex here65–67 through association of the chromodomain-containing Rpd3S subunit ESA1- associated factor 3 (Eaf3) with H3K36me3; this enforces a deacetylated chromatin state in the wake of transcribing RNAPII65,68. Rpd3S preferentially associates with histones containing H3K36me2 and H3K36me3 but not H3K36me1 (REF. 57). The PHD finger of the Rpd3S subunit Rco1 can also cooperate with the Eaf3 chromo-domain to promote these interactions69. This indicates that H3K36me2 and H3K36me3 might act redundantly in the Set2–Rpd3S pathway.
Figure 2. H3K36me3-dependent prevention of aberrant transcription in yeast.
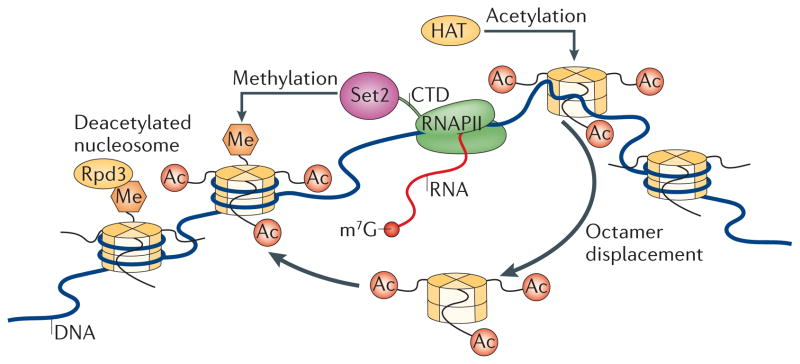
Actively transcribing RNA polymerase II (RNAPII) displaces acetylated nucleosomes. These evicted histones are reincorporated into nucleosomes and chromatin behind the polymerase. As SET domain-containing 2 (Set2) binds RNAPII, this promotes the trimethylation of Lys36 on histone H3 (H3K36me3) in the newly incorporated nucleosomes. H3K36me3 serves as a ‘mark’ that the reduced potassium dependency 3 (Rpd3) deacetylase complex binds; this complex facilitates local nucleosome deacetylation, preventing aberrant, spurious transcription in the wake of RNAPII progression through a region. CTD, carboxy-terminal domain; HAT, histone acetylase; m7G, 7-methylguanosine.
The role of histone Lys methylation in the maintenance of a repressive chromatin environment during transcriptional elongation is also used in humans but may be independent of acetylation70. In this case, Lys demethylase 2 (LSD2; also known as KDM1B) demethylates methylated H3K4 in the intragenic regions of active target genes. LSD2 resides in complexes with NSD3, which acts as a mono- and dimethylase that is specific for H3K36 (REF. 16). Additionally, LSD2 binds the H3K9-specific HMTase G9A (also known as EHMT2). Consistent with this, the LSD2 complex has robust HMTase activity towards H3K9 and, to a lesser extent, H3K36 (REF. 70). Moreover, in chromatin immuno-precipitation followed by microarray (ChIP–chip) experiments, depletion of G9A alters both H3K9 methylation levels and transcriptional regulation at LSD2 targets. In light of these observations and the fact that LSD2 resides in complexes with elongation factors such as cyclin T1 (CCNT1; also known as PTEFb) and the Ser2-phosphorylated form of RNAPII, it appears that the methylation-dependent enforcement of a repressive chromatin state through H3K9 and possibly H3K36 methylation at actively transcribed genes is evolutionarily conserved. Additionally, it seems that NSD3 can participate in transcriptional elongation70. Interestingly, the LSD2 complex contains multiple proteins with PWWP domains, including NSD3. Originally identified in NSD2, PWWP domains also bind H3K36me3 to act as reader proteins7,8,71. In particular, the PWWP domain of DNA methyltransferase 3A (DNMT3A) binds H3K36me3 and methylates nearby DNA, demonstrating a link between H3K36me3 and DNA methylation that could represent a new mechanism for H3K36 methylation-mediated repression of gene expression.
H3K36 methylation in dosage compensation
Coordination between methylation and acetylation also occurs during dosage compensation in D. melanogaster72. To compensate for having only one X chromosome, male flies use the Male-specific lethal (MSL) complex to upregulate the expression of X-linked genes by a factor of two in order to achieve the same level of transcription as females, which have two X chromosomes. This may occur through a two-step model requiring H3K36me3 and increased elongation73–75. The MSL complex is initially recruited to X-linked genes through a GA-rich recognition site and subsequently facilitates transcriptional elongation through an enhancement of RNAPII activity. Next, the MSL3 subunit (an orthologue of yeast Eaf3) is thought to promote spreading of the upregulated state through interactions that involve the chromodomain of MSL3 and H3K36me3 (REF. 73). Additional active marks, such as H4K16 acetylation, also affect X-linked gene expression, requiring the Males absent on the first (MOF) subunit of MSL. Thus, a collection of activating marks together achieves chromosome-wide gene expression74. Although interactions between chromodomain proteins and H3K36me3 marks regulate acetylation in both yeast and D. melanogaster, they have opposite effects: transcription is repressed in yeast but upregulated in flies. This clearly illustrates that the function of a given histone modification, such as H3K36me3, is context-dependent and is much more complicated than a simple code.
NSD enzymes and transcriptional initiation
NSD1 regulation of H3K36 methylation appears to affect transcriptional initiation17. For example, NSD1 binds upstream of the promoter of BMP4 and regulates the levels of H3K36me1, H3K36me2 and H3K36me3 within the body of the gene. This seems to be required for the recruitment of RNAPII to the BMP4 promoter, an observation that links NSD1 to initiation events through RNAPII. NSD3 can also bind promoters and then influence the levels of H3K36 methylation within the body of a gene40. For example, the extraterminal domains of bromodomain-containing 4 (BRD4) specifically recruit NSD3 to the promoter region of the gene encoding CCND1 and the decapping enzyme DCPS, and loss of either BRD4 or NSD3 reduces the levels of H3K36me3, particularly within the body of the gene. This is consistent with the NSD1-dependent regulation of BMP4, during which promoter-proximal bound NSD1 regulates H3K36me3 levels within the body of the gene17. Although NSD3 localization is biased towards the promoter of CCND1, it also can be found, albeit at reduced levels, at the 3′ end of the gene40, suggesting that NSD3 participates in initiation and elongation events. Moreover, NSD3 levels were found to be higher in the coding regions than in the promoter of the proto-oncogene Ser/Thr kinase PIM2 (REF. 40). Thus, NSD3 appears to reside in multiple complexes (including LSD2 and BRD4 complexes) and can localize to either promoter or internal regions to promote H3K36 methylation and thereby influence initiation and elongation processes. This is in apparent contrast to NSD1, which has been reported to localize primarily near the 5′ ends of its targets17; it will be important to confirm this using high-resolution ChIP followed by sequencing (ChIP–seq) analysis. However, previous ChIP–seq data show that NSD2 is enriched near transcription start sites30.
H3K36 methylation in transcriptional repression
Fascinating clues have emerged regarding the relationship between methylation at H3K36, transcriptional repression and worm development26,27. In worms, maternally provided MES-4 is essential for germ cell viability and is involved in X-chromosome silencing in the germline26,27. Early primordial germ cells (PGCs) do not engage in transcription as they lack the active hyperphosphorylated CTD form of RNAPII. But, in the absence of mes-4, active RNAPII persists, indicating that MES-4 is important for the establishment and/or maintenance of transcriptional repression in late stage PGCs26,27. Loss of MES-4 also reduces the levels of H3K36me3, independently of RNAPII function. Thus, MES-4 maintains the H3K36me3 state in germline precursors independently of transcription — an observation that has not been made in other organisms so far.
Beyond H3K36 methylation-mediated regulation of spurious initiation, H3K36 methylation-mediated repression has also been reported. Yeast Set2 represses transcription from a lacZ reporter and, in Set2-deletion mutants, the basal levels of GAL4 transcription are increased76,77; however, in these studies, Set2 is artificially recruited to the promoter, so it will be important to confirm whether this reflects the true nature of Set2 activity. Initial studies with tethered reporters in mammalian cells have implicated NSD1 in both transcriptional activation and repression13, although its targets have not yet been determined. In addition, an RNA interference study has shown that NSD1 represses the expression of the homeobox regulator MEIS1 in neuronal cell lines19. This repressive event is likely to be direct, as NSD1 localizes to the promoter of MEIS1 (REF. 78). Furthermore, NSD2 collaborates with the cardiac-specific factor NKX2-5 to repress targets such as platelet-derived growth factor-α (PDGFRα), probably through modulating H3K36 methylation levels32. Thus, H3K36 methylation appears to act as both an activating and inhibitory signal, and the overall biological readout might depend on the context of additional surrounding marks and their corresponding reader proteins.
H3K36 methylation and exon definition
There has been increasing support for the idea that biased nucleosome positioning favours exonic over intronic sequences79–81. Such a bias seems to be accompanied by distinct histone modifications, including H3K36me3. This should not be surprising, as the 147-base-pair length of a DNA fragment associated with a histone octamer correlates with the average length of an internal exon. This is likely to be more than just mathematical serendipity; it is probably evidence of interplay between chromatin and splicing. Several large-scale bioinformatics studies have analysed both the positions of nucleosomes and their modification status within the genomes of humans, C. elegans, D. melanogaster and mice79–81. In each case, nucleosomes were enriched specifically at exonic sequences. Although the increased deposition of nucleosomes at exons guarantees a bias in histone modifications within exons relative to those within introns, it is also clear that a subset of modifications is specifically enriched here. This is particularly true for H3K36me3 but also includes methylation at H3K79, H4K20 and H2BK5 (REF. 80). Each analysis also found that the H3K36me3 bias is more pronounced within exons further downstream of the transcription start site. This preference may reflect the propensity of RNAPII to abort transcription early in the transcription cycle, thereby reducing the number of nucleosomes that are displaced further downstream. The known association between Set2 and the RNAPII CTD may also further explain the particular increase in H3K36me3 signatures seen at downstream exons61,82. The implication of nucleosome enrichment and the increase in H3K36me3 modifications is twofold. First, nucleosomes probably act as intrinsic pause sites for elongating RNAPII which could alter splice site recognition and, hence, change exon inclusion. Others have found that the introduction of pause sites within minigenes can increase the inclusion of alternatively spliced exons83,84. Furthermore, expression of a ‘slow’ mutant RNAPII in D. melanogaster results in different inclusion patterns of the exons within the Ultrabithorax mRNA85. A second possibility, which is not mutually exclusive with effects on RNAPII pausing, is that the H3K36me3 modification relays a specific signal to the splicing machinery to alter how it defines exons, leading to the specific inclusion or exclusion of particular exons.
Although the global analyses of H3K36me3 positioning in various genomes provide compelling evidence that this modification affects splicing, functional evidence beyond this has been lacking. However, an interesting connection has been made between SETD2, the reader protein MORF-related gene 15 (MRG15; which contains a chromodomain) and polypyrimidine tract-binding protein (PTB), the last of which is a known antagonist of exon definition that affects splicing of fibroblast growth factor receptor 2 (FGFR2) pre-mRNA86,87. FGFR2 contains two mutually exclusive exons (IIIb and IIIc) that encode a region within the extracellular immunoglobulin-like domain and are each responsible for receptor binding to a unique range of FGFs88. Exon IIIb is included in epithelial cells through the action of epithelial splicing regulatory protein (ESRP), whereas exon IIIc is included in cells of mesenchymal origin89 (FIG. 3). Furthermore, the splicing pattern switches from exon IIIb to exon IIIc inclusion as prostate epithelial cells become androgen-independent, an important factor in metastasis. Analysis of nucleosome modifications throughout FGFR2 show that H3K36me3 is specifically enriched within exon IIIb and is restricted to mesenchymal cells, which exclude this exon. This H3K36me3 modification is recognized by MRG15, which also interacts with PTB. Thus, by recruiting PTB to its target exon, these interactions position PTB to bind to its intronic splicing silencer sites, which flank the repressed exon as they emerge from the transcribing RNAPII complex (FIG. 3). PTB repression of this exon can be alleviated by downregulating either MRG15 or SETD2. This regulatory module also exists at other alternatively spliced exons, with a bias towards exons that contain weaker PTB-binding sites86. What remains to be seen is how two distinct cell types achieve this differential methylation of H3K36 within nucleosomes at alternatively spliced exons in order to regulate splicing.
Figure 3. H3K36me3 influences alternative splicing in a cell-type specific manner.

The fibroblast growth factor receptor 2 (FGFR2) locus that undergoes alternative splicing consists of two mutually exclusive exons, IIIb and IIIc, which are located between the constitutive exons 7 and 10. Mesenchymal stem cells favour the inclusion of exon IIIc and achieve this by repressing splicing of exon IIIb. Nucleosomes present near exon IIIb contain the SET domain-containing 2 (SETD2)-dependent trimethylated Lys36 on histone H3 (H3K36me3) ‘mark’ and its reader protein MORF-related gene 15 (MRG15). MRG15 also interacts with polypyrimidine tract-binding protein (PTB), a known repressor of exon inclusion, and this may be the mechanism by which the methylated H3K36 mark can influence splicing at this locus. In epithelial cells, FGFR2 expresses exon IIIb but excludes exon IIIc. Epithelial splicing regulatory protein (ESRP) is expressed and stimulates the inclusion of exon IIIb; reduced levels of H3K36me3 present at this exon, possibly as a result of lower SETD2 levels, allow its derepression. The role of dimethylases, such as the proteins of the nuclear receptor SET domain-containing (NSD) family, in this process has yet to be determined but these enzymes could also influence H3K36 methylation here.
This example of FGFR2 control exemplifies how the methylation status of H3K36 can affect splicing. However, this crosstalk is bidirectional: mutations in splice sites that abrogate intron removal of β-globin reporter genes cause a shift in H3K36me3 signatures towards the 3′ region of genes90. Moreover, others have observed that H3K36me3 signatures are markedly lower in genes without introns91. In both cases, inhibition of splicing with pharmacological agents causes a rapid and global redistribution of H3K36me3 throughout the genome and reduces SETD2 recruitment to these genes90,91. Although it is unclear how the spliceosome regulates H3K36 methylation, these studies suggest that it is a general phenomenon.
H3K36 methylation in DNA replication, recombination and repair
As replication origins initiate at distinct times in S phase, the decision to fire at any particular origin depends on a number of factors, including the transcriptional status of nearby genes. Origins of replication are bound by the origin recognition complex (ORC), which recruits various factors, including CDC6, minichromosome maintenance (MCM) proteins and CDC45, before loading of the DNA polymerase. Studies in budding yeast have revealed a role for H3K36 methylation in regulating replication origin firing, as deletions in SET2 cause a delay in Cdc45 loading at origins92. H3K36 methylation has also been linked to DNA replication checkpoint control in fission yeast93. Furthermore, yeast mutants in the FACT elongation complex are sensitive to the replication inhibitor hydroxyurea, and this sensitivity can be suppressed by mutations in SET2 (REF. 94). How H3K36 methylation influences these S phase events is unknown, but it could keep chromatin in an active state, perhaps through recruiting specific reader proteins that can influence DNA replication control.
Histone methylation is also critical for the maintenance of genomic stability, including the biological response to DSBs39,95. For example, H3K36me2 has been implicated in the repair response to DSBs through the non-homologous end-joining (NHEJ) pathway96. Here, the methylase SETMAR, a SET domain-containing enzyme previously reported to affect NHEJ as well as DNA replication, was shown to directly mediate H3K36me2 formation near sites of DSBs96 (FIG. 4). SETMAR-dependent formation of H3K36me2 was shown to enhance the rate of association of the DNA repair factors KU70 and Nijmegen breakage syndrome 1 (NBS1; also known as nibrin) near DSBs96. However, it is not yet clear how methylated H3K36 acts at the sites of DSBs, particularly with respect to which proteins may respond to the presence of this mark, or what relationship this mark might have with other known histone modifications that are important for DSB repair. Most notably, this may include crosstalk with dimethylation of H4K20, an event that recruits the DNA damage response regulator 53BP1 to sites of damage39. It is also possible that methylated H3K36 in this context mediates its effects by antagonizing transcriptional elongation, as RNAPII is displaced in response to DNA damage97.
Figure 4. Model for H3K36 methylation at sites of DNA damage.

Upon the generation of DNA damage, SET domain and mariner transposase fusion gene- containing (SETMAR) dimethylates Lys36 on histone H3 (H3K36) near sites of DNA double-strand breaks (DSBs), possibly by recruiting currently undefined reader proteins that facilitate the binding of KU70 and the MRE11 RAD50 NBS1 (MRN) complex to facilitate DNA repair. How methylation at H3K36 is coordinated with other chromatin modifications that are known to participate in DNA repair (such as, p53-binding protein 1 (53BP1) binding to dimethylated H4K20 (H4K20me2)) is unknown. NSD2, nuclear receptor SET domain-containing 2.
H3K36 methylation: links with disease
In higher eukaryotes, defects in the genes that maintain the levels of H3K36 methylation cause developmental defects and disease32,33,98 (TABLE 2). Defects in SETD2 are causal for sporadic clear renal cell carcinoma, a disease that is marked by the loss of the short arm of chromosome 3 (REF. 99). SETD2 has also been hypothesized to be a tumour suppressor in breast cancer100. Defects in each member of the NSD family have been implicated in multiple diseases and cancer types31,78,101–106. For example, haploinsufficiency in NSD1 is causal for Sotos overgrowth syndrome98. NSD1 has also been implicated in breast, lung and prostate cancer, acute myeloid leukaemia (AML) and refractory anaemia19,31,78,101–103,107–112. NSD1-defective mice display an embryoniclethal phenotype: the embryos initiate mesoderm formation but fail to complete gastrulation, apparently as a result of apoptosis14. NSD2-defective mice die shortly after birth and display features that are consistent with Wolf–Hirschhorn syndrome32, including facial abnormalities and cardiac defects. Thus, NSD1-knockout mice and NSD2-knockout mice have different phenotypes, despite the fact that both proteins catalyse the formation of H3K36me2. No knockout-mouse phenotype has yet been reported for NSD3.
Table 2.
H3K36 methyltransferases and their roles in diseases
| Gene | Diseases | Molecular defects | Phenotypes | Refs |
|---|---|---|---|---|
| NSD1 | Sotos syndrome | Haploinsufficiency, point mutations, deletions, translocations | Macrocephaly, hypertolerism, cognitive and/or motor skill deficiencies | 98 |
| NSD1 | Myelodysplastic syndrome | Translocations | Anaemia, cytopenia | 111 |
| NSD1 | Cancers: AML, prostate, neuroblastoma, breast | Overexpression, gene silencing, translocations | Numerous tumour types | 19,78,101, 110,112 |
| NSD2 | Wolf–Hirschhorn syndrome | Deletions | Learning difficulties, microcephaly, heart defects | 32 |
| NSD2 | Multiple myeloma | t(4;14)+ translocation | Renal failure, anaemia, bone lesions | 30,115 |
| NSD3 | Breast cancer | Gene amplification at 8p11 | Solid tumours | 107 |
| NSD3 | AML | Translocations | Leukaemic cells in bone marrow | 105 |
| NSD3 | Myelodysplastic syndrome | Translocations | Anaemia, cytopenia | 106 |
| SETD2 | Renal cell carcinoma | Deletions, missense mutations | Haematuria, flank pain | 99 |
The table lists the known diseases resulting from alterations in the genes encoding enzymes that are specific for Lys36 on histone H3 (H3K36). AML, acute myeloid leukaemia; NSD, nuclear receptor SET domain-containing; SETD2, SET domain-containing 2.
Each NSD family member behaves as an oncogene in multiple cancers35,78,105,112. Translocations in NSD1 or NSD3 lead to the development of AML105,112. Moreover, NSD1, an androgen receptor co-regulator113, has been identified as a candidate gene capable of discriminating between malignant and non-malignant prostate tissue101. In addition, silencing of NSD1 has also been shown to cause sensitivity to the oestrogen receptor antagonist tamoxifen114, but how these two observations are linked to the ability of NSD1 to bind and regulate either the androgen or oestrogen receptors is unknown. Overexpression of NSD2 has also been implicated in multiple myeloma and additional cancers, such as neuroblastoma102,115, and overexpression of NSD3 via gene amplification occurs in about 15% of breast cancers107 and correlates with poor prognosis. Therefore, NSD proteins are instrumental in the development of cancer, but their mechanisms of action in this context are only beginning to be elucidated. One possible mechanism underlying NSD-mediated disease is that these proteins enforce H3K36 methylation patterns to change gene expression. This is discussed below in the context of well-known NSD-dependent cancers.
Through overexpression or their incorporation into fusion proteins via translocations, NSD proteins act as potent oncoproteins30,78. For example, fusion proteins between NSD1 and the nucleoporin NUP98 are well established as being causal for a subset of AML112, and they regulate homeobox (Hox) gene expression in a mouse model of AML78. In the case of AML, NSD1–NUP98 fusions enforce an H3K36 methylation signal that contributes to the inappropriate activation of Hox genes during development. These increased H3K36 methylation levels seem to be antagonistic to the repressive methylated H3K27 marks that normally silence Hox gene expression, leading to reduced H3K27 methylation levels and gene activation. As a consequence, unscheduled cell proliferation and the generation of AML are observed in the mouse model78.
Overexpression of NSD2 via a translocation between chromosome 4 and chromosome 14 alters the global profile of H3K36 methylation in KMS11 multiple myeloma cells30,36. Moreover, the catalytic activity of NSD2 is required for tumorigenesis in a mouse xenograft model30. And loss of NSD2 function (through either short hairpin RNA-mediated depletion or homologous recombination) globally decreases H3K36me2 and H3K36me3 levels but has no effect on H4K20 or H3K4 methylation36. It is, however, accompanied by a concomitant decrease in the global levels of H3K27me2 and H3K27me3, and this downregulation results in the expression of target genes that would normally be quiescent. This suggests that the overexpression of NSD2 alters transcriptional programming by promoting the formation of H3K36me2 and thereby changing the chromatin structure. Indeed, ChIP–seq analysis has precisely mapped the distribution of H3K36me2 in KMS11 multiple myeloma cells overexpressing NSD2 (REF. 30). In cells that have one normal allele of NSD2, the H3K36me2 signal revealed by ChIP–seq was preferentially enriched in intragenic regions, which is consistent with previous ChIP–chip results in flies23. However, overexpression of NSD2 disperses an increased H3K36me2 signal throughout the genome, including intergenic regions. Cells expressing one normal allele of NSD2 have a modest H3K36me2 signal in promoter regions, followed by a peak at transcriptional start sites, and then the signal decays downstream to the 3′ end30, and a high signal corresponds with higher transcriptional activation. By contrast, cells overexpressing NSD2 have little variance in H3K36me2 signal intensity across the average gene and the correlation between signal levels and transcription is lost. Instead, as some genes are much more sensitive to H3K36me2 levels than others, this aberrant enrichment of the H3K36me2 signal appears to trigger the expression of quiescent oncogenes, including transforming growth factor alpha (TGFA), MET, p21 activated kinase 1 (PAK1) and RRAS2 (related RAS viral (r-ras) oncogene homologue)30.
H3K36 and H3K27 methylation may be mutually exclusive modifications44. Nucleosomes pre-methylated at H3K27 are largely refractory to enzymatic catalysis at H3K36 and vice versa. As overexpression of NSD3 is causal for the generation of breast cancer through 8p11–12 amplifications107,109, one exciting possibility is that global gene expression profiles are drastically altered through a switch from H3K27 to H3K36 methylation. When considering this and the fact that antagonism between H3K36 and H3K27 methylation has also been observed in worms22, one might anticipate that this could be a general mechanism for the oncogenic behaviour observed for enzymes that promote H3K36 or H3K27 methylation (FIG. 5). But NSD proteins are not solely restricted to acting as oncoproteins; they can also act as tumour suppressors, as a lack of NSD1 expression is observed in neuroblastomas19.
Figure 5. NSD proteins can act as oncoproteins.

Through overexpression and/or translocation events that result in the fusion of nuclear receptor SET domain-containing (NSD) proteins with other proteins, such as the nucleoporin NUP98, NSD proteins can be aberrantly recruited to target loci in various tissues. As a consequence, global levels of dimethylated Lys36 on histone H3 (H3K36me2) increase and are sufficient to activate inappropriate transcription, which contributes to cancer development. In some cases, the increased levels of H3K36me2 are expected to inversely correlate with trimethylated H3K27 (H3K27me3; not shown), altering the balance between competitive activating and repressive ‘marks’. As a consequence, multiple gene sets that are sensitive to the levels of H3K36me2 are turned on, a causal event in oncogenesis. AML, acute myeloid leukaemia; AR, androgen receptor; RNAPII, RNA polymerase II.
Conclusions
Although widely perceived as an activating modification, H3K36 methylation also functions in transcriptional repression as well as in processes such as DNA repair. Thus, H3K36 methylation function extends beyond transcription and is likely to be relevant in a wide range of DNA-based processes. So far, much effort has gone into understanding the nature of the enzymes and their substrate specificities, but little is known about how the enzymes that mediate H3K36 methylation are localized and regulated beyond the well-characterized model of yeast Set2 and its relationship with RNAPII. Given that NSD1 binds various nuclear receptors, it is clear that this H3K36-specific methyltransferase, and probably others, operates in distinct tissues and perhaps in separate complexes. Similarly, NSD2 interacts with different transcription factors in mouse ES cells versus heart cells from embryonic day 12.5 (REF 32). Thus, elucidating the distinct complexes that these enzymes form in different cell types and at different stages of development will be important. As mammalian cells possess multiple, non-redundant enzymes that methylate H3K36 to varying degrees, the roles of this modification are expected to be more complex than in yeast. Despite this, it is apparent that a role for methylated H3K36 in coordinating crosstalk between acetylation and methylation is conserved between flies and higher eukaryotes116. This crosstalk results in a given H3K36 modification that is interpreted by the reader protein in the context of neighbouring histone modifications, potentially changing its meaning. Undoubtedly, this ‘combinatorial histone language’ will be the subject of future studies of methylated H3K36 that will expand its scope of importance to the chromatin landscape.
Acknowledgments
We apologize to those researchers whose work could not be cited owing to space constraints. We thank B. Martinez and K. Eterovic for assistance with the figures, as well as S. Ercan, P. Masamha and A. Sataluri for critical comments regarding the manuscript. Work in the laboratory of P.B.C. has been supported in part by the Robert A. Welch Foundation (AU-1569). Work in the laboratory of E.J.W. is supported by the US National Institutes of Health (5R00GM080447).
Glossary
- Dosage compensation
The mechanism by which expression levels from sex chromosomes are adjusted. In mammalian systems, one copy of the X chromosome is silenced in the female. By contrast, in Drosophila, genes on the male X chromosome are expressed at twofold levels
- SET domain
(Suppressor of variegation 3–9, Enhancer of Zeste and Trithorax domain). A catalytic domain that uses S-adenosylmethionine to transfer methyl groups to substrates
- PWWP domain
(Pro-Trp-Trp-Pro domain). A chromatin-interacting domain that has recently been shown to bind trimethylated Lys36 on histone H3
- Plant homeodomain fingers
(PHD fingers). Motifs that bind zinc and have been shown to bind methylated residues
- Allosteric
Pertaining to the process by which a binding event at one site influences the activity of an enzyme or protein at a second, distant site. This may lead to activation or inhibition by cooperation between ligands, when a ligand bound at one site affects the affinity of another site for its ligand by inducing transitions between distinct conformational states
- Heterogeneous nuclear ribonucleoprotein L
(hnRNPL). A member of a highly abundant family of RNA-binding proteins known to associate with newly synthesized pre-mRNA
- Pro isomerization
Peptide bonds to Pro residues can exist in either a cis or a trans state, and the transitions can be catalysed by prolyl isomerase enzymes
- Anti-silencing function 1
(Asf1). A histone chaperone protein that associates with newly synthesized histones and is involved in chromatin synthesis
- Reader protein
A protein that recognizes and binds post-translational modifications on histones
- Chromodomain
A domain of ~50 residues that has been shown to bind methylated residues
- Polypyrimidine tract-binding protein
(PTB). A protein that has been implicated as an antagonist of exon definition, the action of which results in the repression of exon inclusion
- Fibroblast growth factor receptor 2
(FGFR2). A membrane-bound receptor that undergoes alternative splicing and is subject to regulation by methylation of Lys36 on histone H3
- Epithelial splicing regulatory protein
(ESRP). An alternative splicing factor that is enriched in epithelial tissues and is responsible for enforcing specific exon inclusion
- Non-homologous end joining
(NHEJ). The main pathway that is used primarily in the G1 phase of the cell cycle to repair chromosomal DNA double-strand breaks in somatic cells. In contrast to homologous-recombination repair, NHEJ is error-prone because it leads to the joining of heterologous ends
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Contributor Information
Eric J. Wagner, Email: Eric.J.Wagner@uth.tmc.edu.
Phillip B. Carpenter, Email: Phillip.B.Carpenter@uth.tmc.edu.
References
- 1.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allfrey VG, Mirsky AE. Structural modifications of histones and their possible role in the regulation of RNA Synthesis. Science. 1964;144:559. doi: 10.1126/science.144.3618.559. [DOI] [PubMed] [Google Scholar]
- 3.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nature Rev Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 4.Varier RA, Timmers HT. Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Acta. 2011;1815:75–89. doi: 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Rotili D, Mai A. Targeting histone demethylases: a new avenue for the fight against cancer. Genes Cancer. 2011;2:663–679. doi: 10.1177/1947601911417976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JS, Shilatifard A. A site to remember: H3K36 methylation a mark for histone deacetylation. Mutat Res. 2007;618:130–134. doi: 10.1016/j.mrfmmm.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 7.Dhayalan A, et al. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vezzoli A, et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nature Struct Mol Biol. 2010;17:617–619. doi: 10.1038/nsmb.1797. [DOI] [PubMed] [Google Scholar]
- 9.Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks. Mutat Res. 2008;647:3–12. doi: 10.1016/j.mrfmmm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kizer KO, et al. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eom GH, et al. Histone methyltransferase SETD3 regulates muscle differentiation. J Biol Chem. 2011;286:34733–34742. doi: 10.1074/jbc.M110.203307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SH, et al. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc Natl Acad Sci USA. 2005;102:18075–18080. doi: 10.1073/pnas.0503676102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang N, et al. Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J. 1998;17:3398–3412. doi: 10.1093/emboj/17.12.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rayasam GV, et al. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J. 2003;22:3153–3163. doi: 10.1093/emboj/cdg288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu T, et al. Regulation of NF-κB by NSD1/FBXL11- dependent reversible lysine methylation of p65. Proc Natl Acad Sci USA. 2010;107:46–51. doi: 10.1073/pnas.0912493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem. 2009;284:34283–34295. doi: 10.1074/jbc.M109.034462. Shows, using methyl-Lys analogues in nucleosomal substrates, that proteins of the NSD family are highly specific for the dimethylation of nucleosomal H3K36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucio-Eterovic AK, et al. Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc Natl Acad Sci USA. 2010;107:16952–16957. doi: 10.1073/pnas.1002653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiao Q, et al. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J Biol Chem. 2011;286:8361–8368. doi: 10.1074/jbc.M110.204115. Reveals that NSD1 activation and catalysis occur through an interaction with the nucleosomes, which causes a conformational change to relieve inhibition by the autoinhibitory loop. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berdasco M, et al. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc Natl Acad Sci USA. 2009;106:21830–21835. doi: 10.1073/pnas.0906831106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang H, et al. Preferential dimethylation of histone H4 lysine 20 by Suv4–20. J Biol Chem. 2008;283:12085–12092. doi: 10.1074/jbc.M707974200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simon MD, et al. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. Introduces the technique of generating methyl-Lys analogues as a tool to understand histone methylation and enzyme specificity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bender LB, et al. MES-4: an autosome-associated histone methyltransferase that participates in silencing the X chromosomes in the C. elegans germ line. Development. 2006;133:3907–3917. doi: 10.1242/dev.02584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bell O, et al. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J. 2007;26:4974–4984. doi: 10.1038/sj.emboj.7601926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersen EC, Horvitz HR. Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development. 2007;134:2991–2999. doi: 10.1242/dev.009373. [DOI] [PubMed] [Google Scholar]
- 25.Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27:406–420. doi: 10.1038/sj.emboj.7601967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furuhashi H, et al. Trans-generational epigenetic regulation of C. elegans primordial germ cells. Epigenetics Chromatin. 2010;3:15. doi: 10.1186/1756-8935-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rechtsteiner A, et al. The histone H3K36 methyltransferase MES-4 acts epigenetically to transmit the memory of germline gene expression to progeny. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001091. pii: e1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JY, et al. Multiple-myeloma-related WHSC1/MMSET isoform RE-IIBP is a histone methyltransferase with transcriptional repression activity. Mol Cell Biol. 2008;28:2023–2034. doi: 10.1128/MCB.02130-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim SM, et al. Characterization of a novel WHSC1-associated SET domain protein with H3K4 and H3K27 methyltransferase activity. Biochem Biophys Res Commun. 2006;345:318–323. doi: 10.1016/j.bbrc.2006.04.095. [DOI] [PubMed] [Google Scholar]
- 30.Kuo AJ, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell. 2011;44:609–620. doi: 10.1016/j.molcel.2011.08.042. ChIP–seq data with NSD2 in multiple myeloma cells reveal that H3K36me2 generates a chromatin landscape that is permissive for tumorigenesis by activating oncogenes that are sensitive to H3K36me2 levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marango J, et al. The MMSET protein is a histone methyltransferase with characteristics of a transcriptional corepressor. Blood. 2008;111:3145–3154. doi: 10.1182/blood-2007-06-092122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nimura K, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2–5 to Wolf–Hirschhorn syndrome. Nature. 2009;460:287–291. doi: 10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- 33.Hu M, et al. Histone H3 lysine 36 methyltransferase Hypb/Setd2 is required for embryonic vascular remodeling. Proc Natl Acad Sci USA. 2010;107:2956–2961. doi: 10.1073/pnas.0915033107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan W, et al. Heterogeneous nuclear ribonucleoprotein L is a subunit of human KMT3a/ Set2 complex required for H3 Lys-36 trimethylation activity in vivo. J Biol Chem. 2009;284:15701–15707. doi: 10.1074/jbc.M808431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chesi M, et al. The t(4;14) translocation in myeloma dysregulates both FGFR3 and a novel gene, MMSET, resulting in IgH/MMSET hybrid transcripts. Blood. 1998;92:3025–3034. [PubMed] [Google Scholar]
- 36.Martinez-Garcia E, et al. The MMSET histone methyl transferase switches global histone methylation and alters gene expression in t(4;14) multiple myeloma cells. Blood. 2011;117:211–220. doi: 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pei H, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124–128. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hajdu I, Ciccia A, Lewis SM, Elledge SJ. Wolf–Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc Natl Acad Sci USA. 2011;108:13130–13134. doi: 10.1073/pnas.1110081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Botuyan MV, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahman S, et al. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol. 2011;31:2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka Y, Katagiri Z, Kawahashi K, Kioussis D, Kitajima S. Trithorax-group protein ASH1 methylates histone H3 lysine 36. Gene. 2007;397:161–168. doi: 10.1016/j.gene.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 42.Abu-Farha M, et al. The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol Cell Proteomics. 2008;7:560–572. doi: 10.1074/mcp.M700271-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Brown MA, Sims RJ, 3rd, Gottlieb PD, Tucker PW. Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer. 2006;5:26. doi: 10.1186/1476-4598-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan W, et al. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem. 2011;286:7983–7989. doi: 10.1074/jbc.M110.194027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang J, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 46.Saddic LA, et al. Methylation of the retinoblastoma tumor suppressor by SMYD2. J Biol Chem. 2010;285:37733–37740. doi: 10.1074/jbc.M110.137612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diehl F, et al. Cardiac deletion of Smyd2 is dispensable for mouse heart development. PLoS ONE. 2010;5:e9748. doi: 10.1371/journal.pone.0009748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.An S, Yeo KJ, Jeon YH, Song JJ. Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. J Biol Chem. 2011;286:8369–8374. doi: 10.1074/jbc.M110.203380. Shows that ASH1L is a dimethylase that is highly specific for nucleosomal H3K36 and that histone tails pre-methylated at H3K36 are refractory to methylation at H3K27 and vice versa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gregory GD, et al. Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol Cell Biol. 2007;27:8466–8479. doi: 10.1128/MCB.00993-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du HN, Fingerman IM, Briggs SD. Histone H3 K36 methylation is mediated by a transhistone methylation pathway involving an interaction between Set2 and histone H4. Genes Dev. 2008;22:2786–2798. doi: 10.1101/gad.1700008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson CJ, Santos-Rosa H, Kouzarides T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell. 2006;126:905–916. doi: 10.1016/j.cell.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 52.Psathas JN, Zheng S, Tan S, Reese JC. Set2-dependent K36 methylation is regulated by novel intratail interactions within H3. Mol Cell Biol. 2009;29:6413–6426. doi: 10.1128/MCB.00876-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chu Y, Sutton A, Sternglanz R, Prelich G. The BUR1 cyclin-dependent protein kinase is required for the normal pattern of histone methylation by SET2. Mol Cell Biol. 2006;26:3029–3038. doi: 10.1128/MCB.26.8.3029-3038.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin LJ, Minard LV, Johnston GC, Singer RA, Schultz MC. Asf1 can promote trimethylation of H3 K36 by Set2. Mol Cell Biol. 2010;30:1116–1129. doi: 10.1128/MCB.01229-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Youdell ML, et al. Roles for Ctk1 and Spt6 in regulating the different methylation states of histone H3 lysine 36. Mol Cell Biol. 2008;28:4915–4926. doi: 10.1128/MCB.00001-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chu Y, Simic R, Warner MH, Arndt KM, Prelich G. Regulation of histone modification and cryptic transcription by the Bur1 and Paf1 complexes. EMBO J. 2007;26:4646–4656. doi: 10.1038/sj.emboj.7601887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li B, et al. Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J Biol Chem. 2009;284:7970–7976. doi: 10.1074/jbc.M808220200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bannister AJ, et al. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem. 2005;280:17732–17736. doi: 10.1074/jbc.M500796200. [DOI] [PubMed] [Google Scholar]
- 59.Wu SF, Zhang H, Cairns BR. Genes for embryo development are packaged in blocks of multivalent chromatin in zebrafish sperm. Genome Res. 2011;21:578–589. doi: 10.1101/gr.113167.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li B, Howe L, Anderson S, Yates JR, 3rd, Workman JL. The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. J Biol Chem. 2003;278:8897–8903. doi: 10.1074/jbc.M212134200. [DOI] [PubMed] [Google Scholar]
- 61.Li J, Moazed D, Gygi SP. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J Biol Chem. 2002;277:49383–49388. doi: 10.1074/jbc.M209294200. [DOI] [PubMed] [Google Scholar]
- 62.Xiao T, et al. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 2003;17:654–663. doi: 10.1101/gad.1055503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Phatnani HP, Greenleaf AL. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 2006;20:2922–2936. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 64.Sun XJ, et al. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem. 2005;280:35261–35271. doi: 10.1074/jbc.M504012200. [DOI] [PubMed] [Google Scholar]
- 65.Carrozza MJ, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 66.Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell. 2005;20:971–978. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 67.Keogh MC, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 68.Kaplan CD, Laprade L, Winston F. Transcription elongation factors repress transcription initiation from cryptic sites. Science. 2003;301:1096–1099. doi: 10.1126/science.1087374. [DOI] [PubMed] [Google Scholar]
- 69.Li B, et al. Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science. 2007;316:1050–1054. doi: 10.1126/science.1139004. [DOI] [PubMed] [Google Scholar]
- 70.Fang R, et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell. 2010;39:222–233. doi: 10.1016/j.molcel.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bartke T, et al. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143:470–484. doi: 10.1016/j.cell.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bell O, et al. Transcription-coupled methylation of histone H3 at lysine 36 regulates dosage compensation by enhancing recruitment of the MSL complex in Drosophila melanogaster. Mol Cell Biol. 2008;28:3401–3409. doi: 10.1128/MCB.00006-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alekseyenko AA, et al. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008;134:599–609. doi: 10.1016/j.cell.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gelbart ME, Larschan E, Peng S, Park PJ, Kuroda MI. Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nature Struct Mol Biol. 2009;16:825–832. doi: 10.1038/nsmb.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Larschan E, et al. X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature. 2011;471:115–118. doi: 10.1038/nature09757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Strahl BD, et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol. 2002;22:1298–1306. doi: 10.1128/mcb.22.5.1298-1306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Landry J, et al. Set2-catalyzed methylation of histone H3 represses basal expression of GAL4 in Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:5972–5978. doi: 10.1128/MCB.23.17.5972-5978.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nature Cell Biol. 2007;9:804–812. doi: 10.1038/ncb1608. Shows, using a mouse model system, that a NUP98–NSD1 translocation enforces an activating H3K36 methylation profile at a subset of Hoxa genes, causing uncontrolled cell proliferation and tumorigenesis. [DOI] [PubMed] [Google Scholar]
- 79.Kolasinska-Zwierz P, et al. Differential chromatin marking of introns and expressed exons by H3K36me3. Nature Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schwartz S, Meshorer E, Ast G. Chromatin organization marks exon–intron structure. Nature Struct Mol Biol. 2009;16:990–995. doi: 10.1038/nsmb.1659. [DOI] [PubMed] [Google Scholar]
- 81.Spies N, Nielsen CB, Padgett RA, Burge CB. Biased chromatin signatures around polyadenylation sites and exons. Mol Cell. 2009;36:245–254. doi: 10.1016/j.molcel.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krogan NJ, et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2003;23:4207–4218. doi: 10.1128/MCB.23.12.4207-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roberts GC, Gooding C, Smith CW. Smooth muscle alternative splicing induced in fibroblasts by heterologous expression of a regulatory gene. EMBO J. 1996;15:6301–6310. [PMC free article] [PubMed] [Google Scholar]
- 84.Robson-Dixon ND, Garcia-Blanco MA. MAZ elements alter transcription elongation and silencing of the fibroblast growth factor receptor 2 exon IIIb. J Biol Chem. 2004;279:29075–29084. doi: 10.1074/jbc.M312747200. [DOI] [PubMed] [Google Scholar]
- 85.de la Mata M, et al. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell. 2003;12:525–532. doi: 10.1016/j.molcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 86.Luco RF, et al. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. Demonstrates that H3K36me3 modification is capable of regulating alternative splicing events, notably those regulated by PTB, through the direct recruitment of splicing regulators to nucleosomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wagner EJ, Garcia-Blanco MA. Polypyrimidine tract binding protein antagonizes exon definition. Mol Cell Biol. 2001;21:3281–3288. doi: 10.1128/MCB.21.10.3281-3288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carstens RP, McKeehan WL, Garcia-Blanco MA. An intronic sequence element mediates both activation and repression of rat fibroblast growth factor receptor 2 pre-mRNA splicing. Mol Cell Biol. 1998;18:2205–2217. doi: 10.1128/mcb.18.4.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim S, Kim H, Fong N, Erickson B, Bentley DL. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc Natl Acad Sci USA. 2011;108:13564–13569. doi: 10.1073/pnas.1109475108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Almeida SF, et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nature Struct Mol Biol. 2011;18:977–983. doi: 10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- 92.Pryde F, et al. H3 k36 methylation helps determine the timing of Cdc45 association with replication origins. PLoS ONE. 2009;4:e5882. doi: 10.1371/journal.pone.0005882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim HS, Rhee DK, Jang YK. Methylations of histone H3 lysine 9 and lysine 36 are functionally linked to DNA replication checkpoint control in fission yeast. Biochem Biophys Res Commun. 2008;368:419–425. doi: 10.1016/j.bbrc.2008.01.104. [DOI] [PubMed] [Google Scholar]
- 94.Biswas D, et al. A role for Chd1 and Set2 in negatively regulating DNA replication in Saccharomyces cerevisiae. Genetics. 2008;178:649–659. doi: 10.1534/genetics.107.084202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sanders SL, et al. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 96.Fnu S, et al. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc Natl Acad Sci USA. 2011;108:540–545. doi: 10.1073/pnas.1013571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chou DM, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci USA. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kurotaki N, et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nature Genet. 2002;30:365–366. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- 99.Duns G, et al. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010;70:4287–4291. doi: 10.1158/0008-5472.CAN-10-0120. [DOI] [PubMed] [Google Scholar]
- 100.Newbold RF, Mokbel K. Evidence for a tumour suppressor function of SETD2 in human breast cancer: a new hypothesis. Anticancer Res. 2010;30:3309–3311. [PubMed] [Google Scholar]
- 101.Bianco-Miotto T, et al. Global levels of specific histone modifications and an epigenetic gene signature predict prostate cancer progression and development. Cancer Epidemiol Biomarkers Prev. 2010;19:2611–2622. doi: 10.1158/1055-9965.EPI-10-0555. [DOI] [PubMed] [Google Scholar]
- 102.Hudlebusch HR, et al. The histone methyltransferase and putative oncoprotein mmset is overexpressed in a large variety of human tumors. Clin Cancer Res. 2011;17:2919–2933. doi: 10.1158/1078-0432.CCR-10-1302. [DOI] [PubMed] [Google Scholar]
- 103.Hudlebusch HR, et al. MMSET is highly expressed and associated with aggressiveness in neuroblastoma. Cancer Res. 2011;71:1226–4235. doi: 10.1158/0008-5472.CAN-10-3810. [DOI] [PubMed] [Google Scholar]
- 104.Kassambara A, Klein B, Moreaux J. MMSET is overexpressed in cancers: link with tumor aggressiveness. Biochem Biophys Res Commun. 2009;379:840–845. doi: 10.1016/j.bbrc.2008.12.093. [DOI] [PubMed] [Google Scholar]
- 105.Rosati R, et al. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11) (p11.2; p15) Blood. 2002;99:3857–3860. doi: 10.1182/blood.v99.10.3857. [DOI] [PubMed] [Google Scholar]
- 106.Taketani T, et al. NUP98–NSD3 fusion gene in radiation-associated myelodysplastic syndrome with t(8;11)(p11; p15) and expression pattern of NSD family genes. Cancer Genet Cytogenet. 2009;190:108–112. doi: 10.1016/j.cancergencyto.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 107.Angrand PO, et al. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics. 2001;74:79–88. doi: 10.1006/geno.2001.6524. [DOI] [PubMed] [Google Scholar]
- 108.Job B, et al. Genomic aberrations in lung adenocarcinoma in never smokers. PLoS ONE. 2010;5:e15145. doi: 10.1371/journal.pone.0015145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang H, et al. Forkhead transcription factor foxq1 promotes epithelial–mesenchymal transition and breast cancer metastasis. Cancer Res. 2011;71:1292–1301. doi: 10.1158/0008-5472.CAN-10-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhao Q, et al. Transcriptome-guided characterization of genomic rearrangements in a breast cancer cell line. Proc Natl Acad Sci USA. 2009;106:1886–1891. doi: 10.1073/pnas.0812945106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.La Starza R, et al. Cryptic insertion producing two NUP98/NSD1 chimeric transcripts in adult refractory anemia with an excess of blasts. Genes Chromosomes Cancer. 2004;41:395–399. doi: 10.1002/gcc.20103. [DOI] [PubMed] [Google Scholar]
- 112.Jaju RJ, et al. A novel gene, NSD1, is fused to NUP98 in the t(5;11)(q35; p15.5) in de novo childhood acute myeloid leukemia. Blood. 2001;98:1264–1267. doi: 10.1182/blood.v98.4.1264. [DOI] [PubMed] [Google Scholar]
- 113.Wang X, et al. Identification and characterization of a novel androgen receptor coregulator ARA267-α in prostate cancer cells. J Biol Chem. 2001;276:40417–40423. doi: 10.1074/jbc.M104765200. [DOI] [PubMed] [Google Scholar]
- 114.Mendes-Pereira AM, et al. Genome-wide functional screen identifies a compendium of genes affecting sensitivity to tamoxifen. Proc Natl Acad Sci USA. 2011 Apr 11; doi: 10.1073/pnas.1018872108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Keats JJ, et al. Overexpression of transcripts originating from the MMSET locus characterizes all t(4;14)(p16; q32)-positive multiple myeloma patients. Blood. 2005;105:4060–4069. doi: 10.1182/blood-2004-09-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morris SA, et al. Identification of histone H3 lysine 36 acetylation as a highly conserved histone modification. J Biol Chem. 2007;282:7632–7640. doi: 10.1074/jbc.M607909200. [DOI] [PMC free article] [PubMed] [Google Scholar]