

Abstract
Halisphingosines A (1) and B (2), modified long-chain sphingoid bases, from the marine sponge Haliclona tubifera collected in Brazil, were characterized after conversion to their N-Boc derivatives. The 2R,3R,6R configuration of halisphingosine A, a compound first reported from Haliclona sp. South Korea, was confirmed using a novel CD approach: deconvolution of exciton coupling from mono- and tri-naphthoyl derivatives obtained by derivatization of the natural product. The sensitive CD deconvolution method, applicable to sub-milligram samples, simultaneously predicted the relative and absolute configuration of three stereocenters in halisphingosine A with precision and accuracy. Halisphingosine B was assigned by correlation to halisphingosine A.
Diverse variants of the C18 long-chain bases sphingosine and phytosphingosine and the corresponding ceramides occur throughout the five Kingdoms1 including marine phyla2 such as sponges, tunicates and macroalgae. Modifications include variable chain lengths, chain branching by methyl groups, hydroxylation, and polar head-group modifications. Linear and branched aminoalkanols from sponges and tunicates exhibit substantial antifungal activity. ‘Two-headed’ sphingolipids, constituting C28 or C30 chains functionalized at both termini as aminoalkanols or aminoalkane-diols with four stereocenters,3 show activity against the fungal pathogens Candida albicans, C. glabrata and other species. Heterocyclic analogs derived from linear sphingoid bases have also been characterized from sponges and tunicates; these include azirines4 and piperidines (e.g. pseudodistomins A and B, from Pseudodistoma sp.5). Other heterocyclic sphingoids reported from marine invertebrates are the so-called ‘anhydrophytosphingosines’ including azetidines (penaresidines A and B from Penares sp.6) and tetrahydrofurans (pachastrissamine from Pachastrissa sp.7 – synonymous with jaspine B8 – and penasins A-E from Penares sp.9).
Although mammalian sphingosines are almost always of the 2S,3R configuration (D-erythro), invertebrate-derived sphingoid bases exhibit broad stereochemical heterogeneity. Examples of α,ω-bifunctionalized sphingolipids are known with almost all possible permutations of the four stereocenters (N = 16).3 While the modified Mosher’s method has been the applied method for assignment of absolute configuration of sphingoid bases, the method suffers from the usual limitations; requirement of sufficient sample for preparation of S- and R-MTPA derivatives and NMR analysis, and equivocal interpretations for molecules with multiple contiguous –NH2 and –OH groups. Other approaches based on degradation have similar drawbacks and erroneous assignments have been made in the past.10 Here, we report the N-Boc derivative 1a of halisphingosine A (1), a C18 sphingoid base first isolated from a Haliclona sp. collected off Ulleung Island, South Korea,11 and reisolated in this work from a Brazilian marine sponge, Haliclona tubifera collected off Ilha do Arvoredo, Santa Catarina.12 A new derivative, the N-Boc analog 2a of halisphingosine B (2), is also described and characterized by NMR and MS. Unlike common marine-derived sphingolipids related to D-ribo-phytosphingosine (i) the new compounds are L-threo. The configuration of 1a was verfied at the nanomole scale as 2R,3R,6R [L-threo at the head group], by interpretation of the CD spectra of mono- and tetra-naphthoyl derivatives of 1; specifically, deconvolution of the Cotton effect (CE) arising from different molecular exciton couplings through application of van’t Hoff’s principle of optical superposition.13
Results and Discussion
Ethyl acetate soluble extracts of the sponge Haliclona tubifera were fractionated by flash chromatography (NH3-saturated MeOH: CH2Cl2) to obtain a polar ninhydrin-positive fraction containing a mixture of long-chain bases. In order to simplify the purification, the long-chain base mixture was converted to the corresponding N-Boc derivatives (di-tert-butylcarbonate, NaHCO3, aq. THF), and then separated by multiple rounds of reversed-phase HPLC (C18-bonded silica), leading to pure N-Boc derivatives 1a and 2a. Characterization of 1a by MS and NMR (Table 1) demonstrated that it was the N-Boc derivative of 1 (2-aminooctadec-7-ene-1,3,6-triol), a compound first reported by Mansoor and coworkers in 2007.11 Analysis of the COSY spectrum (CD3OD, 500 MHz) assigned downfield signals to a secondary allylic alcohol (H-6, δ 4.37, dt, J = 9.0, 5.7; H-7, δ 5.31, dddd J = 9.3, 7.8, 1.5, 1.5; H-8, δ 5.45, dt, J = 9.3, 6.4) and a contiguous spin system associated with the terminal head group: CH2OH (H-1a, δ 3.55, m; H1b, δ 3.60, m) adjacent to C-2 CH-NH-(t-Boc) (H-2, δ 3.54, m) which in turn was coupled to a hydroxymethine signal H-3 (δ 3.75, m). The C-1–C–3 segment was separated from the C-6 allylic CH-OH by two CH2 groups and the remainder of the molecule was a linear n-alkyl chain. Taken all together, the constitution of 1a was identical to that published, 11 except for the presence of the N-Boc group.
Table 1.
1H and 13C NMR Data of Halisphingosine A (1a, CD3OD, 500 and 125 MHz) and Per-acetyl Halisphingosine B (8, CDCl3, 600 MHz).
| Position | 1a | 8 | ||
|---|---|---|---|---|
|
| ||||
| δC, typeb | δH, (J in Hz)a | δC, typeb | δH, (J in Hz) a | |
| 1 | 62.8, CH2 | 3.55 m, 3.60 m | 63.1, CH2 | 4.03 ddd (15.9, 11.2, 6.2) |
| 2 | 56.5, CH | 3.54 m | 49.8, CH | 4.41 m |
| 2-NH | – | – | – | 5.63 br t (5.8) |
| 3 | 70.7, CH | 3.75 m | 71.9, CH | 5.06 m |
| 4 | 34.7, CH2 | 1.45 m | 30.5, CH2 | 1.54 m |
| 5 | 38.5, CH2 | 1.38 m, 1.56 m | 34.0, CH2 | 1.50 m |
| 6 | 67.9, CH | 4.37 dt (9.0, 5.7) | 73.8, CH | 4.84 p (12.5, 6.0) |
| 7 | 133.7, CH | 5.31 dddd (9.3, 7.8, 1.5, 1.5) | 30.5, CH2 | 1.54 m |
| 8 | 131.8, CH | 5.45 dt (9.3, 6.4) | ||
| 9 | 28.4, CH2 | 2.09 m | 22.6–31.5, CH2 | 1.20–1.32 m |
| 10–17 | 23.5–32.5, CH2 | 1.29–1.41 m | ||
| 18 | 14.1, CH3 | 0.91 t (6.7) | 14.2, CH3 | 0.88 t (6.7) |
| N-Boc CH3 | 28.5, CH3 | 1.45 s | – | – |
| Acc | – | – | 23.0, CH3 | 2.08 s |
| Acc | – | – | 21.0, CH3 | 2.06 s |
| Acc | – | – | 21.0, CH3 | 2.04 s |
| Acc | – | – | 23.0, CH3 | 2.03 s |
br=broad; s=singlet; d=doublet; dt=doublet of triplets; t=triplet; m=multiplet; p=pentet.
Multiplicities of 13C NMR signals established by edited HSQC and DEPT.
Acetyl 1H and 13C signals cross correlated by HSQC but not assigned.
The challenges of assignment of multiple stereocenters in acyclic natural products can be formidable.14 The original configurational analysis of 1 was carried out by comparison of 1H NMR spectra of the tetra-Mosher’s ester derivative of 111 with those of the corresponding four synthetic stereoisomers of sphinganine.15 Although, strictly not an accurate comparison (1 contains an additional allylic OH at C-6), the conclusion of the analysis suggested a likely match for the (2R,3R,6R) isomer. In order to verify the configuration of 1 and refine methods for sub-milligram stereoanalysis of complex sphingolipids, we adapated a variant of Nakanishi and Berova’s method16 for phytosphingosines based on CD of naphthimide-naphthoate derivatives obtained by two-step sequential derivatization. Nakanishi’s method introduces a C-2 naphthimide group selectively by conversion of the primary NH2 in the first step by condensation with naphthalene-2,3-dicarboxylic acid, followed by per-O-naphthoylation of the remaining OH groups. Degeneracy is avoided in the superposition of pairwise exciton couplings because of the different UV λmax of naphthimide-naphthoate chromophore pairs. This choice provides ‘fingerprint’ CEs that sufficiently discriminate all possible diastereomers and their corresponding enantiomers. We favored, instead, a single-step exhaustive naphthoylation of 1a that we expected would provide the same discrimination of erythro and threo diastereomers by pairwise-interactions of naphthamide-naphthoate pairs, but overlayed with an additional CE from the distal allylic 2-naphthoate at C-6. Application of the latter CE for assignment of allylic alcohols – arising from helicity of the exciton coupled 1Lb transition of the 2-naphthoate chromophore with the ene π-π* transition (Figure 2) – was first introduced by Nakanishi and coworkers17 for secondary alcohols from their allylic benzoates, and successfully validated by others18 to allylic 2-naphthoate esters. Because the exciton coupled chromophores at the head-group C-1–C-3 and the C-6–C-8 are separated by two CH2 groups in an acylic chain backbone, little interaction is expected between the two stereoclusters. In other words, the ensemble should be interpretable as the superposition of each set of independent exciton coupled systems according to van’t Hoff’s principle of optical superposition.13 This was shown to be the case and the configuration of 1 was verified as follows.
Figure 2.

Newman diagrams of major contributing conformers and predicted signs of exciton coupled CD spectra in (a), (b) S and R secondary allylic 2-naphthoate esters,16,18 (c), (d) and N,O,O-tris-naphthoyl derivatives of 2-aminoalkane-1,3-diols, (R,R)-i, and (R,S)-ii, and (e), (f), (S,S)-i, and (S,R)-ii (contributions from 1-naphthoate-2-naphthamide pairs are not shown, but their Cotton effects have the same signs with amplified magnitude A). Np = 2-naphthoyl. Blue bar = 2-naphthoate, red bar = 2-naphthamide.
The N-Boc derivative 1a (ca. 1 mg) was converted to the acetonide 3 (Scheme 1) under standard conditions. Analysis of the 1H NMR (500 MHz, CDCl3) spectrum of 3, in particular the vicinal coupling constants, verified a chair conformation of the 1,3-dioxane ring, possessing an axial C-2 N-Boc group and an equatorial C-3 substituent. It follows from the small vicinal coupling observed between H-2 and H-3 (J = 1.9 Hz) that 3 has the threo relative configuration at C-2–C-3 using arguments similar to those applied by Braekman to the acetonide of a related threo sphingolipid from Haliclona vansoesti.19 Attempted naphthoylation of 3 was sluggish and incomplete under standard conditions (2-naphthoyl chloride, pyridine, 100 °C, 24 h), however, smooth conversion to mononaphthoate 4 was achieved with the alternative acylating reagent, N-(2′-naphthoyl)imidazole (DBU, CH3CN, 80 °C, ~4 h).20 The CD spectrum of 4 (CH3CN, Figure 4b) revealed the long-wavelength half of the expected split CE as a simple broad negative band [λ 236 (Δε −21.9] rising to a weak, positive CE (Δε ~8, λ = 190 nm, truncated). Thus the C-6 configuration of 4 and 1b is verified as R.
Scheme 1.
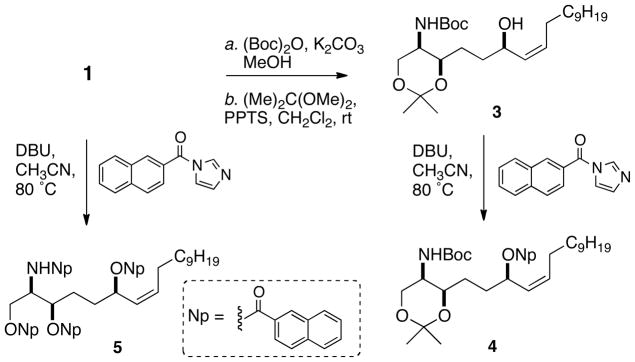
Synthesis of acetonide 3 and mono- and tri-naphthoyl derivatives 4 and 5 from 1.
Figure 4.

Measured circular dichroism spectra (CD, CH3CN, 24 °C) and hybrid CD spectra. (a) N,O,O′,O″-tetranaphthoyl compound (5) prepared from natural product 1. (b) Mononaphthoate, 4, prepared from 3 (see Scheme 1) and N,O,O′-trinaphthoyl compound threo-7b (see Scheme 2). (c) CD of 5 (solid line) overlayed with hybrid CD spectrum (dashed) = [CD(4) − CD(7b), dashed]. (d) CD of 2 (solid line) overlayed with hybrid CD spectrum (dashed) = [CD(4) + CD(7b)]. See Supporting Information for comparisons of the CD of 5 with hybrid spectra derived from 4 and erythro-6b.
Naphthoylation of a fraction enriched in 1 gave, after HPLC purification, the tetra-(2′-naphthoyl) derivative 5. In contrast to 4, the CD spectrum (CH3CN) of 5 (Figure 4c) exhibited expectedly more complex CEs [λ 243 (Δε −20.1), 235 (+17.4), 224 (−12.5)] generated by superposed exciton coupling contributions of the stereoclusters at C-1–C-3 and C-6–C7-C8.
For the purposes of CD stereoanalysis of the head group in 5, model compounds of known configuration were required. The erythro and threo tri-(2′-naphthoyl) diastereomers 6b and 7b were prepared (Scheme 2) from L-serine using a variation of the method reported earlier.3c The CD spectrum of threo-7b (Figure 3) showed a strong negative split CE [λ 237 (Δε −19.3), 221 (+7.2), peak to trough, A = 26.5] while the CD of erythro-6b (Figure 3) was distinctly different: although the sign of the split CE was still negative the magnitude was far larger [λ 226 (Δε +56), 242 (−68.6), A = 124]. Both erythro-6b and threo-7b displayed biphasic CEs that were higher in magnitude than their corresponding tri-benzoyl derivatives3c and, as with the latter compounds, easily discriminated the four possible stereoisomers with the added advantage of approximately five- to ten-fold higher sensitivity.
Scheme 2.

Synthesis of model compounds erythro-6b and threo-7b.
Figure 3.

Measured circular dichroism spectra (CH3CN, 24 °C) of per-naphthoyl derivatives prepared from L-serine (see Scheme 2). (a) erythro-6b ((2S,3R), dashed line, and (b) threo-7b (2S,3S), solid line.
The CD spectrum of 5 was compared with ‘hybrid CD’ spectra generated by simple linear combinations of the CD spectra of 7b, 4 (Figure 4) and their mirror images (not shown; see Supporting Information for hybrid CD of 4 and erythro-6b). The only combination (Figure 3c) that matched the observed CEs of 5 corresponds with the 2R,3R,6R configuration, thereby confirming the assignment for 1 proposed by Mansoor and coworkers.11
Halisphingosine B (2a) was isolated in low yield (~0.4 mg) from an earlier-eluting fraction that yielded 1a. The formula C23H47NO5Na established from ESI HRMS suggested one double bond equivalent less than 1a. Inspection of the 1H NMR spectrum of 2a (Table 1) showed absence of the signals of the disubstituted olefin in 1a but preservation of the spin system corresponding to H-1–H-6, suggesting the new compound was 7,8-dihydro-halisphingosine A. Halisphingosine derivatives 1a and 2a were correlated as follows (Scheme 3). Compound 1a was hydrogenated (H2, Pd-C, MeOH) and the product converted to the per-acetyl derivative 8 by removal of the Boc group (TFA, CH2Cl2) and acetylation (Ac2O, pyridine, rt) followed by purification by HPLC. Compound 8 was also obtained by two step conversion of 2a (~0.1 mg, TFA, CH2Cl2; Ac2O, pyridine). Both samples of 8 were identical by HPLC (rt = 34.5 min, RP C18, 7:3 CH3CN-H2O; one peak by coinjection), HRMS and 1H NMR (CDCl3, 500 MHz, See Supporting Information), in particular, by comparison of the chemical shifts of the four narrow-line CH3(C=O) signals and H-1–H–3 (Table 1). Thus, the configurations of 1a and 2a are the same and given that the two natural products co-occur in Haliclona tubifera, it’s highly likely the absolute configurations are also identical.
Scheme 3.

Conversion of 1a and 2a to peracetyl-compound 8.
The application of CD combined with deconvolution of superposed CEs is exceedingly sensitive; in the case of 5 we estimate the limits of detection (LOD) to be approximately 30 pmol. The method should be applicable to the structures of other sphingoid base natural products containing similar remotely separated stereoelements.
The pathogenic yeast Candida albicans is modestly inhibited by sphingosine or phytosphingosine, however, many modified sphingolipids possess significantly more potent antifungal activity.21 Unfortunately, homogeneous samples of 1 for testing could not be purified from the limited amounts of sponge material available, or by deprotection of pure 1a,22 and insufficient amounts of 2a were available for assay. Efforts in our laboratory to procure additional samples of 1 by total synthesis are underway.
In conclusion, a new sphingolipid halisphingosine B was characterized as its per-acetyl derivative 8 along with 1a, the N-Boc derivative of halisphingosine A (1). A variant of Nakanishi’s CD protocol16 for assignment of sphingolipids, combined with van’t Hoff’s principle of optical superposition, was applied to verify the absolute configuration of the known compound 1. These tools should find general application for configurational analysis of sphingoid base natural products.
Experimental Section
General Experimental Procedures
General experimental procedures are described elsewhere.23 CD spectra were recorded on a Jasco J810 spectropolarimeter in 1 mm pathlength under the following parameters: slit 2 nm, scan speed 100 nm/min, digital resolution 1 nm, 8–16 accumulations 1H NMR spectra were recorded on a dual channel Jeol ECA 500 MHz spectrometer (1H NMR, 500.1599 MHz), an Agilent 500 MHz spectrometer equipped with an XSens 13C{1H} cryoprobe, or a dual channel Bruker 600 MHz spectrometer (1H NMR, 599.5560 MHz) equipped with an Avance III dual channel console and a 1.7 mm 1H{13C,15N} microcryoprobe. Spectra are referenced to residual solvent signals for CD3OD or CDCl3 (1H, δ 3.31 ppm or δ 7.26 ppm; 13C, δ 49.0 or 77.0 ppm, respectively). Accurate mass measurements were carried out on an Agilent 6230 ESI-TOF mass spectrometer, equipped with an Agilent 1200 capillary HPLC system. Solvents for sub-milligram preparative HPLC were distilled from commercial ‘HPLC-grade’ stock.
Animal Material
Specimens of Haliclona tubifera were collected by hand with scuba (−15 to −20 m) in 2010 at Ilha do Arvoredo, Santa Catarina, Brazil. The samples were freeze-dried and stored at −18 °C for 4 months before use. The sponge was identified by João Luis de Fraga Carraro, and specimens are deposited in the Museu de Ciências Naturais–Porifera (MCNPOR) collection of the Fundação Zoobotânica do Rio Grande do Sul, Brazil.
Purification of Halisphingosines A and B: N-Boc derivatives, 1a, 2a
A sample of Haliclona tubifera (56.1g wet wt.) was extracted with MeOH (3 × 500 mL). The resulting extract was adjusted to 9:1 MeOH/H2O and partitioned with hexanes (3 × 300 mL). The aqueous MeOH layer was removed and the H2O layer was extracted with EtOAc (3 × 300 mL). The EtOAc soluble fraction was partially (500 mg) purified by flash chromatography on a silica column using a gradient of MeOH (NH3 satd.):CH2Cl2 (5:95–10:90, v/v). The ninhydrin positive bands were combined resulting in seven fractions. The sixth fraction (46.8 mg) was subjected to a reversed-phase (C18) cartridge with a solvent gradient of MeOH:H2O (50:50–100:0, v/v). The fractions eluted with 80% and 100% of MeOH (29.3 mg and 10 mg, respectively) were treated with excess (Boc)2O and K2CO3 in THF:H2O (4:1) and the mixture stirred for 2 h. After filtration of the solids and concentration, the entire filtrate, was subjected to RP-HPLC (Luna C18, 10 × 250 mm, 5 μm; gradient elution, 65:45 to 100:0 CH3CN:H2O over 35 min, ELSD detection) to give N-Boc derivative 1a (8.0 mg). The entire fourth N-Boc fraction was subjected to RP-HPLC (Luna C18, 10 × 250 mm, 5 μm; 7:3 CH3CN:H2O, 2.5mL min−1) to give N-Boc-halisphingosine B (2a, 0.4 mg).
N-Boc-halisphingosine A (1a)
Colorless oil. [α]D +12.7 (c, 0.20, CHCl3); 1H NMR (CD3OD, 500 MHz), see Table 1; 13C NMR (CDCl3) 156.4 (NH-(C=O)), 132.5* (C-7), 132.3* (C-8), 79.6 (O-C(CH3)3, 73.2 (C-3), 67.7 (C-6), 65.6 (C-1), 54.2 (C-2), 37.4 (C-4§), 34.2 (C-5§), 31.7 (C-16), 29.7 (C-9), 29.41 (C-15§), 29.38 (C-10§), 28.94 (C-11§), 28.35 (3xC, OC(CH3)3§), 27.7 (C-12§), 25.41 (C-13§), 25.23 (C-14§), 22.6 (C-17), 14.1 (C-18). (*,§ interchangeable); HRMS m/z 438.3192 [M+Na]+ (calcd for C23H45NO5Na, 438.3195).
N-Boc-halisphingosine B (2a)
Colorless oil.; 1H NMR (CD3OD, 600 MHz) δ 3.75 (1H, t, J=5.6), 3.60 (1H, m), 3.50–3.54 (3H, m), 1.45 (9H, s), 1.27–1.38 (30H, m), 0.90 (3H, t, J=6.7); HRMS m/z 440.3343 [M+Na]+ (calcd for C23H47NO5Na, 440.3352).
Preparation of Acetonide 3
A solution of N-Boc halisphingosine A (1a) (2.0 mg) in acetone and 2,2-dimethoxypropane (12 μL), CH2Cl2 (0.2 mL) and pyridinium p-toluenesulfonate (1.2 mg, 4.8 μmol) was stirred at room temperature overnight. The mixture was treated with solid K2CO3, filtered and concentrated under a stream of N2. The residue was resuspended and the soluble-portition separated by column chromatography (oversized pipet, silica, 4:1 hexanes:EtOAc) to obtain 3 as a colorless oil pale yellow oil (1.3, 60%). 1H NMR (CDCl3, 500 MHz) δ 5.48 (1H, dt, J = 9.3, 6.4 Hz), 5.36 (1H, dddd, J = 9.3, 7.8, 1.5, 1.5 Hz), 5.29 (1H, d, J = 10.0 Hz), 4.41 (1H, dt, J = 7.6, 6.0 Hz), 4.05 (1H, dd, J = 10.2, 1.7 Hz), 3.89 (1H, m), 3.75 (1H, dd, J = 10.2, 1.7 Hz), 3.5 (1H, dddd, J = 10.0, 1.9, 1.7, 1.7 Hz), 2.08 (2H, m), 1.45 (6H, s, C(CH3)2), 1.40 (2H, t-BuO, s), 1.35-1.24 (21H, m), 0.88 (3H, t, J = 7.0 Hz); ESI HRMS m/z 478.3504 [M+Na]+ (calcd for C26H49NO5Na, 478.3503).
Preparation of Mononaphthoate Ester 4
A mixture of acetonide 3 (0.6 mg), DBU 3.0 μL, 20 μmol) and N-(2′-naphthoyl)imidazole20 (1.4 mg, 6.5 μmol) were dissolved in CH3CN (0.2 mL), sealed in a vial and heated at 60 °C overnight. After cooling, the volatiles were removed under a stream of N2, and the residue purified by HPLC (Luna, 3 μm silica, gradient 65:35 hexanes:EtOAc to 100:0 over 60 min, 0.5 mL.min−1) to give the mononaphthoate ester 4 (0.6 mg, 76%) as a colorless oil. UV (CH3CN) λmax (log ε) 290 (3.67), 280 (3.83), 270 (3.76), 237 (4.76) nm; CD: See Figure 4 and Supporting Information; 1H NMR (CDCl3) 8.58 (s, H-1′), 8.05 (d, J = 8.6 Hz, H-3′), 7.95 (1H, d, J = 8.1 Hz, H-8′*), 7.880 (1H, d, J = 8.6 Hz, H-4′), 7.875 (1H, d, J = 8.1 Hz, H-5′*), 5.83 (m, H-6), 5.61 (m, H-8), 5.47 (1H, bd, J = 9.9 Hz, H-7,), 5.28 (1H, d, J, 10.0 Hz, NH), 4.02 (1H, d, J = 12 Hz, H1a) 3.87 (1H, bt, J = 6.7 Hz, H-3), 3.73 (1H, d, J = 12 Hz, H-1b), 3.47 (1H, bd, J = 10.4 Hz, H-2), 2.23 (H-9, m), 1.85 (m), 1.65 (m), 1.43 (s, t-BuO), 1.42 (s, CCH3), 1.38 (s, CCH3), 1.45-1.23 (m), 0.87 (3H, t, J = 6.9 Hz, H-18) *interchangeable. HRMS m/z 632.3919 [M+Na]+ (calcd for C37H55NO6Na, 632.3922).
Preparation of N,O′,O″,O‴-Tetranaphthoyl Derivative 5
A partially purified fraction (0.5 mg, ~1.2 μmol) containing largely 1 in a solution of DBU (5.4 μL, 36 μmol) and excess N-(2′-naphthoyl)imidazole (2.7 mg, 12.0 μmol) in CH3CN (0.2 mL) and heated in a sealed vial at 70 °C overnight. After cooling, the volatiles were removed under a stream of N2 and the residue purified by analytical HPLC (silica, Luna 3 μm, hexanes to EtOAc over 60 min, 0.5 mL.min−1) to give the tetranaphthoyl derivative 5 (0.3 mg, 27%). UV (CH3CN) λmax (log ε) 290 (4.10), 280 (4.26), 270 (4.20), 233 (5.14) nm. CD: See Figure 4 and Supporting Information; 1H NMR (CDCl3) 8.56 (1H, s, H-1′), 8.56 (H-1′, s), 8.51 (1H, s, H-1′), 8.27 (H-1′, s), 8.2 – 7.2 (m), 6.90 (1H, d, J = 9.5 Hz, NH), 5.78 (1H, m), 5.67 (1H, m), 5.55 (1H, m), 5.42 (1H, m), 5.03 (1H, m), 4.71 (1H, s, m, H-1a), 4.63 (1H, s, m, H-1b), 2.18 (2H, m), 1.95 (2H, m), 1.81 (2H, m), 1.25-1.65 (m), 0.83 (3H, t, J = 6.7 Hz, H-18). HRMS m/z 932.4517 [M+H]+ (calcd 932.4521 for C62H62NO7, 932.4521).
Preparation of L-erythro- and L-threo-Trinaphthoyl Model Compounds 6a and 7a
The starting aminodiols L-erythro-6a and L-threo-7a were prepared from 8 derived from L-serine as described previously3c and used without futher purification. A solution of crude 6a (5.0 mg, 0.0257 mmol), DBU (117 mg, 0.771 mmol) and N-(2′-naphthoyl)imidazole (57 mg, 0.257 mmol) in CH3CN (1.0 mL) was sealed in a vial and heated at 70 °C overnight. After removal of the volatiles, the residue was first purified by TLC (4:1 hexanes:EtOAc) and then semipreparative HPLC (RP C18 Luna column, gradient elution with 60:40 to 100:0 CH3CN:H2O over 15min, 4.0 mL min−1) to give erythro-6b as a white powder (0.7 mg, 5%). Conversion of 7a into threo-7b was achieved in a similar fashion (2.9 mg, 14%).
L-erythro-6a
UV (CH3CN) λmax (log ε) 233 (5.14), 280 (4.52) nm; [α]D23 −67.0 (c 0.060, CHCl3); CD: see Figure 3 and Supporting Information; 1H NMR (600 MHz, CDCl3) δ 8.58 (1H, s), 8.49 (1H, s), 8.35 (1H, s), 8.03 (1H, dd, J=8.5, 1.7 Hz), 7.94 (1H, dd, J=8.6, 1.7 Hz), 7.92 – 7.85 (4H, m), 7.85 – 7.76 (5H, m), 7.73 (1H, d, J=8.6 Hz), 7.59 – 7.42 (5H, m), 7.39 (1H, d, J=8.6 Hz), 5.55 (1H, dt, J=8.6, 4.3 Hz), 5.01 (1H, ddt, J=8.6, 6.0, 4.3 Hz), 4.84 (1H, dd, J=11.8, 6.0 Hz), 4.76 (1H, dd, J=11.8, 4.3 Hz), 2.15 – 2.05 (1H, m), 2.00 – 1.90 (1H, m), 1.69 – 1.60 (2H, m), 1.02 (3H, t, J=7.4 Hz). 13C NMR (126 MHz, CDCl3) δ 167.39, 167.35, 167.1, 135.8, 135.7, 135.0, 132.8, 132.51, 132.47, 131.6, 131.51, 131.50, 129.51, 129.50, 129.2, 128.7, 128.6, 128.5, 128.3, 127.93, 127.87, 127.85, 127.84, 127.80, 126.92, 126.87, 126.84, 126.7, 125.3, 125.2, 123.7, 76.0, 63.3, 52.2, 34.6, 19.2, 14.0. HRMS m/z 618.2250 [M+Na]+ (calcd for C39H33NO5Na, 618.2251).
L-threo-7b
UV (CH3CN) λmax (log ε) 231 (5.22), 280 (4.61) nm; [α]D23 +29.6 (c 0.071, CHCl3); CD: see Figure 3 and Supporting Information; 1H NMR (500 MHz, CDCl3) δ 8.58 (1H, s), 8.53 (1H, s), 8.27 (1H, s), 8.02 (1H, dd, J=8.8, 1.6 Hz), 7.98 (1H, dd, J=8.5, 1.6 Hz), 7.92 – 7.73 (10H, m), 7.62 – 7.49 (5H, m), 7.46 (1H, dd, J=8.4, 6.7 Hz), 6.92 (N-H, 1H, d, J=9.2 Hz), 5.71 (1H, dt, J=8.9, 4.8 Hz), 5.03 (1H, ddt, J=8.9, 5.8, 4.8 Hz), 4.72 (1H, dd, J=11.6, 5.8 Hz), 4.65 (1H, dd, J=11.6, 4.8 Hz), 2.05 – 1.95 (1H, m), 1.95 – 1.86 (1H, m), 1.64 – 1.49 (2H, m), 1.00 (3H, t, J=7.3 Hz). 13C NMR (126 MHz, CDCl3) δ 167.8, 166.9, 166.8, 135.8, 135.7, 134.9, 132.7, 132.55, 132.49, 131.54, 131.50, 131.47, 129.52, 129.50, 129.1, 128.8, 128.6, 128.51, 128.49, 128.36, 127.90, 127.89, 127.87, 127.82, 127.78, 127.02, 126.95, 126.92, 126.86, 126.7, 125.21, 125.16, 123.5, 73.8, 64.9, 51.9, 34.1, 18.9, 14.0. HRMS m/z 618.2254 [M+Na]+ (calcd for C39H33NO5Na, 618.2251).
Conversion of 1a and 2a to Per-acetyl Derivative 8
A sample of 1a (0.5 mg) and Pd/C (1.0 mg) was taken up in MeOH (0.4 mL) and purged with H2 for 2 min. The heterogeneous mixture was allowed to stir at rt under H2 (1 atm) for 4 h. Complete hydrogenation was verified by LCMS. The mixture was then filtered (0.45 μm syringe filter), and then concentrated under a stream of N2. The resultant oil (0.5 mg) was then reconstituted in CH2Cl2:TFA (3:1, 0.2 mL) and stirred at rt for 30 min. The solvent was then removed under a stream of N2, and the oil was reconstituted in pyridine (0.1 mL) and acetic anhydride (0.1 mL). The reaction was allowed to stir at 35 °C under N2 for 3 hr. The volatile reagents were removed, and the crude oil was purified by RP-HPLC (Luna C18, 4.6 × 250 mm, 5 μm; 7:3 CH3CN:H2O, 0.85 mL min−1) to give 7,8-dihydro-peracetate-halisphingosine A (8, 0.4 mg). Conversion of 2a using the conditions of the last two of the three preceding reactions followed by purification gave 8 identical (HRMS, 1H NMR, LC) with the product obtained from 1a. 1H NMR (CDCl3, 600 MHz) see Table 1. 1H NMR (C6D6, 600 MHz) δ 5.16 (1H, m), 5.08 (2H, m), 4.61 (1H, dddd J=13, 9.7, 6.6, 3.1 Hz), 4.05 (1H, ddd, J=11, 6.5, 1.7 Hz), 3.94 (1H, ddd, J=11, 6.5, 1.7 Hz), 1.79 (3H, s), 1.70 (3H, s), 1.69 (3H, s), 1.55 (2H, m), 1.51 (3H, s); 1.15–1.47 (24H, m), 0.91 (3H, t, J=6.7 Hz); HRMS, m/z 508.3244 [M+Na]+ (calcd for C26H47NO7Na, 508.3245). Separate and co-injections of 8 derived from 1a or 2a gave a single peak (LCMS, rt 13.38 min, linear gradient from 60%–100% CH3CN aqu: 0.1 % formic acid, over 15 min; Kinetex C18, 4.6 × 150 mm, 2.7 μm).
Supplementary Material
Figure 1.

Sphingoid bases from Haliclona tubifera and erythro-D-ribosphingosine.
Acknowledgments
We thank Y. Su and Anthony Mrse (UCSD) for assistance with HRMS and NMR measurements, respectively, and to João Luís de Fraga Carraro for collection of the sponge and the photograph used in the graphic abstract. Purchases of the Agilent TOF mass spectrometer and the Jeol 500 MHz NMR spectrometer were made possible with funds from the NIH Shared Instrument Grant program (S10RR025636) and the NSF Chemical Research Instrument Fund (CHE0741968), respectively. While at UCSD, R. B. was supported by a travel fellowship (PDSE-CAPES 8301/11-5, Brazil) and E. P. S was supported by Ruth L. Kirschstein National Research Service Awards (NIH T32 CA009523). We are grateful for funding for this work from NIH (AI100776 to T. F. M).
Footnotes
Dedicated to Dr. Lester A. Mitscher, of the University of Kansas, for his pioneering work in the discovery of bioactive natural products and their derivatives.
Supporting Information Available. Additional experimental details and 1H NMR NMR data for compounds 1b, 2b, 6a, 7a, 12, 13 are available free of charge via Internet at http://pubs.acs.org.
References
- 1.Pruett ST, Bushnev A, Hagedorn K, Adiga M, Haynes CA, Sullards MC, Liotta DC, Merrill AH., Jr J Lipid Res. 2008;49:1621–1639. doi: 10.1194/jlr.R800012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter GT, Rinehart KL. J Am Chem Soc. 1978;100:7441–2.Cuardos R, Montejo de Garcini E, Wandosell F, Faircloth G, Fernández-Sousa JM, Avila J. Cancer Lett. 2000;152:23–29. doi: 10.1016/s0304-3835(99)00428-0.For a review of bioactive aminoalcohols from marine organisms, see Molinski T. Curr Med Chem: Anti-Infect Agents. 2004;3:197–220.
- 3.(a) Makarieva TN, Denisenko VA, Stonik VA. Tetrahedron Lett. 1989;30:6581–6584. [Google Scholar]; (b) Molinski TF, Makarieva TN, Stonik VA. Angew Chem Intl Ed. 2000;39:4076–4079. [PubMed] [Google Scholar]; (c) Nicholas GM, Molinski TF. J Am Chem Soc. 2000;122:4011–4019. [Google Scholar]; (d) Makarieva TN, Zakharenko AM, Dmitrenok PS, Guzii AG, Denisenko VA, Savina AS, Dalisay DS, Molinski TF, Stonik VA. Lipids. 2009;44:1155–1162. doi: 10.1007/s11745-009-3360-0. [DOI] [PubMed] [Google Scholar]; (e) Makarieva TN, Guzii A, Denisenko VA, Dmitrenok PS, Santalova EA, Pokanevich EV, Molinski TF, Stonik VA. J Nat Prod. 2005;68:255–257. doi: 10.1021/np049710z. [DOI] [PubMed] [Google Scholar]; (f) Makarieva TN, Dmitrenok PS, Zakarenko AM, Denisenko VA, Guzzi AG, Li R, Skepper CK, Molinski TF, Stonik VA. J Nat Prod. 2007;70:1991–1998. doi: 10.1021/np0704811. [DOI] [PubMed] [Google Scholar]; (g) Zhou BN, Mattern MP, Johnson RK, Kingston DGI. Tetrahedron. 2001;57:9549–9554. [Google Scholar]
- 4.(a) Molinski TF, Ireland CM. J Org Chem. 1988;53:2103–5. [Google Scholar]; (b) Salomon CE, Williams DH, Faulkner DJ. J Nat Prod. 1995;58:1463–1466. doi: 10.1021/np50123a021. [DOI] [PubMed] [Google Scholar]; (c) Skepper CK, Molinski TF. J Org Chem. 2008;73:2592–2597. doi: 10.1021/jo702435s. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kobayashi J, Naitoh K, Doi Y, Deki K, Ishibashi M. J Org Chem. 1995;60:6941–6945. [Google Scholar]; (b) Kiguchi T, Yuumoto Y, Ninomiya I, Naito T. Chem Pharm Bull. 1997;45:1212–1215. and key references within. [Google Scholar]
- 6.Kobayashi Ji, Cheng J-F, Ishibashi M, Walchli MR, Yamamura S, Ohizumi Y. J Chem Soc Perkin Trans. 1991;1:1135–1137. [Google Scholar]
- 7.Kuroda I, Musman M, Ohtani II, Ichiba T, Tanaka J, Gravalos DG, Higa T. J Nat Prod. 2002;65:1505–1506. doi: 10.1021/np010659y. [DOI] [PubMed] [Google Scholar]
- 8.Ledroit V, Debitus C, Lavaud C, Massiot G. Tetrahedron Lett. 2003;44:225–258. [Google Scholar]
- 9.Ando H, Ueoka R, Okada S, Fujita T, Iwashita T, Imai T, Yokoyama T, Matsumoto Y, van Soest RWM, Matsunaga S. J Nat Prod. 2010;73:1947–1950. doi: 10.1021/np1003565. [DOI] [PubMed] [Google Scholar]
- 10.The absolute configurations of diastereomeric erythro and threo 2-aminotetradec-5,7-dien-3-ols from a Papua New Guinea sponge, Haliclona sp. were first misassigned as (2S,3R) and (2S,3S), respectively, Gulavita NK, Scheuer PJ. J Org Chem. 1989;54:366–369.and revised upon total synthesis. Mori K, Matsuda H. Liebigs Ann Chem. 1992;2:131–137.
- 11.Mansoor TA, Park T, Luo X, Hong J, Lee CO, Jung JH. Nat Prod Sci. 2007;13:247–250. [Google Scholar]
- 12.Compound 1 (Ref. 11) ((2R,3R,6R,Z)-2-aminooctadec-7-ene-1,3,6-triol) had not been assigned a trivial name. Here, the name ‘halisphingosine A’ is coined for 1 for convenience of referencing and relational properties to the co-occurring new compound, halisphingosine B (2).
- 13.van’t Hoff JH. Die Lagerung der Atome im Raume. chp 8. Vieweg: Braunschweig; 1908. pp. 95–97. [Google Scholar]
- 14.For a contemporary review of integrated approaches to assignment of configuration in marine natural products, see Morinaka BI, Molinski TF. Tetrahedron. 2012;68:9307–9343. doi: 10.1016/j.tet.2011.12.070.
- 15.Li S, Wilson WK, Schroepfer GJ., Jr J Lipid Res. 1999;40:117–1125. [PubMed] [Google Scholar]
- 16.(a) Shirota O, Nakanishi K, Berova N. Tetrahedron. 1999;55:13643–13658. [Google Scholar]; (b) Kawamura A, Berova N, Dirsch V, Mangoni A, Nakanishi K, Schwartz G, Bielawska A, Hannun Y, Kitagawa I. Bioorg Med Chem Lett. 1996;4:1035–1043. doi: 10.1016/0968-0896(96)00092-2. [DOI] [PubMed] [Google Scholar]
- 17.Gonnella NC, Nakanishi K, Martin VS, Sharpless KB. J Am Chem Soc. 1982;104:3775–1376. [Google Scholar]
- 18.(a) Schneider C, Schreier P, Humpf HU. Chirality. 1997;9:563–567. [Google Scholar]; (b) Molinski TF, Brzezinski LJ, Leahy JW. Tetrahedron-Asymmetry. 2002;13:1013–1016. [Google Scholar]
- 19.Devijver C, Salmoun M, Daloze D, Braekman JC, De Weerdt WH, De Kluijver MJ, Gomez R. J Nat Prod. 2000;63:978–980. doi: 10.1021/np000081c. [DOI] [PubMed] [Google Scholar]
- 20.Ikemoto N, Lo LC, Nakanishi K. Angew Chem Int Ed. 1992;31:890–891. [Google Scholar]
- 21.Nicholas GN, Li R, MacMillan JB, Molinski TF. Bioorg Med Chem Lett. 2002;12:2159–2162. doi: 10.1016/s0960-894x(02)00367-0. [DOI] [PubMed] [Google Scholar]
- 22.Attempted removal of the N-Boc group in 1a using the standard protocol (50% CF3COOH in CH2Cl2, rt) or milder conditions (10 equiv, −20 °C) resulted in decomposition, most likely through SN1 solvolysis of the allylic OH. More benign conditions for deprotection of N-Boc-alkanamines (H2O, 100 °C, overnight, reported by Wang J, Liang YL, Qu J. J C S Chem Commun. 2009:5144–5146. doi: 10.1039/b910239f.when applied to 1a, returned only unreacted starting material.
- 23.Dalisay DS, Rogers EW, Edison A, Molinski TF. J Nat Prod. 2009;72:732–738. doi: 10.1021/np8007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.