

Abstract
Calcified dental plaque (dental calculus) preserves for millennia and entraps biomolecules from all domains of life and viruses. We report the first high-resolution taxonomic and protein functional characterization of the ancient oral microbiome and demonstrate that the oral cavity has long served as a reservoir for bacteria implicated in both local and systemic disease. We characterize: (i) the ancient oral microbiome in a diseased state, (ii) 40 opportunistic pathogens, (iii) the first evidence of ancient human-associated putative antibiotic resistance genes, (iv) a genome reconstruction of the periodontal pathogen Tannerella forsythia, (v) 239 bacterial and 43 human proteins, allowing confirmation of a long-term association between host immune factors, “red-complex” pathogens, and periodontal disease, and (vi) DNA sequences matching dietary sources. Directly datable and nearly ubiquitous, dental calculus permits the simultaneous investigation of pathogen activity, host immunity, and diet, thereby extending the direct investigation of common diseases into the human evolutionary past.
Unlike other human microbiomes, the oral microbiome will cause disease in a majority of people during their lifetime, suggesting that it is currently in a state of dysbiosis rather than symbiosis.1,2 The human oral microbiome comprises more than 2,000 bacterial taxa, including a large number of opportunistic pathogens involved in periodontal, respiratory, cardiovascular, and systemic disease.3-7 Dental calculus, a complex, calcified bacterial biofilm formed from dental plaque, saliva, and gingival crevicular fluid,8 is emerging as a potential substrate for the direct investigation of oral microbiome evolution and associated measures of oral health and diet.9,10 Recently, a DNA-based, 16S rRNA phylotyping study identified the major bacterial phyla in dental calculus and argued for shifts in microbial diversity associated with the origins of agriculture and industrialization,11 and to date five common oral bacteria have been identified in historic and prehistoric dental calculus using targeted PCR,12 qPCR,11 and immunohistochemistry.13 However, phylum-level community analysis and single species targeted amplification are insufficient to characterize oral health and disease states, as this requires a deeper taxonomic and functional understanding of microbiome ecology.14
We present the first detailed analysis of ancient oral microbiome ecology and function at the genus and species levels, leading to a deeper understanding of recent human oral microbiome evolution. Focusing on the dental tissues of four adult human skeletons (G12, B17, B61, and B78) with evidence of mild to severe periodontal disease from the medieval monastic site of Dalheim, Germany (ca. 950-1200 CE) (Supplementary Fig. 1), as well as modern dental calculus from nine patients with known dental histories, we demonstrate for the first time that the human oral microbiome has long served as a reservoir for a broad suite of opportunistic pathogens implicated in both local and systemic disease and harbored a diverse range of putative antibiotic resistance genes. We confirm the long-term role of host immune activity and “red-complex” pathogen virulence in periodontal pathogenesis, despite major changes in lifestyle, hygiene, and diet over the past millennium. We reconstruct the genome of a major periodontal pathogen, and we present the first evidence of dietary biomolecules to be recovered from ancient dental calculus. Finally, we further validate our findings by applying multiple microscopic, genetic, and proteomic analyses in parallel, providing a systematic biomolecular evaluation of ancient dental calculus preservation, taphonomy, and contamination.
The ancient oral microbiome
Applying shotgun DNA sequencing to dental calculus for the first time, we find that it is strongly dominated by bacterial DNA, with minor contributions from human, viral, dietary, and fungal sources (Fig. 1a). Using both targeted and shotgun 16S rRNA sequences (n=509,067), we identified a total of 2,699 microbial OTUs in the ancient dental calculus, with the top 100 most abundant taxa accounting for 86.6% of the total reads (Fig. 1b, Supplementary Fig. 2). One archaeal and nine bacterial phyla are dominant in ancient dental calculus (Supplementary Table 1): Firmicutes (49.5± 10.6%), Actinobacteria (12.0±6.1%), Proteobacteria (11.5±8.6%), Bacteroidetes (6.6±3.6%), TM7 (4.6±4.0%), Synergistetes (3.3±2.6%), Chloroflexi (2.7±1.5%), Fusobacteria (2.1±1.8%), Spirochaetes (0.6±0.3%), and Euryarchaeota (0.4±0.6%), all of which are also dominant in the human oral microbiome today.4 Notably rare in ancient dental calculus is Acidobacteria, a ubiquitous and abundant bacterial phylum in soil.15
Figure 1. Taxonomic and phylogenetic characterization of ancient dental calculus.
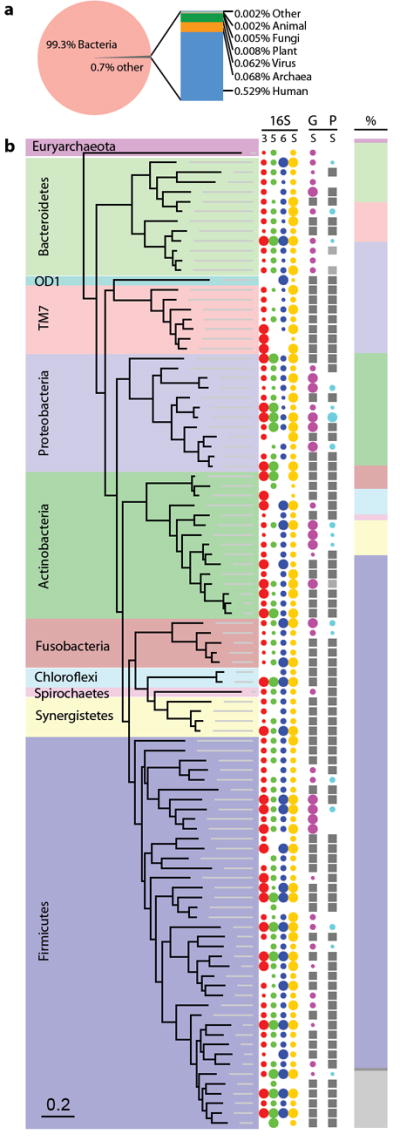
a, Relative proportion of bacterial, archaeal, eukaryotic, and viral DNA in ancient calculus estimated from assembled whole metagenome shotgun sequences of two individuals. b, Phylogenetic tree of the 100 most abundant OTUs in ancient dental calculus samples of four pooled individuals. Evidence for the presence and abundance of each microbial OTU is represented by colored, size-scaled circles for each targeted 16S rRNA region (V3, V5, V6), shotgun 16S rRNA sequences, and other genes and proteins assigned to that OTU. OTUs for which no reference genome exists or for which insufficient proteome data has been validated for inclusion in the protein search databases are marked with a gray square, as no hits could ever be matched to that OTU. Relative phylum abundance (normalized mean of all genetic data generated from 16S sequences) is represented by a column chart showing phyla represented in top 100 OTUs (color), remaining phyla (dark gray), and unidentified OTUs (light gray).
To address biases resulting from sequencing approach and primer choice (Supplementary Fig. 3), evidence for each OTU was visualized separately for each targeted and shotgun 16S rRNA detection method, as well as for shotgun metagenomic and metaproteomic data (Fig. 1b). Most OTUs were detected using multiple methods. OTUs detected from targeted V3 and shotgun data generally showed good agreement, while V5 and V6 primers show clear evidence of primer bias and OTU dropout. Shotgun metagenomic data show excellent agreement with consensus 16S rRNA OTUs when reference genomes are available. Shotgun metaproteomic data also showed good agreement with OTUs identified on the basis of genetic data, and agreement is expected to improve as protein databases grow to include more predicted proteins and epigenetic variants. Because ancient DNA and proteins undergo different taphonomic processes and have different contamination risks, the high degree of phylogenetic consensus observed from data generated from independent extractions, using different methods, and targeting different biomolecular types demonstrates that an endogenous oral microbiome can be robustly and reliably recovered from ancient dental calculus.
Carriage of specific pathogens
The normal human oral flora includes a large number of endogenous cariogenic, periodontal, and other opportunistic pathogens. Although these taxa generally do not cause extra-oral disease in healthy subjects, they nevertheless pose a serious risk for the elderly and immunocompromised16,17 and are known to be involved in the etiology of chronic systemic diseases, including cardiovascular disease.18 As detection of particular species from metagenomic sequence data is an open area of research, we applied a conservative contig assembly and BLAST strategy and screened our results against the Pathosystems Resource Integration Center (PATRIC) database19 to identify 40 putative opportunistic pathogens in ancient dental calculus (Table 1), of which only 5 had been previously reported in ancient samples.11-13 We also identified phage DNA sequences specific to particular bacteria (Table 1), including Streptococcus mitis phage SM1, which has been previously shown to mediate S. mitis attachment to platelets and increase bacterial virulence in the endocardium.20
Table 1.
Putative pathogens identified from assembled metagenomic and metaproteomic sequences in ancient dental calculus
| Pathogensa | Genes (contigs) | Proteins (peptides) | Virulence | Drug resistanceb | Plasmid | CTn/Phage |
|---|---|---|---|---|---|---|
| Actinomyces odontolyticusd | 3 (4) | 3 (34) | ||||
| Aggregatibacter actinomycetemcomitans | 50 (68) | 0 | + | + | ||
| Campylobacter concisus | 10 (20) | 0 | + | |||
| Campylobacter curvus | 12 (11) | 0 | ||||
| Campylobacter rectusd | 3 (9) | 3 (15) | ++ | |||
| Campylobacter showaed | 3 (13) | 1 (2) | ||||
| Capnocytophaga gingivalisd | 2 (11) | 3 (7) | + | |||
| Capnocytophaga ochracea | 938 (4909) | 0 | + | + | + | + |
| Capnocytophaga sputigenad | 2 (2) | 0 | + | |||
| Clostridium difficilee,f | 30 (76) | 0 | + | + | ||
| Corynebacterium matruchotiid | 2 (15) | 12 (89) | ||||
| Eikenella corrodensd | 11 (38) | 2 (11) | + | |||
| Fusobacterium nucleatum | 656 (1525) | 4 (21) | ++ | + | + | + |
| Fusobacterium periodonticumd | 3 (6) | 3 (8) | + | |||
| Gemella morbillorumd | 9 (38) | 0 | ||||
| Gordonibacter pamelaee | 3 (30) | 0 | ||||
| Haemophilus influenzae | 19 (43) | 1 (4) | + | |||
| Histophilus somnie,g | 9 (18) | 0 | + | |||
| Leptotrichia buccalis | 492 (1104) | 0 | + | + | + | + |
| Neisseria gonorrhoeae | 127 (250) | 1 (2) | + | + | ||
| Neisseria meningitidis | 336 (821) | 1 (2) | + | + | + | |
| Neisseria siccad | 3 (8) | 4 (35) | ||||
| Neisseria subflavad | 4 (12) | 0 | ||||
| Porphyromonas gingivalis | 802 (2588) | 7 (72) | ++ | + | + | |
| Rothia mucilaginosa | 24 (17) | 1 (2) | + | |||
| Streptobacillus moniliformise,g | 8 (23) | 0 | ||||
| Streptococcus agalactiae | 7 (27) | 0 | + | + | ||
| Streptococcus dysgalactiaee | 2 (8) | 0 | + | |||
| Streptococcus equie,g | 29 (101) | 0 | + | + | ||
| Streptococcus gallolyticuse,g | 8 (11) | 0 | + | |||
| Streptococcus gordonii | 882 (3397) | 1 (8) | + | + | + | + |
| Streptococcus mitis | 88 (161) | 1 (37) | ++ | + | + | |
| Streptococcus mutans | 21 (67) | 0 | ||||
| Streptococcus pneumoniae | 144 (339) | 1 (8) | + | + | + | |
| Streptococcus pyogenes | 14 (32) | 1 (8) | + | + | ||
| Streptococcus sanguinis | 850 (3272) | 1 (4) | + | + | ||
| Streptococcus suise,g | 2 (3) | 0 | + | |||
| Tannerella forsythia | 1099 (11279) | 10 (137) | ++ | + | + | |
| Treponema denticola | 917 (6106) | 3 (15) | ++ | + | + | + |
| Veillonella parvula | 96 (109) | 0 | + |
Notes
Metagenomic data from G12 and B61; proteomic data from G12, B17, B61, and B78.
+, gene(s) detected; ++, gene(s) and protein(s) detected
Includes only pathogens with valid entries in the PATRIC database. Only taxa represented by >1 DNA contig are shown. All pathogens are known inhabitants of the human oral cavity, as confirmed by cross-referencing with the Human Microbiome Project (HMP) data for supragingival dental plaque.
Putative function based on gene homology and NCBI annotation; functionality not independently validated.
Annotations retrieved from the PATRIC database.
Reference genome sequencing and annotation incomplete.
Not a prevalent inhabitant of the oral cavity; not present in the Human Oral Microbiome Database (HOMD).
Tentative identification; sequences correspond almost exclusively to mobile genetic elements.
Putative zoonosis.
Both DNA and proteins from the periodontal pathogens Tannerella forsythia, Porphyromonas gingivalis, and Treponema denticola are particularly abundant in our ancient dental calculus samples, demonstrating that these so-called “red-complex” bacteria21 were strongly associated with periodontal disease during the medieval period, just as they are today, despite significant changes in oral hygiene, diet, and lifestyle. Additionally, all three of these pathogens are found at substantially higher frequency in our ancient dental calculus samples than in the Human Microbiome Project (HMP)3 healthy cohort (Supplementary Figure 4a-c), consistent with expectations for periodontal disease. We also identified several oral taxa (e.g., Aggregatibacter actinomycetemcomitans, Streptococcus mutans, and S. mitis) that have been shown to cause bacteremia and infective endocarditis,7,18 demonstrating that the human oral microbiome has long harbored pathogens that contribute to cardiovascular disease risk. Additional pathogens include those implicated in acute dental infections (e.g., Actinomyces odontolyticus), caries (S. mutans), and opportunistic upper and lower respiratory illness (e.g., S. pneumoniae, S. pyogenes, and Haemophilus influenzae). Of interest, all ancient dental calculus samples were also found to contain disordered carbon (micro-charcoal), a respiratory irritant. Two obligate human taxa, Neisseria meningitidis and N. gonorrhoeae, causative agents of bacterial meningitis and gonorrhea, respectively, were also observed. N. meningitidis and N. gonorrhoeae form a recently diverged pathogenic clade of Neisseria, a genus comprising many commensal species inhabiting the mucosa and dental surfaces of animals,22 and both are prevalent members of the human oral microbiome. Genital N. gonorrhoeae strains can infect the pharynx and engage in genetic exchange with other Neisseria species;23 however, oral strains are not known to cause genital infection. Oral N. meningitidis is a leading cause of bacterial meningitis, although disease susceptibility is determined by a combination of host genetics and strain virulence.24 Finally, we observed two additional oral taxa present at substantially higher frequency in at least one ancient dental calculus sample compared to the HMP healthy cohort: Filifactor alocis and Olsenella uli (Supplementary Figure 4e-f). Although not classified as pathogens in the PATRIC database, these bacteria have recently been associated with periodontitis and endodontic infections, respectively.25,26
Virulence
To further characterize the pathogens detected in ancient human dental calculus, functional feature information for putative virulence-, drug resistance-, plasmid-, transposon-, and phage-associated genes and proteins were compared to NCBI records. While not exhaustive, a preliminary list of well-supported virulence genes and proteins was compiled using this method (Table 1), revealing a wide range of virulence factors associated with adhesion/aggregation (e.g., adhesins and lectins) and parasitism (e.g., phospholipases, hemagglutinins, and hemolysins), as well as extensive machinery for horizontal gene transfer (e.g., pilin, CTn, and phage sequences). In several cases, we detected both the virulence gene and its protein product, e.g., Msp/major sheath protein in T. denticola and Rgp/Arg-gingipain in P. gingivalis. Arg-gingipain and Lys-gingipain, another extracellular cysteine proteinase identified by proteomic evidence, are highly antigenic and extremely abundant in P. gingivalis, accounting for 10% w/w of the total proteins produced by the organism.27 Notably, we also detected Type IV fimbrilin, an outer membrane protein variant associated with virulent P. ginvialis strains.28
Antibiotic resistance
The human microbiome is an important site of horizontal gene transfer and a potential reservoir of antimicrobial resistance.29 Metagenomic studies of modern dental plaque have found a wide range of predicted genes related to resistance to diverse antibiotics and toxic compounds.30 The antiquity of bacterial antibiotic resistance genes has recently been tested in permafrost soils dating to the Pleistocene,31 but until now, the antiquity of antibiotic resistance in human microbiota prior to the use of therapeutic antibiotics has not been investigated.
Using both automated and manual searching strategies, we identified within ancient dental calculus numerous DNA sequences with homology to antibiotic resistance genes found in oral and pathogenic bacteria, including multi-drug efflux pumps and native resistance genes to aminoglycosides, β-lactams, bacitracin, bacteriocins, and macrolides, among others, as well as a near complete plasmid-encoded conjugative transposon carrying efflux pump genes with high homology to CTn5 of Clostridium difficile (Supplementary Table 2). Although the exact function of these genes in our samples is unclear, their presence nevertheless demonstrates that the biomolecular machinery for broad-spectrum, low-level antibiotic resistance has long been present in the human microbiome, illustrating how the oral microbiome functions as both a source and a reservoir of novel antibiotic resistance.23
Pathogen genome reconstruction: Tannerella forsythia
T. forsythia (formerly Bacteroides forsythus and T. forsythensis) is an anaerobic, gram-negative member of the phylum Bacteroidetes and a known inhabitant of supragingival and subgingival plaque.32 It is associated with advanced forms of periodontal disease and has been reported in atherosclerotic lesions.7 Based on 16S rRNA gene data, T. forsythia was observed to be at moderate abundance (0.09-0.84%) in the dental calculus of one individual (G12) and, as a pathogen of interest, was selected for genome reconstruction.
Using a conservative mapping strategy, a total of 10,991 contigs were recruited to the ancient T. forsythia genome reconstruction, at a mean nucleotide depth of coverage of 5.7 (Fig. 2a). Ninety-one percent of T. forsythia genes (n=2799) were mapped by at least one contig, and unmapped genes included 94 transposases, transfer factors, and other mobilization genes that may be specific to the T. forsythia ATCC 43037 reference genome strain used for alignment. The largest gap in our genome reconstruction, which spans ~48,000 bp and 53 genes, corresponds to a complete conjugative transposon carrying putative tetracycline resistance genes that is absent in our reconstructed ancient T. forsythia genome. In addition to genetic sequences, MS/MS identified 118 peptides belonging to ten T. forsythia proteins (Fig. 2a). Of these proteins, nine are outer membrane or S-layer proteins, seven have known function, and four are antigenic: T. forsythia surface protein A (TF2661-2, tfsA), T. forsythia surface protein B (TF2663, tfsB), outer membrane protein 41 (TF1331, omp41), and one hypothetical protein (TF2339).33
Figure 2.
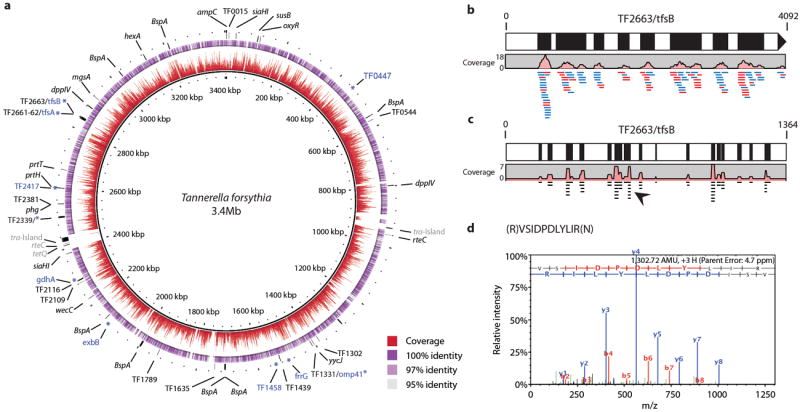
Genomic coverage plot for the periodontal pathogen Tannerella forsythia, with details of gene and protein coverage of the virulence factor TF2663/tfsB, from medieval human dental calculus (G12). a, Plot of the T. forsythia genome with depth of coverage (0 to 30-fold shown) in red and identity in purple. Gene locations of major virulence factors present (black) or absent (gray) in the assembly are indicated in the outer ring. Notably absent are two transposon-related tra pathogenicity islands containing putative tetracycline resistance genes in the T. forsythia ATCC 43037 reference strain. Proteins identified by MS/MS are indicated in blue by an asterisk (*) at the corresponding gene locus. b, Enlarged view of forward (blue) and reverse (red) DNA reads mapped to gene TF2663, which encodes the surface layer B protein, tfsB. c, Enlarged view of peptide coverage for tfsB, a species-diagnostic virulence factor involved in haemagglutination, adherance, and host tissue invasion. d, Detail of a representative unique tfsB peptide (arrowhead) with corresponding spectrum.
Several virulence factors and antigenic proteins have been identified in T. forsythia to date (Fig. 2a), including Bacteroides surface protein A (BspA), dipeptidyl peptidase-4 (dppIV), tfsA, and tfsB, among others.27,33 The genes encoding each of these virulence factors are present in our reconstruction. The glycosylated T. forsythia S-layer proteins tfsA and tfsB are directly involved in hemagglutination, adhesion, and tissue invasion.34 They are also unique and species-diagnostic, as they have no homology to other known S-layer proteins or glycoproteins.35 DNA and protein coverage of tfsA and tfsB was high in our dataset; for example, ten contigs comprising 116 reads mapped to the TF2663/tfsB gene (Fig. 2b), and we identified 65 spectra belonging to 27 unique tfsB peptides (Fig. 2c-d). Given that a functional T. forsythia S-layer is essential for host immune evasion and biofilm co-aggregation,34 the discovery of abundant, well-preserved S-layer gene and protein sequences makes ancient dental calculus an excellent candidate for investigating the evolution of periodontal pathogenesis in humans.
MS/MS analysis of host immunity and disease pathogenesis
Despite dense microbial colonization and the regular introduction of foreign substances, the oral cavity is effective at preventing most infections. At least 45 antimicrobial gene products acting as early responders of the innate immune system have been identified in saliva and gingival crevicular fluid.36 We identified 43 human proteins within ancient dental calculus, of which 25 are involved in the innate immune system (Fig. 3a). Eight of these proteins have demonstrated antimicrobial properties and include cationic peptides (α-defensin, azurocidin), metal ion chelators (calgranulin A, calgranulin B, lactoferrin), protease inhibitors (myeloperoxidase), and bactericidal proteins (bactericidal permeability-increasing protein, lysozyme C, peptidoglycan recognition protein 1). Expression of many of these proteins is specific to a particular cell type and even sub-cellular component (e.g., azurocidin is specific to neutrophil lysozomic azurophil granules), allowing highly resolved characterization of immune system response. Approximately one-third of identified human proteins were shared between ancient and modern calculus (Fig. 3b), and functional profiles were highly similar (Fig. 3a). By contrast ancient tooth roots were distinct both in protein composition and function, being dominated by collagens and other proteins involved in mineralized tissue (biglycan, periostin) and vascular (prothrombin) development and maintenance.
Figure 3. Metaproteomic comparison of human proteins in modern and ancient dental samples.
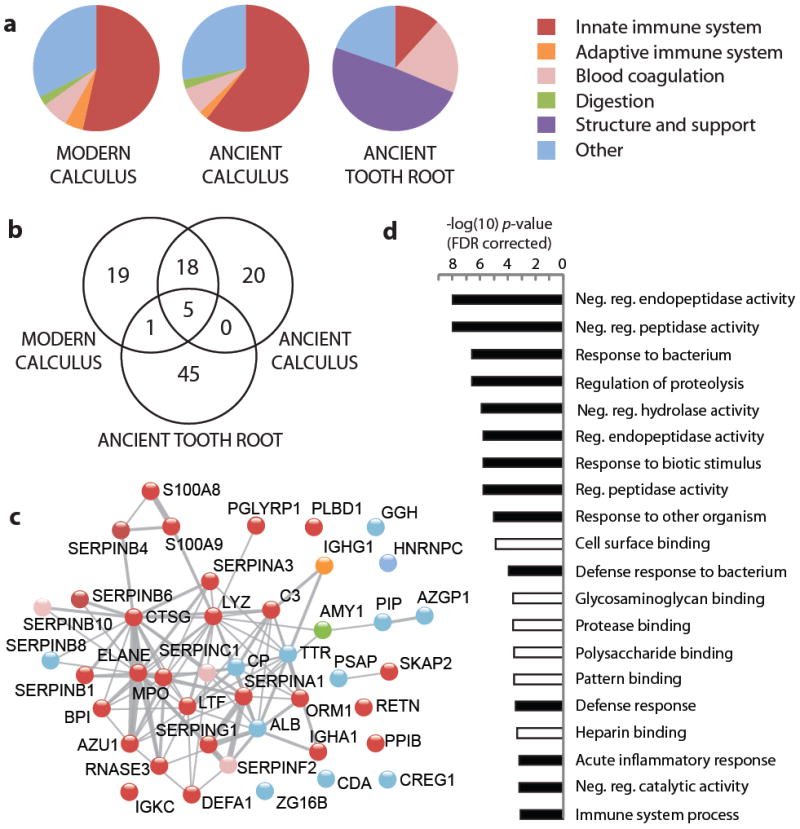
a, Functional characterization of human proteins identified in modern dental calculus (two individuals), ancient dental calculus (four individuals), and ancient tooth roots (four individuals). b, Venn diagram of shared human proteins by sample type. c, STRING network representation of human proteins identified in ancient dental calculus; nodes are labeled by gene name and colored in accordance with functional categories and connections (gray) to predicted functional partners. The network is set to medium confidence (0.4) for all active prediction methods. d, Gene ontology categories with significant enrichment (p < 0.001, FDR corrected) in ancient dental calculus for biological process (black) and molecular function (white) relate to proteinase regulation, substrate binding, and innate immune function. Enrichment calculated relative to the human genome using the STRING-embedded AmiGO term enrichment tool.
The STRING resource37 was used to investigate functional interaction networks among the ancient dental calculus human proteins. A large number of functional interactions were predicted (Fig. 3c), and 79% of proteins (n=34) are functionally connected to at least one other protein in the network. Immunoglobulin heavy chain (IgA, IgG) and light chain (kappa) peptides were detected in ancient calculus, as was α-amylase, a salivary enzyme that breaks down dietary starch; however, the majority of proteins were related to the innate immune system. Ancient dental calculus human proteins are strongly enriched in extracellular (p-value 3.2e-12, FDR-corrected) and secretory (p-value 4.3e-9, FDR-corrected) proteins, mostly of neutrophil origin. Extravasated neutrophils are recruited to sites of injury by IgG and have a life span of less than 24 hours;38 thus, neutrophil proteins are only released into calcifying dental plaques during active infection and inflammation. Relatively few human cellular proteins were found, suggesting that immune cells do not invade the calcifying plaque but rather release antimicrobial substances from the junctional and pocket epithelia, a process that is consistent with neutrophil “frustrated phagocytosis”39 and NETosis.40 Ancient calculus human proteins were significantly enriched in biological processes related to inflammation, innate immunity, and host defense, as well as molecular functions such as cell surface, protease, and glycosaminoglycan binding (Fig. 3d). The observation of an abundance of inflammatory (myeloperoxidase, azurocidin, lysozyme, calprotectin, elastase) and anti-inflammatory (α-1-antitrypsin and α-1-antichymotrypsin) innate immune system proteins in ancient dental calculus, coupled with morphological evidence of attachment loss and alveolar recession, is strongly supportive of active periodontal inflammation and disease.
Coupled to this immunological data, we identify oral pathogens and bacterial virulence proteins in ancient and modern dental calculus known to provoke strong immunological reaction and contribute to periodontal pathogenesis, most notably P. gingivalis (gingipains), T. forsythia (S-layer proteins), and T. denticola (major sheath protein). P. gingivalis has recently been shown to stimulate neutrophils to release resistin, a protein implicated in acquired insulin resistance.41 Resistin may exacerbate the progression of type II diabetes,42 and interestingly, we identified resistin on the basis of reasonably abundant evidence (36 spectra, 9 unique peptides) in ancient dental calculus. Resistin was also identified in modern calculus (7 spectra, 5 unique peptides), but not ancient tooth roots.
Ancient dietary reconstruction
Given current challenges in nutritional health and obesity,43 a growing interest in dietary aspects of the hygiene hypothesis,44 and a recent study suggesting ancient oral microbiome shifts associated with periods of agricultural transition,11 there is great interest in better understanding the evolutionary history of human diet; however, paleodietary reconstruction is made difficult by the generally poor preservation of plants and small animals in the archaeological record. Human bone stable isotope analysis and dental calculus-based plant microfossil research have broadened our knowledge of past dietary practices, but these tools are insufficient to characterize many major dietary components at high taxonomic resolution. Ancient DNA-based approaches offer great advantages and have been used to identify dietary components from archaeological feces (coprolites), as well as to investigate plant remains directly.45 However, as coprolites and preserved plant remains are relatively rare, we sought to characterize dietary information from dental calculus using both biomolecular and conventional methods.
From our metagenomic sequence reads, a total of 487 reads (0.0003%) were confidently identified as Eukaryotic organelle sequences; of these, 266 were assigned to the kingdom Viridiplantae and 21 were assigned to the kingdom Animalia. Within these kingdoms, most of the organelle reads mapped ambiguously to multiple organisms/genera, leaving only 20 reads that could be positively identified at a sub-family level. Of these 20 reads, 17 are of host origin, and the remaining three reads matched diagnostic mitochondrial sequences for pig/boar (Sus sp.), crucifer (Brassica sp.), and bread wheat (Triticum aestivum). Analysis of assembled contigs additionally revealed one putative sheep (Ovis sp.) and several human (n=326) nuclear genomic sequences (Fig. 4a-d). Although previous studies have reported trace animal domesticate DNA contamination (cattle, pig, and chicken) in some PCR reagents,46 we found no evidence of such contamination, and additionally wheat, crucifers, and sheep are not part of this supply chain. The discovery of preserved dietary biomolecules is consistent with previous observations of intact dietary microfossils, such as starch grains, in archaeological dental calculus10 and with reports of wheat and cassava (tapioca) chloroplast DNA in the dental plaque of living subjects.4 Turning to proteins, we identified one putative dietary plant protein, chloroplast glyceraldehyde 3-phosphate dehydrogenase (GAPDH), in ancient calculus, but disambiguation below the phylum Viridiplantae was not possible. Faunal proteins were not confidently identified within ancient dental calculus, but we did identify bovine β-lactoglobulin, a milk protein, in modern dental calculus, demonstrating that recovery of dietary animal proteins from dental calculus is possible.
Figure 4. Genetic, microfossil, zooarchaeological, and stable isotopic evidence for medieval human diet at Dalheim.
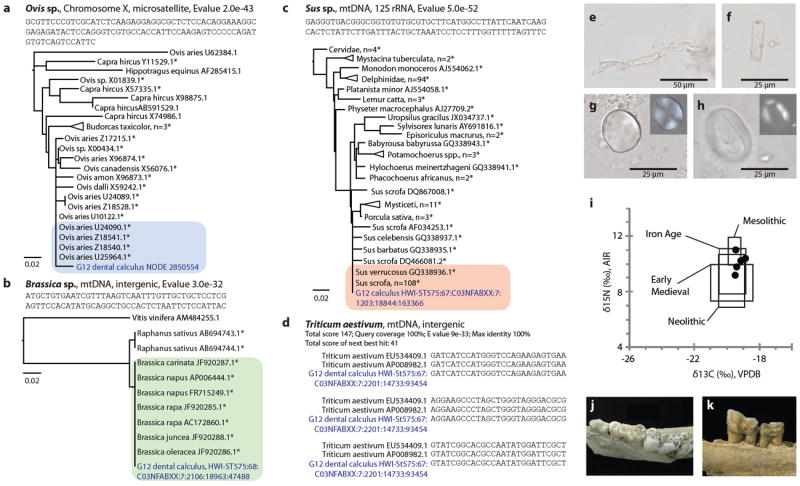
Neighbor joining trees for Genbank sequences aligning to putative dietary sheep (a), crucifer (b), and pig/boar (c) sequences. Trees include accessions with alignment scores >45, except for (c), which is limited to the top 250 alignments; highly significant alignments (E-value < 1e-30) are starred (*). BLAST top hits for each dietary sequence are highlighted. Maximum fraction of mismatched bases is 0.75 for tree generation, and distance was calculated using a Jukes-Cantor substitution model. d, One sequence aligned to two accessions of bread wheat only. e-h, Microfossils recovered from ancient human dental calculus yielded morphological matches to animal collagen fibers (e), a monocot phytolith (f), and starch grains of the grass tribe Triticeae (g) and the legume family Fabaceae (h). i, C and N stable isotopic values of human bone collagen (black circles) fall within two standard deviations (boxes) of those measured for other Central European populations and are consistent with a diet of mixed C3 terrestrial plant and animal resources. j-k, Recovered food waste includes skeletal material from Sus sp. (j) and Caprinae (k).
Because our discovery of dietary biomolecules in dental calculus is novel, we sought to validate our results using independent paleodietary methods. Microfossil analysis of ancient dental calculus yielded morphological matches to animal connective tissue fragments (Fig. 4e, n=2), an unidentified monocot phytolith (Fig. 4f), plant bast fibers (n=3), and starch grains consistent with the cereal tribe Triticeae (Fig. 4g, n=27) and the legume family Fabaceae (Fig. 4h, n=1), among other debris (Supplementary Fig. 5). Stable isotope analysis of human bone collagen (Fig. 4i) from the four ancient human individuals indicated a mixed diet of C3 terrestrial plant and animal resources typical of Central European populations from the late Mesolithic through the medieval period.47-50 Zooarchaeological analysis of food waste at the site confirmed the presence of pig/boar (Sus sp., Fig. 4j) and sheep/goat (Caprinae, Fig. 4k), as well as cattle (Bos sp.) and equids (Equus sp).
Biomolecular analysis of dental calculus thus yields complementary dietary information compared to conventional methods, as well as novel finds. The high taxonomic precision of genetic approaches allows closely related taxa (e.g., Caprinae) to be distinguished in the absence of diagnostic skeletal elements, and underrepresented plant taxa, such as Brassica, can be identified without the biological and taphonomic biases that compromise macro- and microfossil preservation of leafy greens and vegetables.
Taphonomy and Contamination
Postmortem taphonomy and contamination pose challenges in ancient biomolecular research. To address these potential problems in our dataset, we employed multiple protocols for authenticating our data, including scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS), optical microscopy, Raman spectroscopy, protein damage analysis, genetic network analysis, and probabilistic genetic source tracking.
After death, environmental microbes are known to infiltrate the dentition, causing substantial tissue degradation, loss of organic matter, and altered mineralization patterns in dentine and cementum (Supplementary Fig. 6).51 We observe, however, little evidence of postmortem alteration in ancient dental calculus samples (Fig. 5a). EDS imaging reveals a thin deposit of silicon-rich soil matrix only on the dental calculus surface (Fig. 5b), and no evidence of altered mineralization was observed within ancient dental calculus, a finding that we confirmed by Raman spectroscopic comparison with modern controls (Supplementary Fig. 7). During life, growth of dental calculus is appositional,8,52 resulting in a laminar cross-sectional structure characterized by alternating bands of Gram-positive and Gram-negative bacteria (Supplementary Fig. 8), a pattern we also observe in ancient calculus (Fig. 5c-d). DNA fluorescent dye reveals a similar distribution of dsDNA in ancient and modern calculus, in many cases resolving to individual cells (Fig. 5e, Supplementary Fig. 9) corresponding to a diverse range of in situ bacteria embedded within undisturbed dental calculus matrix (Fig. 5f-g).
Figure 5. Evidence of microscopic and biomolecular preservation of ancient dental calculus.

a, Labio-lingual section of a mandibular incisor with dental calculus (arrow) on the labial crown surface; both dentine and cementum within the tooth root show extensive evidence of postmortem alteration. b, EDS visualization of Ca (red) and Si (green) shows Si is restricted to the surface except for one biogenic Si inclusion (arrow). c, Detail of dental calculus which exhibits a layered structure suggesting outward-downward incremental growth. d, Detail of a stained section showing Gram-negative (red) and Gram-positive (blue) bacteria. e, Detail of Hoechst stained section showing abundant in situ dsDNA. f, g, The calculus matrix contains numerous lacunae of microorganisms (arrows), some of which are mineralized (arrow-heads). h, Proportions of human and bacterial proteins identified in ancient and modern samples. i, DNA extraction yields from modern (M) and ancient dental calculus, dentine, and alveolar bone samples. j, Comparison of microbial communities in ancient dental samples (squares) to those in diverse publicly available samples (circles). 16S rRNA data was generated by shotgun sequencing (S) and targeted amplification and sequencing of hypervariable regions (V3, V5, V6), followed by OTU clustering. Ancient metagenomes (n=38) were plotted within a network where distance scales with OTU community similarity. Modern metagenomes with >20% shared community are shown connected by black lines; ancient metagenomes with >20% shared community are connected by green lines. A total of 315 Genbank studies were recruited to the network. Ancient metagenomes segregate into two distinct groups: ancient dental calculus samples cluster tightly together, are connected by thick lines, and show similarity to modern metagenomes of primarily human and oral origin; ancient dentine and abscessed bone tissue samples form a more diffuse cluster and recruit primarily soil and environmental metagenomes. Carious dentine forms an intermediate cluster that shares OTUs with both human-associated and environmental sources.
Ancient dental calculus yields microbial (n=239) and human (n=43) proteins in the same relative proportion and with similar functions as compared to modern controls (Fig. 3a, Fig. 5h, Supplementary Fig. 10), while only human proteins (n=53) were confidently identified from tooth roots and bone. Damage analysis of dental calculus proteins reveals a higher proportion of spontaneous, non-enzymatic post-translational modifications in ancient samples compared to modern controls; however, both modern and ancient dental calculus peptides exhibited relatively high proportions of non-tryptic cleavages (>10%), an observation consistent with in vivo exposure to bacterial and immune system proteases (Supplementary Fig. 11).
Total DNA recovery from ancient dental calculus (5-437 ng DNA/mg calculus) is comparable to modern calculus and 1-3 orders of magnitude greater than from paired dentine (0.3-0.5 ng/mg), carious dentine (0.2 ng/mg) and abscessed bone (0.4 ng/mg) (Fig. 5i). Analysis of 16S rRNA phylotypes using a novel network analysis tool developed for this study reveals that the bacterial communities within ancient dental calculus closely resemble published human oral microbiomes, and are distinct from the communities observed in ancient dentine and bone, which cluster primarily with published soil samples, indicating environmental contamination after death (Fig. 5j, Supplementary Fig. 12). This pattern was found to be robust to extraction method, decontamination method, primer selection, sequencing method, and inter-individual variation. Reanalysis of our data using the methods employed by the HMP3 yields equivalent results (Supplementary Fig.13-15) that were also confirmed using the Bayesian tool SourceTracker53 (Supplementary Fig. 16). Ancient dental calculus is thus revealed to be a remarkably well-preserved biological material that allows direct and detailed investigations of the ancient oral microbiome.
Conclusions
Dental calculus is among the richest biomolecular sources yet identified in the archaeological record. Given the exceptional preservation of DNA within dental calculus (5-437 ng/mg), next generation shotgun sequencing libraries can be built from milligrams of material, thereby reducing sample requirements typically required for ancient DNA analysis by two orders of magnitude. We demonstrate that the “red-complex” pathogens Tannerella forsythia, Porphyromonas gingivalis, and Treponema denticola have long-been associated with periodontal disease, despite changes in lifestyle, hygiene, and diet since the medieval period. We confirm the long-term carriage of opportunistic pathogens in the human oral cavity, including the causative agents of oral and respiratory diseases, as well as bacteria implicated in the progression of cardiovascular disease and the formation of arterial plaques. We find genetic evidence that the human oral cavity has long harbored genes with homology to putative antibiotic resistance genes, the first such demonstration in an ancient human-associated sample. We reconstruct the genome of the periodontal pathogen Tannerella forsythia without prior enrichment and identify the absence of a complete conjugative transposon carrying putative tetracycline resistance genes found in the reference strain. We report for the first time the presence of well-preserved proteins within ancient dental calculus and show that although the dental calculus metagenome is dominated by bacterial DNA (>99%), the dental calculus metaproteome contains high proportions of both host and microbial proteins of clinical significance. Because the growth of calculus is appositional without remodeling, it may offer a potential solution to the “osteological paradox” in studies of ancient disease,54 and given that proteins are known to survive longer in the archaeological record than DNA, dental calculus may allow the recovery of valuable proteomic data from deep time periods that are out of reach using genomic technologies. Finally, we report the first plant and animal DNA sequences recovered from ancient dental calculus; these sequences allow greater taxonomic precision than currently possible using microfossil or stable isotope paleodietary techniques. Dental calculus is a robust, long-term biomolecular reservoir of ancient disease and dietary information, and it has important implications for the fields of medicine, microbiome research, archaeology, and human evolutionary studies.
Methods
Study design and samples
Narrative and graphical overviews of the study design are provided in the Supplementary Note and Supplementary Figure 17. Archaeological material was obtained from the medieval St. Petri church and convent complex in Dalheim, Germany (Supplementary Figure 18) and radiocarbon dated to c. 950-1200 CE (Supplementary Table 3). The assemblage was evaluated for pathologies (Supplementary Table 4), and dental tissues from four well-preserved adult skeletons (G12, B17, B61, B78) and two fauna (F1, F5) were selected for further analysis (Supplementary Figures 1, 19-20). Additionally, dental tissues from nine modern controls (P1-P5, P7, P8, P10, P13) with known dental health histories (Supplementary Table 5) were obtained under informed consent and protocols were approved by the Zürich Ethics Commission (KEK ZH-Nr. 2012-0119).
Microscopy and spectroscopy
A mandibular incisor from B78 was sectioned longitudinally and examined according to standard protocols with a Tescan VEGA scanning electron microscope using backscattered electron (BSE) imaging and energy-dispersive X-ray spectroscopy (EDS) with a Si(Li) detector. Dental calculus deposits from B78 and P3 were fixed, decalcified, and prepared into serial thin sections using modified standard protocols, followed by Gram and Hoechst staining and visualization using a Zeiss Axio Imager M2 and a Leica DMI6000 B microscope. Microfossils were obtained from dental calculus (G12, B17, B61, B78) and dental calculus/crown cementum (F5) deposits (Supplementary Table 6) using an incremental HCl decalcification protocol (Supplementary Note) and visualized using a Zeiss compound microscope under white and polarized light to identify pollen, phytoliths, starch grains, and other debris (Supplementary Table 7) by comparison to reference collections. To evaluate mineralogical composition, Raman spectroscopy was applied to six calculus (G12, B17, B61, B78, P3, P13), nine dentine (G12, B17, B61, B78, P4, P5, P7, P8, P10), and five soil matrix (M1-M5) specimens using a HORIBA XploRA instrument (100x magnification and 532nm laser wavelength) and analyzed for the main PO4 3- peak position and peak area, as well as the peak intensity ratios of C-H (~2940 cm-1) I(CH) and main phosphate peak I(P) (Supplementary Table 8).
Isotope ratio mass spectrometry
Rib specimens from G12, B17, B61, and B78 were cleaned by abrasion and collagen was extracted after the method of Richards and Hedges57 with an additional ultrafiltration step. Carbon and nitrogen isotopic values were measured in duplicate using a Thermo-Finnighan Delta XP continuous-flow isotope-ratio mass spectrometer following combustion in an elemental analyzer FLASH EA 2112 (Supplementary Table 9).
DNA extraction
Ancient samples were extracted in a dedicated ancient DNA laboratory at the ZEM/University of Zürich in accordance with established contamination control precautions and workflows. DNA was extracted from dental calculus (G12, B17, B61, B78, P2), dentine (G12, B17, B61, B78), carious dentine (B17), abscessed alveolar bone (B78), and burial matrix (M1-M5) by phenol:chloroform extraction followed by Qiagen MinElute column purification (Supplementary Tables 10-15). Burial matrix and NaOCl-decontaminated dentine were tested for the presence of endogenous human DNA using targeted PCR and qPCR (Supplementary Tables 16-17). To optimize DNA extraction from dental calculus, five extraction buffers (A-E) and three decontamination methods were tested and compared. Two extraction buffers (A: 0.45M EDTA, 10% proteinase K; B: 0.1M EDTA, 10% proteinase K, 10mM Tris-HCl, 10mM NaCl, 2% w/v SDS, 5mM CaCl2, 40mM DTT) and three decontamination methods (2% NaOCl, 0.5M EDTA wash, none) were selected for further analysis and used in combination to produce nine DNA extracts from B61 and G12 dental calculus.
DNA library construction and sequencing
DNA extracts from the optimization experiment were built into nine shotgun libraries using a NEBNext Quick DNA Library Prep Master Mix Set (e6090) with DNA oligos containing a sample-specific multiplex index sequence (Supplementary Figure 21, Supplementary Table 18). The libraries were amplified with Phusion HS II enzyme and sequenced on one lane of an Illumina HiSeq 2000 using single-end 1×100bp chemistry, resulting in 93,677,545 reads after removal of low-quality sequences (Illumina CASAVA 1.8.0, default settings, sequences <25 bp and/or with Phred scores <35 removed; Supplementary Table 19). Separately, thirty 16S rRNA gene amplicon libraries were generated from dental calculus (G12, B17, B61, B78), dentine (G12, B17, B61, B78), carious dentine (B17), and alveolar bone abscess (B17) ancient DNA extracts generated using extraction buffer A and without prior decontamination. Universal primers targeting variable regions V3, V5, and V6 of the 16S rRNA gene were developed and tested in silico (Supplementary Tables 20-21) and in vitro (Supplementary Figure 20). Each library was generated from a minimum of three amplifications (30-35 cycles) using Phusion HS II enzyme and 454 amplicon Fusion primers with multiplex identifiers (MIDs), and the pooled 454 libraries were sequenced with a Roche GS Junior, resulting in 170,807 reads after removal of low-quality sequences (Roche GS RunProcessor, default settings; Supplementary Table 22).
16S rRNA taxonomic classification
A reference dataset containing full length 16S ribosomal RNA sequences was constructed from the NCBI Genbank database, whereby all publicly available 16S ribosomal gene sequences found in the NCBI Genbank database were downloaded, screened for chimeras using uchime,58 aligned using the INFERNAL aligner59 v1.0.2, trimmed and clustered at a sequence identity cutoff of 98% with a hierarchical clustering algorithm using sequence identity as the measure of distance and single linkage as the cluster metric. This dataset has high overlap with both the Greengenes60 (90%) and RDP61 (92%) databases and was constructed in order to standardize filtering and alignment methods, as well as streamline Genbank data retrieval for network analysis. Amplicon and shotgun sample reads were aligned to the reference OTU dataset, and reads with a bit score <40 or negative structure score were discarded. Sample reads were mapped to the reference OTUs by assigning the OTU ID of the most similar reference sequences. Conflicting OTU IDs were discarded. OTUs containing 16S rRNA gene sequences belonging to a reference genome or culture collection were assigned the consensus taxonomy of all such sequences in the OTU. In the case of OTUs that contained no reliable source of taxonomy, the taxonomy of the OTU was inferred by decreasing the clustering threshold until the point at which the OTU was merged with another in which sequences with reliable taxonomy existed.
Network analysis
Network analysis of community similarity was performed to compare the microbial communities of ancient dental samples to each other and to environmental samples deposited in Genbank and MG-RAST (Project 128). Only environmental samples with at least 20 OTUs were considered (1,818 out of 37,689), and only samples with at least 20% similarity to one of the ancient samples are shown in the network (315 out of 1,818). The similarity between a pair of samples was calculated as the number of shared OTUs divided by the total number of different OTUs found in both samples. The network was rendered using the neato program from the graphviz package (http://www.graphviz.org/).
Phylogenetic tree
Ancient dental calculus, amplicon and shotgun OTU tables were merged and a full-length 16S sequence representative for each OTU was chosen. Phylogenetic relationships were inferred with FastTree62 v2.1.3 (generalized time-reversible model).
Validation of results using RDP and QIIME pipelines
To confirm that the taxonomic characterization of ancient dental samples is robust to database choice and clustering parameters, the 16S rRNA amplicon data was reanalyzed using the Greengenes database (v.4Feb2011) and RDP Pyrosequencing61 and QIIME63 pipelines. Only reads ≥70bp with 100% identity to both forward and reverse primers were analyzed. OTUs were clustered at 97% identity, and singleton OTUs were discarded (Supplementary Table 23). The OTU table was rarefied to 1,265 sequences/sample and analyzed at the L2, L5, and L6 levels. Alpha and beta diversity were calculated using QIIME default parameters. The BIOM file for this data is available as Supplementary Data 1. This OTU table was merged with an OTU table generated from the HMP dataset using the same parameters, and the two data sets were compared using Principal Coordinates Analysis; the BIOM file for this data is available as Supplementary Data 2.
Source Tracking
To test for contamination in the ancient dental samples, Bayesian microbial source tracking53 was performed (1,000 “burn-in” iterations using Gibbs sampling with 25 random restarts) on the merged OTU file using HMP plaque, HMP skin, HMP gut, and ancient tooth root (environmental proxy) as sources.
Dietary DNA analysis
Shotgun reads ≥75 bp in length were searched against a complete collection of full mitochondrial and chloroplast genome sequences published as of 07/2012 (>6,000 organelle genomes) using BLASTn. Results were accepted only if they exhibited 100% query coverage and 100% sequence identity, were not hits to 16S or 23S rRNA genes, did not match more than one genus perfectly, and any secondary its outside the genus of the first hit had to show at least two diagnostic point mutations relative to the perfect hit.
Total taxonomic characterization of dental calculus
Library reads were pooled by individual (B61, S1-S4; G12, S5-S8) and de novo assembled into 2,005,273 contigs using Velvet64 v.1.02.3 (k-mer length 29 bp, min 100 bp contig length) (Supplementary Table 24). Contigs were searched against the NCBI nr and gss databases available as of 07/2012 using Megablast, filtered for highly unique, high-scoring top hits (>95 bp alignment, >97% identity, <1e-14 e-value). A total of 61,584 contigs passing these filters were assigned taxonomy.
Pathogen analysis
The contigs were further filtered to remove contigs with second hits of comparable quality and >90% identity to other taxa, resulting in 53,924 highly unique contigs that can be reasonably assigned to a single species. The species-level assignments were then cross-referenced against the PATRIC database,19 resulting in 40 putative pathogen identifications. To determine if these species assignments are reasonable for the oral cavity, we applied the same BLAST and conditional filter approach to shotgun metagenomic contigs reported for 109 HMP supragingival dental plaque samples and compared the results. Feature information for each ancient contig was retrieved from the top hit BLAST results and manually screened for putative virulence-, drug resistance-, plasmid-, transposon-, and phage-associated genes with annotations in PubMed records.
Antibiotic resistance analysis
Sequences for all identified taxa were screened for putative antibiotic resistance elements using three methods: 1) BLASTx search against the Antibiotic Resistance Database (ARDB),65 2) BLASTx search against the NCBI nr database followed by keyword search of translated gene function, and 3) manual search of gene annotations assigned to pathogens.
Genome reconstruction
All G12 contigs ≥100 bp were searched against the NCBI nt and gss databases using Megablast and filtered for contigs aligning to T. forsythia strain ATCC 43037 with an e-value ≤ 1e-6 within the top 100 hits. The filtered contigs were pooled and submitted to the BLAST Ring Image Generator (BRIG)66 tool for mapping. Using BRIG, the contigs were aligned to the T. forsythia strain ATCC 43037 using the Megablast search option and a sequence identity cutoff ≥95%. In cases where a contig aligned to the T. forsythia genome more than once, the alignment with the highest bit score was mapped. In cases where multiple alignments with identical top bit scores were observed, the contig was mapped to all top bit score loci, but the depth of coverage for each locus was divided by the number of loci. Genes not mapped in the assembly and large gaps (Supplementary Figure 23) were analyzed for function.
Protein analysis
Total proteins were extracted from dental calculus (G12, B17, B61, B78, P1, P2), dentine (G12, B17, B61, B78), carious dentine (B17), abscessed alveolar bone (B78), and dental calculus/crown cementum (F1, F5), and four negative extraction controls using a modified filter-aided sample preparation (FASP)67 protocol. A total of 290,466 MS/MS spectra were generated using three instruments (LTQ-Orbitrap Velos, Q-Exactive Hybrid Quadrupole Oritrap, and MaXis UHR-Qq-TOF) (Supplementary Table 25). Tandem mass spectra were converted to Mascot generic format using proteowizard v.2.2.3101 with vendor peak picking option for MS level 2 and deisotoped and deconvoluted using the H-Scorer script.68 ProteinPilot v.4 was used to analyze protein modification and damage patterns (Supplementary Table 26). MS/MS peak lists were searched using Mascot v.2.3.02 against all proteins in UniProtKB/Swiss-Prot as of 2012/10/31 and two custom protein databases built from the Human Oral Microbiome Database (HOMD)69 as of 2012/10/11 and all complete soil bacterial genomes in Genbank as of 2012/02/22. The results were further validated using Scaffold v.4.0.5, resulting in 12,609 unique peptide identifications resolving to 589 proteins identified with >99% confidence and ≥2 unique peptides. Contaminants were identified and removed (Supplementary Table 27). Metadata for human proteins was retrieved using the GeneCards v.3 GeneALaCart tool70 and used to manually classify each protein into six categories: innate immune system, adaptive immune system, blood coagulation, digestion, structure and support, and other. Protein interaction and gene ontology (GO) information was obtained using STRING 9.037 in protein mode. Bacterial proteins were binned by length (Group 1: <15 residues; Group 2: >15 residues) and searched against the NCBI database using BLASTp (Group 1, expect value 20000, PAM30 Score Matrix; Group 2 expect value 1000, BLOSUM62 Score Matrix). The resulting BLASTp files were then parsed using MEGAN71 and analyzed for protein function using SEED hierarchy.72
Supplementary Material
Acknowledgments
We thank the Kantonalen Ethik-Kommission Zürich, Functional Genomics Center Zürich, Center for Microscopy and Image Analysis and the Institute of Oral Biology at University of Zürich; the PRIDE Team; G. Akgül, K. Alt, D. Ashford, P. Ashton, H. Barton, A. Bouwman, C. Burger, D. Coulthard, J. Hublin, V. Meskenaite, F. Najar, M. Richards, K. Sankaranarayanan, R. Schlapbach, L. Shillito, T. Stöllner, and H. Zbinden for assistance with data collection, analysis, and management; M. Carver, F. Dewhirst, A. Tanner, K. Hardy, and A. Henry for helpful comments on early drafts and data analyses. This work was supported by the Mäxi Foundation Zürich, Swiss Foundation for Nutritional Research, Danish Research Foundation grant 29396, Danish Council for Independent Research grant 10-081390, Lundbeck Foundation grant R52-A5062, NIH grants R01-HG005172, R01-GM089886, R01-DE018499, R21-DE018310, ERC grant UMICIS/242870, Marie Curie grants EUROTAST FP7 PEOPLE-2010 MC ITN, PALIMPSEST FP7-PEOPLE-2011-IEF 299101, ORCA FP7-PEOPLE-2011-IOF 299075, Wellcome Trust C2D2 Research Priming Fund grant 097829, Novartis Foundation, Novo Nordisk Foundation, Max Planck Society, and the University of York.
Footnotes
Author Contributions C.W. conceived the project. R.S. and F.R. contributed samples. C.W., E.C., M.J.C., M.T.P.G., C.M., A.R. and Y.H. designed the experiments. C.W., E.C., N.S., C.T., A.R., Y.H., D.S.G., S.C., S.F., H.L., P.N., C.K., J.O., K.Y.T., and E.E. performed the experiments. J.R., R.V., C.W., C.M., J.G., A.R., Y.H., R.Y.T., S.F., C.S., S.C., D.S.G., J.H., J.S.C., LH, and TK analyzed the data. S.B.O., Y.H., E.W., C.M.L., M.T.P.G., M.J.C., and F.R. contributed material support to the project. Y.H. wrote the supplementary Raman section. C.W. wrote the paper, with critical input from C.M.L., M.T.P.G., M.J.C., C.M., E.W., E.C., and the remaining authors.
Author Information Reprints and permissions information is available at www.nature.com/reprints. The authors declare no conflicts of interest. The authors declare no competing financial interests.
Database accession numbers Illumina and 454 genetic data have been deposited to the NCBI Short Read Archive (SRA) under the project accession SRP029257, sample accessions SRS473742-SRS473771 and SRS480529-SRS480539; and to MG-RAST55 under Project 365, accessions 4486524.3, 4486533.3, 4486537.3, 4486539.3, 4486540.3, 4486544.3, 4486613.3, 4486614.3, 4486617.3, 4487224.3-4487231.3, 4487233.3-4487235.3, 4487237.3-4487248.3, 4488534.3-4488536.3, 4488542.3, 4517539.3, 4530391.3, 4530438.3, 4530439.3, 4530473.3-4530475.3. Proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository56 with the dataset identifier PXD000412, accessions 34605-34628. Computer source code for the network analysis in Figure 5j is deposited to GitHub (https://github.com/jfmrod/metagenome-sample-network-generator).
Supplementary Information is linked to the online version of the paper.
References
- 1.Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology. 2003;149:279–294. doi: 10.1099/mic.0.26082-0. [DOI] [PubMed] [Google Scholar]
- 2.Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 3.Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dewhirst FE, et al. The human oral microbiome. Journal of bacteriology. 2010;192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hujoel P. Dietary carbohydrates and dental-systemic diseases. J Dent Res. 2009;88:490–502. doi: 10.1177/0022034509337700. [DOI] [PubMed] [Google Scholar]
- 6.Kuo LC, Polson AM, Kang T. Associations between periodontal diseases and systemic diseases: a review of the inter-relationships and interactions with diabetes, respiratory diseases, cardiovascular diseases and osteoporosis. Public health. 2008;122:417–433. doi: 10.1016/j.puhe.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Leishman SJ, Do HL, Ford PJ. Cardiovascular disease and the role of oral bacteria. Journal of oral microbiology. 2010;2 doi: 10.3402/jom.v2i0.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin Y, Yip HK. Supragingival calculus: formation and control. Critical reviews in oral biology and medicine : an official publication of the American Association of Oral Biologists. 2002;13:426–441. doi: 10.1177/154411130201300506. [DOI] [PubMed] [Google Scholar]
- 9.Hardy K, et al. Starch granules, dental calculus and new perspectives on ancient diet. J Archaeol Sci. 2009;36:248–255. [Google Scholar]
- 10.Henry AG, Brooks AS, Piperno DR. Microfossils in calculus demonstrate consumption of plants and cooked foods in Neanderthal diets (Shanidar III, Iraq; Spy I and II, Belgium) Proceedings of the National Academy of Sciences of the United States of America. 2011;108:486–491. doi: 10.1073/pnas.1016868108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adler CJ, et al. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nature genetics. 2013 doi: 10.1038/ng.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De La Fuente CP, Flores SV, Moraga ML. Human bacterial DNA from dental calculus: a new source of genetic material. Am J Phys Anthropol. 2012;147:127–127. [Google Scholar]
- 13.Linossier A, Gajardo M, Olavarria J. Paleomicrobiological study in dental calculus: Streptococcus mutans. Scanning microscopy. 1996;10:1005–1013. discussion 1014. [PubMed] [Google Scholar]
- 14.Wang J, et al. Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Scientific reports. 2013;3:1843. doi: 10.1038/srep01843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fierer N, Bradford MA, Jackson RB. Toward an ecological classification of soil bacteria. Ecology. 2007;88:1354–1364. doi: 10.1890/05-1839. [DOI] [PubMed] [Google Scholar]
- 16.Munro CL, Grap MJ. Oral health and care in the intensive care unit: state of the science. American journal of critical care: an official publication, American Association of Critical-Care Nurses. 2004;13:25–33. discussion 34. [PubMed] [Google Scholar]
- 17.Shay K. Infectious complications of dental and periodontal diseases in the elderly population. Clin Infect Dis. 2002;34:1215–1223. doi: 10.1086/339865. [DOI] [PubMed] [Google Scholar]
- 18.Nakano K, et al. Detection of oral bacteria in cardiovascular specimens. Oral microbiology and immunology. 2009;24:64–68. doi: 10.1111/j.1399-302X.2008.00479.x. [DOI] [PubMed] [Google Scholar]
- 19.Gillespie JJ, et al. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun. 2011;79:4286–4298. doi: 10.1128/IAI.00207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willner D, et al. Metagenomic detection of phage-encoded platelet-binding factors in the human oral cavity. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4547–4553. doi: 10.1073/pnas.1000089107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontology 2000. 2005;38:135–187. doi: 10.1111/j.1600-0757.2005.00107.x. [DOI] [PubMed] [Google Scholar]
- 22.Marri PR, et al. Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PloS one. 2010;5:e11835. doi: 10.1371/journal.pone.0011835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deguchi T, Yasuda M, Ito S. Management of pharyngeal gonorrhea is crucial to prevent the emergence and spread of antibiotic-resistant Neisseria gonorrhoeae. Antimicrob Agents Chemother. 2012;56:4039–4040. doi: 10.1128/AAC.00505-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emonts M, Hazelzet JA, de Groot R, Hermans PW. Host genetic determinants of Neisseria meningitidis infections. Lancet Infect Dis. 2003;3:565–577. doi: 10.1016/s1473-3099(03)00740-0. [DOI] [PubMed] [Google Scholar]
- 25.Goker M, et al. Complete genome sequence of Olsenella uli type strain (VPI D76D-27C(T)) Stand Genomic Sci. 2010;3 doi: 10.4056/sigs.1082860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palmer RJ. Composition and development of oral bacterial communities. Periodontol 2000. 2014;64:20–39. doi: 10.1111/j.1600-0757.2012.00453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Brien-Simpson NM, Veith PD, Dashper SG, Reynolds EC. Antigens of bacteria associated with periodontitis. Periodontology 2000. 2004;35:101–134. doi: 10.1111/j.0906-6713.2004.003559.x. [DOI] [PubMed] [Google Scholar]
- 28.Amano A, Nakagawa I, Okahashi N, Hamada N. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. Journal of periodontal research. 2004;39:136–142. doi: 10.1111/j.1600-0765.2004.00719.x. [DOI] [PubMed] [Google Scholar]
- 29.Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science. 2009;325:1128–1131. doi: 10.1126/science.1176950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie G, et al. Community and gene composition of a human dental plaque microbiota obtained by metagenomic sequencing. Molecular oral microbiology. 2010;25:391–405. doi: 10.1111/j.2041-1014.2010.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Costa VM, et al. Antibiotic resistance is ancient. Nature. 2011;477:457–461. doi: 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- 32.Tanner AC, Izard J. Tannerella forsythia, a periodontal pathogen entering the genomic era. Periodontology 2000. 2006;42:88–113. doi: 10.1111/j.1600-0757.2006.00184.x. [DOI] [PubMed] [Google Scholar]
- 33.Sharma A. Virulence mechanisms of Tannerella forsythia. Periodontology 2000. 2010;54:106–116. doi: 10.1111/j.1600-0757.2009.00332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimotahira N, et al. The S-layer of Tannerella forsythia contributes to serum resistance and oral bacterial co-aggregation. Infection and immunity. 2013 doi: 10.1128/IAI.00983-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SW, et al. Identification and characterization of the genes encoding a unique surface (S-) layer of Tannerella forsythia. Gene. 2006;371:102–111. doi: 10.1016/j.gene.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 36.Gorr SU. Antimicrobial peptides of the oral cavity. Periodontology 2000. 2009;51:152–180. doi: 10.1111/j.1600-0757.2009.00310.x. [DOI] [PubMed] [Google Scholar]
- 37.Szklarczyk D, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic acids research. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coxon A, Tang T, Mayadas TN. Cytokine-activated endothelial cells delay neutrophil apoptosis in vitro and in vivo. A role for granulocyte/macrophage colony-stimulating factor. J Exp Med. 1999;190:923–934. doi: 10.1084/jem.190.7.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryder MI. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontology 2000. 2010;53:124–137. doi: 10.1111/j.1600-0757.2009.00327.x. [DOI] [PubMed] [Google Scholar]
- 40.Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? The Journal of cell biology. 2012;198:773–783. doi: 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furugen R, Hayashida H, Saito T. Porphyromonas gingivalis and Escherichia coli lipopolysaccharide causes resistin release from neutrophils. Oral diseases. 2012 doi: 10.1111/odi.12027. [DOI] [PubMed] [Google Scholar]
- 42.Kusminski CM, McTernan PG, Kumar S. Role of resistin in obesity, insulin resistance and Type II diabetes. Clin Sci (Lond) 2005;109:243–256. doi: 10.1042/CS20050078. [DOI] [PubMed] [Google Scholar]
- 43.Gracia-Arnaiz M. Fat bodies and thin bodies. Cultural, biomedical and market discourses on obesity. Appetite. 2010;55:219–225. doi: 10.1016/j.appet.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 44.Frei R, Lauener RP, Crameri R, O’Mahony L. Microbiota and dietary interactions: an update to the hygiene hypothesis? Allergy. 2012;67:451–461. doi: 10.1111/j.1398-9995.2011.02783.x. [DOI] [PubMed] [Google Scholar]
- 45.Palmer SA, Smith O, Allaby RG. The blossoming of plant archaeogenetics. Anatomischer Anzeiger. 2012;194:146–156. doi: 10.1016/j.aanat.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 46.Leonard JA, et al. Animal DNA in PCR reagents plagues ancient DNA research. J Archaeol Sci. 2007;34:1361–1366. [Google Scholar]
- 47.Bocherens H, Grupe G, Mariotti A, Turban-Just S. Molecular preservation and isotopy of Mesolithic human finds from the Ofnet cave (Bavaria, Germany) Anthropologischer Anzeiger. 1997;55:121–129. [PubMed] [Google Scholar]
- 48.Oelze VM, et al. Multi-isotopic analysis reveals individual mobility and diet at the early iron age monumental tumulus of magdalenenberg, germany. Am J Phys Anthropol. 2012;148:406–421. doi: 10.1002/ajpa.22063. [DOI] [PubMed] [Google Scholar]
- 49.Oelze VM, et al. Early Neolithic diet and animal husbandry: stable isotope evidence from three Linearbandkeramik (LBK) sites in Central Germany. J Archaeol Sci. 2011;38:270–279. [Google Scholar]
- 50.Schutkowski H, Herrmann B, Wiedemann F, Bocherens H, Grupe G. Diet, status and decomposition at Weingarten: Trace element and isotope analyses on early mediaeval skeletal material. J Archaeol Sci. 1999;26:675–685. [Google Scholar]
- 51.Turner-Walker G. In: Advances in Human Paleopathology. Pinhasi R, Mays S, editors. Ch. 1. John Wiley & Sons, Ltd.; 2008. p. 29. [Google Scholar]
- 52.Zijnge V, et al. Oral Biofilm Architecture on Natural Teeth. PloS one. 2010;5:e9321. doi: 10.1371/journal.pone.0009321. ARTN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knights D, et al. Bayesian community-wide culture-independent microbial source tracking. Nature methods. 2011;8:761–763. doi: 10.1038/nmeth.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wood JW, Milner GR, Harpending HC, Weiss KM. The Osteological Paradox - Problems of Inferring Prehistoric Health from Skeletal Samples. Curr Anthropol. 1992;33:343–370. [Google Scholar]
- 55.Meyer F, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vizcaino JA, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic acids research. 2013;41:D1063–1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richards MP, Hedges REM. Stable isotope evidence for similarities in the types of marine foods used by late mesolithic humans at sites along the Atlantic coast of Europe. J Archaeol Sci. 1999;26:717–722. [Google Scholar]
- 58.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–1337. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McDonald D, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. Isme Journal. 2012;6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cole JR, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic acids research. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Molecular biology and evolution. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuczynski J, et al. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Current protocols in bioinformatics. 2011;Chapter 10(Unit 10):17. doi: 10.1002/0471250953.bi1007s36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome research. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu B, Pop M. ARDB Antibiotic Resistance Genes Database. Nucleic acids research. 2009;37:D443–447. doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cappellini E, et al. Resolution of the type material of the Asian elephant, Elephas maximus Linnaeus, 1758 (Proboscidea, Elephantidae) Zoological Journal of the Linnean Society. 2013;170:222–232. [Google Scholar]
- 68.Savitski MM, Mathieson T, Becher I, Bantscheff M. H-score, a mass accuracy driven rescoring approach for improved peptide identification in modification rich samples. Journal of proteome research. 2010;9:5511–5516. doi: 10.1021/pr1006813. [DOI] [PubMed] [Google Scholar]
- 69.Chen T, et al. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010 doi: 10.1093/database/baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stelzer G, et al. In-silico human genomics with GeneCards. Human genomics. 2011;5:709–717. doi: 10.1186/1479-7364-5-6-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome research. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mitra S, et al. Functional analysis of metagenomes and metatranscriptomes using SEED and KEGG. BMC bioinformatics. 2011;12(Suppl 1):S21. doi: 10.1186/1471-2105-12-S1-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.