

Abstract
Chimaerins are a family of diacylglycerol- and phorbol ester-regulated GTPase activating proteins (GAPs) for the small G-protein Rac. Extensive evidence indicates that these proteins play important roles in development, axon guidance, metabolism, cell motility, and T cell activation. Four isoforms have been reported to-date, which are products of CHN1 (α1- and α2-chimaerins) and CHN2 (β1- and β2-chimaerins) genes. Although these gene products are assumed to be generated by alternative splicing, bioinformatics analysis of the CHN2 gene revealed that β1- and β2-chimaerins are the products of alternative transcription start sites (TSSs) in different promoter regions. Furthermore, we found an additional TSS in CHN2 gene that leads to a novel product, which we named β3-chimaerin. Expression profile analysis revealed predominantly low levels for the β3-chimaerin transcript, with higher expression levels in epididymis, plasma blood leucocytes, spleen, thymus, as well as various areas of the brain. In addition to the prototypical SH2, C1, and Rac-GAP domains, β3-chimaerin has a unique N-terminal domain. Studies in cells established that β3-chimaerin has Rac-GAP activity and is responsive to phorbol esters. The enhanced responsiveness of β3-chimaerin for phorbol ester-induced translocation relative to β2-chimaerin suggests differential ligand accessibility to the C1 domain.
Keywords: Chimaerin, CHN2, Rac-GAP, C1 domain, Phorbol esters
Introduction
Rac, a small GTPase that belongs to the Rho family, controls numerous cellular functions, including actin cytoskeleton reorganization, motility, and cell cycle progression. Rac activation status is controlled primarily by guanine nucleotide exchange factors (GEFs) that promote GTP loading and thereby activate Rac, and by GTPase activating proteins (GAPs) that inactivate Rac by accelerating GTP hydrolysis. Among the several Rac-GAP families, chimaerins represent a unique class due to their direct regulation by diacylglycerol (DAG), a lipid second messenger that binds to the C1 domain in chimaerins which is highly homologous to C1 domains in protein kinase C (PKC) isozymes. It has been widely recognized that chimaerins play important roles in development, axon guidance, cell migration, and T-cell activation [1-4]. Early studies determined the existence of four chimaerin isoforms (α1, α2, β1, and β2), reported to be alternative splicing products of the CHN1 (α) and CHN2 (β) genes [5-8].
In addition to the C1 domain, all chimaerin isoforms have a C-terminal catalytic Rac-GAP domain. An SH2 domain in the N-terminal region of α2- and β2-chimaerins is possibly involved in interactions with phosphotyrosine proteins, but its function remains essentially unknown. Binding of DAG or DAG mimetics (such as phorbol esters) to the C1 domain leads to allosteric activation of the protein which, in response to various stimuli, results in deactivation of the Rac GTPase [9-11]. The crystal structure of β2-chimaerin revealed that the protein is in an autoinhibited state. The N-terminal region of the protein forms a 33 amino acid alpha-helix that rests against the lipid binding site in the C1 domain and occludes the Rac interaction surface in the GAP domain [12]. We have previously identified residues in α2- and β2-chimaerins involved in intramolecular hydrophobic interactions that keep the protein in a “closed” conformation. When these residues are mutated, the protein becomes more sensitive to lipidand phorbol ester-induced activation [10, 12]. The fact that chimaerins activity and localization are controlled by different mechanisms as protein–protein or lipid–protein interaction, and phosphorylation [13, 14] strongly suggests that these Rac-GAPs are subject to complex regulatory mechanisms.
It is well established that many eukaryotic genes possess alternative promoters regulating distinct expression patterns, probably because one single transcription start site (TSS) or regulatory region may not always be sufficient to accommodate all the required gene expression regulations (e.g. different tissues, different stages of development). Although alternative TSSs have been reported for many genes, this phenomenon still remains poorly understood. A recent report shows that more than 40 % of developmentally expressed genes in Drosophila melanogaster have at least two alternative promoters generally involved in distinct regulatory programs [15]. The usage of alternative promoters could generate protein variants, differentially regulated 5′ UTRs, or a combination of both. In yeast, the 5′ UTR in mRNAs can regulate translation efficiency, and this accounts for large changes in protein expression levels [16]. Regulation of mRNA localization and transport could also depend on 5′ UTR sequences. At the present time there is no information on whether β-chimaerin isoforms could be generated as a consequence of alternative transcription mechanisms. Here we carried out a thorough analysis of the CHN2 gene, which led us to the identification of a novel β-chimaerin isoform, β3-chimaerin, that is the product of an alternative TSS in the CHN2 gene.
Methods
Materials
Cell culture reagents were obtained from Invitrogen (Carlsbad, CA). Reagents for the expression and purification of recombinant glutathione S-transferase (GST) fusion proteins and Gammabind G-Sepharose were purchased from Amersham Biosciences, Inc. (Sunnyvale, CA). Phorbol 12-myristate 13-acetate (PMA) and GF109203X were purchased from LC Laboratories (Woburn, MA).
Cloning of β3-chimaerin and plasmids
β3-Chimaerin was amplified by PCR using commercial cDNA from human brain and kidney (PrimerDesign, Southampton, UK) and a Labnet MultiGene™ 96-well gradient thermal cycler. As primers we used 5′-ctcgagggatccatgacccagacccacagg (sense) and 5′-acgcgtgcggccgcggattagaataaaacgtcttcg (antisense) (Fig. 1c). The same primers were used to clone the entire β3-chimaerin cDNA from A-172 and U-373 human cell lines.
Fig. 1.

CHN2 gene structure. a β3-chimaerin sequence (accession no: ADK47390.1). Red, N-terminal region encoded by exons 1a and 2a; green, SH2 domain; blue, C1 domain; yellow, Rac-GAP domain. b Structure of the CHN2 gene. The figure shows the exon arrangements leading to the different β-chimaerin isoforms. Red, exons that code for the β3-chimaerin N-terminal domain; green, exon 1b that is specific for the β2-chimaerin isoform. Yellow, β1-chimaerin’s exclusive 5′ sequence on exon 7b; gray, promoter sequences for the different isoforms. c Schematic view of the primers used to amplify full-length (primers 1 and 2) or N-terminal region of β3-chimaerin (primers 3 and 4). (Color figure online)
PCR products were ligated into pCRII using the TA cloning kit (Invitrogen) or pCR2.1-TOPO using the TOPO TA cloning kit (Invitrogen). EcoRI–BamHI or BamHI–BamHI fragments were isolated from these plasmids and subcloned into pEGFP-C3 or pEGFP-C1. All constructs were verified by sequencing.
Cell culture and transfections
Cell culture reagents were obtained from Invitrogen (Carlsbad, CA). COS-1 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10 % FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified 5 % CO2 atmosphere at 37 °C. Cells in 6-well plates at 50 % confluence were transfected with different mammalian expression vectors (1–2 μg) using polyethylenimine (CELLnTEC, Bern, Switzerland) according to the manufacturer’s protocol.
Determination of Rac-GTP levels
Rac-GTP levels were determined using a pull-down assay, as previously reported [17]. Briefly, cells were lysed in a buffer containing 8 μg of GST-PBD domain, 20 mM Tris–HCl, pH 7.5, 1 mM dithiothreitol, 5 mM MgCl2, 150 mM NaCl, 0.5 % Nonidet P-40, 5 mM glycerophosphate, and protease inhibitors (5 μg/ml 4-(2-aminoethyl) benzenesulfonyl fluoride, 5 μg/ml leupeptin, 5 μg/ml aprotinin and 1 μg/ml pepstatin A). Lysates were centrifuged at 14,000×g (4 °C, 10 min) and then incubated with glutathione-Sepharose 4B beads (4 °C, 1 h). After extensive washing, the beads were boiled in loading buffer. Samples were resolved in 12 % SDS-polyacrylamide gels and transferred to PVDF membranes for Western blot analysis using an anti-Rac1 antibody (Upstate Biotechnology, Lake Placid, NY).
Translocation assays
Experiments were carried out as previously described [18]. Briefly, COS-1 cells (2 × 105) in six-well plates were transfected with pEGFP-β2-chimaerin or pEGFP-β3-chimaerin. After 24 h, cells were treated with different concentrations of PMA for 20 min. To avoid PKC activation by PMA, experiments were performed in the presence of the PKC inhibitor GF109203X (5 μM), added 30 min before and during PMA stimulation. For fractionation assays, cells were harvested into lysis buffer (20 mM Tris–HCl, pH 7.5, 5 mM EGTA, and protease inhibitor cocktail for mammalian cell and tissue extract, 1:500, Sigma). Separation of cytosolic (soluble) and particulate fractions was performed by ultracentrifugation [19]. Equal amounts of protein were subjected to SDS-polyacrylamide gel electrophoresis, transferred to PVDF membranes, and immunostained with an anti-GFP antibody (Santa Cruz Biotechnology, Dallas, TX). For fluorescence microscopy visualization, cells were washed with cold PBS, immediately fixed in 4 % PFA and visualized in a Olympus IX71 fluorescence microscope.
Tissue arrays
96-Well TissueScan Human Major Tissue qPCR Array (HMRT102) and the TissueScan Human Brain Tissue qPCR Array (HBRT101) from OriGene (Rockville, MD) were used. Primers for β3-chimaerin were as follows: 5′-cattcaggacttacttgcaagccca (sense) and 5′-tcttcagcatcgctagt gcagc (antisense) (Fig. 1c). PCR was performed using the qSTAR SYBR Master Mix kit (OriGene) and an ABI Prism 7300 thermocycler.
Results
Identification of β3-chimaerin
Mammalian β1- and β2-chimaerins have been previously identified as important regulators of the small GTPase Rac. To investigate whether other products of the CHN2 gene might exist, we carried out a screening of EST databases. This led to the identification of a third gene product, which we named β3-chimaerin. β3-Chimaerin was cloned using a PCR approach from two different commercial human cDNAs samples (total brain and kidney) as templates. The PCR products were sequenced, and the full-length β3-chimaerin sequence (Fig. 1a) was reported to GeneBank (accession ADK47390.1). The β3-chimaerin cDNA was also cloned from A-172 cells (a human glioblastoma cell line) and U-373 cells (a cell line derived from human astrocytoma) (data not shown).
Sequence analysis revealed that β3-chimaerin has a novel N-terminal domain that is encoded by two unique exons located 48 kb upstream from the β2-chimaerin open reading frame (Fig. 1b). Comparison of protein sequences from β1-chimaerin (NP_001035025.1), β2-chimaerin (NP_004058.1) and β3-chimaerin isoforms revealed in all cases unique N-terminal regions. β1-chimaerin, an isoform only found in testis [5], is the product of an alternative TSS located on intron 6, ~250 kb downstream of the β2-chimaerin TSS. This isoform lacks the SH2 domain and N-terminal α-helix present in β2-chimaerin, which are coded by a cluster of exons 160 kb downstream from the first exon and 80 kb upstream of the β1-chimaerin TSS. On the other hand, β3-chimaerin N-terminal region lacks key residues involved in autoinhibition of β2-chimaerin GAP activity, as also reported for β1-chimaerin [12], but maintains both SH2 domain and α-helix. The Rac-GAP and C1 domains are encoded by a cluster of exons common to all β-chimaerins. The unique N-terminal region in β3-chimaerin, coded by two exons exclusive of primates, is 91 amino acids long and does not share any sequence identity with other known proteins.
As indicated above, the unique N-terminal domain in β3-chimaerin is encoded by two exons (1a and 2a) located 48 kb upstream from the β2-chimaerin open reading frame, which are followed by exon 2b. Canonical donor sequence of exon 2a binds to the first available acceptor sequence that corresponds with exon 2b. The absence of the canonical acceptor sequence in exon 1b explains why this exon is excluded in β3-chimaerin or other transcripts similar to β3-chimaerin (ENSMBL transcript id: ENST00000539406, ENST00000474070; ENS00000439384). Therefore, β2- and β3-chimaerin cannot be alternative splicing products of the CHN2 gene, supporting the idea that alternate TSSs are responsible for the control of β2-chimaerin and β3-chimaerin expression.
As described previously [5], the testis-specific β1-chimaerin isoform is the product of an alternative TSS located on intron 6. As shown in Fig. 1b, exon 7b is used as the first exon in β1-chimaerin transcription. This is the seventh exon in β2-chimaerin as a result of a canonical splicing process due to an acceptor sequence located inside the exon which generates a shorter exon (exon 7a) (Fig. 1b) [20]. FirstEF software localized a non-CpG related promoter region 479 bp upstream of the exon 7b start site where a CCAAT box is localized. This CCAAT box in β1-chimaerin is similar to the GGCCAATC box located at nucleotides −519 to −512 in α1-chimaerin. Point-mutation experiments revealed that this sequence is essential for α1-chimaerin expression, and its presence in β1-chimaerin suggests that a similar mechanism to α1-chimaerin may control β1-chimaerin transcription [20].
Analysis of the CHN2 gene promoters
Although never formally proven, β1- and β2-chimaerins were proposed to be alternative splicing products of the CHN2 gene in chromosome 7 [5, 6, 8]. However, when we carried out bioinformatics analysis of mRNA, ESTs and genomic sequences available in public databases, we found that these β-chimaerin isoforms are products of alternate TSSs regulated by different promoter regions. Sequence comparison between β1-chimaerin (NM_001039936.1) and β2-chimaerin (NM_004067.2) mRNAs, revealed unique 5′ untranslated regions (UTRs). Alternative TSSs are common features in protein-coding genes and frequently generate alternative N termini [21]. Alternative TSSs usually require distinct promoters that allow RNA polymerase to bind and initiate polymerization of ribonucleotides according to their regulation [21-24].
In order to determine if β3-chimaerin is also a product of a new alternative TSS controlled by a new promoter, we used. The encyclopedia of DNA Elements (ENCODE) to identify putative promoter regions flanking the β2- and β3-chimaerin TSSs. This analysis indicated that β2- and β3-chimaerins are the product of alternative TSSs rather than alternative splicing products. As shown in Fig. 2a (track 5), promoter prediction software FirstEF revealed a CpG-related promoter in the 5′ UTR region of both β2- and β3-chimaerins [25]. We also found that both regions showed enrichment for enhancer- and promoted-associated histone marks (H3K4Me1, H3K4Me3, H3ac, H3K27Ac and H3 acetylation), which are usually linked to promoter regions [26] (Fig. 2a, tracks 2, 3, 4 and 9). In addition, binding sites for the promoter associated transcription factors USF1/USF2 and c-Myc located in β2- and β3-chimaerin promoters support our hypothesis (Fig. 2a, track 7, 8 and 9) [27, 28]. Moreover, Fig. 2a (track 6) shows that these regions presented high promoter activity in HepG2 human cells and poised state promoter in H1-hESC human cells. Finally, analysis of regulatory potential in these regions using ESPERR, an algorithm that discriminate between regulatory elements and neutral DNA, considered this DNA region as a regulatory element (Fig. 2a, track 11) [29]. Using the software methyl primer express (Applied Biosystems, Foster City, CA) we also identified highly dense CpG islands, which are typically associated with promoter regions and transcriptional regulation [30], in both β2-chimaerin and β3-chimaerin promoter regions (Fig. 2b). In addition, the Hudson Alpha Methyl-seq consortium reported that these CpG islands are methylated in several human cell lines (Fig. 2a, track 10). Taken together, results from CHN2 promoter analysis strongly support the idea that alternative TSSs, rather than alternative splicing, controls the generation of β2- and β3-chimaerins.
Fig. 2.

Analysis of the CHN2 gene promoter. a Putative promoter regions of β3-chimaerin (left panel) and β2-chimaerin (right panel) (UCSC Genome Browser). Track #1 CHN2 gene plot; Track #2, enhancer- and promoter-associated histones marks, H3K4Me1, from 9 human cell lines; Track #3, promoter-associated histones marks, H3K4Me3, from 8 human cell lines; Track #4, GIS ChIP-PET showing H3K4me3 and H3K27me3 histone modifications from 4 human cell lines; Track #5, first-exon promoter prediction; Track #6, ENCODE chromatin state segmentation by HMM from Broad Institute of 2 human cell types (Bright Red active promoter/Light Red weak promoter/Purple inactive-poised promoter/Orange strong enhancer/Yellow weak-poised enhancer/Light Green weak transcribed/Gray polycomb-repressed/Light Gray heterochromatin); Track #7, ENCODE transcription factor ChIP-seq; Track #8 GIS ChIP-PET: binding sites for c-Myc; Track #9, Uppsala University ChIP-chip Signal, showing localization of transcription factors (USF1 and USF2) and acetylated histone H3 (H3ac) in a liver cell line (HepG2); Track #10 ENCODE HudsonAlpha CpG methylation showing methylation status of specific CpG dinucleotides in 3 human cell types (orange = methylated; blue = unmethylated); Track #11 ESPERR regulatory potential (RP) compare frequencies of known regulatory elements and neutral DNA from 7 species. b CpG sites (purple bars along the axis) and CpG islands (purple bars below the axis) near β2-chimaerin and β3-chimaerin promoter regions. Red asterisks indicate translation start sites (ATGs). (Color figure online)
Expression of β3-chimaerin
To determine the relative expression of β3-chimaerin in tissues we used a commercial cDNA array (TissueScan Human Major Tissue qPCR Array) and specific β3-primers. Although levels were generally moderate, most tissues expressed β3-chimaerin. The highest levels of mRNA were found in epididymis, plasma blood leucocytes, spleen, and thymus (Fig. 3a). Given the important function of chimaerins in nervous tissues [1, 31], we also examined the expression pattern of β3-chimaerin using a human brain cDNA array (Tissue Scan Human Brain Tissue qPCR Array). High β3-chimaerin levels were detected in the pituitary gland, gray cerebellum, white cerebellum and vermis (Fig. 3b). β3-chimaerin expression was also detected in human cell lines A-172 (glioblastoma) and U-373 (astrocytoma) by RT-PCR (Fig. 3c) and by Western blot using a monoclonal anti-chimaerin antibody (Fig. 3d). Quantification of chimaerin expression by Western blot showed that β2-chimaerin is 3.1 ± 0.6 times more abundant than β3-chimaerin in A-172 and 1.4 ± 0.3 times more in U-373.
Fig. 3.

β3-Chimaerin expression analysis. β3-chimaerin mRNA levels were determined by Q-PCR using cDNA arrays using specific primers against β3-chimaerin. Expression levels are shown relative to the mean value of the array (indicated by the dotted line). a TissueScan human major tissue qPCR array. b TissueScan human brain tissue qPCR array. c RT-PCR showing expression of β3-chimaerin in U-373 and A-172 cell lines. Molecular weight of the amplified insert and a positive control are shown. d Western blot showing relative expression of β2-chimaerin and β3-chimaerin in U-373 and A-172 cell lines (Molecular weight marker is shown in kD)
Responsiveness of β3-chimaerin to PMA
In previous studies we established that the C1 domain in β2-chimaerin binds phorbol esters with an affinity similar to C1 domains in PKC isozymes. The C1 domain is responsible for the redistribution of the protein from the cytosol to the plasma membrane where it binds its target Rac [9, 18, 19, 32]. As C1 domains in β2- and β3-chimaerins are identical, we expected that β3-chimaerin should be responsive to phorbol esters. To address this issue we examined the intracellular redistribution of these chimaerins in response to different concentrations of the phorbol ester PMA using a subcellular fractionation approach. COS-1 cells were transfected with a β3-chimaerin mammalian expression vector and treated with different concentrations of PMA. Cell lysates were prepared, and soluble (cytosolic) and particulate fractions were separated by ultracentrifugation. Western blot analysis revealed that PMA caused significant translocation of β3-chimaerin to the particulate fraction (Fig. 4a). Densitometric analysis of the immunoreactivity in the soluble fraction allowed us to calculate the EC50 for PMA-induced β3-chimaerin translocation (180 ± 1 nM, n = 6). In comparison, the EC50 for PMA-induced translocation of β2-chimaerin was approximately sevenfold higher (1202 ± 1 nM, n = 5).
Fig. 4.
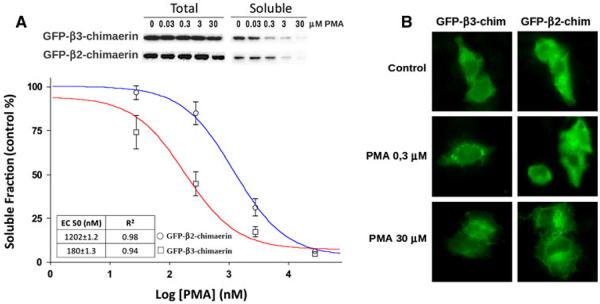
β3-Chimaerin translocation after PMA treatment. a COS-1 cells expressing either β2-chimaerin or β3-chimaerin were treated with different concentrations of PMA for 20 min and subject to cellular fractionation. Upper panel representative western blots. Bottom panel densitometric analysis of immunoreactivity in the soluble fraction. Results are expressed as percentage of the values observed in control (non-treated) cells and represent the mean standard error of 5 (β2-chimaerin) or 6 (β3-chimaerin) independent experiments. The corresponding EC50 values are shown in the inset. b GFP-β2-chimaerin or GFP-β3-chimaerin expressing cells were treated with vehicle, 0.3 or 30 μM PMA for 20 min, fixed and visualized by fluorescence microscopy
Translocation of β3-chimaerin was also evaluated using fluorescence microscopy. COS-1 cells expressing GFP-β3-chimaerin, or GFP-β2-chimaerin as a control, were treated with vehicle, 0.3 or 30 μM of PMA for 20 min, fixed and visualized for GFP localization. Figure 4b, shows that at 0.3 μM β3-chimaerin accumulates in the perinuclear region. Thus, β3-chimaerin translocates more efficiently than β2-chimaerin in response to activation via the C1 domain.
Analysis of β3-chimaerin Rac-GAP activity
Next, we examined the ability of β3-chimaerin to inactivate Rac. COS-1 cells were transfected with mammalian expression vectors for either β2-chimaerin or β3-chimaerin, and Rac-GTP levels were determined using a pull-down assay. Figure 5 shows that, although both β2-chimaerin and β3-chimaerin were able to reduce Rac-GTP levels, β3-chimaerin was more efficient than β2-chimaerin.
Fig. 5.

β3-Chimaerin has Rac-GAP activity. Rac-GTP levels were measured in COS-1 cells expressing either β2-chimaerin or β3-chimaerin. Upper panel representative experiment. Bottom panel densitometric analysis of Rac-GTP levels normalized to total Rac levels in each case. Values are expressed as % of control (C, non-transfected cells) and they represent the mean ± SEM of seven independent experiments
Discussion
In this study we identified a novel product of the CHN2 gene, β3-chimaerin. Like β2-chimaerin, β3-chimaerin has the SH2-C1-Rac-GAP domains tandem. The human CHN2 gene possesses at least three alternative TSSs with promoter regions driving the expression of β1-, β2- and β3-chimaerins. β1-Chimaerin, the smallest member of this family, lacking the SH2 domain, was originally described as a splicing variant in the rat testis [5]. However, our analysis showed that this chimaerin isoform is rather the product of an alternative TSS located on intron 6. β2-Chimaerin is the most abundant product of the CHN2 gene. It is highly expressed in the cerebellum and ubiquitously expressed at low levels in various tissues. There are also reports that β2-chimaerin is down-regulated in several cancer types, suggesting a potential role for this protein as a tumor suppressor [8, 17]. Transcription of β2-chimaerin is driven by a promoter region located on intron 2a of the CHN2 gene. β2-chimaerin possesses all three chimaerin domains, an SH2 domain presumably involved in phosphotyrosine binding, a DAG/phorbol ester-responsive C1 domain, and a Rac-specific GAP domain [4, 6]. Additional 5′ region present in β3-chimaerin mRNA is encoded by two exons located 48 kb upstream of the β2-chimaerin coding sequence. These additional exons generate a unique 5′ UTR region and an additional N-terminus with no obvious sequence identity to any other protein. The sequence coding for this N-terminal region does not display any known domain or motif, and it was found only in primates, suggesting that it came late in the evolutionary process and that it may be involved in a highly specialized function.
Previous studies have assumed that β1- and β2-chimaerins represent alternative splicing products of the CHN2 gene. Although alternative splicing is responsible for a large fraction of protein variants, it is now recognized that other mechanisms exist, such as the usage of alternative TSSs or alternative polyadenylation signals. These mechanisms produce mRNAs with different 5′ UTR or 3′ UTR regions, respectively, which have differential mRNA stability, localization, or translation efficiency [33]. Additionally, these mechanisms could generate protein variants differing in their N- or C-termini. In this paper we show that the different isoforms encoded by the CHN2 gene (β1-, β2- and β3-chimaerin) are not generated by alternative splicing, as suggested previously, but rather through alternative TSSs.
Based on this study and our previous observations [13, 34, 35], and taking into consideration the information provided by the 3-D structure of β2-chimaerin [12], we postulate that different members of the chimaerin family are subject to distinct mechanisms of control. Crystallo-graphic evidence points to the N-terminal region of β2-chimaerin as a major determinant for autoinhibition. This region forms hydrophobic bonds with residues in the C1 domain that occlude the groove where ligands (DAG and phorbol esters) bind to induce allosteric activation. Specifically, Leu28 in the N-terminal region makes contact with Trp234 and Phe232 in the C1 domain, which prevents the access of ligands. In addition, hydrophobic amino acids in the Rac-GAP domain interact with residues in the C1 domain, thus contributing to the stabilization of the tertiary structure in the autoinhibited conformation. In support of this model, disruption of intramolecular contacts by site-directed mutagenesis (such as mutation of Leu28 to Ala) reduces the requirement for acidic phospholipids necessary for phorbol ester binding in vitro and sensitizes β2-chimaerin translocation to plasma membrane by phorbol esters and its subsequent association with Rac [12]. Notably, somatic mutations in Leu20 in α2-chimaerin (equivalent to Leu28 in β2-chimaerin), which leads to Rac-GAP hyperactivation, have been found in patients with Duane’s retraction syndrome, arguing for a critical role of these internal contacts in keeping the protein in a closed conformation [36]. More importantly, these studies support the concept that the structurally related isoforms α2- and β2-chimaerins are highly regulated proteins and possibly require modifications to destabilize the inactive structure and expose the catalytic Rac-GAP domain [10]. Unlike β2-chimaerin, β1-chimaerin lacks the N-terminal region and therefore is not subject to autoinhibition and allosteric activation. Consistent with this, β1-chimaerin is more sensitive than β2-chimaerin to PMA-induced translocation [32]. Chimaerins are responsible for the reduction in Rac-GTP levels after PMA treatment in the presence of PKC inhibitor GF 109203X [37]. Based on the data presented here, β3-chimaerin resembles β1-chimaerin in its hyper-sensitivity to PMA-induced translocation. As a potential explanation, β3-chimaerin residues that mask the C1 domain, such as Leu28 in β2-chimaerin, may be displaced or engaged in other interactions within the N-terminus domain, allowing a conformation that is energetically favorable for ligand binding to the C1 domain. Although the N-terminal domain in β3-chimaerin may not be implicated in autoinhibition, we speculate that this region may be involved in some type of protein–protein as recently described for β2-chimaerin N-terminus [38]. In fact, the atypic proline rich motif P(R/H)P(KR) implicated in the interaction with Nck1 is present in β3-chimaerin. More over, the additional N-terminal region present in β3-chimaerin may be subject to post-translational modifications that modulate activation or intracellular localization. Further studies would be required to address this issue.
In summary, here we identified β3-chimaerin as a novel member of the chimaerin family of Rac-GAPs. Our results suggest that individual chimaerin isoforms may be subject to different mechanisms of regulation. It remains to be determined if β3-chimaerin has specialized functions in any given cell type or under different physiopathological conditions.
Acknowledgments
This work is supported by grants PICT-2008-260 (ANCyP, Argentina), UBACyT 20020090200714 (UBA, Argentina), and PIP 11220110100573 (CONICET, Argentina) to Federico Coluccio Leskow., and Grants R01-CA74197 and R01-CA139120 (NIH) to Marcelo G. Kazanietz.
Footnotes
Lautaro Zubeldia-Brenner and Alvaro Gutierrez-Uzquiza have contributed equally to this work.
References
- 1.Riccomagno MM, Hurtado A, Wang H, Macopson JGJ, Griner EM, et al. The RacGAP β2-chimaerin selectively mediates axonal pruning in the hippocampus. Cell. 2012;149:1594–1606. doi: 10.1016/j.cell.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leskow FC, Holloway BA, Wang H, Mullins MC, Kazanietz MG. The zebrafish homologue of mammalian chimerin Rac-GAPs is implicated in epiboly progression during development. Proc Natl Acad Sci USA. 2006;103:5373–5378. doi: 10.1073/pnas.0508585103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siliceo M, García-Bernal D, Carrasco S, Díaz-Flores E, Coluccio Leskow F, et al. Beta2-chimaerin provides a diacylglycerol-dependent mechanism for regulation of adhesion and chemotaxis of T cells. J Cell Sci. 2006;119:141–152. doi: 10.1242/jcs.02722. [DOI] [PubMed] [Google Scholar]
- 4.Yang C, Kazanietz MG. Chimaerins: GAPs that bridge diacylglycerol signalling and the small G-protein Rac. Biochem J. 2007;403:1–12. doi: 10.1042/BJ20061750. [DOI] [PubMed] [Google Scholar]
- 5.Leung T, How BE, Manser E, Lim L. Germ cell beta-chimaerin, a new GTPase-activating protein for p21rac, is specifically expressed during the acrosomal assembly stage in rat testis. J Biol Chem. 1993;268:3813–3816. [PubMed] [Google Scholar]
- 6.Leung T, How BE, Manser E, Lim L. Cerebellar beta 2-chimaerin, a GTPase-activating protein for p21 ras-related rac is specifically expressed in granule cells and has a unique N-terminal SH2 domain. J Biol Chem. 1994;269:12888–12892. [PubMed] [Google Scholar]
- 7.Hall C, Michael GJ, Cann N, Ferrari G, Teo M, et al. alpha2-chimaerin, a Cdc42/Rac1 regulator, is selectively expressed in the rat embryonic nervous system and is involved in neuritogenesis in N1E-115 neuroblastoma cells. J Neurosci. 2001;21:5191–5202. doi: 10.1523/JNEUROSCI.21-14-05191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan S, Miller DW, Barnett GH, Hahn JF, Williams BR. Identification and characterization of human beta 2-chimaerin: association with malignant transformation in astrocytoma. Cancer Res. 1995;55:3456–3461. [PubMed] [Google Scholar]
- 9.Wang H, Yang C, Leskow FC, Sun J, Canagarajah B, et al. Phospholipase Cc/diacylglycerol-dependent activation of beta2-chimaerin restricts EGF-induced Rac signaling. EMBO J. 2006;25:2062–2074. doi: 10.1038/sj.emboj.7601098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colón-González F, Leskow FC, Kazanietz MG. Identification of an autoinhibitory mechanism that restricts C1 domain-mediated activation of the Rac-GAP alpha2-chimaerin. J Biol Chem. 2008;283:35247–35257. doi: 10.1074/jbc.M806264200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 12.Canagarajah B, Leskow FC, Ho JYS, Mischak H, Saidi LF, et al. Structural mechanism for lipid activation of the Racspecific GAP, beta2-chimaerin. Cell. 2004;119:407–418. doi: 10.1016/j.cell.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 13.Griner EM, Caino MC, Sosa MS, Colón-González F, Chalmers MJ, et al. A novel cross-talk in diacylglycerol signaling: the Rac-GAP beta2-chimaerin is negatively regulated by protein kinase Cdelta-mediated phosphorylation. J Biol Chem. 2010;285:16931–16941. doi: 10.1074/jbc.M109.099036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siliceo M, Mérida I. T cell receptor-dependent tyrosine phosphorylation of beta2-chimaerin modulates its Rac-GAP function in T cells. J Biol Chem. 2009;284:11354–11363. doi: 10.1074/jbc.M806098200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batut PJ, Dobin A, Plessy C, Carninci P, Gingeras TR. High-fidelity promoter profiling reveals widespread alternative promoter usage and transposon-driven developmental gene expression. Genome Res. 2013;23(1):169–180. doi: 10.1101/gr.139618.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rojas-Duran MF, Gilbert WV. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA. 2012;18:2299–2305. doi: 10.1261/rna.035865.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang C, Liu Y, Leskow FC, Weaver VM, Kazanietz MG. Rac-GAP-dependent inhibition of breast cancer cell proliferation by {beta}2-chimerin. J Biol Chem. 2005;280:24363–24370. doi: 10.1074/jbc.M411629200. [DOI] [PubMed] [Google Scholar]
- 18.Caloca MJ, Garcia-Bermejo ML, Blumberg PM, Lewin NE, Kremmer E, et al. Beta2-chimaerin is a novel target for diacylglycerol: binding properties and changes in subcellular localization mediated by ligand binding to its C1 domain. Proc Natl Acad Sci USA. 1999;96:11854–11859. doi: 10.1073/pnas.96.21.11854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caloca MJ, Fernandez N, Lewin NE, Ching D, Modali R, et al. Beta2-chimaerin is a high affinity receptor for the phorbol ester tumor promoters. J Biol Chem. 1997;272:26488–26496. doi: 10.1074/jbc.272.42.26488. [DOI] [PubMed] [Google Scholar]
- 20.Dong JM, Smith P, Hall C, Lim L. Promoter region of the transcriptional unit for human alpha 1-chimaerin, a neuron-specific GTPase-activating protein for p21rac. Eur J Biochem. 1995;227:636–646. doi: 10.1111/j.1432-1033.1995.tb20183.x. [DOI] [PubMed] [Google Scholar]
- 21.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 22.Ayoubi TA, Van De Ven WJ. Regulation of gene expression by alternative promoters. FASEB J. 1996;10:453–460. [PubMed] [Google Scholar]
- 23.Ayoubi TA, Creemers JW, Roebroek AJ, Van de Ven WJ. Expression of the dibasic proprotein processing enzyme furin is directed by multiple promoters. J Biol Chem. 1994;269:9298–9303. [PubMed] [Google Scholar]
- 24.Schibler U, Sierra F. Alternative promoters in developmental gene expression. Annu Rev Genet. 1987;21:237–257. doi: 10.1146/annurev.ge.21.120187.001321. [DOI] [PubMed] [Google Scholar]
- 25.Davuluri RV, Grosse I, Zhang MQ. Computational identification of promoters and first exons in the human genome. Nat Genet. 2001;29:412–417. doi: 10.1038/ng780. [DOI] [PubMed] [Google Scholar]
- 26.ENCODE Project Consortium A user’s guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rada-Iglesias A, Ameur A, Kapranov P, Enroth S, Komorowski J, et al. Whole-genome maps of USF1 and USF2 binding and histone H3 acetylation reveal new aspects of promoter structure and candidate genes for common human disorders. Genome Res. 2008;18:380–392. doi: 10.1101/gr.6880908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rada-Iglesias A, Wallerman O, Koch C, Ameur A, Enroth S, et al. Binding sites for metabolic disease related transcription factors inferred at base pair resolution by chromatin immuno-precipitation and genomic microarrays. Hum Mol Genet. 2005;14:3435–3447. doi: 10.1093/hmg/ddi378. [DOI] [PubMed] [Google Scholar]
- 29.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 31.Beg AA, Sommer JE, Martin JH, Scheiffele P. alpha2-chimaerin is an essential EphA4 effector in the assembly of neuronal locomotor circuits. Neuron. 2007;55:768–778. doi: 10.1016/j.neuron.2007.07.036. [DOI] [PubMed] [Google Scholar]
- 32.Caloca MJ, Wang H, Delemos A, Wang S, Kazanietz MG. Phorbol esters and related analogs regulate the subcellular localization of beta 2-chimaerin, a non-protein kinase C phorbol ester receptor. J Biol Chem. 2001;276:18303–18312. doi: 10.1074/jbc.M011368200. [DOI] [PubMed] [Google Scholar]
- 33.Penas-Steinhardt A, Barcos LS, Belforte FS, De Sereday M, Vilariño J, et al. Functional characterization of TLR4 +3725 G/C polymorphism and association with protection against overweight. PLoS One. 2012;7:e50992. doi: 10.1371/journal.pone.0050992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Kazanietz MG. p23/Tmp21 differentially targets the Rac-GAP beta2-chimaerin and protein kinase C via their C1 domains. Mol Biol Cell. 2010;21:1398–1408. doi: 10.1091/mbc.E09-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menna PL, Skilton G, Leskow FC, Alonso DF, Gomez DE, et al. Inhibition of aggressiveness of metastatic mouse mammary carcinoma cells by the beta2-chimaerin GAP domain. Cancer Res. 2003;63:2284–2291. [PubMed] [Google Scholar]
- 36.Miyake N, Chilton J, Psatha M, Cheng L, Andrews C, et al. Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane’s retraction syndrome. Science. 2008;321:839–843. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caloca MJ, Wang H, Kazanietz MG. Characterization of the Rac-GAP (Rac-GTPase-activating protein) activity of beta2-chimaerin, a “non-protein kinase C” phorbol ester receptor. Biochem J. 2003;375(Pt 2):313–321. doi: 10.1042/BJ20030727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutierrez-Uzquiza A, Colon-Gonzalez F, Leonard TA, Canagarajah BJ, Wang H, Mayer BJ, Hurley JH, Kazanietz MG. Coordinated activation of the Rac-GAP β2-chimaerin by an atypical proline-rich domain and diacylglycerol. Nat Commun. 2013;4:1849. doi: 10.1038/ncomms2834. [DOI] [PMC free article] [PubMed] [Google Scholar]